

Abstract
The physiological significance of the renal tubular prorenin receptor (PRR) has been difficult to elucidate due to developmental abnormalities associated with global or renal-specific PRR knockout (KO). We recently developed an inducible renal tubule-wide PRR KO using the Pax8/LC1 transgenes and demonstrated that disruption of renal tubular PRR at 1 mo of age caused no renal histological abnormalities. Here, we examined the role of renal tubular PRR in blood pressure (BP) regulation and Na+ excretion and investigated the signaling mechanisms by which PRR regulates Na+ balance. No detectable differences in BP were observed between control and PRR KO mice fed normal- or low-Na+ diets. However, compared with controls, PRR KO mice had elevated plasma renin concentration and lower cumulative Na+ balance with normal- and low-Na+ intake. PRR KO mice had an attenuated hypertensive response and reduced Na+ retention following angiotensin II (ANG II) infusion. Furthermore, PRR KO mice had significantly lower epithelial Na+ channel (ENaC-α) expression. Treatment with mouse prorenin increased, while PRR antagonism decreased, ENaC activity in isolated split-open collecting ducts (CD). The prorenin effect was prevented by protein kinase A and Akt inhibition, but unaffected by blockade of AT1, ERK1/2, or p38 MAPK pathways. Taken together, these data indicate that renal tubular PRR, likely via direct prorenin/renin stimulation of PKA/Akt-dependent pathways, stimulates CD ENaC activity. Absence of renal tubular PRR promotes Na+ wasting and reduces the hypertensive response to ANG II.
Keywords: prorenin receptor, blood pressure, sodium transport, angiotensin II
the prorenin receptor (PRR) is a recently discovered component of the renin-angiotensin system (RAS) (27). The PRR is a multifunctional protein; binding of prorenin/renin to the PRR not only increases catalytic conversion of angiotensinogen to angiotensin I but it also activates angiotensin II (ANG II)-independent intracellular cell signaling pathways (27). Furthermore, the PRR functions as an accessory protein for the vacuolar ATPase and is involved in lysosomal acidification (1).
Recent studies propose a role for renal tubule-derived PRR in regulation of blood pressure (BP) and Na+ transport (8, 10, 13, 25, 31, 32, 36). Within the kidney, the PRR has been localized to the mesangium, podocytes, macula densa, proximal tubule, and the collecting duct (CD) (1, 13, 27, 32, 40). Increased renal medullary PRR expression was described in rats with chronic angiotensin II-infused hypertension or 2-kidney 1-clip Goldblatt hypertension (8, 31). Augmented renal medullary PRR expression has also been reported with low-Na+ intake (10, 13, 25). In addition, activation of PRR by prorenin in cultured CD cells induced both ANG II-dependent and ANG II-independent signaling (1, 9, 27, 36). Thus, renal tubular PRR might regulate tubular Na+ reabsorption and BP via ANG II-dependent and -independent mechanisms. Notably, transgenic rats with ubiquitous human PRR overexpression remained normotensive despite proteinuria and progressive nephropathy (14); in contrast, rats with selective PRR overexpression in vascular smooth muscle cells manifested hypertension and tachycardia without any renal effects (3). Thus, the physiological role of renal tubule-derived PRR in BP regulation remains an open question.
The functional significance of renal PRR was attempted to be examined in podocyte-specific or CD-specific knockout (KO) mice (29, 44). Unfortunately, both these models suffered from early lethality and malformed organ development most likely due to abnormal lysosomal acidification. We recently developed an inducible renal tubule- wide KO of PRR such that PRR deletion occurs in early adulthood, avoiding the effects of PRR disruption on organ development; these mice have an impaired renal concentrating ability associated with reduced CD arginine vasopressin (AVP) responsiveness (37). In the current study, we took advantage of this inducible renal tubular-specific PRR KO mouse model to examine the role of renal tubular PRR in the regulation of renal Na+ excretion and BP. In addition, we describe the effects of prorenin, independent of ANG II, on renal Na+-reabsorptive pathways.
MATERIALS AND METHODS
Animal care.
All animal studies were conducted with the approval of the Universities of Utah and Texas San Antonio Animal Care and Use Committees in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Inducible renal tubular-specific PRR KO mice.
Details on generation of renal tubular-wide PRR KO mice have been published (37). In brief, loxP-flanked (floxed) PRR mice were bred with mice containing Pax8-rtTA and LC-1 transgenes. The Pax8-rtTA transgene contains the Pax8 gene promoter driving expression of the reverse tetracycline transactivator (rtTA) (48). The LC-1 transgene encodes tetracycline-inducible bicistronic Cre recombinase and luciferase (43). In the presence of tetracycline or doxycycline, rtTA binds and activates the LC1 transgene leading to expression of luciferase and Cre recombinase in renal tubular cells. To induce renal tubular-wide KO, mice hemizygous for Pax8-rtTA and LC-1, and homozygous for floxed PRR genes, were treated with 2 mg/ml doxycycline in 2% sucrose drinking water for 12 days at 1 mo of age. Floxed PRR mice without the Pax8-rtTA or LC-1 transgenes were used as controls. All mice were bred on a C57BL/6J background, and control and PRR KO mice aged 3–4 mo of both sexes were used for all studies.
BP monitoring.
BP was recorded via telemetry (TA11-PAC10, Data Sciences International, St. Paul, MN). Control and PRR KO mice were anesthetized with 2% isoflorane, implanted with radio transmitters with the catheter in the carotid artery, and allowed to recover for 5 days. Mice were maintained on a normal-Na+ pellet diet (0.26% Na+ Harlan Teklad #2920X, Indianapolis, IN) for 4 days and low-Na+ diet (0.01% Na+ Test Diet #7472, St. Louis, MO) for 7 days. Mice were not handled during BP recording period since even small stimuli can markedly affect BP in mice.
Metabolic balance studies.
Control and PRR KO mice were placed in metabolic cages for 12 consecutive days for measurement of food and water intake, body weight, and 24-h urine collection. Mice were given 9 ml of a gelled normal (3 days, Test Diet #7551)- or low-Na+ (7 days, Test Diet #7286) diet with free access to water. On days 3 and 10, 35 μl of blood were collected for assay of plasma renin concentration. Plasma renin concentration (PRC) was measured as the amount of angiotensin I (ANG I) generated after incubation with excess porcine angiotensinogen using the ANG I enzyme immunoassay (EIA) kit (Peninsula Laboratories, San Carlos, CA). Urinary Na+ and K+ were determined using the EasyVet Analyzer (Medica, Bedford, MA). Daily Na+ and K+ balance was calculated as the difference between intake (Na+/K+ content in gelled diet × amount consumed/day) and output (24-h urinary excretion). Cumulative Na+ and K+ balance was calculated as the continuous summation by each consecutive day. At the conclusion of metabolic balance studies, mice were killed and kidneys were isolated for further analyses.
Western blotting.
Whole kidneys were homogenized in ice-cold isolation buffer (50 mM Tris, 5 mM EDTA, 1% Triton, 1 mM PMSF) and Complete protease inhibitors (Roche, Pleasanton, CA). Protein content was determined using the modified Lowry assay and samples were solubilized with Laemmli loading buffer containing 0.5% lithium dodecyl sulfate. Loading control gels were initially run on 12% Bis-Tris gels, stained with Coomassie blue and random bands quantified by densitometry to assess equal loading. Equal amounts of protein (20 μg/lane) were run on a denaturing NUPAGE 4–12% Bis-Tris minigel (Invitrogen) and transferred to a polyvinylidene difluoride plus nylon membrane. Membranes were incubated with specific antibodies against NHE3 (1:1,000, Millipore, Bedford, MA), NKCC2 (1:20,000, gift from Dr. Pablo Ortiz, Henry Ford Hospital), NCC (1:1,000, gift from Dr. David Ellison, Oregon Health Sciences University), ENaC-α, -β, or -γ (1:1,000, StressMarq, Victoria, BC), and mouse monoclonal β-actin (1:1,000, Life Technologies, Carlsbad, CA). Secondary horseradish peroxidase-conjugated antibodies (goat anti-mouse for NHE3 and goat anti-rabbit for other transporters, Santa Cruz Biotech, Santa Cruz, CA) were used at a dilution of 1:5,000. Immunoblots were visualized with the Advance ECL system (GE Healthcare, Piscataway, NJ). Densitometry was performed with a Bio-Rad gel documentation system (Hercules, CA).
Angiotensin II infusion.
Control and PRR KO mice were implanted with radio transmitters. After 3 days of continuous BP monitoring, mini-osmotic pumps (Alzet model 1002, Durect, Cupertino, CA) were placed subcutaneously in between the scapulas under isoflurane anesthesia. ANG II was infused for 14 days at 600 ng·kg−1·min−1 and BP was recorded continuously for 13 days on a normal-Na+ diet. At the end of the BP studies, mice were killed and kidneys were isolated for Western blot analyses as described above. In a separate experiment, immediately following mini-pump placement, mice were placed in metabolic cages for 10 days for urine and plasma collection. Metabolic balance studies were limited to the first 10 days of ANG II infusion so that any differences in electrolyte excretion are apparent before the mice reach steady state. Cumulative Na+ and K+ balance was calculated as described above. Urinary ANG II was measured using EIA kit (Peninsula Laboratories, San Carlos, CA).
Isolated, split-open CD preparation and electrophysiological analyses.
The following studies were performed at University of Texas Health Sciences Center at San Antonio. As previously described, CDs were microdissected from kidney slices (<1 mm) and split open to gain access to the apical membrane of principal cells (2, 46). ENaC activity was quantified in cell-attached patches of the apical membrane made under voltage-clamp conditions (−Vp = −60 mV) using standard procedures (2, 46). For the current experiments, the typical bath and pipette solutions were as follows (values shown in mM): 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, 10 HEPES (pH 7.4), 140 LiCl, 2 MgCl2, and 10 HEPES (pH 7.4), respectively. For each experimental condition, CDs from at least three different mice were assayed.
Separate groups of control and PRR KO mice were maintained on a normal (0.32% Na+; TD 7912; Harlan Teklad)-, low (<0.01%; TD 90228, Harlan Teklad)-, or high (2% Na+ TD 92034, Harlan Teklad)-Na+ pellet diet for 7 days with ad libitum access to water in regular cages. ENaC activity was measured in control and PRR KO mice under basal conditions and after pretreatment with recombinant mouse prorenin (10 nM; Anaspec, Fremont, CA) or PRO20, a specific PRR inhibitor (2 mM, gift from Dr. Yumei Feng, University of Nevada, Reno) for 30 min. ENaC activity was also determined in control mice fed a high-Na+ diet for 7 days after simultaneous treatment of isolated CDs with mouse prorenin (10 nM) and one of the following inhibitors for 30 min: 10 μM losartan (AT1 receptor blocker), 10 μM SB203580 (p38 MAPK inhibitor), 5 μM U0126 (MEK1/2 inhibitor), 20 μM H89 (PKA inhibitor), or 10 μM A6730 (Akt inhibitor). All reagents were obtained from Sigma (St. Louis, MO).
Statistical analysis.
All results are expressed as means ± SE. The Student's t-test was used to compare differences in discrete parameters (weight, food intake, water intake, Na+ balance, densitometry) between KO and control animals. For parameters measuring changes over time (BP), repeated-measures ANOVA with Scheffé's post hoc test was used. The criterion for significance was P ≤ 0.05.
RESULTS
Effect of renal tubular PRR KO on BP and Na+ balance.
The renal tubule-wide PRR KO mice were recently characterized (37) and demonstrate normal survival and no gross anatomical or renal histological abnormalities up to 10 mo after doxycycline induction. Compared with control mice, PRR KO mice had significantly reduced medullary PRR mRNA, protein, and immunostaining as reported previously (37). In addition, PRR KO mice demonstrate a nephrogenic diabetes insipidus phenotype (37).
There were no detectable differences in 24-h systolic or diastolic BP between control and PRR KO mice fed normal- or low-Na+ diets (Fig. 1). Although both PRR KO and control mice had similar food intake and body weight, water intake and urine volume were significantly higher in the PRR KO mice on both diets (Table 1), consistent with our previous findings (37).
Fig. 1.

Daily blood pressure (BP) in control and prorenin receptor (PRR) knockout (KO) mice with varying Na+ intake. BP was measured via telemetry and mice were fed normal (0.26%)- and low (0.03%)-Na+ diets (n = 8–10/group). Top 2 lines show systolic, and bottom 2 lines show diastolic, BP. Analysis by ANOVA shows no difference between the 2 groups.
Table 1.
Food intake, water intake, urine volume, and body weight in control and PRR
| Normal-Na+ Diet |
Low-Na+ Diet |
Post ANG II Infusion |
||||
|---|---|---|---|---|---|---|
|
Day 2 |
Day 2 |
Day 2 |
||||
| Control | KO | Control | KO | Control | KO | |
| Food intake, g/day | 4.9 ± 0.4 | 4.4 ± 0.2 | 4.0 ± 0.3 | 3.8 ± 0.2 | 4.7 ± 0.3 | 5.1 ± 0.2 |
| Water intake, ml/day | 3.6 ± 0.3 | 8.6 ± 0.8* | 3.9 ± 0.4 | 7.2 ± 0.8* | 7.2 ± 1.3 | 12.4 ± 0.5* |
| Body weight, g | 28.0 ± 2.9 | 25.1 ± 1.4 | 26.3 ± 2.7 | 23.3 ± 1.4 | 24.1 ± 2.1 | 24.5 ± 0.9 |
| Urine volume, ml/day | 1.4 ± 0.2 | 5.2 ± 0.8* | 1.3 ± 0.4 | 4.4 ± 0.7* | 4.7 ± 1.1 | 8.3 ± 0.3* |
Data are means ± SE. Knockout (KO) mice, n = 8–10 per group.
PRR, prorenin receptor.
P < 0.05 compared with control within each diet.
Compared with controls, cumulative Na+ balance was lower in PRR KO mice on both normal- and low-Na+ diets (P < 0.05 on each day; Fig. 2). Potassium balance was similar between the two groups on normal- or low-Na+ diets.
Fig. 2.

Cumulative Na+ and K+ balance on normal- and low-Na+ diets. Cumulative Na+ balance (A) and cumulative K+ balance (B) in control and PRR KO mice fed normal (0.26%)- or low (0.03%)-Na+ diets. Compared with controls, PRR KO mice had lower cumulative Na+ balance (*P < 0.05, n = 8–10/group).
The expression of Na+ transporters in control and PRR KO mice was examined using Western blotting. On a normal-Na+ diet, no differences were observed in expression of Na+/H+ exchanger (NHE3), Na+-K+-Cl− cotransporter (NKCC2), and Na+/Cl− cotransporter (NCC; Fig. 3) between control and PRR KO mice. Epithelial Na+ channel (ENaC)-α subunit expression was markedly lower while ENaC-β and γ expressions were significantly higher in the PRR KO mice (Fig. 3). The trends in ENaC expression remained unchanged when gels were run with half the amount of protein to address linearity.
Fig. 3.

Renal Na+ transporter expression in control and PRR KO mice on a normal-Na+ diet. A: representative Western blots of whole kidney lysates. B: densitometry of Na+ transporter expression adjusted to GAPDH and represented as % control (n = 10/group). *P < 0.05 vs. control.
ANG II infusion.
Since chronic ANG II infusion increases expression of renal tubular PRR (8, 10, 49), BP and Na+ balance were evaluated following ANG II infusion. As noted earlier, BP was similar between control and KO mice preinfusion (Fig. 4). Following ANG II infusion at 600 ng·kg−1·min−1, systolic BP rose in control mice starting on day 2 (control −151 ± 4 vs. KO −136 ± 4 mm) and remained elevated throughout the study period (day 7: control −152 ± 10 vs. KO −133 ± 10 mm; day 12: control −162 ± 5 vs. KO −142 ± 5 mm; Fig. 4). In contrast, PRR KO mice had an attenuated hypertensive response throughout the infusion. Similar trends were also observed in diastolic BP (Fig. 4).
Fig. 4.

Renal tubular-wide deletion of PRR leads to attenuated hypertensive response following 14-day ANG II infusion (600 ng·kg−1·min−1). Twenty four-hour systolic (A) and diastolic BP (B) measured via radiotelemetry in control and PRR KO mice (n = 6–8/group). P < 0.001 for systolic and diastolic BP by ANOVA. *P < 0.05 vs. control for each day.
While body weight and food intake were comparable between the two groups following ANG II infusion (Table 1), PRR KO mice had significantly higher water intake and urine volume compared with controls. Notably, cumulative Na+ balance was significantly lower in PRR KO mice following ANG II infusion (Fig. 5; P < 0.05). No differences in cumulative K+ balance were observed between the two groups following ANG II infusion (Fig. 5). Additionally, compared with controls, whole kidney expression of ENaC-α subunit was lower in PRR KO mice following ANG II infusion (Fig. 6). In contrast, expression of ENaC-β and γ subunits demonstrated increased expression, while NHE3, NKCC2, and NCC were similar in the PRR KO mice compared with controls (Fig. 6).
Fig. 5.

Cumulative Na+ and K+ balance following ANG II infusion (600 ng·kg−1·min−1). Cumulative Na+ balance (A) and cumulative K+ balance (B) in control and PRR KO mice fed normal (0.26%)-Na+ diet. Compared with controls, PRR KO mice had lower cumulative Na+ balance (*P < 0.05, n = 6–8/group).
Fig. 6.

Renal Na+ transporter expression in control and PRR KO mice following ANG II infusion. A: representative Western blots of whole kidney lysates isolated from control and PRR KO mice 14 days after ANG II infusion (600 ng·kg−1·min−1). B: densitometry of Na+ transporter expression adjusted to GAPDH and represented as % control (n = 4/group). *P < 0.05 vs. control.
Before ANG II infusion, PRC was elevated in the PRR KO mice regardless of Na+ intake (normal-Na+: KO −320 ± 47 vs. control −129 ± 16 ng·ml−1·h−1; low-Na+: KO −390 ± 38 vs. control −184 ± 13 ng·ml−1·h−1; Fig. 7). Low-Na+ intake increased PRC in control mice compared with normal-Na+ diet (P < 0.001 by paired t-test) but did not change PRC in the PRR KO mice. ANG II infusion suppressed PRC in both PRR KO and control mice on day 13 of infusion, with no discernible differences noted between the two groups (Fig. 7). Urinary ANG II excretion was similar between control and PRR KO mice following ANG II infusion (control −1.57 ± 0.9 vs. KO −2.03 ± 0.5 ng/day).
Fig. 7.
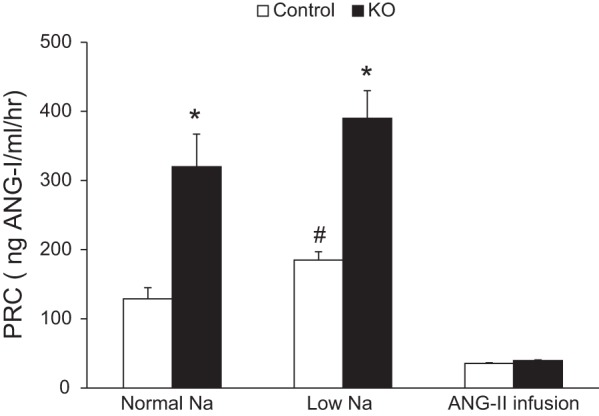
Plasma renin concentration (PRC) in control and PRR KO mice on varying Na+ diets and after ANG II infusion. PRC was measured using ANG I EIA from control and PRR KO mice on normal (0.26%)- and low (0.03%)-Na+ diets and 14 days following ANG II infusion (600 ng·kg−1·min−1; n = 8–10/group). *P < 0.001 vs. control. #P < 0.001 vs. normal-Na+ diet.
Prorenin regulation of ENaC activity.
Since the CD is the predominant site of renal tubular PRR expression (1, 8), and the above findings suggested a role for PRR regulation of ENaC, the effect of prorenin on ENaC activity in isolated split-open cortical CD (CCD) was determined using patch-clamp analyses. Mice were fed a normal-, high-, or low-Na+ diet for 1 wk before these studies were conducted. The rationale was to study the effect of prorenin when ENaC expression was normal, low (high-Na+ diet), or high (low-Na+ diet) since a stimulatory effect, if it occurred, might be most evident when ENaC levels were low, while an inhibitory effect, if it occurred, might be most evident when ENaC levels were high. Incubation of CCD with recombinant mouse prorenin increased the number of channels but not the channel open probability (Fig. 8) in control mice fed a high-Na+ (2% Na) diet for 7 days. ENaC activity was increased in control mice fed a high-Na+ diet following prorenin (Fig. 9, A and B). Addition of mouse prorenin did not alter ENaC activity in PRR KO mice fed a high-Na+ diet (Fig. 9C). Addition of mouse prorenin did not alter ENaC activity in PRR KO and control mice maintained on a normal- or low-Na+ diet (data not shown). Thus, the stimulatory effect of prorenin on ENaC activity was evident when basal ENaC activity was relatively low.
Fig. 8.
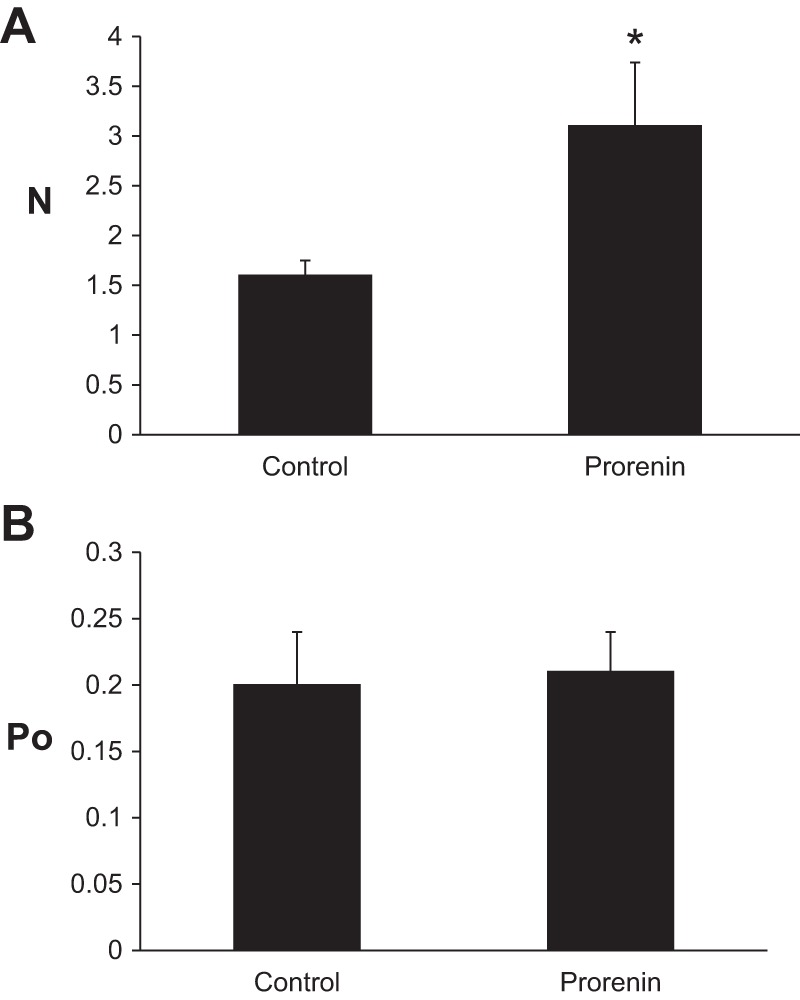
Effect of prorenin on number and activity of epithelial Na+ channels (ENaC) in isolated split-open cortical collecting ducts (CCD). Number of ENaC (N; A) and channel open probability (Po; B) analyzed using patch-clamp analyses in control mice fed a high-Na+ (3.2%) diet. Tubules from control mice were treated with 10 nM recombinant mouse prorenin for 30 min (n = 10). *P < 0.002 vs. control.
Fig. 9.

Prorenin agonist and antagonist regulation of ENaC activity in isolated split-open CCD. Representative gap-free ENaC current traces and summary of ENaC activity from cell-attached patches made on the apical surface of principal cells from control mice (A and B) and PRR KO mice (C) fed a high (3.2%)-Na+ diet and treated with 10 nM recombinant mouse prorenin for 30 min. Similar results are shown for isolated CCD after a low (0.1%)-Na+ diet and treatment with the PRR inhibitor PR020 (2 μM) for 30 min (control mice in D and E; PRR KO mice in F). *P < 0.01 vs. control.
Next, ENaC activity in isolated CCD was assessed in the presence of a novel PRR antagonist (PR020) (19) to evaluate the endogenous activity of the PRR in regulating ENaC. Similar to the prorenin studies, mice were fed a normal-, high-, or low-salt intake for 1 wk before study. Isolated CCD ENaC activity was reduced following treatment with PRO20 in control mice fed a low-Na+ diet (Fig. 9, D and E). In contrast, PRO20 treatment did not change ENaC activity in PRR KO mice fed a low-Na+ diet (Fig. 9F). PRO20 did not decrease ENaC activity in CD from mice fed normal- or high-Na+ diets. Thus, the inhibitory effect of PRO20 was evident when basal ENaC activity was relatively high.
Studies were then undertaken to examine the signaling pathways by which prorenin regulated ENaC activity. The following studies were performed in control mice fed a high-Na+ diet for 7 days to reproduce the optimal conditions identified above. Treatment with losartan (AT1 receptor blocker) did not affect prorenin-stimulated ENaC activity in acutely isolated CD (Fig. 10), suggesting that prorenin can modulate ENaC in an ANG II-independent manner. Since PRR has been reported to affect MEK1/2 and p38 MAPK signaling pathways (6, 27, 41), and PKA, Akt, and MEK1/2 pathways have been implicated in the regulation of ENaC (15, 18, 45), ENaC activity was examined in the presence of inhibitors of these signaling pathways (Fig. 10). Addition of H89 (PKA inhibitor) or A6730 (Akt inhibitor) significantly decreased prorenin-stimulated ENaC activity. In contrast, treatment with SB203580 (p38MAPK inhibitor) or U0126 (MEK1/2 inhibitor) did not alter prorenin-stimulated ENaC activity.
Fig. 10.
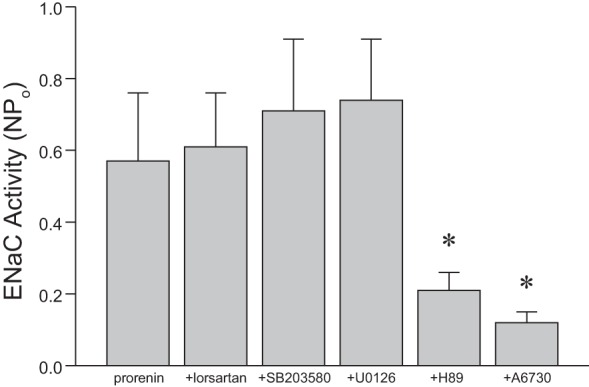
Prorenin regulation of ENaC activity is dependent on PKA and Akt signaling pathways. ENaC activity in isolated split-open CCD following treatment with 10 nM prorenin and either losartan (AT1 blocker, 10 μM), SB203580 (p38 MAPK inhibitor, 10 μM), U0126 (MEK1/2 inhibitor, 5 μM), H89 (PKA inhibitor, 20 μM), or A6730 (Akt inhibitor, 10 μM) for 30 min; n = 15–20 for each condition. *P < 0.05 vs. control.
DISCUSSION
The current study provides evidence that renal tubular PRR is capable of regulating BP. No detectable differences were noted in BP between control and renal tubular PRR KO mice during normal- or low-Na+ intake, suggesting that renal tubular PRR does not play a substantial role in regulating BP under physiological conditions. This supposition is supported by our previous study (38) wherein we ablated renin specifically in the CD–compared with controls, CD-specific renin KO mice did not have altered BP with varying Na+ intake. However, PRC was elevated in the PRR or CD renin KO mice fed a normal- or low-salt diet compared with controls, suggesting that loss of renal tubular PRR or CD renin evokes a compensatory systemic RAS response. Notably, low-Na+ intake (as compared with normal-Na+ intake) increased PRC in control, but not PRR KO mice; the reason for this is not fully known; however, the already substantially elevated PRC in PRR KO mice on a normal-Na+ diet (above that seen in control mice on a low-Na+ diet) may have obviated or masked any further increases in PRC in PRR KO mice. Taken together, these results suggest that under physiological conditions, compensatory systems can raise BP sufficiently to alleviate any differences in BP caused by PRR deletion. In contrast, absence of renal tubular PRR attenuated, but did not completely prevent, the hypertensive response to ANG II infusion, suggesting that renal tubule PRR contributes to the development of ANG II-dependent hypertension.
The mechanism by which renal tubular PRR KO leads to reduced BP and/or elevated systemic PRC is conceivably related to reduced renal tubular Na+ reabsorption. Cumulative Na+ balance was lower in renal tubular PRR KO mice with low-Na+ intake and following ANG II infusion. Renal expression of NHE3, NKCC2, and NCC in the PRR KO mice was similar to control mice under normal physiological conditions and with ANG II infusion. In contrast, before and following ANG II infusion, ENaC-α expression was significantly reduced while ENaC-β and -γ expression was significantly increased in the PRR KO mice. While these findings do not rule out reductions in cell surface expression and/or activity of other transporters in PRR KO mice, they do indicate that at least ENaC expression is altered in PRR KO mice. It is unknown why ENaC-β and -γ expressions were increased in PRR KO mice. Although speculative, one reason for this expression pattern could be related to differential processing and activation of ENaC subunits in the PRR KO mice. Previous studies have reported differential and noncoordinated expression of ENaC subunits in response to salt depletion and aldosterone, particularly with β- and γ-subunits (7, 50). Clearly, additional studies are needed to evaluate the changes in ENaC subunit expression in the PRR KO mice.
This notion that ENaC is affected in PRR KO mice is consistent with observations that PRR expression is greatest in the CD (1, 31, 32) and that the CD is the major renal tubular site of prorenin/renin synthesis (33, 34, 37, 39). Such a scenario raises the possibility that prorenin/renin can act through autocrine and/or paracrine pathways to regulate ENaC activity. The finding that exogenous prorenin, at least under acute conditions, increased isolated CD ENaC activity (via increasing channel number but not open probability) supports the potential for such regulation. Furthermore, the finding that PRR antagonism, in the absence of exogenously added prorenin, inhibited isolated CD ENaC activity suggests that endogenous prorenin and/or renin can tonically stimulate CD ENaC activity. Finally, addition of exogenous prorenin or PRO20 did not change ENaC activity in PRR KO mice, suggesting that prorenin effects on ENaC activity are likely to be mediated via PRR. Of note, the stimulatory effect of exogenous prorenin on ENaC activity was only detected under conditions when baseline ENaC activity was suppressed (high-salt diet), while the inhibitory effect of PRO20 was only observed when baseline ENaC activity was augmented (low-salt diet); i.e., under conditions that maximized the chances of observing these effects. Interestingly, these results are consistent with the findings that renal tubular prorenin/renin production and PRR expression are enhanced during a low-Na+ diet (10, 13, 17, 25, 36); PRO20 effects would be most evident under these low-Na+ diet conditions. Consistent with our results, a recent study using cultured CD cell line demonstrated that prorenin increased ENaC activity via NADPH-oxidase 4-derived H2O2, which was attenuated by the PRR inhibitor, PRO20 (22). Similarly, mIMCD3 cells exposed to low salt/ANG II and PRR siRNA had reduced ENaC mRNA and protein levels compared with vehicle or scramble siRNA (35).
Prorenin could potentially regulate ENaC activity through ANG II-dependent or -independent mechanisms. Binding of prorenin to PRR activates prorenin to full enzymatic activity (27). Since all components necessary for ANG II synthesis and action are present in the CD lumen (angiotensinogen, prorenin, PRR, angiotensin-converting enzyme, AT1 receptor) (1, 4, 11, 16, 17, 20, 28, 34), and since luminal ANG II can potently stimulate CD Na+ reabsorption (23, 24, 30), it is possible that prorenin regulation of ENaC is, at least in part, ANG II dependent. The finding that AT1 receptor blockade did not inhibit prorenin-stimulated ENaC activity in isolated CD indicates that prorenin can regulate ENaC in an ANG II-independent manner, but does not rule out the possibility that ANG II-dependent mechanisms are operative in vivo (since the in vitro preparation does not contain all RAS components necessary for ANG II production). However, urinary ANG II excretion was similar between control and PRR KO mice, indicating that PRR was more likely to mediate direct prorenin/renin effects rather than increasing formation of ANG II. Independent of ANG II, binding of prorenin to the PRR has been reported to activate MEK1/2 and p38 MAPK signaling pathways (6, 27, 41); however, inhibition of MEK1/2 or p38 MAPK did not alter prorenin-stimulated CD ENaC activity. In contrast, administration of PKA or Akt inhibitors decreased prorenin-stimulated ENaC activity. Both signaling pathways are known to enhance ENaC cell surface expression effects (3, 5, 15, 18, 26, 42) similar to those observed with prorenin. The PKA pathway has primarily been implicated in vasopressin-mediated ENaC regulation (47) while the Akt pathway has been primarily implicated in insulin-stimulated ENaC activity (18). Importantly, both kinases are downstream targets of cyclic AMP (cAMP) (18, 26). Of note, AVP-stimulated cAMP accumulation in acutely isolated inner medullary CD was markedly reduced in renal tubular PRR KO mice compared with controls (37). Similarly, the AVP-stimulated increase in cAMP accumulation was reduced in MDCK.C11 cells (a distal nephron cell line) transfected with PRR siRNA (21). Thus, renal tubular prorenin/PRR is capable of stimulating CD Na+ reabsorption via ANG II-independent, cAMP-dependent cell signaling pathways that ultimately regulate ENaC apical membrane expression.
A key remaining question is which cell types are involved in PRR regulation of Na+ reabsorption. Immunostaining studies involving colocalization with intercalated and principal cell-specific markers have localized renal tubular PRR exclusively to the luminal membrane of type A intercalated cells in rats fed a normal-Na+ diet (1, 8). In contrast, diffuse and intense PRR staining was observed throughout all cells of the CD in rats fed a low- but not a normal- or high-Na+ diet (13). Additionally, diffuse PRR immunostaining was described in CD of mice deficient in the Ceacam1 gene (12). Hence, it may be that prorenin/renin stimulates ENaC via PRR activation on principal cells; however, it is also possible that PRR activation on intercalated cells could alter ENaC activity (how such an effect would occur is unknown). Ideally, determination of whether PRR regulation of ENaC is mediated via principal and/or intercalated cells could be accomplished with principal or intercalated cell-specific PRR KO models. Furthermore, such KO models may need to be inducible in adulthood to avoid developmental effects occurring during embryogenesis and early postnatal life. Accurate and reliable inducible principal or intercalated cell-specific KO models have not yet been developed, hence determination of the in vivo role of principal and intercalated cell PRR remains a challenge.
In summary, findings in the current study indicate that renal tubular PRR can potentially regulate BP and urinary Na+ excretion and that this effect may be most evident under conditions characterized by elevated systemic ANG II (low-salt diet and particularly pathologically excessive ANG II). This effect of the PRR is likely mediated, at least in part, by prorenin/renin stimulation of ENaC activity via PKA/Akt-dependent pathways. Future research will need to focus on PRR regulation of intracellular signaling mechanisms and cellular subtypes by which renal tubular PRR mediates its effects.
GRANTS
The research in this study was supported in part by research grants from American Society of Nephrology (to N. Ramkumar), National Institutes of Health (DK097007) and Veterans Affairs Merit Review (to D. E. Kohan), American Heart Association (22930030; to J. D. Stockand), and Ministry of Education, Science, and Culture of Japan (25293198; to A. Ichihara).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.R., J.D.S., and D.E.K. conception and design of research; N.R., D.S., E.M., V.B., S.W., and N.A. performed experiments; N.R., D.S., E.M., V.B., and N.A. analyzed data; N.R., D.S., E.M., V.B., J.D.S., and D.E.K. interpreted results of experiments; N.R., E.M., and D.E.K. prepared figures; N.R. and D.E.K. drafted manuscript; N.R., A.I., J.D.S., and D.E.K. edited and revised manuscript; N.R., D.S., E.M., V.B., S.W., N.A., A.I., J.D.S., and D.E.K. approved final version of manuscript.
REFERENCES
- 1.Advani A, Kelly DJ, Cox AJ, White KE, Advani SL, Thai K, Connelly KA, Yuen D, Trogadis J, Herzenberg AM, Kuliszewski MA, Leong-Poi H, Gilbert RE. The (Pro)renin receptor: site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension 54: 261–269, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Bugaj V, Pochynyuk O, Stockand JD. Activation of the epithelial Na+ channel in the collecting duct by vasopressin contributes to water reabsorption. Am J Physiol Renal Physiol 297: F1411–F1418, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burckle CA, Jan Danser AH, Muller DN, Garrelds IM, Gasc JM, Popova E, Plehm R, Peters J, Bader M, Nguyen G. Elevated blood pressure and heart rate in human renin receptor transgenic rats. Hypertension 47: 552–556, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Casarini DE, Boim MA, Stella RC, Krieger-Azzolini MH, Krieger JE, Schor N. Angiotensin I-converting enzyme activity in tubular fluid along the rat nephron. Am J Physiol Renal Physiol 272: F405–F409, 1997. [DOI] [PubMed] [Google Scholar]
- 5.Ecelbarger CA, Kim GH, Terris J, Masilamani S, Mitchell C, Reyes I, Verbalis JG, Knepper MA. Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. Am J Physiol Renal Physiol 279: F46–F53, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Feldt S, Batenburg WW, Mazak I, Maschke U, Wellner M, Kvakan H, Dechend R, Fiebeler A, Burckle C, Contrepas A, Jan Danser AH, Bader M, Nguyen G, Luft FC, Muller DN. Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension 51: 682–688, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Frindt G, Palmer LG. Surface expression of sodium channels and transporters in rat kidney: effects of dietary sodium. Am J Physiol Renal Physiol 297: F1249–F1255, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez AA, Lara LS, Luffman C, Seth DM, Prieto MC. Soluble form of the (pro)renin receptor is augmented in the collecting duct and urine of chronic angiotensin II-dependent hypertensive rats. Hypertension 57: 859–864, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez AA, Luffman C, Bourgeois CR, Vio CP, Prieto MC. Angiotensin II-independent upregulation of cyclooxygenase-2 by activation of the (Pro)renin receptor in rat renal inner medullary cells. Hypertension 61: 443–449, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez AA, Womack JP, Liu L, Seth DM, Prieto MC. Angiotensin II increases the expression of (Pro)Renin receptor during low-salt conditions. Am J Med 348: 416–422, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA. The absence of intrarenal ACE protects against hypertension. J Clin Invest 123: 2011–2023, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, Ledford KJ, Pitkin WB, Russo L, Najjar SM, Siragy HM. Targeted deletion of murine CEACAM 1 activates PI3K-Akt signaling and contributes to the expression of (Pro)renin receptor via CREB family and NF-κB transcription factors. Hypertension 62: 317–323, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J, Siragy HM. Sodium depletion enhances renal expression of (pro)renin receptor via cyclic GMP-protein kinase G signaling pathway. Hypertension 59: 317–323, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaneshiro Y, Ichihara A, Sakoda M, Takemitsu T, Nabi AH, Uddin MN, Nakagawa T, Nishiyama A, Suzuki F, Inagami T, Itoh H. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol 18: 1789–1795, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Kleyman TR, Ernst SA, Coupaye-Gerard B. Arginine vasopressin and forskolin regulate apical cell surface expression of epithelial Na+ channels in A6 cells. Am J Physiol Renal Fluid Electrolyte Physiol 266: F506–F511, 1994. [DOI] [PubMed] [Google Scholar]
- 16.Kobori H, Harrison-Bernard LM, Navar LG. Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production. Kidney Int 61: 579–585, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lantelme P, Rohrwasser A, Gociman B, Hillas E, Cheng T, Petty G, Thomas J, Xiao S, Ishigami T, Herrmann T, Terreros DA, Ward K, Lalouel JM. Effects of dietary sodium and genetic background on angiotensinogen and renin in mouse. Hypertension 39: 1007–1014, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Lee IH, Dinudom A, Sanchez-Perez A, Kumar S, Cook DI. Akt mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J Biol Chem 282: 29866–29873, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Li W, Sullivan MN, Zhang S, Worker CJ, Xiong Z, Speth RC, Feng Y. Intracerebroventricular infusion of the (pro)renin receptor antagonist PRO20 attenuates deoxycorticosterone acetate-salt-induced hypertension. Hypertension 65: 352–361, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu L, Gonzalez AA, McCormack M, Seth DM, Kobori H, Navar LG, Prieto MC. Increased renin excretion is associated with augmented urinary angiotensin II levels in chronic angiotensin II-infused hypertensive rats. Am J Physiol Renal Physiol 301: F1195–F1201, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu X, Garrelds IM, Wagner CA, Danser AH, Meima ME. (Pro)renin receptor is required for prorenin-dependent and -independent regulation of vacuolar H+-ATPase activity in MDCK.C11 collecting duct cells. Am J Physiol Renal Physiol 305: F417–F425, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Lu X, Wang F, Liu M, Yang KT, Nau A, Kohan DE, Reese VR, Richardson RS, Yang T. Activation of ENaC in collecting duct cells by prorenin and its receptor PRR: involvement of Nox4-derived hydrogen peroxide. Am J Physiol Renal Physiol (December 23, 2015). doi: 10.1152/ajprenal.00492.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem 287: 660–671, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mamenko M, Zaika O, Prieto MC, Jensen VB, Doris PA, Navar LG, Pochynyuk O. Chronic angiotensin II infusion drives extensive aldosterone-independent epithelial Na+ channel activation. Hypertension 62: 1111–1122, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matavelli LC, Huang J, Siragy HM. In vivo regulation of renal expression of (pro)renin receptor by a low-sodium diet. Am J Physiol Renal Physiol 303: F1652–F1657, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris RG, Schafer JA. cAMP increases density of ENaC subunits in the apical membrane of MDCK cells in direct proportion to amiloride-sensitive Na+ transport. J Gen Physiol 120: 71–85, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109: 1417–1427, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oliverio MI, Kim HS, Ito M, Le T, Audoly L, Best CF, Hiller S, Kluckman K, Maeda N, Smithies O, Coffman TM. Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci USA 95: 15496–15501, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oshima Y, Kinouchi K, Ichihara A, Sakoda M, Kurauchi-Mito A, Bokuda K, Narita T, Kurosawa H, Sun-Wada GH, Wada Y, Yamada T, Takemoto M, Saleem MA, Quaggin SE, Itoh H. Prorenin receptor is essential for normal podocyte structure and function. J Am Soc Nephrol 22: 2203–2212, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT(1) receptors. J Am Soc Nephrol 13: 1131–1135, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Prieto MC, Botros FT, Kavanagh K, Navar LG. Prorenin receptor in distal nephron segments of 2-kidney, 1-clip goldblatt hypertensive rats. Ochsner J 13: 26–32, 2013. [PMC free article] [PubMed] [Google Scholar]
- 32.Prieto MC, Williams DE, Liu L, Kavanagh KL, Mullins JJ, Mitchell KD. Enhancement of renin and prorenin receptor in collecting duct of Cyp1a1-Ren2 rats may contribute to development and progression of malignant hypertension. Am J Physiol Renal Physiol 300: F581–F588, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prieto-Carrasquero MC, Botros FT, Pagan J, Kobori H, Seth DM, Casarini DE, Navar LG. Collecting duct renin is upregulated in both kidneys of 2-kidney, 1-clip goldblatt hypertensive rats. Hypertension 51: 1590–1596, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, Ozawa Y, Hering-Smith KS, Hamm LL, Navar LG. Enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Hypertension 44: 223–229, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quadri S, Siragy HM. (Pro)renin receptor contributes to regulation of renal epithelial sodium channel. J Hypertens 34: 486–494, 2016. doi: 10.1097/HJH.0000000000000825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quadri S, Siragy HM. Regulation of (pro)renin receptor expression in mIMCD via the GSK-3beta-NFAT5-SIRT-1 signaling pathway. Am J Physiol Renal Physiol 307: F593–F600, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramkumar N, Stuart D, Calquin M, Quadri S, Wang S, Van Hoek AN, Siragy HM, Ichihara A, Kohan DE. Nephron-specific deletion of the prorenin receptor causes a urine concentration defect. Am J Physiol Renal Physiol 309: F48–F56, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramkumar N, Stuart D, Rees S, Hoek AV, Sigmund CD, Kohan DE. Collecting duct-specific knockout of renin attenuates angiotensin II-induced hypertension. Am J Physiol Renal Physiol 307: F931–F938, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramkumar N, Ying J, Stuart D, Kohan DE. Overexpression of renin in the collecting duct causes elevated blood pressure. Am J Hypertens 26: 965–972, 2013. [DOI] [PubMed] [Google Scholar]
- 40.Riquier-Brison AV, Burford JL, Peti-Peterdi J. The macula densa (pro)renin receptor functions as an amplifier of renin synthesis and release. In: American Society of Nephrology Kidney Week. San Diego, CA: 2013. [Google Scholar]
- 41.Saris JJ, Hoen PA, Garrelds IM, Dekkers DH, den Dunnen JT, Lamers JM, Jan Danser AH. Prorenin induces intracellular signaling in cardiomyocytes independently of angiotensin II. Hypertension 48: 564–571, 2006. [DOI] [PubMed] [Google Scholar]
- 42.Sauter D, Fernandes S, Goncalves-Mendes N, Boulkroun S, Bankir L, Loffing J, Bouby N. Long-term effects of vasopressin on the subcellular localization of ENaC in the renal collecting system. Kidney Int 69: 1024–1032, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Schonig K, Schwenk F, Rajewsky K, Bujard H. Stringent doxycycline dependent control of CRE recombinase in vivo. Nucleic Acids Res 30: e134, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song R, Preston G, Ichihara A, Yosypiv IV. Deletion of the prorenin receptor from the ureteric bud causes renal hypodysplasia. PLos One 8: e63835, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soundararajan R, Zhang TT, Wang J, Vandewalle A, Pearce D. A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem 280: 39970–39981, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Staruschenko A, Pochynyuk O, Vandewalle A, Bugaj V, Stockand JD. Acute regulation of the epithelial Na+ channel by phosphatidylinositide 3-OH kinase signaling in native collecting duct principal cells. J Am Soc Nephrol 18: 1652–1661, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Stockand JD. Vasopressin regulation of renal sodium excretion. Kidney Int 78: 849–856, 2010. [DOI] [PubMed] [Google Scholar]
- 48.Traykova-Brauch M, Schonig K, Greiner O, Miloud T, Jauch A, Bode M, Felsher DW, Glick AB, Kwiatkowski DJ, Bujard H, Horst J, von Knebel Doeberitz M, Niggli FK, Kriz W, Grone HJ, Koesters R. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med 14: 979–984, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang F, Lu X, Peng K, Zhou L, Li C, Wang W, Yu X, Kohan DE, Zhu SF, Yang T. COX-2 mediates angiotensin II-induced (pro)renin receptor expression in the rat renal medulla. Am J Physiol Renal Physiol 307: F25–F32, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weisz OA, Wang JM, Edinger RS, Johnson JP. Noncoordinate regulation of endogenous epithelial sodium channel (ENaC) subunit expression at the apical membrane of A6 cells in response to various transporting conditions. J Biol Chem 275: 39886–39893, 2000. [DOI] [PubMed] [Google Scholar]