

Abstract
There is an alarming global increase in the incidence of end-stage kidney disease, for which early biomarkers and effective treatment options are lacking. Largely based on the histology of the end-stage kidney and on the model of unilateral ureteral obstruction, current investigation is focused on the pathogenesis of renal interstitial fibrosis as a central mechanism in the progression of chronic kidney disease (CKD). It is now recognized that cumulative episodes of acute kidney injury (AKI) can lead to CKD, and, conversely, CKD is a risk factor for AKI. Based on recent and historic studies, this review shifts attention from the glomerulus and interstitium to the proximal tubule as the primary sensor and effector in the progression of CKD as well as AKI. Packed with mitochondria and dependent on oxidative phosphorylation, the proximal tubule is particularly vulnerable to injury (obstructive, ischemic, hypoxic, oxidative, metabolic), resulting in cell death and ultimately in the formation of atubular glomeruli. Animal models of human glomerular and tubular disorders have provided evidence for a broad repertoire of morphological and functional responses of the proximal tubule, revealing processes of degeneration and repair that may lead to new therapeutic strategies. Most promising are studies that encompass the entire life cycle from fetus to senescence, recognizing epigenetic factors. The application of techniques in molecular characterization of tubule segments and the development of human kidney organoids may provide new insights into the mammalian kidney subjected to stress or injury, leading to biomarkers of early CKD and new therapies.
Keywords: acute kidney injury, atubular glomeruli, chronic kidney disease, proximal tubule, unilateral ureteral obstruction
The Challenge: a Unifying Concept of AKI and CKD
acute kidney injury (aki) is the consequence of a series of biological processes that follow a host response to an initial renal insult (9). In the past 20 yr, the incidence of AKI has increased in parallel with an increase in end-stage kidney disease (ESKD) (99). Of greater concern, between 1976 and 1994, the prevalence of ESKD has exceeded that predicted from the prevalence of chronic kidney disease (CKD) by 70% (76). This trend has led to the recent formulation of the hypothesis that cumulative episodes of AKI culminate in CKD, and that preexisting CKD increases the risk for AKI (99). In fact, a dose-response relationship can be derived between the severity of AKI and the risk of progression of CKD (82). Moreover, the link between AKI and CKD has been established for children as well as adults (109).
Current diagnosis of AKI and CKD is limited by generally available clinical measures of kidney structure (renal ultrasound and biopsy) and function (serum creatinine concentration and urine albumin excretion). Reliance on the clearance of creatinine [or other marker of glomerular filtration rate (GFR)] as the mainstay of renal functional status is undermined by the well-established process of nephron loss accompanied by adaptive hypertrophy of remaining nephrons in CKD: a rise in serum creatinine concentration does not occur until nearly 50% of nephrons are nonfunctional (181). The observation that renal tissue from virtually all patients with ESKD is characterized by glomerulosclerosis and renal interstitial fibrosis has led to the conclusion that fibrosis plays a primary role in the process of nephron loss (37, 104, 196). As a consequence, measures of interstitial fibrosis have become the established end point for clinical studies and animal models of CKD (25, 42). As is the case for the reliance on serum creatinine concentration to detect reduced renal function, the major disadvantage of depending on renal interstitial collagen deposition to measure tissue injury is that it develops late in the response to injury. As a corollary to the unsatisfactory available biomarkers for diagnosis of kidney disease, antifibrotic therapies to prevent or to slow CKD progression have proved equally disappointing (29).
This review is an attempt to redirect the focus of renal translational research from the glomerulus and renal interstitium to the proximal tubule. First, the history leading to the current emphasis on fibrosis is examined. The central role played by unilateral ureteral obstruction (UUO) as a model of fibrogenesis in CKD is then considered, followed by the discovery of the generation of atubular glomeruli in this model, thereby shifting attention from the interstitium to the proximal tubule. Animal models reveal that glomerulotubular disruption and the ultimate formation of atubular glomeruli develop not only in tubular disorders, such as polycystic kidney disease (PKD) and nephropathic cystinosis, but also in “glomerular” disorders (congenital nephrotic syndrome and diabetic nephropathy). Due to its high rates of oxygen consumption and relative paucity of endogenous antioxidant defenses, the proximal tubule is particularly vulnerable to injury. Of the greatest significance, clinically relevant studies of kidney injury reveal that molecular targeting of the proximal tubule is sufficient to induce AKI, progressing to CKD when the injury is repeated. Taken together, this evidence points to the proximal tubule as the central link between AKI and CKD, and the progression of CKD is molded by the life cycle from fetus to senescence.
Focus on the Glomerulus and the Interstitium: How Did We Get Here?
Hyperfiltration: the glomerulocentric era.
Recognition of the association of renal pathological characteristics with clinical signs of CKD date back to the work of Richard Bright in the early nineteenth century (16). A century later, Thomas Addis developed a clinical classification of “Bright's disease” with the collaboration of Jean Oliver, a brilliant morphologist whose elegant microdissections of nephrons from patients with CKD remain unrivaled today (2, 128). Oliver emphasized the marked heterogeneity of nephrons from patients with ESKD, with intermingling of atrophied and hypertrophied tubules, aglomerular tubules, and atubular glomeruli (Fig. 1) (128). At this time, Homer Smith (154) was making major advances in renal physiology, applying the clearance concept to elucidate glomerular and tubule function: his approach was to treat the kidney as a “black box” rather than as a collection of individual nephrons. This set the stage for the development of animal models of CKD in the second half of the twentieth century, and the formulation of the “intact nephron hypothesis” by Neal Bricker (15). Notably, Bricker concluded from glucose titration curves that there is no evidence of functional nephron heterogeneity in the diseased kidney (15). New insights were provided by the use of renal micropuncture techniques by Barry Brenner and his associates, leading to a landmark paper in 1981 (75). This study revealed that, following unilateral nephrectomy and partial ablation of the remaining kidney, remaining nephrons undergo hyperfiltration with glomerular epithelial and mesangial injury: these changes are markedly attenuated by a low-protein diet (75). The authors concluded that sustained glomerular hyperfiltration is a maladaptive response, preserving whole-kidney GFR at the expense of progressive nephron injury. This work launched decades of studies of mechanisms underlying glomerular hyperfiltration and hypertrophy, as well as resulting glomerulosclerosis: the glomerulocentric era was born.
Fig. 1.

Measurements of microdissected nephrons from patients with chronic Bright's disease (chronic kidney disease). Left, top: schematic normal and hypertrophied nephrons with the relative volume of glomerulus and tubular segments represented by the area of each segment. Virtually all of the adaptive growth is contributed by the proximal tubule. Left, bottom: relative proximal tubular diameter; represented by marks reflecting varying cross-sectional area along the length of the tubule. Representative hypertrophied and atrophied proximal tubular segments are compared with a normal one. Right: atubular glomerulus and aglomerular tubule. [Reproduced with permission from J. Oliver (128) and Wolters Kluwer.]
The concurrent development of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers resulted in their use as therapeutic agents to prevent or slow the hyperfiltration response in animal models of CKD (142). The United States Food and Drug Administration approved the use of this therapy in diabetic nephropathy, and it has found widespread use in attempting to slow virtually all forms of CKD, although with limited effectiveness (19). The hyperfiltration theory predicted that reduction in nephron number, regardless of etiology, is a major risk factor for progression of CKD (13).
Unilateral ureteral obstruction: focus on the interstitium.
Over the past 3 decades, complete UUO has become the most widely used animal model of progressive CKD, with the annual number of publications increasing from 10 in 1986 to 100 in 2015 (Fig. 2) (38). Surgical ligation of a ureter is a simple procedure: it rapidly results in nephron injury with characteristics of human ESKD, and its application to murine genetically engineered mutants can be used to target the roles of specific genes and cell types in the kidney (8, 25). This work has been fueled by the rapid development of molecular techniques and reagents that led to the identification of many cellular pathways and interactions that play a role in injury to the obstructed kidney (22). As noted above, because renal interstitial fibrosis is the hallmark of ESKD, the end point of most of the studies targeted renal interstitial cellular infiltrates [monocytes/macrophages and α-smooth muscle actin (a marker of myofibroblasts)] and interstitial collagen deposition (25). Our laboratory has used rodent models of UUO for over 30 yr, with a primary focus on the neonatal period, seeking insight into the pathogenesis of congenital urinary tract obstruction, the primary identifiable cause of CKD in the pediatric population (119). These studies revealed tubule cell death as well as altered renal expression of growth factors and cytokines. These include components of the renin-angiotensin system, transforming growth factor-β (TGF-β), and adhesion molecules (26–28, 40, 44, 95, 96, 126, 189, 190, 193). Although the model of unilateral ureteral ligation provides a convenient assay for quantitation of interstitial fibrosis, complete UUO rarely occurs in the clinical setting and represents an extreme insult to the kidney. To better reproduce congenital obstructive nephropathy, we have developed models of reversible variable partial UUO, which allows independent manipulation of the timing, severity, and duration of obstruction (150, 167, 168, 191, 192).
Fig. 2.
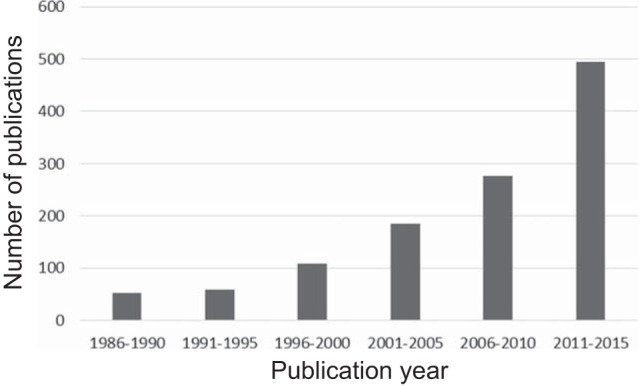
Number of papers containing the key words “unilateral ureteral obstruction” published between 1986 and 2015. Medline search was performed April 2016. There was a 10-fold increase in the annual number of publications between 1986 and 2015.
Shifting the Focus to the Proximal Tubule
Serendipity: the glomerulotubular junction and atubular glomeruli.
In the course of a study of the renal cellular recovery following release of partial UUO in the neonatal mouse (168), we were preparing to investigate the role of endothelial nitric oxide synthase (eNOS) in obstructive injury, using eNOS knockout mice (51). However, kidneys of sham-operated adult eNOS mutant mice exhibited macroscopic indented scars containing crowded small glomeruli that proved by serial sectioning to be disconnected from their proximal tubules (51). Examination of neonatal eNOS knockout mouse kidneys revealed apoptosis and necrosis of focal wedge-shaped areas of renal parenchyma, containing nephrons with narrowing of the glomerulotubular junction (51). This led to the conclusion that endogenous renal eNOS production contributes to the maintenance of nephron maturation and integrity. Of note, human eNOS gene polymorphisms are associated with reduced enzyme activity and the development of ESKD (124). Upon reexamination of kidneys in the neonatal partial UUO study, areas of proximal tubule cell death and glomerulotubular disconnection were discovered; release of UUO arrested this process, with remodeling of the renal architecture (Table 1) (168).
Table 1.
Segmental proximal tubular function and response to injury: animal models and human diseases
| Model/Disease | Glomerulotubular Junction Response To Injury | Proximal Tubule | Proximal Tubule Energy |
|---|---|---|---|
| Renal ablation | ATG early (58) | Progressive proximal tubular atrophy → ↑ ATG from 9 to 48%, 10–25 wk after 5/6 nephrectomy | ↑Qo2, early (121) |
| Unilateral ureteral obstruction | ATG early (50) | Oxidative stress and cell death by autophagy, apoptosis, necrosis | ↓Qo2, ↑glycolysis (158) |
| Polycystic kidney disease | ATG late (56) | Oxidative stress and cell death | |
| Congenital nephrotic syndrome | ATG early (176) | Protein overload proximal tubule injury | |
| Cystinosis | ATG late (55, 97) | Swan neck. Mitoquinone protective (antioxidant) (55) | ↓mitochondria (55) |
| Diabetes | Type 1 → ATG early (117) | 71% GTJ abnormalities in proteinuric type 1 diabetics | ↑catalase protective in mouse (14), mitochondrial uncoupling and ↑Qo2 →hypoxia (52) |
| Type 2 → ATG late (186) | 44% GTJ abnormalities in type 2 diabetic patients with low proteinuria | ||
| Renal artery stenosis | Mild-reversible (73) | Tubular atrophy and thickened TBM reverse with unclipping and stopping ACE inhibitor (73) | ↓mitochondria |
| ↓Na-K-ATPase | |||
| severe → ATG late (110) | 52% ATG, 40% glomeruli connected to atrophic tubules; normal number of glomeruli (110) | ||
| Chronic allograft rejection | ATG late | 18% ATG 7 yr posttransplant (133) | Rat renal transplant → mitochondrial uncoupling, ↑Qo2 → hypoxia (134) |
Nos. in parentheses, reference nos.
ATG, atubular glomeruli; GTJ, glomerulotubular junction; TBM, tubular basement membrane; ACE, angiotensin-converting enzyme; Qo2, oxygen consumption.
These observations prompted a review of the literature, which revealed that injury to the glomerulotubular junction and formation of atubular glomeruli has been described in a wide variety of renal disorders, including glomerular diseases (IgA nephropathy and diabetic nephropathy), tubular diseases (renal allograft rejection, obstructive nephropathy, pyelonephritis, and PKD), and toxic nephropathies (adriamycin, cisplatin, and lithium) (24). Using microdissection in the 1930s, Jean Oliver (128) had described atubular glomeruli (and aglomerular tubules) in kidneys from patients with Bright's disease (Fig. 1). Subsequently, in the 1990s, using serial sectioning, Niels Marcussen (111) described atubular glomeruli in patients with CKD due to a variety of disorders. It is likely that glomerulotubular injury and the formation of atubular glomeruli has remained largely underappreciated due to the labor-intensive nature of microdissection and serial sectioning of tissue. A recent study demonstrated the use of multiphoton microscopy to visualize atubular glomeruli in situ in a murine model of cisplatin nephropathy, an approach that could significantly reduce the time and effort required to quantify the fraction of atubular glomeruli (170).
Unilateral ureteral obstruction revisited.
In light of this information, we implemented histomorphometry to investigate the most commonly used animal model of CKD: 7 and 14 days of complete UUO in the adult mouse (48). This revealed that, compared with sham-operated animals, relative vascular volume fraction remained unchanged, and interstitial matrix components did not exceed 15% of the total volume fraction of the obstructed kidney (50). In contrast, there was a very rapid loss of renal parenchyma in the completely obstructed kidney, due to a 65% reduction in proximal tubule mass (50). Proximal tubules underwent oxidative injury, apoptosis, necrosis, and autophagy, with widespread mitochondrial loss and tubular collapse (50). After 14 days of UUO, this was associated with 46% of glomeruli developing atrophic proximal tubules at the glomerulotubular junction, and 39% becoming atubular (Table 1) (48). Thus, based on sequential quantitative morphology, rather than interstitial expansion or fibrosis, the primary casualty of obstructive injury is massive proximal tubule cell death initially localized to the glomerulotubular junction. In contrast to the formation of collapsed proximal tubule fragments, epithelial cells at the urinary pole of Bowman's capsule undergo transition to a mesenchymal phenotype and extend to seal the capsule while maintaining perfusion of the glomerular tuft, with recruitment of renin-expressing cells down the afferent arteriole (48). Progenitor cells have been identified at the urinary pole of Bowman's capsule, and these may play a role in its repair (139), allowing the glomerulus to continue its endocrine function by production of renin, a homeostatic response (149). These events suggest that, rather than primary interstitial signaling inducing fibrotic pathways, both regenerative and degenerative processes are activated in the proximal tubule by UUO, and that these are drivers of additional nephron loss. This conclusion is supported by a recent study in which two proapoptotic members of the Bcl-2 family were investigated in mice subjected to 7 days of complete UUO (84). Mice with either proximal tubule-specific Bax deletion or systemic deletion of Bak had no effect on obstructive renal injury, whereas combined deletion of Bax and Bak inhibited tubule apoptosis and atrophy, as well as secondary interstitial inflammation and fibrosis: targeting the fate of proximal tubule cells determines the downstream interstitial consequences of UUO (84). Underscoring the importance of long-term follow up in animal models, an earlier study of rats 10 and 25 wk following renal ablation revealed tubule atrophy and formation of atubular glomeruli that preceded the development of glomerulosclerosis; at 25 wk, 48% of glomeruli were atubular, whereas only 14% were globally sclerotic (Table 1) (58).
Allometry of the proximal tubule: limits of growth and senescence.
Allometry, or the study of differential growth of components of an organism's morphology, tends to be forgotten in the pursuit of cellular and molecular mechanisms. However, the functional consequences of changes in nephron size are of central importance: metabolic rate is the primary determinant of GFR (151). This becomes particularly relevant in relating the myriad metabolic activities of the proximal tubule to segments of the nephron, as well as individual cells (185). For example, in addition to proliferation and hypertrophy, the proximal tubule cells must elaborate a brush border to maximize reabsorptive surface area, a feature that is evolutionarily conserved from the pronephros of fish to the metanephros of mammals (140, 185). At present, the gene regulatory networks responsible for orchestrating proximal tubular size remain largely unknown.
The proximal tubular mass constitutes over 50% of the volume of the normal kidney, and growth of the proximal tubule accounts for the increasing size of the kidney from late gestation through early childhood (23). Proximal tubular length increases from 2 mm at birth to 12 mm in the adult, and its rate of growth outstrips that of the glomerulus (Figs. 3 and 4) (45). Because nephron maturation proceeds centrifugally, the longest proximal tubules lie in the inner cortex, whereas the shortest are in the subcapsular region: nephron heterogeneity is extreme in the neonate, with the ratio of longest to shortest being greater than 10:1 (45). By 1 mo of age, internephron variation decreases with a ratio of only 3.5:1, and by adulthood decreases further to 1.5:1 (45). The reduction in proximal tubule mass with aging is due not only to nephron loss (127), but also to increasing nephron heterogeneity, with the formation of two populations of nephrons, those that are atrophied and those with adaptive hypertrophy, a pattern also characteristic of CKD (Figs. 1 and 3) (128, 132). Proximal tubule hypertrophy can be a homeostatic response to reduced nephron number due to genetic disorders (144), to nephron loss (as in CKD) (130), or to chronic hyperglycemia (as in diabetes, see below) (175).
Fig. 3.

Microdissected human nephrons. A and B: glomerulus and proximal tubule from term neonate and adult, respectively. C: complete atrophic nephron from aging adult. D: complete hypertrophic nephron from aging adult. Arrows indicate glomeruli. [Reproduced with permission from J. Oliver (129; 132) and Wolters Kluwer.]
Fig. 4.

A: length of human proximal tubule from birth to 90 yr. Data are shown from measurements of microdissected nephrons from 105 cadavers undergoing routine necropsy, who had suffered acute death. Steady growth from birth to 20 yr is followed by a plateau and a gradual decline after the 3rd to 4th decade. Red box = 20–30 yr of age, peak reproductive years. B: lifetime risk for diagnosis of ESKD from birth to 90 yr, from Grams et al. (68). [Graphic representation of data in A reproduced with permission from Darmady et al. (33) and Wiley.]
Maximal tubular size is limited by the dimensions (luminal diameter and tubular length) that create low enough resistance to allow tubular fluid flow for adequate reabsorption. This is solved in kidneys of the whale by nephrons being packed in many small clusters (renicules) (108). These considerations become important in understanding the response to injury of the newborn kidney, or in devising strategies to enhance regeneration in the mature kidney: physical constraints dictate the minimum number of nephrons that can maintain homeostasis. As noted above, congenital obstructive nephropathy is the primary cause of CKD in children and is often accompanied by reduced nephron number, an independent risk factor for adult CKD (105). To determine the role of nephron number on the long-term recovery following the release of obstruction in neonatal mice, we compared wild-type mice to Os/+ mice that are born with a 50% reduction in number of nephrons (150). Whereas release of obstruction allowed proximal tubular growth to resume in wild-type mice, growth was not restored in Os/+ mice, which also underwent additional nephron loss (150). These findings suggest that infants with low nephron number (such as very-low-birth-weight infants) are at increased risk for progressive CKD due to obstructive nephropathy.
A provocative study of renal allograft mass-to-recipient size ratio in pediatric transplanted kidneys revealed that 24 mo posttransplantation, transplants of lower graft mass demonstrated increasing renal volume, whereas those of higher graft mass actually decreased by a similar amount (43). The resistive index decreased throughout early follow-up in patients receiving transplants of lower graft mass, whereas an increase was noted in those receiving organs of high graft mass, suggesting inadequate perfusion of the larger grafts (43). These observations suggest that, in addition to immunological incompatibilities, a mismatch of physical organ size can influence graft outcome. Despite decades of investigation, the determinants of compensatory renal growth remain to be elucidated.
Following very rapid growth in proximal tubular length in the first 2 decades of life (45), tubule length gradually begins to shrink after 40 yr of age (Fig. 4A) (33). In a recent study, kidney volume was measured by computed tomography in 1,344 potential kidney donors 18–75 yr of age (182). Consistent with the earlier microdissection study (33), reduction in renal parenchymal volume with age was correlated with decreasing GFR and decreasing cortical volume (a reflection of proximal tubular mass), but slightly increasing medullary volume (182). Notably, albuminuria was associated with increasing parenchymal volume in apparently healthy adults, suggesting hypertrophy of some proximal tubules in hyperfiltering nephrons.
Responses of the Proximal Tubule to Injury: a Varied Repertoire
The cumulative incidence of ESKD remains below 1% until after 40 yr of age, with significantly greater subsequent increases in men than women and in black than white patients (Fig. 4B) (68). Race and sex disparities in CKD remain important epidemiological concerns and have been recently reviewed (125). Unlike men or postmenopausal women, in naturally ovulating women, urinary excretion of proximal tubule-specific enzymes peaks within 7 days after ovulation or onset of menses (148). These findings indicate novel proximal tubular adaptive responses to the normal human female reproductive hormone cycle, consistent with recurring increases of tubular cell turnover that may confer resistance to tubular injury (148). Notable is the increase in CKD after the peak reproductive period (20–30 yr) (Fig. 4B), which coincides with the decrease in proximal tubular length (Fig. 4A). There is a shift in the primary diagnoses for CKD in adults compared with children: congenital anomalies of the kidneys and urinary tract contribute >50% of cases in children (with obstructive nephropathy the leading cause) (119), whereas diabetes and hypertension predominate in adults (174). It is now apparent, however, that, for most children with congenital anomalies progressing to ESKD, renal replacement therapy is delayed until adulthood (188). There is marked heterogeneity in the rate of progression of kidney disease over the life span, and the proximal tubule displays a variety of responses to injury, ultimately leading to tubule atrophy, glomerulotubular disruption, and formation of atubular glomeruli in glomerular, tubular, and toxic nephropathies (see Table 1 in Ref. 24). Renal morphometric analysis by serial sectioning reveals that, in nephropathic cystinosis, a rare inherited disorder of cystine transport that leads to ESKD in the second decade of life, there is a preponderance of atubular glomeruli by 10–24 yr of age (Fig. 5) (97). In contrast, diabetic nephropathy progresses less rapidly, develops later in life, and results in a greater proportion of atubular glomeruli in type I than type II, which spans later adulthood (Fig. 5) (117, 186).
Fig. 5.

Fractional distribution of glomerulotubular junction integrity in serial sections of glomeruli from patients with nephropathic cystinosis and diabetic nephropathy. Data are arranged by age group, with age range for patients in each diagnostic group. “Atubular glomeruli” are those with no tubular connection; “Tubular atrophy” indicates glomeruli connected to atrophic tubules; and “Normal” indicates glomeruli connected to normal tubules. DM, diabetes mellitus. Data were derived from Larsen et al. (97), Najafian et al. (117), and White et al. (186).
Recent experimental studies have elucidated a remarkable variety of mechanisms leading to proximal tubular injury and progression of CKD. To illustrate these, four disorders are examined below: PKD and nephropathic cystinosis (“tubular” disorders), and congenital nephrotic syndrome and diabetic nephropathy (“glomerular” disorders).
Polycystic kidney disease: the proximal tubule as collateral damage.
The leading monogenic cause of CKD, autosomal dominant PKD is characterized by the slowly progressive increase in the size of scattered cysts that arise from proximal and distal tubules (171). Expanding cysts encroach on neighboring intact nephrons, and, with continued exponential enlargement, remaining proximal tubular volume is progressively reduced, leading to ESKD (20). The pcy mutant mouse develops progressive PKD with markedly increasing cyst size by 30 wk of age. Morphometric study of pcy compared with wild-type mice revealed a linear relationship between fractional cyst area and damage to the glomerulotubular junction (56), a measure of severity of urinary tract obstruction (see above). This is paralleled by a progressive increase in the fraction of glomeruli attached to atrophic tubules, as well as atubular glomeruli (56). Tubule apoptosis is present in patients with PKD and pcy mice, as in mice subjected to UUO (48, 187). Examination of kidneys from patients with early- and late-stage PKD revealed a progressive increase in the fraction of glomeruli attached to atrophic tubules and atubular glomeruli (Table 1) (56). These findings demonstrate important parallels between the UUO model and PKD and support efforts to develop therapies to slow the growth in cysts in patients with PKD. Superior to available molecular markers, total cyst volume is currently the best predictor of progression of the disease and may serve as a surrogate for nephron loss due to tubular obstruction (20).
Nephropathic cystinosis: the “swan-neck lesion” as an adaptive strategy.
Nephropathic cystinosis is a rare congenital metabolic disorder resulting from a mutation in cystinosin, a lysosomal cystine carrier, which leads to severe Fanconi syndrome and progressive CKD (122). An attenuated glomerulotubular junction (“swan-neck lesion”), the characteristic renal histological feature, develops between 6 and 12 mo of age (Fig. 6) (107). By the onset of renal failure between 10 and 25 yr of age, most glomeruli are atubular, the result of chronic proximal tubular injury due to cystine accumulation (Fig. 5) (97). An animal model of nephropathic cystinosis, the CTNS knockout mouse, develops glucosuria, phosphaturia, and progressive CKD, all of which mirror features of human cystinosis (123). Detailed studies of proximal tubular changes during the life cycle of CTNS−/− mice revealed that, rather than representing atrophy, epithelial cells at the leading edge of the swan-neck lesion (beginning at the glomerulotubular junction) undergo progressive loss of mitochondria and expression of megalin Transfer as shown, consistent with dedifferentiation, and undergo extreme flattening with thickening of the underlying tubule basement membrane (54, 55). Whereas flattened cells of the swan-neck lesion resist uptake of filtered cystine (due to loss of megalin and mitochondria), the narrower tubular diameter and thickened basement membrane resist distension that would result from high hydrostatic pressure present in the S1 segment (the glomerular tuft remains normal) (55, 143, 184). Notably, epithelial cells of the downstream S3 segment compensate for the thinning S1 segment by increasing endocytosis with subsequent lysosomal clearance of cystine into tubular fluid (54). Initiation of the swan-neck lesion is at least in part dependent on proximal tubule oxidative injury resulting from cystine accumulation: administration of mitoquinone, an antioxidant targeted to mitochondria, significantly delays extension of the lesion (55). The adaptive responses of the proximal tubule ultimately fail, with clusters of proximal tubules exhibiting oxidative stress, upregulation of kidney injury molecule-1 (KIM-1), and activation of inflammasomes (55, 136). As is the case in human cystinosis, the terminal event in CTNS−/− mice is the formation of atubular glomeruli and interstitial fibrosis (55) (Table 1, Figs. 5 and 6).
Fig. 6.

Progression of cystinosis in mouse and man. A: “swan neck deformity” of the proximal tubule in human cystinosis develops after 5 mo of age is shown: normal, cystinosis 5 mo, and cystinosis 14 mo. B: scheme showing nephron changes with age in murine and human cystinosis. Fanconi syndrome generally develops in the first 6 mo of life, with significant decrease in GFR by 2–5 yr, and renal failure in the 2nd decade. There is marked interindividual variability in the rate of progression of renal lesions, and treatment with cysteamine can significantly delay progression to renal failure. The evolution of renal lesions in the mouse is more gradual, with GFR decreasing only later in adulthood. The period of proximal tubular maturation extends through the first 6 mo in humans and the first 3 mo in the Ctns−/− mouse; the “swan-neck lesion” is initiated in the first 3 mo in the mouse, and between 6 and 12 mo in humans. The development of irreversible renal lesions in both species includes loss of functional proximal tubular mass and the development of tubular atrophy, interstitial fibrosis, and formation of atubular glomeruli. Therapy should be directed to the early period of adaptation (potentially reversible changes) rather than the late (destructive) phase. [Reproduced with permission from Mahoney and Striker (107) and Springer, and Galarreta et al. (55).]
Congenital nephrotic syndrome: overwhelming proximal tubule albumin reabsorption.
Congenital nephrotic syndrome usually results from mutations coding for proteins essential to the podocyte slit diaphragm, resulting in massive proteinuria and hypoalbuminemia (138). While generally regarded as a purely glomerular disorder, microdissection of nephrons from a 2-mo-old infant with congenital nephrotic syndrome revealed narrowed glomerulotubular junctions, with cystic dilatation of the downstream portion of the proximal tubule (46). Examination of serial histological sections of kidneys from seven additional patients with congenital nephrotic syndrome revealed 30% atubular glomeruli, which were ascribed to heavy proteinuria (Table 1) (176). Kriz and LeHir (93) propose two mechanisms for the proximal tubular response to proteinuria: 1) direct encroachment of extracapillary lesions, resulting from primary glomerular injury, formation of crescents, and misdirected glomerular filtrate and obstruction of the glomerulotubular junction; and 2) leakage of proteins through injured glomeruli, leading to excessive protein reabsorption by proximal tubules and secondary tubular degeneration. Contrary to long-held views, it appears that the proximal tubule can reclaim large amounts of filtered albumin by transcytosis mediated by the neonatal Fc receptor pathway in concert with megalin, a receptor in the tubulovesicular system below the apical membrane of proximal tubule cells that serves as an endocytic scavenger of filtered proteins (36). Comparison of two models of protein overload reveals a surprising capacity of the proximal tubule to regulate uptake of albumin (180). Acute exogenous albumin overload is followed by a significant decrease in proximal tubular albumin uptake, whereas targeted deletion of podocytes leads to hypoalbuminemia and markedly increased proximal tubule uptake (180). The latter response presumably contributes to progressive proximal tubule injury in patients with congenital nephrotic syndrome. The role of megalin in this context has been revealed in double transgenic mice lacking proximal tubule megalin and podocyte depletion that develop massive proteinuria but mild glomerular and tubule injury (114). Comparison of megalin-containing to megalin-deficient proximal tubule cells showed that albumin and immunoglobulins were preferentially accumulated in megalin-containing cells, which also preferentially expressed heme oxygenase-1 (HO-1) and monocyte chemotactic protein-1, and underwent apoptosis (114). A prior study using a similar experimental approach, but in which glomerular injury was induced by anti-glomerular basement membrane antibody, concluded that tubular lesions arise from direct extension of glomerular lesions down the glomerulotubular junction (166). These two studies garner support for both mechanisms proposed by Kriz and LeHir (93), which are not mutually exclusive (30). Although inhibition of megalin-mediated proximal tubular transcytosis would appear to be an effective therapy to reduce proximal tubular injury due to glomerular protein leakage, megalin is necessary for proximal tubular uptake of survivin, an antiapoptotic molecule that regulates cell division and survival (85). There is clearly much to be learned regarding the role of the proximal tubule in maintaining albumin homeostasis: this function must be balanced with multiple other vital homeostatic functions, such as reclamation of sodium, bicarbonate, phosphorus, amino acids, and glucose (118).
Diabetic nephropathy: a disorder of the proximal tubule.
Diabetes is the major cause of CKD in adults, and diabetic nephropathy has been viewed primarily as a glomerular disorder, resulting in glomerulosclerosis. There is evidence, however, for the earliest responses of the nephron being centered in the proximal tubule rather than the glomerulus. Proximal tubule cells cannot decrease glucose transport rates enough to prevent increased intracellular glucose in the face of hyperglycemia (175). This results in hypertrophy of proximal tubule cells, hyper-reabsorption, and hyperfiltration due to tubuloglomerular feedback, and increased production of reactive oxygen species leads to oxidative stress and increased production of angiotensinogen (6, 175). Initially, proximal tubule cells undergo hyperplasia with increased production of TGF-β, leading to G1 cell cycle arrest, hypertrophy, and a senescence phenotype (175). Transformed proximal tubule cells, in turn, stimulate interstitial inflammation with myofibroblast-initiated collagen deposition (175). Of note, morphometric study of kidneys from patients with type 1 or type 2 diabetes reveal widespread glomerulotubular junction abnormalities and formation of atubular glomeruli (Table 1, Fig. 5) (116, 186). Nondiabetic transgenic mice overexpressing angiotensinogen in proximal tubules develop albuminuria, tubular apoptosis, and interstitial fibrosis (64). These sequelae are prevented in double-transgenic mice that overexpress both catalase and angiotensinogen in proximal tubule cells (64). Streptozotocin-induced diabetic mice transgenically overexpressing catalase in proximal tubule cells reveal that proximal tubular oxidative stress plays a central role in the generation of angiotensinogen and apoptosis, hallmarks of diabetic nephropathy (6, 14).
Proximal Tubule Function, Injury, and Energy Conservation
Overlapping mechanisms in AKI and CKD.
Abnormalities of renal perfusion have been shown to play a significant role in both AKI and CKD. The interplay between impaired production of vascular endothelial growth factor by damaged proximal tubules and peritubular rarefaction likely plays an important role in AKI-CKD transition (163). Four weeks following microembolisms induced by intra-arterial injection of microspheres in intact adult rats, foci of atrophic tubules appear, some of which contain a mosaic distribution of both atrophic and normal-appearing epithelial cells (159). The tubules are surrounded by damaged peritubular capillaries and interstitial cells expressing platelet-derived growth factor receptor-β and α-smooth muscle actin (159). These data indicate that ischemia-induced hypoxia can impair the renal microcirculation, leading to focal proximal tubular injury that, in turn, results in interstitial inflammation and fibrosis (18). In progression of CKD, injured tubule cells with impaired production of vascular endothelial growth factor are more likely to be the cause rather than the consequence of capillary rarefaction (177).
As is the case for CKD, the pathophysiology of AKI is complex and involves all components of the nephron. Ischemia-reperfusion is a common cause of AKI, and micropuncture studies have shown that tubular obstruction is a major factor in its pathogenesis, whereas tubular repair with relief of obstruction are early events in the recovery from injury (5, 47). However, in contrast to the UUO model, in which the glomerulotubular junction appears to be a major target of injury and repair, the proximal tubule S3 segment is most susceptible in AKI (Table 2). Targeted ablation of this segment in transgenic mice results in polyuria followed by oliguria, with exfoliation of S3 segment cells into the tubular lumen (147). It appears that S1 proximal segments act as sensors of injurious stimuli and modulate the response of S2 and S3 segments to injury by signaling (cross talk) between segments (39). Unlike S2 and S3 segments, the S1 segment lacks peroxisomes, organelles that detoxify accumulated fatty acids but can be a source of reactive oxygen species in injured cells (39). By contrast, like macrophages, S1 segment cells protect themselves from oxidative injury by upregulation of HO-1 and sirtuin 1 (Table 2) (39, 87). Administration of an inhibitor of heme oxygenase aggravates kidney dysfunction in a model of glycerol-induced AKI, revealing the importance of endogenous HO-1 in proximal tubule protection (120). A subfamily of nuclear receptor transcription factors, peroxisome proliferator-activated receptors (PPARs), appear to play a significant role in the proximal tubular response to injury. Chronic UUO reduces kidney PPAR-α expression in mice, and transgenic expression of proximal tubule PPAR-α reduces production of TGF-β and proinflammatory cytokines following UUO, and also reduces oxidative injury and S3 segment necrosis following cisplatin or ischemia-reperfusion injury (100, 101).
Table 2.
Segmental proximal tubular function and response to injury: mechanisms
| Function | Glomerulo-Tubular Junction | S1 | S2, S3 | Proximal Tubule Energy |
|---|---|---|---|---|
| Normal | Site of reabsorption of amino acids, glucose, bicarbonate; lacks peroxisomes (87) |
Peroxisomes (87) | Proximal tubule ATP > distal tubule energy consumption (156); Glycolysis (neonate) → oxidative metabolism (adult) (35) | |
| Response to stress (AKI): Ischemia/reperfusion; metabolic injury; hypoxia; toxins; sepsis | Autophagy, apoptosis → ATG | Fanconi syndrome; “sensor” of stressors in AKI: signals S2 and S3 responses (39); ↑sirtuin and HO-1 (antioxidants) (87); ↑urinary retinol binding protein (34) | ↑TGF-β; apoptosis, necrosis (39) | ↓oxidative metabolism; ↑IF1 (endogenous inhibitor of mitochondrial ATPase) (74) |
| Regenerative response | Tubular progenitor cells seal urinary pole →ATG (48) | Cytoresistance to subsequent insults (194); cells in G1 phase ready to proliferate in response to stress (179); regeneration capacity is limited: AKI → proximal tubule shortening (162) | ||
Nos. in parentheses, reference nos.
AKI, acute kidney injury; ATG, atubular glomeruli; HO-1, heme oxygenase-1; IF1, ATPase inhibitory factor 1; TGF-β, transforming growth factor-β.
Following ischemia-reperfusion injury, KIM-1 is upregulated by proximal tubule cells, which assume a macrophage-like phagocytic phenotype and engulf apoptotic and necrotic cells, favoring tubular repair (81). However, sustained proximal tubular expression of KIM-1 in transgenic mice (in the absence of injury) results in secretion of monocyte chemotactic protein-1, macrophage recruitment, and ultimately leads to interstitial fibrosis (80). In contrast to UUO and ischemia-reperfusion, sepsis-induced AKI is not characterized by tubule cell death, but rather by inflammation and oxidative stress that triggers an adaptive response of proximal tubule cells with downregulation of mitochondrial energy metabolism and cell cycle arrest (Table 2) (65).
Proximal tubule susceptibility to injury increases with maturation.
The phenotypic response of the proximal tubule to injury is molded by the stage of kidney development or aging. Thus it should not be surprising that altered proximal tubular structure and function following obstructive, ischemic, toxic, or metabolic injury during nephrogenesis can differ from those in the mature or the senescent kidney. Either intrinsic renal disease (e.g., PKD, congenital nephrotic syndrome, or nephropathic cystinosis) or reduced nephron number at birth (due to prematurity or intrauterine growth restriction) increases susceptibility to episodes of AKI. The proximal tubule serves as sensor of the insult and as effector of responses for both AKI and CKD (Table 2).
Compared with distal tubules, proximal tubules are more susceptible to ischemic and toxic injury, largely a consequence of their lower capacity for anaerobic glycolytic ATP production and lack of antioxidant and antiapoptotic proteins (Table 2) (63, 89). However, factors secreted by distal tubules can signal proximal tubules through cross talk, thereby enhancing their survival (63). In addition, the proximal tubule is particularly vulnerable to mitochondrial toxicity: the relative activity of ATPase inhibitory factor 1, an endogenous inhibitor of the F1 subunit of mitochondrial ATP synthase, is greater in proximal than distal tubules, and production of reactive oxygen species is greater in proximal tubules (Table 2) (74). In rats 3–6 h following the release of UUO, proximal tubule mitochondria are swollen and vacuolated, and renal ATP production is decreased by 50% (11). Whereas adult proximal tubules have low levels of glycolytic metabolism (7), these levels are fourfold higher in the newborn than the adult rat (Table 2) (35), and mitochondria increase in size and number throughout the first 2 wk of life in the mouse (31). This would account for the relative resistance of the proximal tubule to injury following complete UUO in the newborn mouse: unlike the adult (48, 50), proximal tubule mitochondrial metabolism and glomerulotubular integrity are maintained for 14 days before undergoing mitochondrial loss, cell death, and formation of atubular glomeruli (49).
Energy metabolism.
There may be a threshold of injury that activates the switch between recovery and glomerulotubular disconnection: patients with severe renal artery stenosis develop widespread formation of atubular glomeruli (Table 1) (110). Experimental renal artery stenosis in the rat treated with an angiotensin-converting enzyme inhibitor leads to severe renal ischemia and reduced kidney (proximal tubule) size after 7 wk (73). This is accompanied by a 97% reduction in GFR, atrophic (but not necrotic) tubules with reduced numbers of mitochondria, and 90% reduction in proximal tubule Na-K-ATPase: all of these changes were reversible within 3 wk following removal of the clip and withdrawal of enalapril (72, 73). This study reveals a remarkable adaptation by the proximal tubule to chronic ischemia by downregulation of proximal tubular mass, energy production, and reabsorptive function in concert with decreased GFR, a phenomenon the authors named “renal hibernation.” Complete interruption of renal blood flow, a consequence of renal transplantation, may inflict a more severe tubular insult. Experimental renal transplantation induces mitochondrial uncoupling via uncoupling protein-2, increased kidney oxygen consumption, and decreased oxygen tension before tubular injury is detectable; diabetes results in similar mitochondrial uncoupling (Table 1) (52, 134). These early tubular responses may set the stage for diabetic or chronic allograft nephropathy, which lead to the formation of atubular glomeruli (116, 133, 186).
Urinary tract obstruction also leads to renal ischemia, and partial or complete UUO in the dog reduces renal oxygen consumption by 60%, with a doubling of the respiratory quotient, consistent with a shift to glycolytic metabolism (Table 1) (158). Whereas either partial or complete UUO in weanling rats impairs mitochondrial oxidative metabolism, urinary hippurate and dimethylglycine excretion is increased following partial UUO, but decreased with complete UUO (106). As in renal artery stenosis, downregulation of proximal tubular oxidative metabolism is an adaptive response to UUO, and increased excretion of osmolytes (hippurate and dimethylglycine) may compensate for oxidative stress following partial UUO, a response that is abrogated by the severity of complete UUO (106). Nondiabetic patients with moderate CKD manifest decreased renal oxygen consumption, whereas diabetic patients with equivalent GFR have even lower renal oxygen consumption (94). Interestingly, following renal ablation, oxygen consumption by surviving nephrons more than doubles, but this does not result in oxidative stress unless animals are fed a high-protein diet (Table 1) (121). Taken together, these studies indicate that the proximal tubule can downregulate energy metabolism in response to diffuse chronic renal injury, but upregulate metabolism in intact nephrons in response to renal ablation. In the context of energy conservation, persistent upregulation of oxygen consumption by hypertrophied proximal tubules may not be sustainable, thereby contributing to the progression of CKD or aging.
Ischemic renal injury reduces mitochondrial number in the proximal tubule, which produces 14 μmol ATP/min (distal tubule, 6 μmol/min; remainder of nephron, <1 μmol/min) (156). Proximal tubular ATP sodium transport and gluconeogenesis compete for the same pool of ATP when supply is limited by ischemia (156). Adaptive energy conservation by the proximal tubule has also been demonstrated for AKI resulting from sepsis, which causes hypoxia and oxidative stress (65). In this setting, there is downregulation of mitochondrial metabolism resulting in maintenance of the cell membrane potential and cell cycle arrest at the G1/S checkpoint: this prevents the cell from undertaking a process that would lead to a lethal energy imbalance and cell death (65). Damaged mitochondria are removed by mitophagy, and energy is redirected to essential processes, such as maintenance of ion gradients (65). Thus proximal tubular function is temporarily killed in the benefit of individual cell survival. This is important because even sublethal mitochondrial injury can lead to the generation of reactive oxygen species, inducing irreversible injury and cell death (164). In cultured mouse proximal tubule cells, ATP depletion between 25 and 70% of control results in apoptosis, whereas ATP depletion below 15% leads to necrosis (102). As noted above, mitoquinone, an antioxidant targeted to mitochondria, can significantly delay progressive proximal tubular injury in a mouse model of nephropathic cystinosis (Table 1) (55). The importance of mitochondria in the pathogenesis of kidney disease is underscored by the case of a child with CKD and partial Fanconi syndrome. Renal biopsy revealed tubular atrophy with dysmorphic mitochondria, and focal tubular absence of cytochrome-c oxidase (a respiratory chain enzyme partially encoded by mitochondrial DNA), but preservation of succinate dehydrogenase, which is entirely encoded by nuclear DNA (160).
The kidney constitutes 1% of body weight but utilizes 10% of total body oxygen consumption, primarily devoted to proximal tubule sodium reabsorption (32). As pointed out by Homer Smith in his memorable book From Fish to Philosopher (155), the necessity for reabsorption of 180 liters of glomerular filtrate per day is the result of our evolutionary heritage. Transition from a marine to freshwater environment gave rise to a high filtering glomerulus that was necessary to counteract the osmotic gradient, but adaptation to land and homeothermy required reclamation of 99% of the filtrate, leading to the large energy requirement (69). The formation of atubular glomeruli in CKD may represent an atavism that harks back to a common ancestor with fish that were faced with adaptation from freshwater back to saltwater: aglomerular kidneys evolved independently on three occasions, each separated by about 50 million years (10). The daddy sculpin belongs to one of these groups, and microdissection of kidneys from this fish revealed that, with increasing age, nephrons undergo progressive degeneration of the glomerulotubular junction, resulting in atubular glomeruli and aglomerular tubules (67). It is likely that, with progression of CKD, microenvironments created by proximal tubule responses to injury (e.g., production of cytokines, growth factors, reactive oxygen species) activate this program.
Repair and Regeneration
Homeostasis or failed repair?
Both AKI and CKD are characterized by a heterogeneous population of nephrons: those that undergo regeneration, and those that become atrophied and nonfunctional (86). Current understanding of progression from AKI to CKD is focused on signaling that promotes tubular atrophy, fibroblast proliferation, and collagen deposition, a result of “failed differentiation” of regenerating tubules (99). This has been ascribed to pathological arrest of tubule cells at the G2/M phase in a population of nephrons that is responsible for production of cytokines that stimulate surrounding interstitial inflammatory infiltration and fibroblast proliferation (178). A potential functional role for the proliferation of α-smooth muscle actin-positive myofibroblasts in the interstitium has been demonstrated following complete UUO in the rabbit. On exposure to norepinephrine or angiotensin II, strips of fresh renal cortex from the 8-day obstructed kidney show marked contractility compared with the intact opposite kidney; this response is attenuated in kidneys obstructed for 32 days (115). Peritubular contractile cells would oppose tubular dilation resulting from increased hydrostatic pressure, a response that decreases with decreasing intratubular hydrostatic pressure and increasing collagen deposition in later stages of obstructive nephropathy.
It appears that many differentiated proximal tubule cells are in the G1 phase of the cell cycle and can respond to injury by proliferating and initiating tubular repair (Table 2) (179). Proximal tubule cells that respond to injury by dedifferentiating and proliferating are CD24-positive cells scattered throughout the tubule (153). A novel transgenic mouse strain with timed targeted injury to proximal tubule cells develops massive proliferation of resident proximal tubule cells, but repair is incomplete, resulting in measurable shortening of the proximal tubules associated with interstitial fibrosis (41). This study reveals that there are limits to regeneration and predicts that repeated episodes of proximal tubular injury would result in irreversible CKD, as demonstrated in a separate study, described below (162). Nonetheless, the proximal tubule has developed cytoresistance (inherent in the tubule cells) that increases its resistance to future superimposed episodes of AKI, including uremia-associated compounds that accumulate with renal insufficiency and can exert renoprotective effects (194).
These responses likely result from epigenetic remodeling of proinflammatory and profibrotic genes, such that a “maladaptive” response can develop with an imbalance of cytoprotective defenses (Table 2) (194). Enhanced protein acetylation by inhibition of histone deacetylases (HDAC) impairs epigenetic regulation of recovery from AKI. Inhibition of class I HDAC in mouse models of AKI aggravates renal injury and impairs renal regeneration with suppressed EGF receptor phosphorylation (165). Following ischemia-reperfusion AKI in the mouse, proximal tubule histone acetylation is transiently reduced, accompanied by downregulation of HDAC5 and upregulation of bone morphogenetic protein-7 in the regenerative response (112). Targeted deletion in proximal tubules of dicer, a key enzyme for micro-RNA (miRNA) production, markedly reduces tubule apoptosis and improves renal function and survival following ischemia-reperfusion AKI (183).
The regenerative capacity of the proximal tubule exceeds that of the distal nephron or glomerulus, and there is substantial interest in understanding the biology of renal progenitors (140). Following ischemic AKI, surviving intrinsic tubule epithelial cells are primarily responsible for tubular regeneration (79). Blockade of CXCR4-stromal cell-derived factor-1 reduces a surge in renal stem cells following UUO and impairs parenchyma regeneration after release of obstruction (135). The presence of adult kidney stem cells in the renal papilla may result from a favorable hyperosmolar and hypoxic microenvironment (131). It is tempting to postulate that the destruction of the renal papilla resulting from prolonged complete UUO contributes to irreversible parenchymal damage in this model. There is also growing evidence for the presence of progenitor cells lining Bowman's capsule. Epithelial cells lining the vascular pole of Bowman's capsule have the capacity to differentiate into podocytes, whereas cells lining the urinary pole can differentiate into tubule cells (141). These findings have implications for the process of glomerulotubular disconnection following UUO, and the growth of epithelial cells around the urinary pole of Bowman's capsule, maintaining the integrity of the capsule of the atubular glomerulus (48). The epithelial cells extending across the glomerulotubular junction are flattened and express mesenchymal markers (vimentin and α-smooth muscle actin) (see Fig. 4 in Ref. 48).
Fibrosis as limited repair.
Lineage tracing suggested that proximal tubule cells expressing mesenchymal markers following UUO could undergo “epithelial-mesenchymal transition,” crossing the basement membrane into the interstitium to become transformed into myofibroblasts (83). However, this finding could not be confirmed in subsequent studies, which suggest instead that myofibroblasts derive from resident interstitial fibroblasts or from pericytes (71, 77). While the proximal tubule can clearly promote contiguous interstitial collagen deposition through paracrine signaling (90), migration of transformed epithelial cells remains in doubt. Contrary to prevailing opinion, extracellular matrix deposition around injured tubules has not been proven to be detrimental, but more likely represents a self-limiting repair process that restricts injury, rather than damaging surrounding intact nephrons (86, 178). Notably, peritubular collagen deposition is reversed following recovery from direct proximal tubular injury (162), or following early release of partial ureteral obstruction (150). Treatment of fibrotic pathways with c-ABL tyrosine kinase inhibitors carries the danger of also inhibiting pathways that are renoprotective and contribute to renal repair (172).
The Key Question: Is Proximal Tubular Injury Sufficient to Explain AKI-CKD Progression?
Data reviewed to this point support the temporal primacy of proximal tubular injury, with interstitial fibrosis as a later reparatory response. What is the evidence for proximal tubular injury as the sole initiator of AKI-CKD progression? In vitro studies have demonstrated that human proximal tubule cells exposed to cisplatin can convert fibroblasts to activated myofibroblasts (113). Selective depletion of mouse S3 segment of proximal tubule results in azotemia and albuminuria with polyuria progressing to anuria (147). Histological examination revealed S3 segment tubule cells exfoliated in the tubular lumen (147). Proximal tubule TGF-β is upregulated in AKI and CKD, which contributes to tubular cell injury, cell death, and recruitment of peritubular inflammatory cells with deposition of extracellular matrix (59). Following UUO, proximal tubule TGF-β is selectively upregulated compared with remaining tubule segments (53), and inhibition of TGF-β markedly reduces proximal tubular cell death in the obstructed kidney of adult mice (57). Transgenic mouse overexpression of proximal tubule TGF-β induces proliferation of peritubular cells and deposition of collagen: sustained expression of TGF-β leads to focal degeneration of nephrons, leaving atubular glomeruli (90). These studies show clearly that either direct or secondary injury to proximal tubule cells can recapitulate many of the features of AKI and CKD. Sublethal injury to S1 and S2 proximal tubule segments in transgenic mice results in acute cellular injury followed by inflammatory infiltration and tubular cell proliferation leading to recovery (70). In contrast, three insults at 1-wk intervals leads to interstitial capillary loss, fibrosis, and glomerulosclerosis (70). In a subsequent study using a similar transgenic model, α-smooth muscle actin-positive myofibroblasts emerged around KIM-1-positive injured proximal tubule cells, with formation of glomerulosclerosis and atubular glomeruli (Table 2) (162). When cocultured with fibroblasts, injured tubule cells from the transgenic mice stimulated production of extracellular matrix (162). Based on tissue-specific molecular targeting, these experiments strongly support the conclusion that proximal tubular injury alone can lead to the cascade of events, resulting in peritubular inflammation, interstitial fibrosis, and glomerulosclerosis. The dose-dependent effect of repeated proximal tubular injury also confirms that repeated episodes of AKI result in CKD (70, 162).
New Directions: Proximal Tubular Structure and Function
The anatomy of the kidney is highly complex, and multiple factors are ultimately involved in the progression of kidney disease, including glomerular and microvascular damage, as well as tubular and interstitial damage (145, 146). The clinical challenge is to identify early risk factors for susceptibility to kidney injury, and this review argues for the proximal tubule as a primary source for biomarkers of AKI and CKD. The proximal tubule has multiple functional roles, and impairment of transport functions can result in Fanconi syndrome or selective transport disorders, as well as AKI and CKD (118). In this regard, proximal tubular dysfunction reflected by increased urinary excretion of retinol binding protein is an early predictor of a loss of renal allograft function, preceding structural nephron damage (Table 2) (34). A consensus conference was recently convened to identify promising pathways of kidney repair after AKI and concluded that restoration of structure should be considered in parallel with recovery of function (78). Novel targets in epithelial cells include mitochondrial biogenesis and function, cellular senescence/cell cycle, and miRNAs (3, 78). Deep sequencing in microdissected rat renal tubules has revealed segment-specific transcriptomes that link structure to function (98). This nonbiased approach will allow detection of proteins encoded by proximal tubules at earlier time points and is more specific than markers of inflammation or fibrosis (1, 92). Combining this information with the recently reported generation of kidney organoids from human-induced pluripotent stem cells promises a new era of human disease modeling for screening nephrotoxic drugs (many of which target the proximal tubule) or elucidating monogenic kidney diseases (such as nephropathic cystinosis) (137, 161). Mapping of gene expression in the developing kidney will further enhance interpretation of this data (17). The generation of proximal tubule-like cells from human-induced pluripotent stem cells has been used to evaluate nephrotoxicity of 30 compounds (88). Proximal tubule toxicity in humans could be predicted with 99.8% training accuracy and 87.0% test accuracy and revealed cellular mechanisms of injury were in agreement with animal and human data (88).
As terrestrial homeotherms, mammals evolved energetically expensive nephrons that are highly susceptible to hypoxia and oxidative injury; this “tradeoff” can impair survival and reduce reproductive success. Interpretation of the function of gene regulatory networks will require innovative use of model organisms to understand their role from embryonic life through senescence, as well as responses to environmental changes (61). This approach could explain some of the unique adaptations by mammalian kidneys to organismal life cycle and metabolism. Vampire bats and shrews have a very high protein intake, but do not have an increase in GFR commensurate with the urea excretory load: although azotemic, neither species develops hyperfiltration or CKD (152). In contrast, the hibernating bear undergoes continued urea production, but, through unknown metabolic processes, it can reutilize urea nitrogen for protein synthesis (152). The selection of model species should target mechanisms (conserved transcription factor modules and signaling networks) in independent evolutionary lineages (173), and the appropriate ecological context of the organism must be considered when determining a putative adaptation in the laboratory (60, 157). Some miRNAs are encoded in introns in the Hox gene locus, and some are 100% conserved throughout bilaterian evolution. It is likely that targets of miRNAs maintain constraints and trigger evolution: there has been continual addition of lineage-specific miRNA families throughout metazoan evolution (accrual of miRNAs correlates with increased morphological complexity) (91). On the one hand, miR-127 protects tubule cells from ischemic injury, modulating its targets HIF-1α and kinesin family member 3B (78). By contrast, upregulation of proximal tubule miR-21 aggravates ischemic or obstructive injury by silencing PPAR-α-mediated peroxisome metabolism of fatty acids and silencing genes that inhibit reactive oxygen species generation in mitochondria (66). Chronic UUO in the mouse increases mainly distal tubule miR-21, associated with development of interstitial fibrosis, which is attenuated by blocking miR-21 (195). Following ischemia or UUO in miRNA-21 knockout mice, de-repression of redox metabolic pathways and PPAR-α lipid metabolism pathways is associated with decreased injury (21). Notably, although miR-21 engages target genes in interstitial cells in cardiac disease (169), the tubule epithelial cell is the initial target of miR-21 engagement in the kidney, with subsequent activation of interstitial fibrosis (21).
The life course progression of CKD has been emphasized throughout this review and illustrated in mouse models studied from birth to adulthood (obstructive nephropathy, PKD, and nephropathic cystinosis) (55, 56, 150). This takes into account the now well-established effects of fetal and neonatal environmental factors that are determinants of adult health and disease (62). Based on the hypothesis that urine of preterm neonates born before the completion of nephrogenesis would be rich in stem/progenitor cells, urine was collected from 1-day-old infants born at 31–36 wk gestational age. Cells were characterized for gene expression analysis and flow cytometry and immunofluorescence for protein expression analysis. Isolated cells expressed markers of nephron and stromal progenitors and protected cocultured tubule cells from cisplatin-induced apoptosis (4). This previously untapped source of progenitor cells holds significant promise for kidney regenerative therapies.
The Kidney Research National Dialogue, supported by the National Institute of Diabetes and Digestive and Kidney Diseases, has placed particular emphasis on targeting the early stages of CKD (12, 103). The selection of optimal animal models is equally important to study the loss of homeostasis with aging (103). Senescence reflects a gradual cumulative process not modeled by acute severe injury, such as temporary complete interruption of renal blood flow (a model of AKI), or complete UUO (a model of CKD): new models should more closely parallel clinical disorders. Shifting attention from established interstitial fibrotic processes to early responses by the proximal tubule would significantly expand future research initiatives in the study of progressive kidney disease.
GRANTS
The support of National Institutes of Health Center of Excellence in Pediatric Nephrology DK096373, the American Heart Association, and the Cystinosis Research Foundation is gratefully acknowledged.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
R.L.C. conception and design of research; R.L.C. performed experiments; R.L.C. analyzed data; R.L.C. interpreted results of experiments; R.L.C. prepared figures; R.L.C. drafted manuscript; R.L.C. edited and revised manuscript; R.L.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The author thanks long-term collaborators Barbara Thornhill and Michael Forbes, who were instrumental in completing the studies described herein. Ms. Thornhill developed the technically challenging surgical model of reversible partial UUO in the neonatal mouse, and Dr. Forbes combined expertise in immunohistochemistry and semithin tissue sectioning with visual acumen to elucidate the implications of structural changes in the obstructed kidney. Critical review of the manuscript was provided by John Barcia, Jennifer R. Charlton, Michael S. Forbes, R. Ariel Gomez, Victoria F. Norwood, and Frank T. Saulsbury.
REFERENCES
- 1.Adam B, Mengel M. Molecular nephropathology: ready for prime time? Am J Physiol Renal Physiol 309: F185–F188, 2015. [DOI] [PubMed] [Google Scholar]
- 2.Addis T, Oliver J. The Renal Lesion in Bright's Disease. New York: Paul Hoeber, 1931. [Google Scholar]
- 3.Agarwal A, Dong Z, Harris R, Murray P, Parikh SM, Rosner MH, Kellum JA, Ronco C. Cellular and molecular mechanisms of AKI. J Am Soc Nephrol 27: 1288–1299, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arcolino FO, Zia S, Held K, Papadimitriou E, Theunis K, Bussolati B, Raaijmakers A, Allegaert K, Voet T, Deprest J, Vriens J, Toelen J, van den Heuvel L, Levtchenko E. Urine of preterm neonates as a novel source of kidney progenitor cells. J Am Soc Nephrol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arendshorst WJ, Finn WF, Gottschalk CW. Pathogenesis of acute renal failure following temporary renal ischemia in the rat. Circ Res 37: 558–568, 1975. [DOI] [PubMed] [Google Scholar]
- 6.Bagby SP. Diabetic nephropathy and proximal tubule ROS: challenging our glomerulocentricity. Kidney Int 71: 1199–1202, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Bagnasco S, Good D, Balaban R, Burg M. Lactate production in isolated segments of the rat nephron. Am J Physiol Renal Fluid Electrolyte Physiol 248: F522–F526, 1985. [DOI] [PubMed] [Google Scholar]
- 8.Bascands JL, Schanstra JP. Obstructive nephropathy: insights from genetically engineered animals. Kidney Int 68: 925–937, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH, Kellum JA, Ronco C. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol 27: 687–697, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beyenbach KW. Kidneys sans glomeruli. Am J Physiol Renal Physiol 286: F811–F827, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Blondin J, Purkerson ML, Rolf D, Schoolwerth AC, Klahr S. Renal function and metabolism after relief of unilateral ureteral obstruction. Proc Soc Exp Biol Med 150: 71–76, 1975. [DOI] [PubMed] [Google Scholar]
- 12.Bonventre JV, Boulware LE, Dember LM, Freedman BJ, Furth SL, Holzman LB, Ketchum CJ, Little MH, Mehrotra R, Moe SM, Sands JM, Sedor JR, Somlo S, Star RA, Rys-Sikora KE. The kidney research national dialogue: gearing up to move forward. Clin J Am Soc Nephrol 9: 1806–1811, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more of the other? Am J Hypertens 1: 335–347, 1988. [DOI] [PubMed] [Google Scholar]
- 14.Brezniceanu ML, Liu F, Wei CC, Tran S, Sachetelli S, Zhang SL, Guo DF, Filep JG, Ingelfinger JR, Chan JSD. Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney Int 71: 912–923, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Bricker NS, Morrin PAF, Kime SW. The pathologic physiology of chronic Bright's disease. An exposition of the “intact nephron hypothesis”. Am J Med 28: 77–98, 1960. [DOI] [PubMed] [Google Scholar]
- 16.Bright R. Original Papers of Richard Bright on Renal Disease. London: Oxford University Press, 1937,. [Google Scholar]
- 17.Brunskill EW, Aronow BJ, Georgas K, Rumballe B, Valerius MT, Aronow J, Kaimal V, Jegga AG, Grimmond S, McMahon AP, Patterson LT, Little MH, Potter SS. Atlas of gene expression in the developing kidney at microanatomic resolution. Dev Cell 15: 781–791, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cachat F, Lange-Sperandio B, Chang AY, Kiley SC, Thornhill BA, Forbes MS, Chevalier RL. Ureteral obstruction in neonatal mice elicits segment-specific tubular cell responses leading to nephron loss. Kidney Int 63: 564–575, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Casas JP, Chua W, Loukogeorgakis S, Vallance P, Smeeth L, Hingorani AD, MacAllister RJ. Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: systematic review and meta-analysis. Lancet 366: 2026–2033, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Chapman AB, Bost JE, Torres VE, Guay-Woodford L, Bae KT, Landsittel D, Li J, King BF, Martin D, Wetzel LH, Lockhart ME, Harris PC, Moxey-Mims M, Flessner M, Bennett WM, Grantham JJ. Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 7: 479–486, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, Li J, Tran PT, Kaimal V, Huang X, Chang AN, Li S, Kalra A, Grafals M, Portilla D, MacKenna DA, Orkin SH, Duffield JS. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med 4: 121, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chevalier RL. Obstructive nephropathy: towards biomarker discovery and gene therapy. Nat Clin Prac Nephrol 2: 157–168, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Chevalier RL, Charlton JR. The human kidney at birth: structure and function in transition. In: Kidney Development in Renal Pathology, edited by Faa G, Fanos V. New York: Springer, Humana Press, 2014. [Google Scholar]
- 24.Chevalier RL, Forbes MS. Generation and evolution of atubular glomeruli in the progression of renal disorders. J Am Soc Nephrol 19: 197–206, 2008. [DOI] [PubMed] [Google Scholar]
- 25.Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int 75: 1145–1152, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Chevalier RL, Goyal S, Kim A, Chang AY, Landau D, LeRoith D. Renal tubulointerstitial injury from ureteral obstruction in the neonatal rat is attenuated by IGF-1. Kidney Int 57: 882–890, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Chevalier RL, Goyal S, Wolstenholme JT, Thornhill BA. Obstructive nephropathy in the neonatal rat is attenuated by epidermal growth factor. Kidney Int 54: 38–47, 1998. [DOI] [PubMed] [Google Scholar]
- 28.Chevalier RL, Thornhill BA, Forbes MS, Kiley SC. Mechanisms of renal injury and progression of renal disease in congenital obstructive nephropathy. Pediatr Nephrol 25: 687–697, 2010. [DOI] [PubMed] [Google Scholar]
- 29.Cho ME, Kopp JB. Pirfenidone: an anti-fibrotic and cytoprotective agent as therapy for progressive kidney disease. Expert Opin Investig Drugs 19: 275–283, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christensen EI, Verroust PJ. Interstitial fibrosis: tubular hypothesis vs. glomerular hypothesis. Kidney Int 74: 1233–1236, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Clark SL. Cellular differentiation in the kidneys of newborn mice studied with the electron microscope. J Biophys Biochem Cytol 3: 349–362, 1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen JJ. Relationship between energy requirements for Na+ reabsorption and other renal functions. Kidney Int 29: 32–40, 1986. [DOI] [PubMed] [Google Scholar]
- 33.Darmady EM, Offer J, Woodhouse MA. The parameters of the ageing kidney. J Pathol 109: 195–207, 1973. [DOI] [PubMed] [Google Scholar]
- 34.de Matos ACC, Camara NOS, de Oliveira AFF, Franco MF, Moura LAR, Nishida S, Pereira AB, Pacheco-Silva A. Functional and morphologic evaluation of kidney proximal tubuli and correlation with renal allograft prognosis. Transpl Int 23: 493–499, 2010. [DOI] [PubMed] [Google Scholar]
- 35.Dicker SE, Shirley DG. Rates of oxygen consumption and of anaerobic glycolysis in renal cortex and medulla of adult and new-born rats and guinea-pigs. J Physiol 212: 235–243, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DIckson LE, Wagner MC, Sandoval RM, Molitoris BA. The proximal tuble and albuminuria: really! J Am Soc Nephrol 25: 443–453, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol 15: 290–301, 2000. [DOI] [PubMed] [Google Scholar]
- 38.Eddy AA, Lopez-Guisa JM, Okamura DM, Yamaguchi I. Investigating mechanisms of chronic kidney disease in mouse models. Pediatr Nephrol 27: 1233–1247, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El-Achkar TM, Dagher PC. Tubular cross talk in acute kidney injury: a story of sense and sensibility. Am J Physiol Renal Physiol 308: F1317–F1323, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.El-Dahr SS, Gomez RA, Gray MS, Peach MJ, Carey RM, Chevalier RL. In situ localization of renin and its mRNA in neonatal ureteral obstruction. Am J Physiol Renal Fluid Electrolyte Physiol 258: F854–F862, 1990. [DOI] [PubMed] [Google Scholar]
- 41.Endo T, Nakamura J, Sato Y, Asada M, Yamada R, Takase M, Takaori K, Oguchi A, Iguchi T, Higashi AY, Ohbayashi T, Nakamura T, Muso E, Kimura T, Yanagita M. Exploring the origin and limitations of kidney regeneration. J Pathol 236: 251–263, 2015. [DOI] [PubMed] [Google Scholar]
- 42.Farris AB, Adams CD, Brousaides N, Della Pelle PA, Collins AB, Moradi E, Smith RN, Grimm PC, Colvin RB. Morphometric and visual evaluation of fibrosis in renal biopsies. J Am Soc Nephrol 22: 176–186, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feltran Ld Nogueira PCK, Ajzen SA, Verrastro CGY, Pacheco-Silva A. Does graft mass impact on pediatric kidney transplant outcomes? Pediatr Nephrol 29: 297–304, 2014. [DOI] [PubMed] [Google Scholar]
- 44.Fern RJ, Yesko CM, Thornhill BA, Kim HS, Smithies O, Chevalier RL. Reduced angiotensinogen expression attenuates renal interstitial fibrosis in obstructive nephropathy in mice. J Clin Invest 103: 39–46, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fetterman GH, Shuplock NA, Philipp FJ, Gregg HS. The growth and maturation of human glomeruli and proximal convolutions from term to adulthood. Studies by microdissection. Pediatrics 35: 601–619, 1965. [PubMed] [Google Scholar]
- 46.Fettermann GH, Feldman JD. Congenital anomalies of renal tubules in a case of “infantile nephrosis”. Am J Dis Child 100: 319–332, 1960. [DOI] [PubMed] [Google Scholar]
- 47.Finn WF, Chevalier RL. Recovery from postischemic acute renal failure in the rat. Kidney Int 16: 113–123, 1979. [DOI] [PubMed] [Google Scholar]
- 48.Forbes MS, Thornhill BA, Chevalier RL. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: a new look at an old model. Am J Physiol Renal Physiol 301: F110–F117, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Forbes MS, Thornhill BA, Galarreta CI, Minor JJ, Gordon KA, Chevalier RL. Chronic unilateral ureteral obstruction in the neonatal mouse delays maturation of both kidneys and leads to late formation of atubular glomeruli. Am J Physiol Renal Physiol 305: F1736–F1746, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forbes MS, Thornhill BA, Minor JJ, Gordon KA, Galarreta CI, Chevalier RL. Fight-or-flight: murine unilateral ureteral obstruction causes extensive proximal tubular degeneration, collecting duct dilatation, and minimal fibrosis. Am J Physiol Renal Physiol 303: F120–F129, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Forbes MS, Thornhill BA, Park MH, Chevalier RL. Lack of endothelial nitric-oxide synthase leads to progressive focal renal injury. Am J Pathol 170: 87–99, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Friederich M, Fasching A, Hansell P, Nordquist L, Palm F. Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim Biophys Acta 1777: 935–940, 2008. [DOI] [PubMed] [Google Scholar]
- 53.Fukuda K, Yoshitomi K, Yanagida T, Tokumoto M, Hirakata H. Quantification of TGF-β1 mRNA along rat nephron in obstructive nephropathy. Am J Physiol Renal Physiol 281: F513–F521, 2001. [DOI] [PubMed] [Google Scholar]
- 54.Gaide Chevronnay HP, Janssens V, Van Der Smissen P, N′kuli F, Nevo N, Guiot Y, Levtchenko E, Marbaix E, Pierreux CE, Cherqui S, Antignac C, Courtoy PJ. Time course of pathogenic and adaptation mechanisms in cystinotic mouse kidneys. J Am Soc Nephrol 25: 1256–1269, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Galarreta CI, Forbes MS, Thornhill BA, Antignac C, Gubler MC, Nevo N, Murphy MP, Chevalier RL. The swan-neck lesion: proximal tubular adaptation to oxidative stress in nephropathic cystinosis. Am J Physiol Renal Physiol 308: F1155–F1166, 2015. [DOI] [PubMed] [Google Scholar]
- 56.Galarreta CI, Grantham JJ, Forbes MS, Maser RL, Wallace DP, Chevalier RL. Tubular obstruction leads to progressive proximal tubular injury and atubular glomeruli in polycystic kidney disease. Am J Pathol 184: 1957–1966, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galarreta CI, Thornhill BA, Forbes MS, Simpkins LN, Kim DK, Chevalier RL. Transforming growth factor-beta 1 receptor inhibition preserves glomerulotubular integrity during ureteral obstruction in adults but worsens injury in neonatal mice. Am J Physiol Renal Physiol 304: F481–F490, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gandhi M, Olson JL, Meyer TW. Contribution of tubular injury to loss of remnant kidney function. Kidney Int 54: 1157–1165, 1998. [DOI] [PubMed] [Google Scholar]
- 59.Garcia-Sanchez O, Lopez-Hernandez FJ, Lopez-Novoa JM. An integrative view on the role of TGF-beta in the progressive tubular deletion associated with chronic kidney disease. Kidney Int 77: 950–955, 2010. [DOI] [PubMed] [Google Scholar]
- 60.Garland T Jr, Carter PA. Evolutionary physiology. Annu Rev Physiol 56: 579–621, 1994. [DOI] [PubMed] [Google Scholar]
- 61.Gitler AD, Lehmann R. Modeling human disease. Science 337: 269, 2012. [DOI] [PubMed] [Google Scholar]
- 62.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359: 61–73, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gobe GC, Johnson DW. Distal tubular epithelial cells of the kidney: potential support for proximal tubular cell survival after renal injury. Int J Biochem Cell Biol 39: 1551–1561, 2007. [DOI] [PubMed] [Google Scholar]
- 64.Godin N, Liu F, Lau GJ, Brezniceanu ML, Chenier I, Filep JG, Ingelfinger JR, Zhang SL, Chan JSD. Catalase overexpression prevents hypertension and tubular apoptosis in angiotensinogen transgenic mice. Kidney Int 77: 1086–1097, 2010. [DOI] [PubMed] [Google Scholar]
- 65.Gomez H, Ince C, De Backer D, Pikkers P, Payen D, Hotchkiss J, Kellum JA. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 41: 3–11, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gomez IG, Grafals M, Portilla D, Duffield JS. MicroRNAs as potential therapeutic targets in kidney disease. J Formos Med Assoc 112: 237–243, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grafflin AL. Glomerular degeneration in the kidney of the daddy sculpin (Myoxocephalus scorpius). Anat Rec 57: 59–78, 1933. [Google Scholar]
- 68.Grams ME, Chow EKH, Segev DL, Coresh J. Lifetime incidence of CKD Stages 3–5 in the United States. Am J Kidney Dis 62: 245–252, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grantham JJ, Wallace DP. Return of the secretory kidney. Am J Physiol Renal Physiol 282: F1–F9, 2002. [DOI] [PubMed] [Google Scholar]
- 70.Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 82: 172–183, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grgic I, Duffield JS, Humphreys BD. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr Nephrol 27: 183–193, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grone HJ, Helmchen U. Impairment and recovery of the clipped kidney in two kidney, one clip hypertensive rats during and after antihypertensive therapy. Lab Invest 54: 645–655, 1986. [PubMed] [Google Scholar]
- 73.Grone HJ, Warnecke E, Olbricht CJ. Characteristics of renal tubular atrophy in experimental renovascular hypertension: a model of kidney hibernation. Nephron 72: 243–252, 1996. [DOI] [PubMed] [Google Scholar]
- 74.Hall AM, Unwin RJ, Parker N, Duchen MR. Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J Am Soc Nephrol 20: 1293–1302, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Brenner BM. Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol Renal Fluid Electrolyte Physiol 241: F85–F93, 1981. [DOI] [PubMed] [Google Scholar]
- 76.Hsu C, Vittinghoff E, Lin F, Shlipak MG. The incidence of end-stage renal disese is increasing faster than the prevalence of chronic renal insufficiency. Ann Intern Med 141: 95–101, 2004. [DOI] [PubMed] [Google Scholar]
- 77.Humphreys BC, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epthelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Humphreys BD, Cantaluppi V, Portilla D, Singbarti K, Yang L, Rosner MH, Kellum JA, Ronco C; Acute Dialysis Quality Initiative (ADQI) XIII Work Group . Targeting endogenous repair pathways after AKI. J Am Soc Nephrol 27: 990–998, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV. Intrinsic epithlial cells repair the kidney after injury. Cell Stem Cell 2: 284–291, 2008. [DOI] [PubMed] [Google Scholar]
- 80.Humphreys BD, Xu F, Sabbisetti V, Grgic I, Naini SM, Wang N, Chen G, Xiao S, Patel D, Henderson JM, Ichimura T, Mou S, Soeung S, McMahon AP, Kuchroo VK, Bonventre JV. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest 123: 4023–4035, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ichimura T, Asseldonk JPv Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest 118: 1657–1668, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ishani A, Nelson D, Clothier B, Schult T, Nugent S, Greer N, Slinin Y, Ensrud KE. The magnitude of acute serum creatinine increase after cardiac surgery and the risk of chronic kidney disease, progression of kidney disease, and death. Arch Intern Med 171: 226–233, 2011. [DOI] [PubMed] [Google Scholar]
- 83.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110: 341–350, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jang HS, Padanilam BJ. Simultaneous deletion of Bax and Bak is required to prevent apoptosis and intersitital fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 309: F540–F550, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jobst-Schwan T, Knaup KX, Nielsen R, Hackenbeck T, Buettner-Herold M, Lechler P, Kroening S, Goppelt-Struebe M, Schloetzer-Schrehardt U, Furnrohr BG, Voll RE, Amann K, Eckardt KU, Christensen EI, Wiesener MS. Renal uptake of the antiapoptotic protein survivin is mediated by megalin at the apical membrane of the proximal tubule. Am J Physiol Renal Physiol 305: F734–F744, 2013. [DOI] [PubMed] [Google Scholar]
- 86.Kaissling B, LeHir M, Kriz W. Renal epithelial injury and fibrosis. Biochim Biophys Acta 1832: 931–939, 2013. [DOI] [PubMed] [Google Scholar]
- 87.Kalakeche R, Hato T, Rhodes G, Dunn KW, El-Achkar TM, Plotkin Z, Sandoval RM, Dagher PC. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol 22: 1505–1516, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kandasamy K, Chuah JKC, Su R, Huang P, Eng KG, Xiong S, Li Y, Chia CS, Loo LH, Zink D. Prediction of drug-induced nephrotoxicity and injury mechanisms with human induced pluripotent stem cell-derived cells and machine learning methods. Sci Rep 5: 12337, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kiyama S, Yoshioka T, Burr IM, Kon V, Fogo A, Ichikawa I. Strategic locus for the activation of the superoxide dismutase gene in the nephron. Kidney Int 47: 536–546, 1995. [DOI] [PubMed] [Google Scholar]
- 90.Koesters R, Kaissling B, LeHir M, Picard N, Theilig F, Gebhardt R, Glick AB, Hahnel B, Hosser H, Grone HJ, Kriz W. Tubular overexpression of transforming growth factor-beta 1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol 177: 632–643, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kosik KS. MicroRNAs tell an evo-devo story. Nat Rev Neurol 10: 754–759, 2009. [DOI] [PubMed] [Google Scholar]
- 92.Kretzler M, Ju W. A transcriptional map of the renal tubule: linking structure to function. J Am Soc Nephrol 26: 2603–2605, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases–insights from animal models. Kidney Int 67: 404–419, 2005. [DOI] [PubMed] [Google Scholar]
- 94.Kurnik BRC, Weisberg LS, Kurnik PB. Renal and systemic oxygen consumption in patients with normal and abnormal renal function. J Am Soc Nephrol 2: 1617–1626, 1992. [DOI] [PubMed] [Google Scholar]
- 95.Lange-Sperandio B, Cachat F, Thornhill BA, Chevalier RL. Selectins mediate macrophage infiltration in obstructive nephropathy in newborn mice. Kidney Int 61: 516–524, 2002. [DOI] [PubMed] [Google Scholar]
- 96.Lange-Sperandio B, Schimpgen K, Rodenbeck B, Chavakis T, Bierhaus A, Nawroth P, Thornhill B, Schaefer F, Chevalier RL. Distinct roles of Mac-1 and its counter-receptors in neonatal obstructive nephropathy. Kidney Int 69: 81–88, 2006. [DOI] [PubMed] [Google Scholar]
- 97.Larsen CP, Walker PD, Thoene JG. The incidence of atubular glomerluli in nephropathic cystinosis renal biopsies. Mol Genet Metab 101: 417–420, 2010. [DOI] [PubMed] [Google Scholar]
- 98.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron setment-specific transcriptomes. J Am Soc Nephrol 26: 2669–26677, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leung KCW, Tonelli M, James MT. Chronic kidney disease following acute kidney injury–risk and outcomes. Nat Rev Nephrol 9: 77–85, 2013. [DOI] [PubMed] [Google Scholar]
- 100.Li S, Mariappan N, Megyesi J, Shank B, Kannan K, Theus S, Price PM, Duffield JS, Portilla D. Proximal tubule PPAR-α attenuates renal fibrosis and inflammation caused by unilateral ureteral obstruction. Am J Physiol Renal Physiol 305: F618–F627, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li S, Nagothu KK, Desai V, Lee T, Branham W, Moland C, Megyesi JI, Crew MD, Portilla D. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-α in mice confers protection during acute kidney injury. Kidney Int 76: 1049–1062, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lieberthal W, Menza SA, Levine JS. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am J Physiol Renal Physiol 274: F315–F327, 1998. [DOI] [PubMed] [Google Scholar]
- 103.Little MH, Brown D, Humphreys BD, McMahon AP, Miner JH, Sands JM, Weisz OA, Mullins C, Hoshizaki D. Defining biology to understand disease. Clin J Am Soc Nephrol 9: 809–811, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int 69: 213–217, 2006. [DOI] [PubMed] [Google Scholar]
- 105.Luyckx VA, Bertram JF, Brenner BM, Fall C, Hoy WE, Ozanne SE, Vikse BE. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet 382: 273–283, 2013. [DOI] [PubMed] [Google Scholar]
- 106.MacLellan DL, Mataija D, Doucette A, Huang W, Langlois C, Trottier G, Burton IW, Walter JA, Karakach TK. Alterations in urinary metabolites due to unilateral ureteral obstruction in a rodent model. Mol Biosyst 7: 2181–2188, 2011. [DOI] [PubMed] [Google Scholar]
- 107.Mahoney CP, Striker GE. Early development of the renal lesions in infantile cystinosis. Pediatr Nephrol 15: 50–56, 2000. [DOI] [PubMed] [Google Scholar]
- 108.Maluf NSR, Gassman JJ. Kidneys of the killer whale and significance of reniculism. Anat Rec 250: 34–44, 1998. [DOI] [PubMed] [Google Scholar]
- 109.Mamman C, Al Abbas A, Skippen P, Nadel H, Levine D, Collet JP, Matsell DG. Long-term risk of CKD in children surviving episodes of acute kidney injury in the intensive care unit: a prospective cohort study. Am J Kidney Dis 59: 523–530, 2016. [DOI] [PubMed] [Google Scholar]
- 110.Marcussen N. Atubular glomeruli in renal artery stenosis. Lab Invest 65: 558–565, 1991. [PubMed] [Google Scholar]
- 111.Marcussen N. Atubular glomeruli and the structural basis for chronic renal failure. Lab Invest 66: 265–284, 1992. [PubMed] [Google Scholar]
- 112.Marumo T, Hishikawa K, Yoshikawa M, Fujita T. Epigenetic regulation of BMP7 in the regenerative response to ischemia. J Am Soc Nephrol 19: 1311–1320, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Moll S, Ebeling M, Weibel F, Farina A, del Rosario AA, Hoflack JC, Pomposiello S, Prunotto M. Epithelial cells as active player in fibrosis: findings from an in vitro model. PLoS One 8: e56575, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Motoyoshi Y, Matsusaka T, Saito A, Pastan I, Willnow TE, Mizutani S, Ichikawa I. Megalin contributes to the early injury of proximal tubule cells during nonselective proteinuria. Kidney Int 74: 1262–1269, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nagle RB, Evans LW, Reynolds DG. Contractility of renal cortex following complete ureteral obstruction. Proc Soc Exp Biol Med 148: 611–614, 1975. [DOI] [PubMed] [Google Scholar]
- 116.Najafian B, Crosson JT, Kim Y, Mauer M. Glomerulotubular junction abnormalities are associated with proteinuria in type I diabetes. J Am Soc Nephrol 17: S53–S60, 2006. [DOI] [PubMed] [Google Scholar]
- 117.Najafian B, Kim Y, Crosson JT, Mauer M. Atubular glomeruli and glomerulotubular junction abnormalities in diabetic nephropathy. J Am Soc Nephrol 14: 908–917, 2003. [DOI] [PubMed] [Google Scholar]
- 118.Nakhoul N, Batuman V. Role of proximal tubules in the pathogenesis of kidney disease. Contrib Nephrol 169: 37–50, 2011. [DOI] [PubMed] [Google Scholar]
- 119.NAPRTCS. North American Pediatric Renal Trials and Collaborative Studies Annual Report (Online). https://web.emmes.com/study/ped/annlrept/annualrept2014.pdf [April 25, 2016].
- 120.Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest 90: 267–270, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nath KA, Croatt AJ, Hostetter TH. Oxyten consumption and oxidant stress in surviving nephrons. Am J Physiol Renal Fluid Electrolyte Physiol 258: F1354–F1362, 1990. [DOI] [PubMed] [Google Scholar]
- 122.Nesterova G, Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol 28: 51–59, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nevo N, Chol M, Bailleux A, Kalatzis V, Morisset L, Devuyst O, Gubler MC, Antignac C. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant 25: 1059–1066, 2010. [DOI] [PubMed] [Google Scholar]
- 124.Noiri E, Satoh H, Taguchi J, Brodsky WV, Nakao A, Ogawa Y, Nishijima S, Yokomizo T, Tokunaga K, Fujita T. Association of eNOS Glu298Asp polymorphism with end-stage renal disease. Hypertension 40: 535–540, 2002. [DOI] [PubMed] [Google Scholar]
- 125.Norris K, Nissenson AR. Race, gender, and socioeconomic disparities in CKD in the United States. J Am Soc Nephrol 19: 1261–1270, 2008. [DOI] [PubMed] [Google Scholar]
- 126.Norwood VF, Carey RM, Geary KM, Jose PA, Gomez RA, Chevalier RL. Neonatal ureteral obstruction stimulates recruitment of renin- secreting renal cortical cells. Kidney Int 45: 1333–1339, 1994. [DOI] [PubMed] [Google Scholar]
- 127.Nyengaard JR, Bendtsen TF. Glomerular number and size in relation to age, kidney weight, and body surface in normal man. Anat Rec 232: 194–201, 1992. [DOI] [PubMed] [Google Scholar]
- 128.Oliver J. Architecture of the Kidney in Chronic Bright's Disease. New York: Hoeber, 1939, p. 43–57. [Google Scholar]
- 129.Oliver J. Nephrons and Kidneys. New York: Hoeber, 1968. [Google Scholar]
- 130.Oliver J, Lund EM. Plastic studies in abnormal renal architecture. I. The two architectural units in chronic Bright's disease and their possible functional significance. Arch Pathol 15: 755–774, 1933. [Google Scholar]
- 131.Oliver JA, Maarouf O, Cheema FH, Martens TP, Al-Awqati Q. The renal papilla is a niche for adult kidney stem cells. J Clin Invest 114: 795–804, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Oliver JR. Urinary system. In: Problems of Ageing. Biological and Medical Aspects, edited by Cowdry EV. Baltimore, MD: Williams and Wilkins, 1939. [Google Scholar]
- 133.Pagtalunan ME, Oberbauer R, Haas M, Barlan M, Mayer G, Olson JL, Meyer TW. Atubular glomeruli in patients with chronic allograft rejection. Transplantation 61: 1166–1171, 1996. [DOI] [PubMed] [Google Scholar]
- 134.Papazova DA, Friederich-Persson M, Joles JA, Verhaar MC. Renal transplantation induces mitochondrial uncoupling, increased kidney oxygen consumption, and decreased kidney oxygen tension. Am J Physiol Renal Physiol 308: F22–F28, 2015. [DOI] [PubMed] [Google Scholar]
- 135.Park HC, Yasuda K, Ratliff B, Stoessel A, Sharkovska Y, Yamamoto I, Jasmin JF, Bachmann S, Lisanti MP, Chander P, Goligorsky MS. Postobstructive regeneration of kidney is derailed when surge in renal stem cells during course of unilateral ureteral obstruction is halted. Am J Physiol Renal Physiol 298: F357–F364, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Prencipe G, Caiello I, Cherqui S, Whisenant T, Petrini S, Emma F, De Benedetti F. Inflammasome activation by cystine crystals: implications for the pathogenesis of cystinosis. J Am Soc Nephrol 25: 1163–1169, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Quaggin SE. Kindling the kidney. N Engl J Med 374: 281–283, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rheault MN. Nephrotic and nephritic syndrome in the newborn. Clin Perinatol 41: 605–618, 2014. [DOI] [PubMed] [Google Scholar]
- 139.Romagnani P. Toward the identification of a “renopoietic system”. Stem Cells 27: 2247–2253, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Romagnani P, Remuzzi G. Renal progenitors: an evolutionary conserved strategy for kidney regeneration. Nat Rev Nephrol 9: 137–146, 2013. [DOI] [PubMed] [Google Scholar]
- 141.Ronconi E, Sagrinati C, Angelotti ML, Lazzeri E, Mazzinghi B, Ballerini L, Parente E, Becherucci F, Gacci M, Carini M, Maggi E, Serio M, Vannelli GB, Lasagni L, Romagnani S, Romagnani P. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol 20: 322–332, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ruster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol 17: 2985–2991, 2006. [DOI] [PubMed] [Google Scholar]
- 143.Sakai T, Hijikata T. Structural specializations of the basement membrane and their mechanical relevance to the kidney. Contrib Nephrol 95: 34–47, 1991. [DOI] [PubMed] [Google Scholar]
- 144.Salomon R, Tellier AL, Attie-Bitach T, Amiel J, Vekemans M, Lyonnet S, Dureau P, Niaudet P, Gubler MC, Broyer M. PAX2 mutations in oligomeganephronia. Kidney Int 59: 457–462, 2001. [DOI] [PubMed] [Google Scholar]
- 145.Schlondorff DO. Overview of factors contributing to the pathophysiology of progressive renal disease. Kidney Int 74: 860–866, 2008. [DOI] [PubMed] [Google Scholar]
- 146.Schnaper HW, Hubchak SC, Runyan CE, Browne JA, Finer G, Liu X, Hayashida T. A conceptual framework for the molecular pathogenesis of progressive kidney disease. Pediatr Nephrol 25: 2223–2230, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sekine M, Monkawa T, Morizane R, Matsuoka K, Taya C, Akita Y, Joh K, Itoh H, Hayashi M, Kikkawa Y, Kohno K, Suzuki A, Yonekawa H. Selective depletion of mouse kidney proximal straight tubule cells causes acute kidney injury. Transgenic Res 21: 51–62, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Seppi T, Prajczer S, Dorler MM, Eiter O, Hekl D, Nevinny-Stickel M, Skvortsova I, Gstraunthaler G, Lukas P, Lechner J. Sex differences in renal proximal tubular cell homeostasis. J Am Soc Nephrol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA. Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell 6: 719–726, 2004. [DOI] [PubMed] [Google Scholar]
- 150.Sergio M, Galarreta CI, Thornhill BA, Forbes MS, Chevalier RL. The fate of nephrons in congenital obstructive nephropathy: Adult recovery is limited by nephron number. J Urol 194: 1463–1472, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Singer MA. Of mice and men and elephants: metabolic rate sets glomerular filtration rate. Am J Kidney Dis 37: 164–178, 2001. [DOI] [PubMed] [Google Scholar]
- 152.Singer MA. Vampire bat, shrew, and bear: comparative physiology and chronic renal failure. Am J Physiol Regul Integr Comp Physiol 282: R1583–R1592, 2002. [DOI] [PubMed] [Google Scholar]
- 153.Smeets B, Boor P, Dijkman H, Sharma SV, Jirak P, Mooren F, Berger K, Bornemann J, Gelman IH, Floege J, van der Vlag, Wetzels JF, Moeller MJ. Proximal tubular cells contain a phenotypically distinct, scattered cell population involved in tubular regeneration. J Pathol 229: 645–659, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Smith HW. The Kidney. New York: Oxford University Press, 1951. [Google Scholar]
- 155.Smith HW. From Fish to Philosopher. Boston, MA: Little, Brown, 1953. [Google Scholar]
- 156.Soltoff SP. ATP and the regulation of renal cell function. Annu Rev Physiol 48: 9–31, 1986. [DOI] [PubMed] [Google Scholar]
- 157.Sommer RJ. The future of evo-devo: model systems and evolutionary theory. Nat Rev Genet 10: 416–422, 2009. [DOI] [PubMed] [Google Scholar]
- 158.Stecker JF Jr, Vaughan ED Jr, Gillenwater JY. Alteration in renal metabolism occurring in ureteral obstruction in vivo. Surg Gynecol Obstet 133: 846–848, 1971. [PubMed] [Google Scholar]
- 159.Suzuki T, Kimura M, Asano M, Fujigaki Y, Hishida A. Role of atrophic tubules in development of interstitial fibrosis in microembolism-induced renal failure in rat. Am J Pathol 158: 75–85, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Szabolcs MJ, Seigle R, Shanske S, Bonilla E, DiMauro A, D'Agati V. Mitochondrial DNA deletion: a cause of chronic tubulointerstitial nephropathy. Kidney Int 45: 1388–1396, 1994. [DOI] [PubMed] [Google Scholar]
- 161.Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, Parton RG, Wolvetang EJ, Roost MS, Chuva de Sousa Lopes SM, Little MH. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526: 564–568, 2015. [DOI] [PubMed] [Google Scholar]
- 162.Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takese M, Nameta M, Yamamoto T, Economides AN, Kohno K, Haga H, Sharma K, Yanagita M. Severity and frequency of proximal tubule injury determines renal prognosis. J Am Soc Nephrol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tanaka S, Tanaka T, Nangaku M. Hypoxia as a key player in the AKI-to-CKD transition. Am J Physiol Renal Physiol 307: F1187–F1195, 2014. [DOI] [PubMed] [Google Scholar]
- 164.Tang C, Dong Z. Mitochondria in kidney injury: when the power plant fails. J Am Soc Nephrol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tang J, Yan Y, Zhao TC, Gong R, Bayliss G, Yan H, Zhuang S. Class I HDAC activity is required for renal protection and regeneration after acute kidney injury. Am J Physiol Renal Physiol 307: F303–F336, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Theilig F, Kriz W, Jerichow T, Schrade P, Hahnel B, Willnow T, LeHir M, Bachmann S. Abrogation of protein uptake through megalin-deficient proximal tubules does not safeguard against tubulointerstitial injury. J Am Soc Nephrol 18: 1824–1834, 2007. [DOI] [PubMed] [Google Scholar]
- 167.Thornhill BA, Burt LA, Chen C, Forbes MS, Chevalier RL. Variable chronic partial ureteral obstruction in the neonatal rat: A new model of ureteropelvic junction obstruction. Kidney Int 67: 42–52, 2005. [DOI] [PubMed] [Google Scholar]
- 168.Thornhill BA, Forbes MS, Marcinko ES, Chevalier RL. Glomerulotubular disconnection in neonatal mice after relief of partial ureteral obstruction. Kidney Int 72: 1103–1112, 2007. [DOI] [PubMed] [Google Scholar]
- 169.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo S, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JS, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschi T, Martin GR, Bauersachs MJ, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Sci Transl Med 4: 12, 2012. [DOI] [PubMed] [Google Scholar]
- 170.Torres R, Velazquez H, Chang JJ, Levene MJ, Moeckel G, Desir GV, Safirstein R. Three-dimensional morphology by multiphoton microscopy with clearing in a model of cisplatin-induced CKD. J Am Soc Nephrol 27: 1102–1112, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Torres VE, Harris PC. Polycystic kidney disease in 2011: connecting the dots toward a polycystic kidney disease therapy. Nat Rev Nephrol 82: 66–68, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Torres VE, Leof EB. Fibrosis, regeneration, and aging: playing chess with evolution. J Am Soc Nephrol 22: 1391–1402, 2011. [DOI] [PubMed] [Google Scholar]
- 173.Uhl EW, Warner NJ. Mouse models as predictors of human responses: evolutionary medicine. Curr Pathobiol Rep 3: 219–223, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.USRDS. United States Renal Data System. 2015 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States (Online). http://www.usrds.org/adr.aspx [April 25, 2016]. [DOI] [PMC free article] [PubMed]
- 175.Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 300: R1009–R1022, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Vats AN, Costello B, Mauer M. Glomerular structural factors in progression of congenital nephrotic syndrome. Pediatr Nephrol 18: 234–240, 2003. [DOI] [PubMed] [Google Scholar]
- 177.Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumanr P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol 298: F1078–F1094, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol 26: 1765–1776, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Vogetseder A, Picard N, Gaspert A, Walch M, Kaissling B, LeHir M. Proliferation capacity of the renal proximal tubulle involves the bulk of differentiated epithelial cells. Am J Physiol Cell Physiol 294: C22–C28, 2008. [DOI] [PubMed] [Google Scholar]
- 180.Wagner MC, Campos-Bilderback SB, Chowdhury M, Flores B, Lai X, Myslinski J, Pandit S, Sandoval RM, Wean SE, Wei Y, Satlin LM, Wiggins RC, Witzmann FA, Molitoris BA. Proximal tubules have the capacity to regulate uptake of albumin. J Am Soc Nephrol 27: 482–494, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Waikar SS, Betensky RA, Bonventre JV. Creatinine as the gold standard for kidney injury biomarker studies? Nephrol Dial Transplant 24: 3263–3265, 2009. [DOI] [PubMed] [Google Scholar]
- 182.Wang X, Vrtiska TJ, Avula RT, Walters LR, Chakkera HA, Kremers WK, Lerman LO, Rule AD. Age, kidney function, and risk factors associate differently with cortical and medullary volumes of the kidney. Kidney Int 85: 677–685, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wei Q, Bhatt K, He HZ, Mi QS, Haase VH, Dong Z. Targeted deletion of dicer from proximal tubules protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 21: 756–761, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Welling LW, Grantham JJ. Physical properties of isolated perfused renal tubules and tubular basement membranes. J Clin Invest 51: 1063–1075, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wessely O, Cerqueira DM, Tran U, Kumar V, Hassey JM, Romaker D. The bigger the better: determining nephron size in kidney. Pediatr Nephrol 29: 525–530, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.White KE, Marshall SM, Bilous RW. Prevalence of atubular glomeruli in type 2 diabetic patients with nephropathy. Nephrol Dial Transplant 23: 3539–3545, 2008. [DOI] [PubMed] [Google Scholar]
- 187.Woo D. Apoptosis and loss of renal tissue in polycystic kidney diseases. N Engl J Med 333: 18–25, 1995. [DOI] [PubMed] [Google Scholar]
- 188.Wuhl E, van Stralen KJ, Verrina E, Bjerre A, Wanner C, Heaf JG, Zurriaga O, Hoitsma A, Niaudet P, Palsson R, Ravani P, Jager KJ, Schaefer F. Timing and outcome of renal replacement therapy in patients with congenital malformations of the kidney and urinary tract. Clin J Am Soc Nephrol 8: 67–74, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yoo KH, Norwood VF, El-Dahr SS, Yosipiv I, Chevalier RL. Regulation of angiotensin II AT1 and AT2 receptors in neonatal ureteral obstruction. Am J Physiol Regul Integr Comp Physiol 273: R503–R509, 1997. [DOI] [PubMed] [Google Scholar]
- 190.Yoo KH, Thornhill BA, Chevalier RL. Angiotensin stimulates TGF-beta 1 and clusterin in the hydronephrotic neonatal rat kidney. Am J Physiol Regul Integr Comp Physiol 278: R640–R645, 2000. [DOI] [PubMed] [Google Scholar]
- 191.Yoo KH, Thornhill BA, Forbes MS, Chevalier RL. Compensatory renal growth due to neonatal ureteral obstruction: Implications for clinical studies. Pediatr Nephrol 21: 368–375, 2006. [DOI] [PubMed] [Google Scholar]
- 192.Yoo KH, Thornhill BA, Forbes MS, Chevalier RL. Inducible nitric oxide synthase modulates hydronephrosis following partial or complete unilateral ureteral obstruction in the neonatal mouse. Am J Physiol Renal Physiol 298: F62–F71, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yoo KH, Thornhill BA, Forbes MS, Coleman CM, Marcinko ES, Liaw L, Chevalier RL. Osteopontin regulates renal apoptosis and interstitial fibrosis in neonatal chronic unilateral ureteral obstruction. Kidney Int 70: 1735–1741, 2006. [DOI] [PubMed] [Google Scholar]
- 194.Zager RA. ‘Biologic memory’ in response to acute kidney injury: cytoresistance, toll-like receptor hyper-responsiveness ant the onset of progressive renal disease. Nephrol Dial Transplant 28: 1985–1993, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zarjou A, Yang S, Abraham E, Agarwal A, Liu G. Identification of a microRNA signature in renal fibrosis: role of miR-21. Am J Physiol Renal Physiol 301: F793–F801, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zeisberg M, Kalluri R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specivic mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol 304: C216–C225, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]