

Abstract
Epigenetic mechanisms, including DNA methylation and histone acetylation, regulate gene expression in idiopathic pulmonary arterial hypertension (IPAH). These mechanisms can modulate expression of extracellular superoxide dismutase (SOD3 or EC-SOD), a key vascular antioxidant enzyme, and loss of vascular SOD3 worsens outcomes in animal models of pulmonary arterial hypertension. We hypothesized that SOD3 gene expression is decreased in patients with IPAH due to aberrant DNA methylation and/or histone deacetylation. We used lung tissue and pulmonary artery smooth muscle cells (PASMC) from subjects with IPAH at transplantation and from failed donors (FD). Lung SOD3 mRNA expression and activity was decreased in IPAH vs. FD. In contrast, mitochondrial SOD (Mn-SOD or SOD2) protein expression was unchanged and intracellular SOD activity was unchanged. Using bisulfite sequencing in genomic lung or PASMC DNA, we found the methylation status of the SOD3 promoter was similar between FD and IPAH. Furthermore, treatment with 5-aza-2′-deoxycytidine did not increase PASMC SOD3 mRNA, suggesting DNA methylation was not responsible for PASMC SOD3 expression. Though total histone deacetylase (HDAC) activity, histone acetyltransferase (HAT) activity, acetylated histones, and acetylated SP1 were similar between IPAH and FD, treatment with two selective class I HDAC inhibitors increased SOD3 only in IPAH PASMC. Class I HDAC3 siRNA also increased SOD3 expression. Trichostatin A, a pan-HDAC inhibitor, decreased proliferation in IPAH, but not in FD PASMC. These data indicate that histone deacetylation, specifically via class I HDAC3, decreases SOD3 expression in PASMC and HDAC inhibitors may protect IPAH in part by increasing PASMC SOD3 expression.
Keywords: extracellular superoxide dismutase, idiopathic pulmonary arterial hypertension, DNA methylation, histone deacetylation
extracellular superoxide dismutase (EC-SOD or SOD3) has a prominent role in the protection of the pulmonary circulation against oxidative stress. SOD3 is the extracellular isoform of the superoxide dismutases, an important family of antioxidant enzymes that catalyze the rapid dismutation of superoxide (O2·−) to hydrogen peroxide (H2O2). SOD3 is the most abundant isoform in the vasculature, accounting for 60–70% of total SOD activity (13, 32, 35), and the loss of SOD3 in multiple animal models of lung or vascular injury, including pulmonary hypertension, increases disease severity (5, 11, 12, 19, 28, 31, 36, 47). One clinical study has reported diminished SOD3 protein in the bronchus of patients with idiopathic pulmonary arterial hypertension (IPAH) (28).
The mechanisms responsible for SOD3 expression and activity include posttranslational modifications, genetic polymorphisms, and epigenetic regulation. Posttranslational events include proteolytic cleavage of the COOH-terminal heparin binding domain with loss of binding to the extracellular matrix, altered protein folding, and disulfide bond formation (37, 39). Genetic studies show that polymorphisms in the promoter region or heparin binding domain of SOD3 impact disease outcome for diabetic vasculopathy, ischemic heart disease, and chronic obstructive pulmonary disease (5, 11, 18, 41, 43). There is now strong evidence that epigenetic mechanisms, in particular DNA methylation of the SOD3 promoter and histone deacetylation with change in histone occupancy of the promoter, can also regulate SOD3 expression (20, 25, 44, 46, 55, 57). These data provide a strong rationale to further evaluate the changes in SOD3 expression and study the epigenetic regulation of this important antioxidant enzyme in patients with IPAH.
Epigenetic mechanisms are inheritable factors that regulate genetic expression without changing the DNA sequence; they include DNA methylation, histone modification, and small regulatory RNAs. DNA methylation involves modification of cytosine nucleotides within the promoter region, specifically cytosines adjacent to guanosine (CpG islands). Regulation of histone acetylation/deacetylation influences chromatin structure and the access to transcriptional machinery. The histone acetyltransferases (HATs) and histone deacetylases (HDACs) work in concert to tightly regulate this process. Their activity, critical for normal cellular homeostasis, can be disrupted in disease states, leading to pathological gene expression. Epigenetic mechanisms contribute to the pathogenesis of diseases such as cancer and atherosclerosis, which have features in common with IPAH including proliferation, inflammation, and vascular remodeling (40, 42). Furthermore, epigenetic mechanisms have been implicated in the regulation of the mitochondrial isoform of SOD, SOD2 (2). It is therefore important to identify the epigenetic pattern in these cells, which in turn can be used as an epigenetic biomarker for new and improved therapeutic development. To date, there is limited information on the epigenetic regulation of SOD3. We tested the hypothesis that SOD3 gene expression is silenced in IPAH by epigenetic mechanisms and thus enhances pulmonary artery smooth muscle cell (PASMC) proliferation. We utilized lung tissue and PASMC provided by the Pulmonary Hypertension Breakthrough Initiative (PHBI) obtained from subjects with IPAH at the time of lung transplantation and from failed donors (FD) to measure SOD3 gene and protein expression and to test whether decreased DNA methylation and/or increased histone acetylation regulate SOD3 expression.
METHODS
Human lung tissue and PASMC.
All explanted lungs were collected by the PHBI Research Network. RNA and DNA isolated from lung tissue, flash frozen lung tissue, and PASMC were provided as deidentified samples from lung explants not suitable for lung transplantation (FD) and from IPAH patients at the time of lung transplantation. Human PASMC were isolated from distal muscularized small pulmonary arteries. The study was deemed IRB exempt by all institutions involved in the study. The age, sex, and race for the individuals are shown in Table 1 for lung RNA, lung tissue, and PASMC. Control human PASMC for selected experiments were purchased from Lonza.
Table 1.
Age, sex, and race of subjects
| Lung RNA |
Lung Tissue |
PASMC |
||||
|---|---|---|---|---|---|---|
| FD | IPAH | FD | IPAH | FD | IPAH | |
| Total numbers | 14 | 16 | 6 | 6 | 6 | 6 |
| Mean age, yr ± SD | 39.00 ± 16.55 | 37.75 ± 16.20 | 42.00 ± 18.87 | 38.33 ± 15.40 | 45.67 ± 11.22 | 39.17 ± 10.55 |
| Female, n (%) | 5 (38.5) | 12 (75.0) | 1 (16.67) | 2 (33.33) | 6 (100) | 6 (100) |
| White, n (%) | 13 (100.0) | 10 (62.5) | 6 (100.00) | 3 (50.00) | 5 (83.3) | 3 (50) |
| Black, n (%) | 0 (0) | 2 (12.5) | 0 (0) | 1 (16.67) | 0 (0) | 2 (33.3) |
| Hispanic, n (%) | 0 (0) | 2 (12.5) | 0 (0) | 1 (16.67) | 1 (16.7) | 1 (16.7) |
| Asian American, n (%) | 0 (0) | 2 (12.5) | 0 (0) | 1 (16.67) | 0 (0) | 0 (0) |
FD, failed donor; IPAH, idiopathic pulmonary arterial hypertension; PASMC, pulmonary artery smooth muscle cells.
Cell culture growth conditions and treatments.
Human PASMC were maintained in Sm-GM2 (Lonza, Basel, Switzerland) at 37°C, 5% CO2 in a humidified incubator. PASMC were used between passages 5 and 8. To inhibit DNA methyltransferase activity, cells were seeded into six-well plates at 60,000 cells/well and grown to 80% confluence. Cells were then treated on 4 consecutive days with 5-aza-2′-deoxycytidine (5-aza-dC) (1 μM) (Sigma-Aldrich, St. Louis, MO) or fresh medium and harvested on day 5. To inhibit HDAC activity, cells were treated with the general HDAC inhibitor trichostatin A (TSA) (200 nM) (Sigma-Aldrich), selective class I HDAC 1, 2, and 3 inhibitor mocetinostat (MGCD0103) (1 μM) (Selleck Chemicals), class I HDAC 1 and 3 inhibitor entinostat (MS275) (1 μM) (Selleck Chemicals), class I HDAC 1 and 2 inhibitor biaryl-60 (BA-60) (1 μM), or class IIb inhibitor tubastatin A (TubA) (1 μM). Each HDAC inhibitor was dissolved in dimethyl sulfoxide (DMSO) (Fisher Scientific, Waltham, MA) and diluted 1:1,000 in fresh medium. The cells were treated with medium containing HDAC inhibitors or DMSO alone and harvested 24 h posttreatment. Lonza PASMC were transfected with Silencer Select siRNAs (Life Technologies, Carlsbad, CA) targeting class I HDAC1 (s73), HDAC2 (s6495), HDAC3 (s16877), combinations of the three HDAC siRNAs, or a Silencer Select Negative Control 1 siRNA using Lipofectamine RNAiMAX transfection reagent (Life Technologies) according to manufacturer's instructions and harvested at 48, 72, or 96 h. A second siRNA molecule against class I HDAC3 (s16876) was also tested to confirm results.
Cell proliferation.
Cell growth in PASMC from FD and IPAH subjects was determined after treatment with 200 nM TSA by two methods, manual cell counts and doubling time. For cell counts, 25,000 cells were seeded in six-well plates and allowed to adhere for 24 h. Cells were treated with either TSA or DMSO on day 0. The medium was changed to fresh medium without inhibitors on day 1. Cell counts were performed in triplicate on days 2 and 4 with a hemocytometer. Doubling time was measured with the xCELLigence Real-Time Cell Analyzer (ACEA Biosciences, San Diego, CA). Cells were treated as described above with either DMSO or TSA. Cells were plated at 1,000 cells per well in fresh medium into an E-plate 16 (ACEA Biosciences) 24 h after treatment. Growth was measured in real time over a 48-h period and cell doubling time was determined and plotted (48).
Quantitative real-time PCR.
RNA was isolated from lung and PASMC using RNeasy Kits (Qiagen, Venlo, Limburg), and cDNA was synthesized using Maxima First Strand cDNA Synthesis Kits (Thermo Scientific, Waltham, MA) or iScript (Bio-Rad, Hercules, CA) cDNA Synthesis Kits. RT-qPCR was performed on an Applied Biosystems 7300 Real-Time PCR, StepOnePlus Real-Time PCR or a QuantStudio 6 Real-Time PCR machine (Applied Biosystems, Carlsbad, CA) using TaqMan Universal PCR Master Mix or TaqMan Fast Advanced Master Mix (Life Technologies) and TaqMan Gene Expression Assays (Life Technologies) designed for human SOD3 (Hs00984230_m1), HDAC1 (Hs02621185_s1), HDAC2 (Hs00231032_m1), HDAC3 (Hs00187320_m1), SP1 (Hs00916521), and housekeeping gene β2 microglobin (β2M) (Hs00162090_m1) and rat Sod3 (Rn00563570_m1) and housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (Gapdh) (Rn00563570_m1).
Protein preparation.
Cells and tissue were homogenized in 300 mM NaCl, 0.5% Triton X-100 in phosphate-buffered saline with the addition of Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific). Histone extractions were performed following Abcam's histone extraction protocol. Nuclear proteins were extracted with the EpiQuick Nuclear Extraction Kit (Epigentek, Farmingdale, NY) with the addition of 10 mM sodium butyrate (Sigma-Aldrich) to the lysis buffer. Immunoprecipitation was performed with 125 μg of nuclear extracts and 0.5 μg rabbit polyclonal SP1 (EMD Millipore, Billerica, MA) using the Universal Magnetic Co-IP Kit (Active Motif, Carlsbad, CA). Protein concentration was assayed with Pierce 660 nm Protein Assay Reagent (Thermo Scientific).
Western blot.
We separated 15–20 μg of total protein, 5 μg of histone extracts, or the immunoprecipitated protein by gel electrophoresis using Criterion XT 4–12% Bis-Tris gels (Bio-Rad) with MES SDS running buffer (Life Technologies). Proteins were transferred to polyvinylidene fluoride membranes (Bio-Rad) with NuPAGE transfer buffer using a Novex Semi-Dry Blotter (Life Technologies). Membranes were activated in methanol and blocked in 5% nonfat dry milk in Tris-buffered saline containing 0.05% Tween20 (TBST) for 1 h. Membranes were incubated in the following primary antibodies prepared at 1:1,000 in 5% milk in TBST at 4°C, overnight unless otherwise noted: rabbit polyclonal SOD3, rabbit polyclonal SOD2 (EMD Millipore, Billerica, MA), rabbit polyclonal Calnexin (H-70) (Santa Cruz Biotechnology, Dallas, TX), mouse monoclonal β-actin clone AC-74 (1:10,000 at room temperature for 1 h) (Sigma-Aldrich), rabbit polyclonal histone H3ac (1:1,500) (Active Motif, Carlsbad, CA), rabbit polyclonal histone H4ac (Active Motif), rabbit polyclonal histone H3 (1:10,000) (Active Motif), rabbit polyclonal acetyl-Lysine (Cell Signaling), and rabbit polyclonal SP1 (EMD Millipore) in TBST. The appropriate horseradish peroxidase-conjugated anti-rabbit or mouse secondary antibody (EMD Millipore) was applied at 1:10,000 in TBST for 1 h at room temperature. Detection was accomplished using SuperSignal West Pico or Femto Chemiluminescent substrates (Thermo Scientific). Bands were quantified by densitometry using Image Lab Software (Bio-Rad) or FluorChem HD 9900 Software (ProteinSimple, San Jose, CA).
SOD activity.
Lung tissue was homogenized in SOD assay buffer, containing 50 mM potassium phosphate pH 7.4, 0.3 M potassium bromide, 0.5 mM phenylmethylsulfonylfluoride, and 3 mM diethylenetriaminepentaceic acid. Concanavalin A-Sepharose 4B or Sepharose 4B were equilibrated in 250 mM NaCl in 50 mM HEPES, pH 7.0. Beads were spun and supernatant was removed. Beads were washed in SOD assay buffer and spun, and supernatant was removed. Homogenized tissue was applied to the beads and incubated at 4°C for 30 min. Samples were spun and supernatant was assayed for SOD activity with a SOD assay kit-WST according to instructions (Dojindo, Rockville, MD). The supernatant in the concanavalin A-Sepharose 4B beads reflected intracellular SODs, as SOD3 remained bound to the beads. The supernatant in the Sepharose 4B beads contained total SOD activity. Adequate separation with this protocol was confirmed by Western blot analysis for SOD3 and SOD2 (data not shown). To calculate the SOD activity, several dilutions of the samples were performed to find two dilutions that contained between 0.5 and 2 U/ml SOD activity, which fell within the linear portion of a standard curve. The concentrations were determined by linear regression for the two dilutions and averaged to obtain the final SOD activity level. The activity was expressed as units per milligram protein. Intracellular SOD activity was subtracted from total SOD activity to determine SOD3 activity within each sample.
Bisulfite conversion and sequencing.
Genomic DNA was isolated by use of DNeasy Blood and Tissue Kits (Qiagen). Bisulfite conversion of genomic DNA was performed using EpiTect Bisulfite Kits (Qiagen). The region of interest in the SOD3 promoter, containing the 18 CpG sites, was amplified by using the following primers:
Hs SOD3 NI BS F1: CCATAAACAACCTCACACCCCCATTTTAC.
Hs SOD3 NI BS R2: CCGTATTAATTTTTTAGAGTAGTTAGGGAAAGT.
PCR was performed with EpiMark Hot Start Taq DNA Polymerase (New England Biolabs, Ipswich, MA). Recommended reaction conditions were followed and PCR products were purified using a QiaQuick PCR Purification Kit (Qiagen). The PCR products were inserted into pCR2.1 TOPO vector using a TOPO TA Cloning Kit (Invitrogen, Carlsbad, MA) and transformed using TOP10 cells. Ten to 20 colonies per patient were picked for Miniprep cultures and plasmid DNA was subsequently extracted. DERC Molecular Biology Core (Aurora, CO) sequenced plasmids containing the PCR product using M13F-20 and M13R primers. Sequences were analyzed with CLC Main Workbench software.
HDAC activity.
Class I, IIa, and IIb HDAC activity was measured in lung tissue and PASMC using a previously published protocol (27). Briefly, the HDAC activity was determined by incubating lung or cell extracts with specific synthetic HDAC substrates: class I HDAC substrate (custom synthesis by Genscript, Piscataway, NJ), class IIa HDAC substrate (I-1985) (Bachem, Torrance, CA), class I/IIb substrate (I-1875) (Bachem). The class-specific HDAC substrates are based on ε-N-acylated lysine, derivatized on the carboxyl group with 7-amino-4-methylcoumarin (AMC) (Alfa Aesar). Subsequent to deacylation by HDAC activity, samples were treated with trypsin to release AMC, and the signal was detected as an increase in fluorescence using a BioTek Synergy 2 plate reader, with excitation and emission 360 and 460 nm, respectively, along with a 400-nm dichroic top mirror. Background signals were subtracted from buffer blanks and data were normalized to FD.
HAT activity.
HAT activity was measured in PASMC nuclear extractions using an EpiQuick HAT Activity/Inhibition Assay Kit (Epigentek). Data were expressed as nanograms per minute.
Chronically hypoxic rat model.
RNA was isolated from lung tissue harvested from rats exposed to 3 wk of hypobaric hypoxia from a published study demonstrating protection against chronic hypoxic pulmonary hypertension by daily intraperitoneal injections of the HDAC inhibitor MGCD0103 (10 mg/kg in 50:50 DMSO:PEG-300) (4). The animal experiments were conducted in accordance with the National Institute of Health's “Guide for the Care and Use of Laboratory Animals” and were approved by the University of Colorado Denver Institutional Animal Care and Use Committee.
Statistical analysis.
Data were analyzed by unpaired t-test or one-way or two-way ANOVA followed by multiple comparisons test using Prism software (GraphPad Software, La Jolla, CA). Data are expressed as means ± standard error. Significance was defined as P < 0.05.
RESULTS
Decreased lung and PASMC SOD3 expression and activity in IPAH. Lung SOD3 mRNA expression was significantly decreased in IPAH compared with FD (relative SOD3 expression 1.0 ± 0.09 FD vs. 0.47 ± 0.07 IPAH, P < 0.001, n = 13–14) (Fig. 1A). Lung SOD3 protein expression was variable, tending to decrease in IPAH (1.0 ± 0.40 FD vs. 0.58 ± 0.11 IPAH, n = 6) (Fig. 1B). In these samples, lung SOD3 activity was significantly less in IPAH (SOD3 U/mg protein: 201.7 ± 61.7 FD vs. 42.8 ± 26.2 IPAH, P < 0.05, n = 6) (Fig. 1C). PASMC were also evaluated for SOD3 content at baseline. Both SOD3 mRNA and protein tended to decrease in the untreated IPAH PASMC compared with FD, though the FD values were highly variable and the differences did not reach statistical significance (P > 0.05, n = 6) (Fig. 2, A and B).
Fig. 1.

Decreased lung SOD3 mRNA expression and protein activity in IPAH. A: lung SOD3/β2M mRNA expressed relative to FD (n = 13–16). B: Western blot analysis for lung SOD3 and calnexin, with corresponding densitometry data showing SOD3/calnexin relative to FD (n = 6). C: SOD3 in lung homogenates was separated from the intracellular SODs by using concanavalin A-Sepharose 4B beads to pull down SOD3. SOD3 activity was determined as the difference in activity in supernatant after incubation with concanavalin A-Sepharose 4B (intracellular SOD) or plain Sepharose 4B beads (total SOD) SOD activity was assayed with the SOD assay kit-WST (Dojindo) and expressed as units SOD activity per mg protein (U/mg protein) (n = 6). FD, failed donor; IPAH, idiopathic pulmonary arterial hypertension. *P < 0.05 vs. FD by unpaired t-test.
Fig. 2.

Variable PASMC SOD3 mRNA and protein expression tends to decrease in IPAH. A: PASMC SOD3/β2M mRNA by qPCR expressed relative to FD. B: Western blot for PASMC SOD3 and β-actin, with corresponding densitometry data for SOD3/β-actin relative to FD (n = 6). P > 0.05 vs. FD by unpaired t-test.
No evidence for decreased lung SOD2 expression or activity in IPAH lung. SOD2 expression was evaluated in the same samples used for SOD3 analysis. No change in SOD2 expression was observed (P > 0.05) (Fig. 3A). Furthermore, intracellular SOD activity in the lung, which included SOD1 and SOD2, was similar in IPAH and FD (P > 0.05) (Fig. 3B).
Fig. 3.

No change in lung SOD2 expression or intracellular SOD activity in IPAH. A: Western blot for lung SOD2 and calnexin, with corresponding densitometry data for SOD2/calnexin relative to FD (n = 6). P > 0.05 vs. FD by unpaired t-test. SOD2 expression was tested in the same membrane shown in Fig. 1B. B: intracellular SOD activity in lung expressed as units SOD activity per mg protein (U/mg protein) (n = 6). P > 0.05 vs. FD by unpaired t-test. IC-SOD, intracellular SOD.
No evidence that the low SOD3 gene expression in IPAH is regulated by DNA methylation of the SOD3 promoter. DNA methylation of the SOD3 promoter contributes to low SOD3 mRNA expression in several types of tumors, therefore we examined the lung and PASMC from individuals with IPAH to see whether we could observe a similar increase in DNA methylation of the same 18 CpG sites in the promoter of SOD3. To increase the probability of observing a difference between FD and IPAH, we selected four FD lungs with high SOD3 mRNA expression and four IPAH with low SOD3 mRNA expression. Following bisulfite conversion and DNA sequencing, we found that the overall % methylation of the SOD3 promoter was not different in FD vs. IPAH, with 36.8% methylation in FD and 31.0% in IPAH (P > 0.05) (Fig. 4A). Furthermore, there was no difference at any of the 18 CpG sites between FD and IPAH (P > 0.05), though there was a difference in % methylation at particular CpG sites, ranging from 11 to 65% (P < 0.001) (Fig. 4B). The % methylation of the SOD3 promoter in PASMC was significantly lower than the % methylation observed in the lung samples. Furthermore, in contrast to tumors, there was not an increase in DNA methylation in the IPAH PASMC and, in fact, it was lower than the FD PASMC (7.9 ± 2.0 vs. 2.8 ± 0.6%, P < 0.05) (Fig. 4C). The findings were similar when analyzed for each of the CpG sites, with very low to absent methylation, particularly in IPAH PASMC DNA (P < 0.001) (Fig. 4D). To further evaluate the contribution of methylation on SOD3 expression, cells were treated with 5-aza-dC and data were expressed as change in SOD3 from baseline for each individual. There was no significant increase in SOD3 mRNA expression following treatment with 5-aza-dC and the change from baseline was not different between FD vs. IPAH (Fig. 4E).
Fig. 4.

No evidence that the low SOD3 gene expression in IPAH is regulated by DNA methylation of the SOD3 promoter. Lung genomic DNA was subject to bisulfite conversion and sequencing for the 18 CpG sites in the SOD3 promoter. A: percent methylation in the SOD3 promoter region in FD and IPAH lung (n = 4). P > 0.05 vs. FD by unpaired t-test. B: DNA methylation at each of the 18 CpG sites in the SOD3 promoter in FD and IPAH lung. P > 0.05 between FD and IPAH for each CpG site by 2-way ANOVA. C: percent methylation in the SOD3 promoter region in FD and IPAH PASMC (n = 6); *P < 0.05 vs. FD by unpaired t-test. D: DNA methylation at each of the 18 CpG sites in the SOD3 promoter for FD and IPAH PASMC; n = 6. E: PASMC SOD3/β2M mRNA expression following treatment with DNA methyltransferase inhibitor, 1 μM 5-aza-dC, on days 1–4 with harvesting on day 5 in FD vs. IPAH. Data are expressed as change in SOD3/β2M from baseline for each individual (n = 6). P > 0.05 vs. FD by unpaired t-test.
Blocking class I HDAC activity increased SOD3 mRNA expression and reduced proliferation in IPAH PASMC. We next evaluated whether increased histone deacetylation could contribute to low SOD3 expression in IPAH. We first measured class I, class IIa, and class IIb HDAC activity in lung and PASMC (Fig. 5, A–F). We did not observe a difference in HDAC activity between FD and IPAH lung or PASMC. In PASMC, HAT activity and histone acetylation also did not differ between FD and IPAH (Fig. 5, G–J).
Fig. 5.

No change in HDAC activity, HAT activity, or total histone H3 and H4 acetylation between FD and IPAH. Class-specific HDAC activity was determined by incubating tissue or cell extracts with specific synthetic HDAC substrates against class I, class IIa, or class IIb HDACs. Activity levels were measured in FD and IPAH lung (A, C, E) or PASMC (B, D, F). Data are expressed as the fluorescent signal relative to the FD (n = 14–16 for lung; n = 6 for PASMC). P > 0.05 vs. FD by unpaired t-test. G: HAT activity was measured in nuclear extracts isolated from PASMC and expressed as ng/min (n = 6). P > 0.05 vs. FD by unpaired t-test. H: Western blot analysis for acetylated histone H3 (H3ac), acetylated histone H4 (H4ac), and total histone H3 (H3) in histone extracts from FD and IPAH with corresponding densitometry for H3ac (I) or H4ac (J) expressed relative to total H3 (n = 5–6). P > 0.05 vs. FD by unpaired t-test.
We then tested whether HDAC activity contributed to low EC-SOD mRNA expression in IPAH using a series of specific HDAC inhibitors. We evaluated the SOD3 mRNA expression in PASMC from five different IPAH and FD subjects. One outlier from each group with very high SOD3 content at baseline was excluded from analysis (data not shown). There was an overall significant difference in SOD3 gene expression in IPAH PASMC compared with FD and in response to HDAC inhibitors (P < 0.001) (Fig. 6A). Specifically, IPAH PASMC SOD3 mRNA expression increased following treatment with the selective class I HDAC 1, 2, and 3 inhibitor MGCD0103; or class I HDAC 1 and 3 inhibitor MS275, with no significant response to the class I HDAC 1 and 2 inhibitor BA-60 or class IIb inhibitor TubA. There was also a trend toward an increase in SOD3 expression with the pan-HDAC inhibitor TSA in the IPAH cells (Fig. 6A). Cell proliferation was evaluated by two methods, cell counts and doubling time, following treatment with the pan-HDAC inhibitor, TSA. Overall the IPAH cells showed enhanced growth at baseline and had a more robust response to TSA than FD. By cell counts, we measured a higher number of IPAH cells at 4 days compared with FD. TSA decreased proliferation in IPAH PASMC, with lower cell counts at both 2 and 4 days compared with untreated cells. In FD PASMC, TSA did not significantly decrease proliferation at 2 days but did decrease FD cell counts by 4 days (Fig. 6B). IPAH PASMC also had a shorter doubling time at baseline compared with the FD PASMC, reflecting more rapid growth (26.99 ± 3.29 h in FD vs. 20.82 ± 1.10 h in IPAH). When cells were treated with TSA, only the IPAH PASMC significantly increased the doubling time (43.2 ± 5.4 h) compared with the FD cells (36.9 ± 4.0 h) (P < 0.05) (Fig. 6C).
Fig. 6.

Treatment with HDAC inhibitors increase SOD3 mRNA expression in PASMC and enhanced cell proliferation in IPAH. A: SOD3 mRNA expression following treatment with the following HDAC inhibitors, expressed as SOD3/β2M. Cells were treated for 24 h with the general HDAC inhibitor trichostatin A (TSA) (200 nM); selective class I HDAC 1, 2, and 3 inhibitor MGCD0103 (MGCD) (1 μM); class I HDAC 1, 2, and 3 inhibitor entinostat (MS275) (1 μM); class I HDAC 1 and 2 inhibitor biaryl-60 (BA-60) (1 μM); class IIB HDAC6 inhibitor tubastatin A (TubA) (1 μM); or dimethyl sulfoxide (DMSO) (1:1,000) (n = 5). B: cell counts at 2 days and 4 days after a 24-h treatment with TSA (200 nM) in FD and IPAH PASMC (n = 4–5). Doubling time was measured by using the xCELLigence Real-Time Cell Analyzer (ACEA Biosciences) to provide a real-time measurement of cell proliferation. Cells treated with either TSA (200 nM) or DMSO (1:1,000) were plated 24 h posttreatment in fresh medium on an E-plate 16 (1,000 cells/well) and monitored continuously over a 48-h period (n = 4–5). *P < 0.05 vs. FD DMSO, #P < 0.05 vs. IPAH DMSO by 2-way ANOVA.
Silencing class I HDAC3 increased SOD3 expression in PASMC. To confirm the effect of class I HDAC inhibitors on SOD3 expression, and further define the contribution of specific class I HDACs, we measured SOD3 expression after siRNA knockdown of class I HDAC1, 2, or 3 in human PASMC. In pilot experiments, we observed that class I HDAC1 siRNA decreased HDAC1 protein at 48 and 72 h but increased HDAC2 protein expression, with no change in SOD3 protein (data not shown). Therefore, we tested each siRNA molecule 72 h after transfection individually and in combination to account for potential compensatory responses in HDAC expression. Figure 7, A–C shows the >90% knockdown of class I HDAC1, HDAC2, or HDAC3 mRNA at 72 h with the respective siRNA molecule. SOD3 mRNA expression after class I HDAC knockdown was highly variable and showed a different response and time course each time the experiment was repeated. Overall, we could not demonstrate a consistent or significant change in SOD3 gene expression after siRNA treatments at 48 or 72 h (data not shown). SOD3 protein expression, however, significantly increased 72 h after knockdown of class I HDAC3 siRNA, but not class I HDAC1 or HDAC2 siRNA (Fig. 7D). We pooled the protein expression data into three groups for analysis: 1) siHDAC1 alone; 2) siHDAC2 and siHDAC1&2; and 3) siHDAC3, siHDAC1&3, and siHDAC1,2&3. This approach best demonstrates the increase in SOD3 expression whenever HDAC3 is included in the pool of siRNA (Fig. 7E).
Fig. 7.

siRNA knockdown of class I HDAC3 in PASMC increased SOD3 protein expression. PASMC (Lonza) were transfected with siRNA (Life Technologies) against HDAC1, HDAC2, and/or HDAC3 and combinations of the 3 siRNA molecules. HDAC1-3/β2M mRNA by qPCR expressed relative to siNC (A–C). The data were pooled into 3 groups: 1) HDAC1 alone, 2) HDAC2 and HDAC1&2, and 3) HDAC3, HDAC1&3, and HDAC1,2&3. D: representative Western blot of SOD3 and β-actin for each experimental condition. E: densitometry data for SOD3 expression. Data are expressed as SOD3/β-actin relative to HDAC1. Experiments were repeated at least 3 times; n = 3–4; *P < 0.05 vs. HDAC1 and #P < 0.05 vs. HDAC2 and HDAC1&2 group by 1-way ANOVA. siNC, negative control siRNA.
SP1 acetylation.
SOD3 expression is regulated by the transcription factor SP1, and acetylation of SP1 by HDAC1 or HDAC2 can decrease DNA binding in promoter regions (49, 56). Therefore, we evaluate the nuclear expression of SP1 in PASMC nuclear extracts and immunoprecipitated SP1 to evaluate for lysine acetylation. We observed no difference in total nuclear SP1 expression between FD and IPAH (Fig. 8A). We did detect acetylation in immunoprecipitated SP1 from PASMC, though there was no difference between FD and IPAH (Fig. 8B).
Fig. 8.

Acetylation of SP1 did not differ between FD and IPAH PASMC. A: representative Western blot data of SP1 and total histone 3 expression in nuclear extracts of FD and IPAH PASMC along with densitometry data. B: SP1 was immunoprecipitated from PASMC nuclear extracts and evaluated for protein acetylation. The Western blot and corresponding densitometry are shown for acetylated lysine (Ac-lysine) and SP1 (n = 3). P > 0.05 by unpaired t-test.
Treatment of chronically hypoxic rats with the class I HDAC inhibitor MGCD0103 increased lung Sod3 mRNA expression. To further evaluate the role of histone deacetylation on Sod3 mRNA expression, we examined the lungs of chronically hypoxic rats treated with the HDAC inhibitor MGCD0103. We previously reported that treatment with MGCD0103 protected against chronic hypoxic pulmonary hypertension, including a 30% decrease in pulmonary artery systolic pressures measured by right heart catheterization as well as normalization of the pulmonary artery acceleration time by echocardiography (4). We analyzed lung tissue from these rats for SOD3 mRNA expression. We observed an increase in Sod3 mRNA expression in rats exposed to 3 wk of hypoxia during HDAC inhibitor treatment compared with normoxic or hypoxic rats (P < 0.001) (Fig. 9).
Fig. 9.
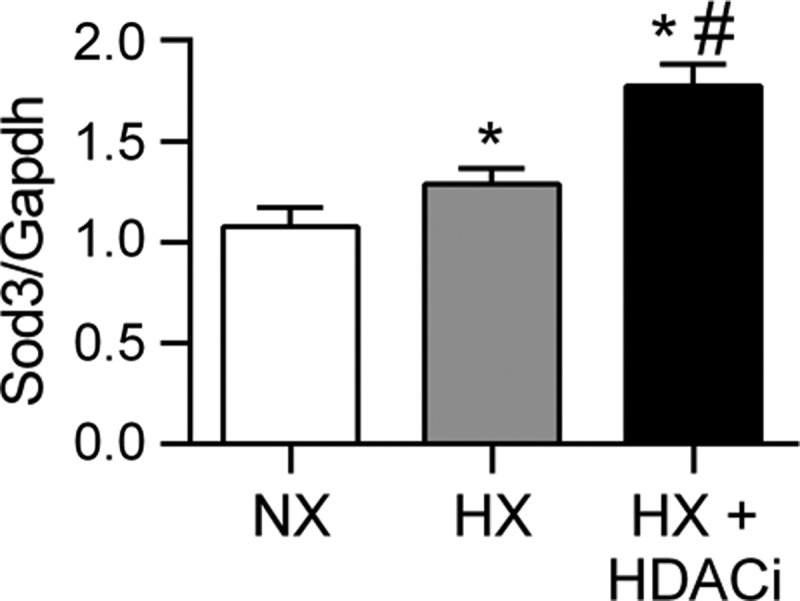
Treatment of chronically hypoxic rats with the HDAC inhibitor MGCD0103 increased lung Sod3 mRNA expression. Sod3 mRNA expression in the lungs of 3-wk chronically hypoxic rats treated daily with the HDAC inhibitor MGCD0103 (10 mg/kg) intraperitoneal injections (HX + HDACi) compared with sham treated normoxic (NX) and hypoxic (HX) rats (2) (n = 6). *P < 0.001 vs. NX, #P < 0.001 vs. HX by 1-way ANOVA.
DISCUSSION
We utilized human lung tissue and PASMC obtained through the PHBI to test whether SOD3 is decreased in IPAH lung due to DNA methylation or histone deacetylation. We report a significant decrease in lung SOD3 gene expression and enzyme activity in lung tissue from individuals with late-stage IPAH at time of lung transplantation compared with lungs from failed donors. Furthermore, SOD3 expression in IPAH PASMC was regulated by class I HDAC3 activity. This conclusion was based on our data showing selective class I HDAC inhibitors with activity against HDAC3 increased SOD3 gene expression and reduced the rate of cell proliferation in IPAH PASMC. Furthermore, siRNA knockdown of class I HDAC3, but not HDAC1 or HDAC2, increased SOD3 gene expression. Finally, SOD3 mRNA expression was increased in the lungs of chronically hypoxic rats treated with MGCD0103, the selective class I HDAC inhibitor with activity against HDAC3. These studies provide new insight in the regulation of SOD3 in the pulmonary circulation and add to the accumulating literature providing a rationale to test the therapeutic role of selective HDAC inhibitors for the treatment of IPAH.
We focused our studies on SOD3 in IPAH because of its function as a major vascular antioxidant enzyme and the published evidence from our group and others implicating a critical role for low SOD3 on disease pathogenesis in animal models of pulmonary hypertension (1, 12, 19, 31, 33, 47, 51). Furthermore, we performed in vitro studies with PASMC because this is the primary cellular source of SOD3 in the vessel wall (33, 57). The observation that SOD3 gene expression is decreased in the lungs of individuals with IPAH is consistent with an earlier report that SOD3 mRNA is decreased in the bronchial tissue of individuals with IPAH (28). We found that the changes in SOD expression and activity were selective for SOD3 isoform. This is in contrast to a previous study that reported that SOD2 gene expression is decreased in the lungs of three individuals who died of IPAH or non-lung-related conditions and was consistent with the loss in SOD2 in the fawn-hooded rat model of pulmonary hypertension (2). One major challenge in the study of PAH in humans is the limited availability of human lung and cell culture samples. Because IPAH specimens are obtained either at lung transplantation, as in our study, or at the time of autopsy, the data reflect the state in end-stage disease. Furthermore, the control population is not uniform; in our study, the lungs provided by the PHBI tissue bank were not accepted for lung transplantation and therefore the reproducibility of the data from control samples may have been confounded by the presence of underlying lung diseases or acute lung injury that could impact SOD expression or activity. We speculate that these factors contributed to the high variability in the outcome measures including SOD3 expression, particularly in FD PASMC. In the face of these limitations, the decrease in lung SOD3 gene expression in IPAH was notable, so we proceeded to evaluate the role of DNA methylation and histone acetylation on the regulation of SOD3.
We first evaluated DNA hypermethylation of the SOD3 promoter because this epigenetic mechanism is responsible for low SOD3 expression in lung, breast, and pancreatic cancers and contributes to enhanced tumor growth, survival, and invasion (30, 34, 44, 45, 55). In addition, DNA methylation of the SOD3 promoter contributes to lower baseline SOD3 expression in normal human PAEC compared with PASMC (57). The study by Zelko et al. (57) reports low levels (<10%) of DNA methylation of the SOD3 promoter in PASMC, similar to what we measured in the FD and IPAH PASMC. The observation that the lung had a higher overall level of DNA methylation of the SOD3 promoter sites compared with PASMC suggests that DNA hypermethylation could regulate low SOD3 in other important cell types in IPAH. The clinical significance of a further decrease in DNA methylation in IPAH from a low baseline level in FD PASMC is unclear. DNA hypermethylation can also lower SOD3 expression in leukocytes in the setting of coronary artery disease, and it regulates the differential expression of SOD3 in different monocyte lines (20, 25). Others have evaluated SOD2 and found that methylation of the SOD2 promoter inhibits SOD2 transcription and contributes to cell proliferation in several cancer lines and in the fawn-hooded rat model of IPAH described above (2, 14, 15, 17). Overall, our results indicate that, in contrast to cancer, low SOD3 expression in IPAH is not regulated by increased DNA methylation. These findings prompted us to consider the contribution of histone deacetylation to low SOD3 expression.
Our data collectively indicate that class I HDAC3 activity regulates SOD3 expression in IPAH PASMC. There is emerging interest in the role of histone deacetylation in the pathogenesis of cardiovascular diseases including human IPAH, and advances in the development of new selective HDAC inhibitors (9, 10, 21, 26, 29, 42, 58). In animal studies, it is clear that HDAC inhibitors may not benefit every model of pulmonary hypertension, and a pan-HDAC inhibitor may even be harmful, both important points necessitating a better understanding of this process in human disease (3, 6). To date, there are few human studies examining histone deacetylation in IPAH, and they are limited to testing HDAC subtype expression, rather than activity (21, 23, 58). Zhao et al. (58), in collaboration with our group, reported an increase in class I HDAC1 and class IIa HDAC4 and HDAC5 protein expression in lung tissue from 12 individuals with IPAH compared with control lobectomy tissue. Interestingly, when Korfei and colleagues (23) examined HDAC expression in lung tissue from individuals with idiopathic pulmonary fibrosis, a lethal lung disease that can be complicated by pulmonary hypertension, they reported an increase in class I HDAC subtypes that included HDAC3, the subtype implicated in this study in SOD3 expression. It is thus possible that different HDAC isoforms are altered in different forms of pulmonary hypertension.
Our observations implicating class I HDAC3 in SOD3 regulation build on a limited but important series of published studies evaluating SOD3 regulation by histone deacetylation. Zelko and Folz (54, 57) were the first investigators to demonstrate that histone deacetylation could regulate SOD3 gene expression in cell lines derived from mouse liver, kidney, and lung fibroblasts, and histone deacetylation contributed to the low expression of SOD3 in PAEC compare to PASMC. They recently also showed histone H3 and H4 acetylation in the SOD3 promoter region in PAEC, further implicating histone acetylation in the normal low PAEC SOD3 expression. They also did not see an increase in SOD3 in commercially available PASMC with two class I and II HDAC inhibitors, scriptaid or HDAC-42, similar to our observation that control FD PASMC did not significantly increase SOD3 expression in response to class I HDAC inhibitors (53). In a published study using neonatal ovine PASMC, class I HDAC inhibition with apicidin increased SOD3 expression, providing further evidence that class I HDACs regulate SOD3 and suggesting that there may be age- or species-dependent variability (52). Though we focused on class I HDACs, because of their recognized role in cardiovascular diseases, there are also a number of recent though discrepant studies examining sirtuins, in particular SIRT1 and SIRT3, in pulmonary hypertension; the role of sirtuins warrants future interrogation (7, 38, 50). To the best of our knowledge, this is the first study to show that class I HDAC3 activity regulates SOD3 expression.
Class I HDACs, including HDAC3, can associate with SP1 and SP3, ubiquitous hypoxia-responsive transcription factors implicated in both repression and activation of genes (16, 20, 22, 24). This has potential relevance in the regulation of SOD3 in IPAH, particularly given the known role of SP1/SP3 in the regulation of SOD3 gene expression and the impact of HDAC inhibitors on SOD3 expression. In addition to modulating HDAC activity, SP1 is also a target of acetylation; acetylation of SP1 decreases its promoter binding affinity and can be reversed by HDAC inhibitors (16, 20, 22, 24, 49). Our data confirm SP1 acetylation in PASMC, indicating that it may be a mechanism involved in SOD3 expression. However, we did not observe SP1 hyperacetylation in IPAH, suggesting that it did not appear to be responsible for the low SOD3 expression in IPAH. The specific mechanism(s) by which HDACs regulate SOD3 in different cell types in IPAH or other forms of pulmonary hypertension will be an important future direction. We propose that selective class I HDAC inhibitors may protect not only by restoring normal PASMC SOD3 expression, but potentially by increasing SOD3 in other cells including PAEC in which expression is repressed by histone acetylation.
Though total HDAC activity, HAT activity, and histone acetylation were similar between FD and IPAH, there are a number of points to consider when interpreting these data. A decrease in specific HDAC isoforms may not be reflected by the measurement of total HDAC activity. Though the substrates are useful to differentiate different classes, they are not specific for the different HDAC subtypes within a class. A similar problem could contribute to the measures of histone acetylation, since site-specific changes in histone acetylation may not be reflected by the total histone measurements. This critical area of inquiry continues to require further investigation.
In conclusion, we provide new evidence that the lung expression and activity of the key vascular antioxidant enzyme, SOD3, is selectively decreased in IPAH. In contrast to a series of cancer studies, we did not find evidence that DNA hypermethylation was responsible for the decrease in SOD3 expression. We provide strong evidence that class I HDAC3 activity contributes to the impaired SOD3 expression and enhanced cell proliferation in IPAH PASMC. Further studies are necessary to establish the mechanisms responsible for cell-specific regulation of SOD3 in different forms of pulmonary hypertension and determine whether selective HDAC inhibitors can improve SOD3 activity and contribute to their therapeutic efficacy to improve outcomes in this devastating disease.
GRANTS
E. Nozik-Grayck was funded by grants from the NIH (R03 HL110783-01, R01 HL110783 and R01 HL086680). K. R. Stenmark was funded by grants from the NIH (1R01HL114887-03 and 5 P01 HL014985-39). B. S. Ferguson was funded by fellowships from the American Heart Association (12POST10680000) and NIH (1F32HL124893-01). T. A. McKinsey was supported by grants from the NIH (R01HL116848, R21AG043822, R01HL127240) and American Heart Association (13GRNT14510001). Human samples were provided by Katherine Sexton in the Tissue Collection and Banking Facility at University of Alabama at Birmingham, Horace DeLisser in the Pulmonary Vascular Cell Core of the University of Pennsylvania, and Mark Geraci in the RNA/DNA core at University of Colorado Denver under the Pulmonary Hypertension Breakthrough Initiative (PHBI). Funding for the PHBI is provided by the Cardiovascular Medical Research and Education Fund (CMREF).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
E.N.-G., S.V., R.P.B., and F.E.D. conception and design of research; E.N.-G., C.W., R.S.S., S.V., B.S.F., and T.A.M. analyzed data; E.N.-G., R.S.S., S.V., B.S.F., K.S., R.P.B., K.I.-S., K.R.S., T.A.M., and F.E.D. interpreted results of experiments; E.N.-G. and C.W. prepared figures; E.N.-G. drafted manuscript; E.N.-G., C.W., R.S.S., S.V., B.S.F., K.S., R.P.B., M.W.G., K.I.-S., K.R.S., T.A.M., and F.E.D. edited and revised manuscript; E.N.-G., C.W., R.S.S., S.V., B.S.F., K.S., R.P.B., M.W.G., K.I.-S., K.R.S., T.A.M., and F.E.D. approved final version of manuscript; C.W., R.S.S., S.V., B.S.F., K.S., and T.A.M. performed experiments.
ACKNOWLEDGMENTS
We thank Ana-Laura Hernandez for excellent technical assistance and Marcia McGowan for assistance with formatting of the manuscript.
REFERENCES
- 1.Ahmed MN, Zhang Y, Codipilly C, Zaghloul N, Patel D, Wolin M, Miller EJ. Extracellular superoxide dismutase overexpression can reverse the course of hypoxia-induced pulmonary hypertension. Mol Med 18: 38–46, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thebaud B, Husain AN, Cipriani N, Rehman J. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121: 2661–2671, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med 183: 1402–1410, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, Weiser-Evans MC, Harral J, Irwin DC, Anwar A, Yeager ME, Li M, Watson PA, Nemenoff RA, Buttrick PM, Stenmark KR, McKinsey TA. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res 110: 739–748, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahl M, Bowler RP, Juul K, Crapo JD, Levy S, Nordestgaard BG. Superoxide dismutase 3 polymorphism associated with reduced lung function in two large populations. Am J Respir Crit Care Med 178: 906–912, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Raaf MA, Hussaini AA, Gomez-Arroyo J, Kraskaukas D, Farkas D, Happe C, Voelkel NF, Bogaard HJ. Histone deacetylase inhibition with trichostatin A does not reverse severe angioproliferative pulmonary hypertension in rats (2013 Grover Conference series). Pulm Circ 4: 237–243, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Onofrio N, Vitiello M, Casale R, Servillo L, Giovane A, Balestrieri ML. Sirtuins in vascular diseases: emerging roles and therapeutic potential. Biochim Biophys Acta 1852: 1311–1322, 2015. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson BS, Harrison BC, Jeong MY, Reid BG, Wempe MF, Wagner FF, Holson EB, McKinsey TA. Signal-dependent repression of DUSP5 by class I HDACs controls nuclear ERK activity and cardiomyocyte hypertrophy. Proc Natl Acad Sci USA 110: 9806–9811, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Findeisen HM, Kahles FK, Bruemmer D. Epigenetic regulation of vascular smooth muscle cell function in atherosclerosis. Curr Atheroscler Rep 15: 319, 2013. [PubMed] [Google Scholar]
- 10.Galletti M, Cantoni S, Zambelli F, Valente S, Palazzini M, Manes A, Pasquinelli G, Mai A, Galie N, Ventura C. Dissecting histone deacetylase role in pulmonary arterial smooth muscle cell proliferation and migration. Biochem Pharmacol 91: 181–190, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Grammer TB, Renner W, Hoffmann MM, Kleber M, Winkelhofer-Roob BM, Boehm BO, Maerz W. SOD3 R231G polymorphism associated with coronary artery disease and myocardial infarction. The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Free Radic Res 43: 677–684, 2009. [DOI] [PubMed] [Google Scholar]
- 12.Hartney JM, Stidham T, Goldstrohm DA, Oberley-Deegan RE, Weaver MR, Valnickova-Hansen Z, Scavenius C, Benninger RK, Leahy KF, Johnson R, Gally F, Kosmider B, Zimmermann AK, Enghild JJ, Nozik-Grayck E, Bowler RP. A common polymorphism in extracellular superoxide dismutase affects cardiopulmonary disease risk by altering protein distribution. Circ Cardiovasc Genet 7: 659–666, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hassoun PM, Mouthon L, Barbera JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, Newman JH, Rabinovitch M, Schermuly R, Stenmark KR, Voelkel NF, Yuan JX, Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 54: S10–S19, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Hitchler MJ, Oberley LW, Domann FE. Epigenetic silencing of SOD2 by histone modifications in human breast cancer cells. Free Radic Biol Med 45: 1573–1580, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hitchler MJ, Wikainapakul K, Yu L, Powers K, Attatippaholkun W, Domann FE. Epigenetic regulation of manganese superoxide dismutase expression in human breast cancer cells. Epigenetics 1: 163–171, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Huang W, Tan D, Wang X, Han S, Tan J, Zhao Y, Lu J, Huang B. Histone deacetylase 3 represses p15(INK4b) and p21(WAF1/cip1) transcription by interacting with Sp1. Biochem Biophys Res Commun 339: 165–171, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Hurt EM, Thomas SB, Peng B, Farrar WL. Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines. Br J Cancer 97: 1116–1123, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Juul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R, Sillesen H, Jensen G, Nordestgaard BG. Genetically reduced antioxidative protection and increased ischemic heart disease risk: The Copenhagen City Heart Study. Circulation 109: 59–65, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Kamezaki F, Tasaki H, Yamashita K, Tsutsui M, Koide S, Nakata S, Tanimoto A, Okazaki M, Sasaguri Y, Adachi T, Otsuji Y. Gene transfer of extracellular superoxide dismutase ameliorates pulmonary hypertension in rats. Am J Respir Crit Care Med 177: 219–226, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Kamiya T, Machiura M, Makino J, Hara H, Hozumi I, Adachi T. Epigenetic regulation of extracellular-superoxide dismutase in human monocytes. Free Radic Biol Med 61: 197–205, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D, Park H, Ju H, McLean DL, Comhair SA, Erzurum SC, Chun HJ. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 131: 190–199, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim Y, Kim K, Park D, Lee E, Lee H, Lee YS, Choe J, Jeoung D. Histone deacetylase 3 mediates allergic skin inflammation by regulating expression of MCP1 protein. J Biol Chem 287: 25844–25859, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korfei M, Skwarna S, Henneke I, MacKenzie B, Klymenko O, Saito S, Ruppert C, von der Beck D, Mahavadi P, Klepetko W, Bellusci S, Crestani B, Pullamsetti SS, Fink L, Seeger W, Kramer OH, Guenther A. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 70: 1022–1032, 2015. [DOI] [PubMed] [Google Scholar]
- 24.Kumar P, Tripathi S, Pandey KN. Histone deacetylase inhibitors modulate the transcriptional regulation of guanylyl cyclase/natriuretic peptide receptor-a gene: interactive roles of modified histones, histone acetyltransferase, p300, and Sp1. J Biol Chem 289: 6991–7002, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lakshmi SV, Naushad SM, Reddy CA, Saumya K, Rao DS, Kotamraju S, Kutala VK. Oxidative stress in coronary artery disease: epigenetic perspective. Mol Cell Biochem 374: 203–211, 2013. [DOI] [PubMed] [Google Scholar]
- 26.Lan B, Hayama E, Kawaguchi N, Furutani Y, Nakanishi T. Therapeutic efficacy of valproic acid in a combined monocrotaline and chronic hypoxia rat model of severe pulmonary hypertension. PLoS One 10: e0117211, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemon DD, Horn TR, Cavasin MA, Jeong MY, Haubold KW, Long CS, Irwin DC, McCune SA, Chung E, Leinwand LA, McKinsey TA. Cardiac HDAC6 catalytic activity is induced in response to chronic hypertension. J Mol Cell Cardiol 51: 41–50, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masri FA, Comhair SA, Dostanic-Larson I, Kaneko FT, Dweik RA, Arroliga AC, Erzurum SC. Deficiency of lung antioxidants in idiopathic pulmonary arterial hypertension. Clin Transl Sci 1: 99–106, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathew OP, Ranganna K, Yatsu FM. Butyrate, an HDAC inhibitor, stimulates interplay between different posttranslational modifications of histone H3 and differently alters G1-specific cell cycle proteins in vascular smooth muscle cells. Biomed Pharmacother 64: 733–740, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naushad SM, Hussain T, Al-Attas OS, Prayaga A, Digumarti RR, Gottumukkala SR, Kutala VK. Molecular insights into the association of obesity with breast cancer risk: relevance to xenobiotic metabolism and CpG island methylation of tumor suppressor genes. Mol Cell Biochem 392: 273–280, 2014. [DOI] [PubMed] [Google Scholar]
- 31.Nozik-Grayck E, Suliman HB, Majka S, Albietz J, Van Rheen Z, Roush K, Stenmark KR. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 295: L422–L430, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nozik-Grayck E, Suliman HB, Piantadosi CA. Extracellular superoxide dismutase. Int J Biochem Cell Biol 37: 2466–2471, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Nozik-Grayck E, Woods C, Taylor JM, Benninger RK, Johnson RD, Villegas LR, Stenmark KR, Harrison DG, Majka SM, Irwin D, Farrow KN. Selective depletion of vascular EC-SOD augments chronic hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307: L868–L876, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Leary BR, Fath MA, Bellizzi AM, Hrabe JE, Button AM, Allen BG, Case AJ, Altekruse S, Wagner BA, Buettner GR, Lynch CF, Hernandez BY, Cozen W, Beardsley RA, Keene J, Henry MD, Domann FE, Spitz DR, Mezhir JJ. Loss of SOD3 (EcSOD) expression promotes an aggressive phenotype in human pancreatic ductal adenocarcinoma. Clin Cancer Res 21: 1741–1751, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oury TD, Day BJ, Crapo JD. Extracellular superoxide dismutase in vessels and airways of humans and baboons. Free Radic Biol Med 20: 957–965, 1996. [DOI] [PubMed] [Google Scholar]
- 36.Oury TD, Day BJ, Crapo JD. Extracellular superoxide dismutase: a regulator of nitric oxide bioavailability. Lab Invest 75: 617–636, 1996. [PubMed] [Google Scholar]
- 37.Oury TD, Schaefer LM, Fattman CL, Choi A, Weck KE, Watkins SC. Depletion of pulmonary EC-SOD after exposure to hyperoxia. Am J Physiol Lung Cell Mol Physiol 283: L777–L784, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Paulin R, Dromparis P, Sutendra G, Gurtu V, Zervopoulos S, Bowers L, Haromy A, Webster L, Provencher S, Bonnet S, Michelakis ED. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab 20: 827–839, 2014. [DOI] [PubMed] [Google Scholar]
- 39.Petersen SV, Oury TD, Valnickova Z, Thogersen IB, Hojrup P, Crapo JD, Enghild JJ. The dual nature of human extracellular superoxide dismutase: one sequence and two structures. Proc Natl Acad Sci USA 100: 13875–13880, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saco TV, Parthasarathy PT, Cho Y, Lockey RF, Kolliputi N. Role of epigenetics in pulmonary hypertension. Am J Physiol Cell Physiol 306: C1101–C1105, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sorheim IC, DeMeo DL, Washko G, Litonjua A, Sparrow D, Bowler R, Bakke P, Pillai SG, Coxson HO, Lomas DA, Silverman EK, Hersh CP. Polymorphisms in the superoxide dismutase-3 gene are associated with emphysema in COPD. COPD 7: 262–268, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stratton MS, McKinsey TA. Acetyl-lysine erasers and readers in the control of pulmonary hypertension and right ventricular hypertrophy. Biochem Cell Biol 93: 149–157, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tamai M, Furuta H, Kawashima H, Doi A, Hamanishi T, Shimomura H, Sakagashira S, Nishi M, Sasaki H, Sanke T, Nanjo K. Extracellular superoxide dismutase gene polymorphism is associated with insulin resistance and the susceptibility to type 2 diabetes. Diabetes Res Clin Pract 71: 140–145, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Teoh ML, Fitzgerald MP, Oberley LW, Domann FE. Overexpression of extracellular superoxide dismutase attenuates heparanase expression and inhibits breast carcinoma cell growth and invasion. Cancer Res 69: 6355–6363, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teoh-Fitzgerald ML, Fitzgerald MP, Jensen TJ, Futscher BW, Domann FE. Genetic and epigenetic inactivation of extracellular superoxide dismutase promotes an invasive phenotype in human lung cancer by disrupting ECM homeostasis. Mol Cancer Res 10: 40–51, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teoh-Fitzgerald ML, Fitzgerald MP, Zhong W, Askeland RW, Domann FE. Epigenetic reprogramming governs EcSOD expression during human mammary epithelial cell differentiation, tumorigenesis and metastasis. Oncogene 33: 358–368, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Rheen Z, Fattman C, Domarski S, Majka S, Klemm D, Stenmark KR, Nozik-Grayck E. Lung extracellular superoxide dismutase overexpression lessens bleomycin-induced pulmonary hypertension and vascular remodeling. Am J Respir Cell Mol Biol 44: 500–508, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venkataraman S, Alimova I, Balakrishnan I, Harris P, Birks DK, Griesinger A, Amani V, Cristiano B, Remke M, Taylor MD, Handler M, Foreman NK, Vibhakar R. Inhibition of BRD4 attenuates tumor cell self-renewal and suppresses stem cell signaling in MYC driven medulloblastoma. Oncotarget 5: 2355–2371, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waby JS, Chirakkal H, Yu C, Griffiths GJ, Benson RS, Bingle CD, Corfe BM. Sp1 acetylation is associated with loss of DNA binding at promoters associated with cell cycle arrest and cell death in a colon cell line. Mol Cancer 9: 275, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waypa GB, Osborne SW, Marks JD, Berkelhamer SK, Kondapalli J, Schumacker PT. Sirtuin 3 deficiency does not augment hypoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol 49: 885–891, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu D, Guo H, Xu X, Lu Z, Fassett J, Hu X, Xu Y, Tang Q, Hu D, Somani A, Geurts AM, Ostertag E, Bache RJ, Weir EK, Chen Y. Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertension 58: 303–309, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Q, Dahl MJ, Albertine KH, Ramchandran R, Sun M, Raj JU. Role of histone deacetylases in regulation of phenotype of ovine newborn pulmonary arterial smooth muscle cells. Cell Prolif 46: 654–664, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zelko IN, Folz RJ. Regulation of oxidative stress in pulmonary artery endothelium. Modulation of extracellular superoxide dismutase and NOX4 expression using histone deacetylase class I inhibitors. Am J Respir Cell Mol Biol 53: 513–524, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zelko IN, Folz RJ. Sp1 and Sp3 transcription factors mediate trichostatin A-induced and basal expression of extracellular superoxide dismutase. Free Radic Biol Med 37: 1256–1271, 2004. [DOI] [PubMed] [Google Scholar]
- 55.Zelko IN, Mueller MR, Folz RJ. CpG methylation attenuates Sp1 and Sp3 binding to the human extracellular superoxide dismutase promoter and regulates its cell-specific expression. Free Radic Biol Med 48: 895–904, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zelko IN, Mueller MR, Folz RJ. Transcription factors sp1 and sp3 regulate expression of human extracellular superoxide dismutase in lung fibroblasts. Am J Respir Cell Mol Biol 39: 243–251, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zelko IN, Stepp MW, Vorst AL, Folz RJ. Histone acetylation regulates the cell-specific and interferon-gamma-inducible expression of extracellular superoxide dismutase in human pulmonary arteries. Am J Respir Cell Mol Biol 45: 953–961, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, Wilkins MR. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 126: 455–467, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]