

Abstract
Agricultural dust exposure results in significant lung inflammation, and individuals working in concentrated animal feeding operations (CAFOs) are at risk for chronic airway inflammatory diseases. Exposure of bronchial epithelial cells to aqueous extracts of hog CAFO dusts (HDE) leads to inflammatory cytokine production that is driven by protein kinase C (PKC) activation. cAMP-dependent protein kinase (PKA)-activating agents can inhibit PKC activation in epithelial cells, leading to reduced inflammatory cytokine production following HDE exposure. β2-Adrenergic receptor agonists (β2-agonists) activate PKA, and we hypothesized that β2-agonists would beneficially impact HDE-induced adverse airway inflammatory consequences. Bronchial epithelial cells were cultured with the short-acting β2-agonist salbutamol or the long-acting β2-agonist salmeterol prior to stimulation with HDE. β2-Agonist treatment significantly increased PKA activation and significantly decreased HDE-stimulated IL-6 and IL-8 production in a concentration- and time-dependent manner. Salbutamol treatment significantly reduced HDE-induced intracellular adhesion molecule-1 expression and neutrophil adhesion to epithelial cells. Using an established intranasal inhalation exposure model, we found that salbutamol pretreatment reduced airway neutrophil influx and IL-6, TNF-α, CXCL1, and CXCL2 release in bronchoalveolar lavage fluid following a one-time exposure to HDE. Likewise, when mice were pretreated daily with salbutamol prior to HDE exposure for 3 wk, HDE-induced neutrophil influx and inflammatory mediator production were also reduced. The severity of HDE-induced lung pathology in mice repetitively exposed to HDE for 3 wk was also decreased with daily salbutamol pretreatment. Together, these results support the need for future clinical investigations to evaluate the utility of β2-agonist therapies in the treatment of airway inflammation associated with CAFO dust exposure.
Keywords: bronchial epithelial cells, β2-agonists, agricultural dusts, concentrated animal feeding operations, lung inflammation
organic dust present in agricultural environments causes significant lung inflammation, and agriculture industry workers experience airway inflammatory diseases associated with these exposures (11, 12, 31). Workers employed in concentrated animal feeding operations (CAFOs) experience a variety of lung diseases, including chronic bronchitis, asthma, and chronic obstructive pulmonary disease (31). Workers initially exposed to CAFOs demonstrate a robust inflammatory response marked by fevers, airway neutrophil influx, and release of proinflammatory mediators, including tumor necrosis factor (TNF)-α, interleukin (IL)-6, and the neutrophil chemoattractant IL-8 (20), observations that have been replicated in animal models (23). However, treatment strategies for these individuals are not well described or effective. To improve treatment strategies for individuals suffering from organic dust-induced lung disease, a better understanding of the biology underlying these exposures is required.
Bronchial epithelial cells (BECs) are a first line of defense against exogenous inflammatory agents in the lung. Several mechanisms are utilized by epithelial cells to respond to potentially injurious environmental stimuli. In addition to orchestrating mucociliary clearance to continually rid the lungs of pathogenic and particulate matter, BECs secrete a variety of proinflammatory cytokines and chemokines that modulate the host immune response (16, 28). Exposure of epithelial cells to aqueous extracts of organic dust derived from hog CAFOs [hog dust extract (HDE)] leads to significant cellular changes, including increased release of inflammatory cytokines/chemokines such as TNF-α, IL-6, and IL-8 (19, 24, 36). These events are propagated by the sequential activation of protein kinase C (PKC) isoforms, whereby early activation of PKCα is required for the release of TNF-α, which stimulates the activation of PKCε and subsequent production of IL-8 (37). Interestingly, PKC and cAMP-dependent protein kinase (PKA) appear to work in opposition: HDE-induced PKC activation leads to reduced epithelial wound-healing capacity and increased IL-8 production, while PKA activation accelerates wound healing and prevents HDE-induced IL-8 release (27, 34).
Based on these findings, we hypothesize that pharmacological activators of PKA would limit the epithelial cell inflammatory response following agricultural dust exposures and, thereby, reduce the severity of dust-induced lung inflammation. β2-Adrenergic receptor agonists (β2-agonists) act on β2-adrenergic receptors, leading to the activation of adenylyl cyclase and the production of cAMP, which activates PKA (29). These compounds are routinely used in the treatment of chronic obstructive pulmonary disease and asthma because of their bronchodilatory actions (26), making them attractive candidates for use in dust-associated inflammatory lung disease. In the studies presented here, we sought to investigate the potential application of β2-agonists in reducing airway inflammatory consequences associated with exposure to agricultural dusts. We treated BECs with the short-acting β2-agonist (SABA) salbutamol or the long-acting β2-agonist (LABA) salmeterol to assess the impact of these compounds on the BEC inflammatory response to HDE. We found that β2-agonist treatment of BECs leads to activation of PKA in vitro and significantly reduces HDE-induced inflammatory cytokine production and intracellular adhesion molecule-1 (ICAM-1) expression. In vivo, mice treated with salbutamol prior to a single HDE exposure exhibit decreased bronchoalveolar lavage fluid (BALF) cytokine levels and reduced inflammatory cell recruitment. After repetitive HDE exposures, mice treated with salbutamol were protected against HDE-induced inflammatory lung pathology. Together, these findings demonstrate a protective role for β2-agonists in reducing airway inflammatory outcomes associated with agricultural dust exposure.
MATERIALS AND METHODS
Reagents.
For cell culture, Laboratory for Human Carcinogenesis (LHC) basal medium was purchased from Life Technologies (Carlsbad, CA) and growth factor supplements, antibiotics, and type I collagen were obtained from Sigma-Aldrich (St. Louis, MO). For enzyme-linked immunosorbent assays (ELISAs), anti-human IL-6 and IL-8 ELISA capture antibodies, streptavidin-horseradish peroxidase, mouse cytokine ELISA kits, and ELISA substrate were obtained from R & D Systems (Minneapolis, MN). ELISA bridge antibodies and phycoerythrin (PE)-conjugated anti-human ICAM-1 were obtained from Biolegend (San Diego, CA), and detection antibodies were purchased from Rockland (Gilbertsville, PA) or Thermo-Fisher (Waltham, MA). All other reagents not specified were purchased from Sigma-Aldrich.
Hog confinement dust extract.
Settled dust collected from hog CAFOs housing 500–1,000 animals was sifted through a coarse 0.25-mm sieve before being stored at −20°C in a desiccator. HDE was prepared as previously described (24). Briefly, saturated aqueous extracts of the dust (100 mg/ml in HBSS) were filter-sterilized (0.22 μm) and stored at −20°C until use. Stock (100% HDE) was diluted to a final concentration of 5% HDE in growth medium for cell culture studies and 12.5% HDE solution in sterile saline for animal model studies. A 5% solution of HDE in culture medium contains 2.2 mg/ml total protein and 40 EU/ml (18 ng/ml) endotoxin (Limulus amebocyte lysate assay, Sigma-Aldrich).
Cell culture.
Immortalized human BECs (BEAS-2B, American Type Culture Collection, Manassas, VA) were used for many of the experiments. Cells were grown on type I collagen-coated dishes and maintained in serum-free medium at 37°C in 5% CO2. For preparation of growth medium, equal volumes of growth factor-supplemented LHC basal medium were mixed with RPMI medium containing 1% penicillin-streptomycin and amphotericin B (LHC-9/RPMI), as previously described (24). For cytokine release and flow cytometry experiments, subconfluent cell monolayers were treated with or without β2-agonists [salmeterol (0.01–10 μM) or salbutamol (0.1–20 μM), 0.5% final DMSO concentration] for 1 h and then exposed to 5% HDE or control medium for 24 h. No cytotoxic effects were detected under these conditions (TOX-7 lactate dehydrogenase release assay, Sigma-Aldrich). Primary human bronchial epithelial (NHBE) cells were used for selected experiments and were isolated as previously described (2). Briefly, normal human lungs that were rejected for transplantation were purchased from the International Institute for the Advancement of Medicine. Cells were isolated from the lumen of sectioned bronchi by gentle overnight protease digestion, washed serially, plated on collagen-coated dishes, and maintained in serum-free medium (BEC growth medium; Lonza, Basel, Switzerland). Cells used for these experiments were passaged fewer than four times and were derived from two different donors.
Cytokine measurements.
For cell culture experiments, cell-free supernates were harvested and either stored at −80°C or assayed immediately using lab-designed immunoassays, as published previously (24). Murine BALF was assessed using commercially available ELISA development antibody sets (Duoset, R & D Systems). Lower limits of detection for sets are as follows: 125 pg/ml for human IL-8, 15 pg/ml for human TNF-α, 60 pg/ml for human IL-6, 15 pg/ml for murine chemokine C-X-C ligand (CXCL) 1, 52 pg/ml for murine CXCL2, 35 pg/ml for murine IL-6, and 25 pg/ml for murine TNF-α. For the detection of IL-6, two IL-6 antibody isolates were utilized due to reagent availability limitations: rabbit anti-human IL-6 polyclonal antibody, IgG fraction (catalog no. P620, Thermo-Fisher), was utilized for data in Figs. 1 and 2B and was found to have less antigen affinity than the no-longer-available rabbit anti-human IL-6 (IgG) polyclonal antibody (catalog no. I2143, Sigma-Aldrich), which was used for experiments depicted in Fig. 2A. All samples were assayed in duplicate or triplicate within an experiment, and experiments were repeated a minimum of three times.
Fig. 1.

Pretreatment of bronchial epithelial cells (BECs) with β2-agonists dose-responsively inhibits hog dust extract (HDE)-induced cytokine release. Cultured immortalized human bronchial epithelial (BEAS-2B; A) and primary human bronchial epithelial (NHBE; B) cells were exposed to various concentrations of the long-acting β2-agonist salmeterol or the short-acting β2-agonist salbutamol for 1 h and then to 5% HDE in the presence or absence of β2-agonist for an additional 24 h. Supernatant medium was analyzed for IL-6 and IL-8 levels by ELISA. Values are means ± SE for 3 (A) or 4 (B) independent experiments. *P < 0.05 vs. HDE alone (by 1-way ANOVA and Tukey's posttest).
Fig. 2.

β2-Agonists significantly attenuate HDE-mediated BEAS-2B cell cytokine release over time. Cultured BEAS-2B cells were treated with salmeterol (A) or salbutamol (B) at 10 μM in the presence or absence of 5% HDE for 2, 6, or 12 h. Supernates were collected for IL-6 and IL-8 measurements by ELISA. Both β2-agonists significantly diminished the dust-induced release of IL-6 by 12 h and IL-8 as early as 6 h following HDE challenge. Values are means ± SE for 3 parallel experiments (12 technical replicates per condition). *P < 0.05 vs. HDE + salmeterol (A) or HDE + salbutamol (B) (by 1-way ANOVA and Tukey's posttest).
PKA activity assay.
BECs grown to 80% confluence on triplicate 60-mm dishes were first treated with salmeterol or salbutamol (10 μM) for 1 h and then stimulated with 5% HDE (in the presence or absence of β2-agonist) for an additional 1 h. PKA activity was determined as previously described (35). Briefly, supernatant media were removed from treated cells, and the cell monolayers were immediately flash-frozen in cell lysis buffer. For measurement of PKA activity, the plates were thawed, and cells were collected, ultrasonically disrupted, and pelleted. The cytosolic fraction was collected, and the pellet was resuspended in cell lysis buffer containing 0.01% Triton X-100 and sonicated again. PKA activity was determined using a previously described method (35). PKA activity was corrected for total protein in the original cell cultures and is expressed as picomoles of phosphate incorporated per minute per milligram of protein.
ICAM-1 expression.
BEAS-2B cells were treated with 10 μM salbutamol for 1 h prior to 5% HDE challenge (in the presence or absence of salbutamol) for an additional 24 h. A prewarmed 2 mM EDTA solution was used to wash and gently remove the cells from the plates. After they were washed, the cells (1 × 106 per condition) were immediately fixed in 1% paraformaldehyde-PBS and stained with a PE-conjugated (mouse) anti-human CD54 antibody or a matched PE-conjugated mouse IgG isotype control antibody. Antibody complexes were cross-linked by fixing a second time, and cells were immediately analyzed. Fluorescein-activated cell-sorting (FACS) analysis was performed on a FACSCalibur dual-laser flow cytometer utilizing BD CellQuest and DeNovo software (Becton-Dickinson, Lincoln Park, NJ). Morphological gating was used to exclude nonviable cells and debris from the analysis, and mean fluorescence intensity was reported for 10,000 gated events for each sample. The ICAM-1 FACS analysis experiment was repeated three times, and the data were pooled.
Neutrophil adhesion.
For the analysis of HDE-mediated neutrophil-BEC adhesion interactions, a modification of the technique reported by Braut-Boucher et al. (6) was employed. Briefly, peripheral blood neutrophils were isolated from healthy volunteers by dextran sedimentation, hypotonic lysis, and Ficoll separation. Isolated neutrophils (15–18 × 106 per experiment) were labeled with the vital dye calcein-AM (10 μg·106 cells−1·ml−1) and incubated for 30 min. Labeled cells (0.5 × 106 per well) were washed three times to remove unbound dye and cocultured with adherent epithelial cell monolayers grown in 24-well cluster plates that had been treated with 10 μM salbutamol, 5% HDE, or salbutamol + 5% HDE for 24 h. Neutrophil-to-BEC ratio was 1.6:1. Neutrophils were allowed to adhere to the epithelial cells for 40 min; then the cocultures were gently washed four times to remove unbound neutrophils. Trypsin-EDTA was used to remove all cells from the plates, and the cells were sonicated in lysing buffer. Lysates were transferred to black 96-well microtiter plates, and fluorescence was measured using a microplate reader (Fluorlite 1000, Dynex Laboratories; excitation at 490 nm and emission at 530 nm). Percent adherence was taken as the ratio of fluorescence intensity for treatment conditions to maximum fluorescence (0.5 × 106 labeled neutrophils). Six replicate wells were recorded for each condition, and experiments were repeated three times. With use of this technique, neutrophils were not exposed to HDE or salbutamol and were handled carefully to avoid activation prior to coculture.
Animals.
Male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility on the University of Nebraska Medical Center campus. At the beginning of the study, mice were 8–10 wk of age and had unrestricted access to standard mouse chow and sterile water. All experimental protocols were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee (protocol no. 10-062-08-EP).
Murine model of HDE exposure.
Animals were first treated with nebulized salbutamol and then with intranasal inhalation of HDE. Mice were treated with 12.5% HDE by intranasal inhalation once (acute, single exposure) or daily for 3 wk (repetitive exposure 1), as described previously (23). For salbutamol delivery, an inhalation chamber (5 liter) containing five conscious mice was fitted with an inner wire cage to ensure that the mice could not escape the nebulized drug by nose-pressing or burrowing. In single-HDE-exposure studies, mice (5 per group) were exposed to aerosolized salbutamol (1 mg/ml in 25 ml at 2 l/min) for 20 min, allowed to rest, and then exposed once to 12.5% HDE (50 μl) by intranasal inhalation under light sedation. For repetitive-exposure experiments, mice were given aerosolized salbutamol and intranasal HDE, as described above, each day for 15 consecutive weekdays. At 5 h following the final HDE exposure, mice were euthanized and tracheas were cannulated for bronchoalveolar lavage (three 1-ml fractions). Cells recovered from the pooled BALF were counted, and slides were prepared for cell differential analysis [Cytopro, ELITech Group (Logan, UT) and protocol HEMA-3 (hematoxylin), Fisher Scientific (Kalamazoo, MI)]. Cytokines were measured in cell-free BALF supernate from the first lavage fraction by ELISA. After lavage, lungs were excised, fixed in 10% formalin solution, and embedded in paraffin for histopathology, as previously described (23). Slides were processed and stained with hematoxylin-eosin and analyzed by a pathologist blinded to treatment conditions for histopathology. A semiquantitative score was assessed for the degree and distribution of inflammatory features reflected in the alveolar compartment, bronchiolar compartments, and lymphoid aggregate development, as previously described (23). The repetitive-exposure study was performed a second time (repetitive exposure 2), and data were pooled for 10 mice per condition.
Statistical analysis.
Values are means ± SE for data pooled from three or more parallel experiments, with the exception of Fig. 3, for which data were pooled from two experiments. All pair-wise comparisons were analyzed by a one-way ANOVA with post hoc analyses or by paired Student's t-test (GraphPad Prism software, San Diego, CA). Statistical significance was accepted when P < 0.05.
Fig. 3.
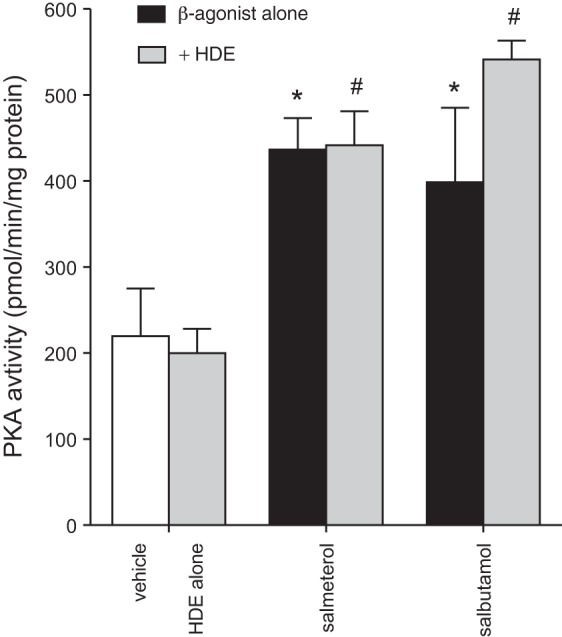
Salbutamol and salmeterol stimulate PKA activity in cultured BEAS-2B cells. Subconfluent BEAS-2B monolayers were pretreated with β2-agonists (10 μM) for 1 h prior to 5% HDE challenge or medium alone for an additional 1 h. Cell lysates were flash-frozen for PKA analysis. Both β2-agonists significantly induced PKA activity in cell lysates. HDE had no effect on PKA activity either alone or in combination with β2-agonist. Values are means ± SE of data pooled from 2 independent experiments. *P < 0.05 vs. vehicle, #P < 0.05 vs. HDE alone (by 1-way ANOVA and Tukey's posttest).
RESULTS
Salmeterol inhibits HDE-induced cytokine release from cultured BECs in a dose- and time-dependent manner.
HDE is a potent stimulus for the release of epithelial cell IL-6 and IL-8 (23, 24). We assessed whether the LABA salmeterol or the SABA salbutamol would suppress this effect. HDE exposure for 24 h increased release of both IL-6 and IL-8 from BEAS-2B cells, and salmeterol dose-dependently inhibited HDE-induced IL-6 and IL-8, reaching significance at 1 and 10 μM (P < 0.05; Fig. 1A). Salbutamol pretreatment also reduced HDE-induced IL-6 and IL-8 production in a similar dose-dependent manner, with significance at salbutamol concentrations of 1 μM for IL-6 and 10 μM for IL-8. In addition, the inhibitory effects of salmeterol and salbutamol pretreatment were demonstrated in primary NHBE cells (Fig. 1B). At 0.1 and 1 μM, salmeterol significantly reduced HDE-induced IL-6 and IL-8 release, respectively, while 10 μM salbutamol significantly blunted HDE-induced IL-6 and IL-8 production in primary cell cultures (P < 0.05; Fig. 1B). On the basis of these findings, salbutamol or salmeterol was used at 10 μM in further experiments for both β2-agonists. Although concentration-dependent responses for salmeterol and salbutamol were similar to the observations in BEAS-2B cells, the HDE-induced IL-6 response was weaker and the β2-agonist-mediated suppression was less robust than the BEAS-2B cell responses.
Next, we determined the time course for the inhibitory β2-agonist effect with HDE-stimulated IL-6 and IL-8 production. As shown in Fig. 2A, HDE-stimulated IL-8 release was significantly attenuated by pretreatment with salmeterol after 6 h of HDE challenge (P < 0.05). Salbutamol pretreatment inhibited IL-6 and IL-8 at 6 h after HDE treatment (Fig. 2B). At 12 h, both β2-agonists significantly decreased HDE-induced IL-6 and IL-8 (P < 0.05). Together, these results indicate that β2-agonists can rapidly inhibit HDE-induced inflammatory cytokine production by BECs.
Both salmeterol and salbutamol stimulate PKA activity in BECs.
When bound to β2-adrenergic receptors, β2-agonists activate adenylyl cyclase, causing a rapid increase in cAMP production, which leads to the activation of PKA (29). We previously showed that activators of PKA can potently inhibit HDE inflammatory responses in epithelial cells (34). We thus sought to determine if the β2-agonists at the doses used in these experiments were able to effectively activate PKA in BECs. Treatment with HDE alone had no effect on baseline PKA activation (Fig. 3). However, salbutamol and salmeterol were effective at increasing baseline levels of PKA activity, and this increased PKA activity remained elevated with HDE treatment (Fig. 3). These results indicate that β2-agonist treatment stimulates PKA activation in BECs. On the basis of results of the preceding experiments showing that salmeterol and salbutamol display very similar patterns of inflammatory modulation, only the SABA salbutamol was used in all subsequent studies.
Salbutamol inhibits HDE-induced BEC-neutrophil interactions.
In addition to stimulating the production of chemotactic factors for neutrophil recruitment to the lungs, HDE induces epithelial cell ICAM-1 expression (15). ICAM-1 expression by BECs facilitates neutrophil-BEC interactions that are known to enhance neutrophil actions (4, 30), and in these studies we sought to investigate whether β2-agonists would reduce HDE-mediated neutrophil adhesion. In control cultures or SABA-only-treated BEC monolayers, <20% of human neutrophils adhered to the BECs (Fig. 4). HDE treatment significantly increased neutrophil adherence, with ∼60% of neutrophils attaching to HDE-stimulated epithelial cells (P < 0.05). Salbutamol pretreatment significantly attenuated HDE induction of neutrophil adherence to treated BECs (P < 0.05; Fig. 4). Again, in these experiments, neutrophils were never directly exposed to HDE or salbutamol. To determine whether these changes in neutrophil-epithelial cell interactions correlated with alterations in salbutamol-regulated ICAM-1 expression, flow cytometry was performed. In epithelial cells treated with 5% HDE, ICAM-1 expression increased significantly compared with control or salbutamol-treated cells (P < 0.05; Fig. 5). Salbutamol pretreatment resulted in a significant decrease in HDE-induced ICAM-1 upregulation (P < 0.05; Fig. 5). Together, these results demonstrate that salbutamol reduces HDE-mediated epithelial cell-neutrophil interactions most likely in response to epithelial cell ICAM-1 expression.
Fig. 4.
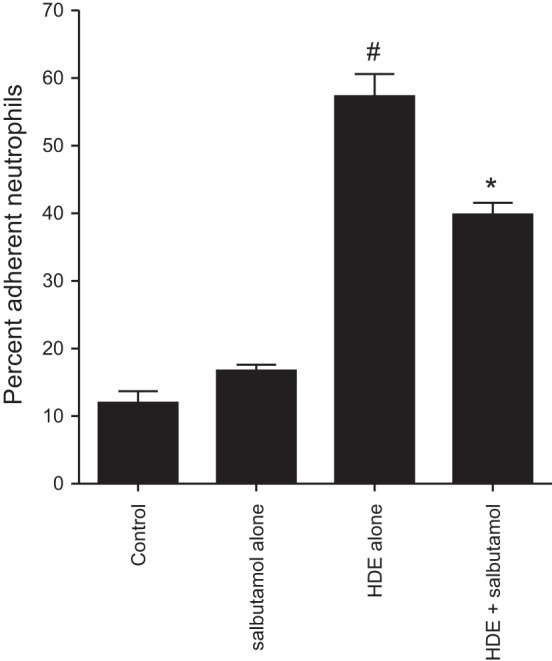
Pretreatment of epithelial cells with the short-acting β2-agonist salbutamol significantly dampens the HDE-induced binding of neutrophils. BEAS-2B monolayers were treated with or without 10 μM salbutamol for 1 h and then with 5% HDE in the presence or absence of salbutamol for 24 h. Freshly isolated human neutrophils (0.5 × 106/well) were labeled with calcein-AM and allowed to attach to the treated epithelial cells for 40 min. Unattached PMNs were removed, and adherent cells were quantified by residual fluorescence as percentage of the initial number of PMNs in the coculture. Values are means ± SE derived from 3 parallel experiments (18 technical replicates per condition). *P < 0.05 vs. HDE alone, #P < 0.05 vs. salbutamol alone (by 1-way ANOVA).
Fig. 5.
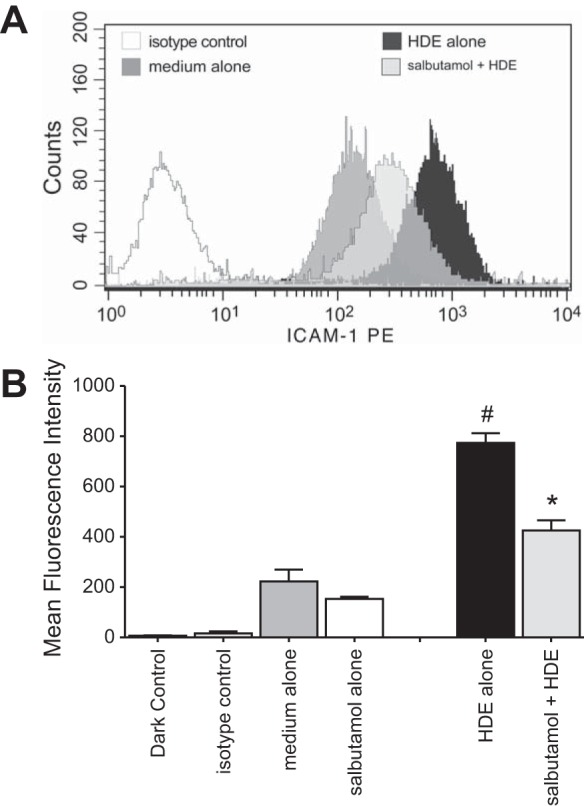
Dust extract-mediated surface expression of ICAM-1 is significantly diminished when BEAS-2B cells are pretreated with salbutamol. Subconfluent BEAS-2B cultures were incubated with 10 μM salbutamol for 1 h prior to 5% HDE challenge for an additional 24 h. Cells were detached from the plates and probed for surface ICAM-1 using a sensitive and specific phycoerythrin-conjugated monoclonal antibody. A total of 10,000 gated events were captured per condition. A: mean fluorescence intensity histograms from 1 representative experiment. B: mean signal intensity derived from pooling results of 3 experiments. *P < 0.05 vs. HDE alone, #P < 0.05 vs. medium alone (by Student's paired t-test).
Salbutamol pretreatment attenuates airway inflammation associated with a single exposure to HDE in mice.
Acute inhalation exposure to CAFO dust extract results in airway inflammatory cytokine production and neutrophil influx (13, 18, 31, 33). Mice exposed to inhaled HDE exhibited a significant accumulation of neutrophils (P < 0.05; Fig. 6A) and increased production of the inflammatory cytokines/chemokines IL-6, TNF-α, CXCL1 (keratinocyte chemoattractant), and CXCL2 (macrophage inflammatory protein-2) (Fig. 6B). However, in mice pretreated with nebulized salbutamol for 30 min prior to HDE exposure, HDE-induced neutrophil and inflammatory mediator levels were significantly reduced (P < 0.05; Fig. 6). In addition, acute HDE exposure increased airway epithelial cell ICAM-1 expression in vivo compared with saline-treated mice (brown staining; Fig. 7, A and B). However, nebulized salbutamol pretreatment reduced HDE-induced ICAM-1 expression in the airway epithelium (Fig. 7C). These results complement the in vitro findings indicating that β2-agonist treatment inhibited HDE-induced ICAM-1 expression in BECs (Fig. 5).
Fig. 6.

Nebulized salbutamol pretreatment attenuates HDE-mediated inflammatory cell influx and cytokine release in the airways of acutely exposed mice. Mice (n = 5 per condition) were exposed to nebulized salbutamol in an exposure chamber 30 min before a single challenge with 12.5% intranasal HDE or saline. At 5 h following the HDE exposure, mice were euthanized, and bronchoalveolar lavage was performed. Total cells and differential counts in bronchoalveolar lavage fluid (BALF) were quantified (A), and cytokine concentrations were determined by ELISA (B). CXCL, chemokine C-X-C motif ligand; MIP-2, macrophage inflammatory protein-2; KC, keratinocyte chemoattractant. *P < 0.05 vs. HDE alone (by 1-way ANOVA and Tukey's posttest).
Fig. 7.

HDE-induced ICAM-1-specific immunostaining on airway luminal epithelial cells is abrogated when mice are pretreated with salbutamol. Paraffin-embedded lung sections from mice exposed a single time to 12.5% HDE (B) demonstrate prominent epithelial ICAM-1 immunostaining compared with mice given saline alone (A). Tissues from mice pretreated with nebulized salbutamol and then challenged with HDE showed a marked reduction in ICAM-1 staining (C). Micrographs are representative of lung sections from 3 mice per condition. Arrows indicate localization of ICAM-1 on the apical surface of the epithelium. Parenchymal alveolar cells stained equally positive in all groups.
Salbutamol exposure reduces airway inflammation associated with repetitive exposure to HDE in mice.
To ascertain whether β2-agonist treatment alters the airway inflammatory response to repetitive HDE treatment, mice were treated with HDE daily for 3 wk with salbutamol delivered prior to each HDE treatment. Consistent with previously published studies (23), we demonstrated that neutrophil recruitment and cytokine/chemokine production in repetitively treated mice are dampened compared with a one-time HDE exposure, yet levels are increased compared with saline-treated mice (Fig. 8). In mice receiving nebulized salbutamol prior to each HDE exposure, neutrophil infiltration was reduced (Fig. 8A). Correspondingly, BALF inflammatory cytokine/chemokine levels were significantly reduced in salbutamol-treated mice receiving HDE treatment compared with those receiving HDE alone (P < 0.05; Fig. 8B). Moreover, repetitive HDE exposure resulted in increased lymphoid aggregates as well as bronchiolar and alveolar inflammation (Fig. 9, E, F, and I). Importantly, salbutamol pretreatment reduced the HDE-mediated adverse lung pathology changes (Fig. 9, C, D, and I). Together, these results suggest that salbutamol is effective in reducing the airway inflammatory consequences associated with repetitive HDE exposure in vivo.
Fig. 8.

Inflammatory cell recruitment and cytokine/chemokine production are reduced in airways of mice repeatedly exposed to HDE following β2-agonist treatments. Each day for 15 consecutive weekdays, mice (5 per group) were given nebulized salbutamol 30 min prior to intranasal HDE challenge. At 5 h following the final HDE exposure, mice were euthanized, BALF was collected, and lungs were removed for histological analysis. Inflammatory cells were recovered from BALF and quantified (A), and cytokines were measured in the BALF supernates (B). The entire experiment was performed twice, resulting in 10 mice per condition. *P < 0.05 vs. HDE alone (by 1-way ANOVA and Tukey's posttest).
Fig. 9.

Salbutamol improves the histopathology of mice repeatedly exposed to HDE. Paraffin-embedded hematoxylin-eosin-stained thin sections of inflated lungs removed from mice exposed to HDE for 15 days exhibit both diffuse parenchymal inflammatory infiltrates (E and F) and focal mononuclear aggregates (E and F insets) compared with control mice (A and B) and mice treated with salbutamol alone (C and D). In mice pretreated with salbutamol prior to HDE, inflammatory cell influx is markedly blunted (G and H). The entire experiment was performed twice, resulting in 10 mice per condition. Micrographs are representative of 8 slides per condition. In I, inflammatory scores [from 0 (no inflammation) to 3 (severe inflammation)] were assigned to each treatment group by a clinical pathologist blinded to study parameters. *P < 0.05 vs. HDE alone (by 1-way ANOVA and Bonferroni's posttest).
DISCUSSION
Agriculture workers may experience debilitating airway diseases as a result of their exposures (11). In particular, CAFO workers, including nonsmokers, are susceptible to several lung ailments, including organic dust toxic syndrome, asthma-like illness, and chronic bronchitis (31, 32). Defining the mechanistic basis of these inflammatory events is critical for developing optimal therapies for treating these individuals. Airway epithelial cells are first responders to environmental insults and are important in orchestrating the immune responses to these exposures (16, 28). Previous investigations have demonstrated that PKC activation in the lung following organic dust exposures drives the inflammatory response in BECs (21, 24, 37). Our current findings indicate that PKA-activating β2-agonists counterbalance organic dust-induced PKC activation; β2-agonists may therefore be effective drugs for treating organic dust-induced airway inflammation.
In individuals with acute exposures to CAFO environments, serum inflammatory cytokines, including IL-6 and TNF-α, are significantly elevated above unexposed individuals, and BALF and nasal lavage fluids exhibit significantly increased IL-8 levels and immune cell recruitment comprising predominantly neutrophilic granulocytes (13, 32, 33). While an apparent chronic inflammatory adaptation response occurs in individuals working in CAFO environments, these individuals may experience an accelerated decline in forced expiratory volume and are at increased risk for chronic lung disease (10, 18, 32). In vitro culture systems, as well as in vivo murine studies, have been used to develop models of organic dust exposure that replicate many clinical features of CAFO dust-mediated pathologies (15, 23, 24). These model systems have identified critical regulators of the inflammatory responses to organic dusts (1, 3, 19, 22, 25). In particular, the sequential activation of PKCα and PKCε is central to the response of BECs following HDE exposure (37). PKCα inhibition in epithelial cells results in reductions in IL-6, TNF-α, and IL-8 in response to HDE, whereas PKCε inhibition results in IL-8 downregulation (37). Thus, attenuation of HDE-induced PKC activation presents a promising approach for the prevention of organic dust-induced airway inflammation and disease.
Previous reports indicate a protective role for PKA activation in HDE-stimulated BECs, in that PKA-activating agents are able to modulate HDE-induced inflammatory cytokine production (7, 34). Mechanistic studies indicate that PKA activation interferes with BEC inflammatory responses by blocking the TNF-α production and release that are required for subsequent PKCε activation (34). β2-Agonists are potent activators of adenylyl cyclase, leading to increased cellular cAMP levels and PKA activation (29). Our in vivo investigations reported here reveal that exposure to the SABA salbutamol prior to a single HDE exposure or repetitive HDE exposures is sufficient to reduce lung inflammatory responses. At the level of the bronchial epithelium, we have also shown that SABA and LABA treatments result in increased PKA activity and attenuation of HDE-induced inflammatory cytokine production while reducing ICAM-1 expression and interactions with neutrophils. Neutrophilic recruitment is a hallmark feature of CAFO dust-induced airway inflammation in patients and in rodent studies (13, 23, 32, 33). BEC-neutrophil interactions facilitated by ICAM-1 lead to enhanced neutrophilic functioning (4, 30). The ability of β2-agonists to inhibit both the production of neutrophil-attracting chemokines and BEC-neutrophil interactions thus likely accounts for much of the inhibition of the inflammatory response seen in our in vivo modeling system.
Although β2-agonists are used clinically for their actions as bronchodilators targeting airway smooth muscle cells, new treatment designs are testing their pharmacological utility as adenylyl cyclase modulators in other cell types (29). Published studies using animal models indicate that β2-agonists may be beneficial in reducing lung inflammatory consequences following cigarette smoke exposure (17) and acute lung injury (5). In addition, recent work suggests that the β2-agonist procaterol protects airway epithelial cells from oxidative stress (9). In contrast, studies have also found that β2-agonist therapy can worsen lung inflammation. In an acute particulate matter exposure study, β2-agonists were found to augment production of IL-6 in response to particulate matter exposure, while β2-adrenergic receptor-knockout mice were unaffected by β2-agonist treatment (8). In a murine asthma model, chronic albuterol use was found to exacerbate airway inflammatory responses (14). These contradictory findings highlight the physiological complexity of β2-adrenergic receptor signaling. Thus it is important to identify the mechanisms underlying inflammation in specific settings leading to airway diseases to ensure appropriate therapy choices in a clinical setting. For example, β2-agonists are utilized in numerous airway diseases that are of diverse etiologies, including eosinophilc atopic/allergic asthma as well as neutrophilic obstructive pulmonary diseases, owing to their bronchodilator actions in both settings (26). Our findings would be applicable to neutrophilic obstructive pulmonary diseases as opposed to eosinophilic atopic/allergic asthma. Moreover, the PKA-mediating actions of the β2-agonists in these different inflammatory settings could lead to alternative disease outcomes.
As another possible explanation for these contradictory findings, it is important to note that proinflammatory regulators not only initiate damaging inflammation but control prohealing/proresolution responses as well. For example, our previous work demonstrates that PKCε is an inflammatory driver of HDE responses; yet we have also found that PKCε-knockout mice exhibit a heightened airway inflammatory response to HDE in vivo (21). It is therefore prudent to recognize that treatment modalities should possibly allow for productive inflammatory responses while limiting deleterious, nonresolving inflammation. Because of the contradictory/complex nature of β2-adrenergic receptor signaling in inflammatory responses, future investigations are warranted to investigate how the entire immune response is regulated by β2-agonist treatment, including not only effects on inflammatory drivers like PKCε, but also downstream effects on inflammation resolution processes. Investigations into the effects of β2-agonists on other cells within the lung milieu will also be vital to understanding the complete modulatory activities of β2-agonists in the lung. Lastly, we have investigated the efficacy of β2-agonist treatment prior to organic dust exposure, while clinical use of the pharmacological agents would likely begin following an inflammatory exposure. Postexposure treatment investigations are thus warranted to ascertain the mechanisms underlying the efficacy of β2-agonist therapy in patients with airway inflammatory diseases.
Our previous studies demonstrate an important role for PKC activation in the propagation of the inflammatory responses to HDE (34, 37). Our present findings suggest that PKA-activating agents such as β2-agonists might provide a promising therapeutic tool for the prevention and/or treatment of CAFO dust-induced airway inflammation. β2-Agonist treatment significantly reduced BEC and airway inflammatory consequences associated with HDE exposure, with no identified detriment to the cells or host. Future studies are warranted to determine the specific utility of this drug class in the clinical management of lung disease in CAFO workers.
GRANTS
The investigations reported in this manuscript were supported by the following funding agencies: Centers for Disease Control National Institute for Occupational Safety and Health Grant 2R01 OH-008539 (D. J. Romberger), National Institute of Environmental Health Sciences Grants R01 ES-019325 (J. A. Poole) and F32 ES-022913 (T. M. Nordgren), Department of Veterans Affairs Grant IO1BX000728 (T. A. Wyatt), and Central States Center for Agricultural Safety and Health Grant U54OH010162 (J. A. Pool and T. A. Wyatt).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.J.R., A.J.H., T.M.N., J.A.P., M.L.T., and T.A.W. developed the concept and designed the research; D.J.R., T.M.N., W.W.W., and T.A.W. analyzed the data; D.J.R. and T.M.N. interpreted the results of the experiments; D.J.R. and T.M.N. drafted the manuscript; D.J.R., A.J.H., T.M.N., J.A.P., M.L.T., W.W.W., and T.A.W. edited and revised the manuscript; D.J.R., A.J.H., T.M.N., J.A.P., M.L.T., W.W.W., and T.A.W. approved the final version of the manuscript; A.J.H., T.M.N., and T.A.W. performed the experiments; A.J.H. prepared the figures.
ACKNOWLEDGMENTS
The authors thank Lisa Chudomelka for expert assistance in manuscript preparation.
REFERENCES
- 1.Bailey KL, Poole JA, Mathisen TL, Wyatt TA, Von Essen SG, Romberger DJ. Toll-like receptor 2 is upregulated by hog confinement dust in an IL-6-dependent manner in the airway epithelium. Am J Physiol Lung Cell Mol Physiol 294: L1049–L1054, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey KL, Robinson JE, Sisson JH, Wyatt TA. Alcohol decreases RhoA activity through a nitric oxide (NO)/cyclic GMP (cGMP)/protein kinase G (PKG)-dependent pathway in the airway epithelium. Alcohol Clin Exp Res 35: 1277–1281, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauer C, Kielian T, Wyatt TA, Romberger DJ, West WW, Gleason AM, Poole JA. Myeloid differentiation factor 88-dependent signaling is critical for acute organic dust-induced airway inflammation in mice. Am J Respir Cell Mol Biol 48: 781–789, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bloemen PG, van den Tweel MC, Henricks PA, Engels F, Wagenaar SS, Rutten AA, Nijkamp FP. Expression and modulation of adhesion molecules on human bronchial epithelial cells. Am J Respir Cell Mol Biol 9: 586–593, 1993. [DOI] [PubMed] [Google Scholar]
- 5.Bosmann M, Grailer JJ, Zhu K, Matthay MA, Sarma JV, Zetoune FS, Ward PA. Anti-inflammatory effects of β2-adrenergic receptor agonists in experimental acute lung injury. FASEB J 26: 2137–2144, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braut-Boucher F, Pichon J, Rat P, Adolphe M, Aubery M, Font J. A non-isotopic, highly sensitive, fluorimetric, cell-cell adhesion microplate assay using calcein AM-labeled lymphocytes. J Immunol Methods 178: 41–51, 1995. [DOI] [PubMed] [Google Scholar]
- 7.Burvall K, Palmberg L, Larsson K. Influence of 8-bromo-cyclic AMP on interleukin-6 and -8 mRNA levels in A549 human lung epithelial cells exposed to organic dust: a time-kinetic study. Life Sci 75: 2733–2749, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Chiarella SE, Soberanes S, Urich D, Morales-Nebreda L, Nigdelioglu R, Green D, Young JB, Gonzalez A, Rosario C, Misharin AV, Ghio AJ, Wunderink RG, Donnelly HK, Radigan KA, Perlman H, Chandel NS, Budinger GR, Mutlu GM. β2-Adrenergic agonists augment air pollution-induced IL-6 release and thrombosis. J Clin Invest 124: 2935–2946, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng Z, Zhou JJ, Sun SY, Zhao X, Sun Y, Pu XP. Procaterol but not dexamethasone protects 16HBE cells from H2O2-induced oxidative stress. J Pharm Sci 125: 39–50, 2014. [DOI] [PubMed] [Google Scholar]
- 10.Iversen M, Dahl R. Working in swine-confinement buildings causes an accelerated decline in FEV1: a 7-yr follow-up of Danish farmers. Eur Respir J 16: 404–408, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Kirkhorn SR, Garry VF. Agricultural lung diseases. Environ Health Perspect 108 Suppl 4: 705–712, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langley RL. Consequences of respiratory exposures in the farm environment. N C Med J 72: 477–480, 2011. [PubMed] [Google Scholar]
- 13.Larsson BM, Palmberg L, Malmberg PO, Larsson K. Effect of exposure to swine dust on levels of IL-8 in airway lavage fluid. Thorax 52: 638–642, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin R, Degan S, Theriot BS, Fischer BM, Strachan RT, Liang J, Pierce RA, Sunday ME, Noble PW, Kraft M, Brody AR, Walker JK. Chronic treatment in vivo with β-adrenoceptor agonists induces dysfunction of airway β2-adrenoceptors and exacerbates lung inflammation in mice. Br J Pharmacol 165: 2365–2377, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathisen T, Von Essen SG, Wyatt TA, Romberger DJ. Hog barn dust extract augments lymphocyte adhesion to human airway epithelial cells. J Appl Physiol 96: 1738–1744, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Message SD, Johnston SL. Host defense function of the airway epithelium in health and disease: clinical background. J Leukoc Biol 75: 5–17, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montalbano AM, Anzalone G, Albano GD, Sano CD, Gagliardo R, Bonanno A, Riccobono L, Nicolini G, Ingrassia E, Gjomarkaj M, Profita M. Beclomethasone dipropionate and formoterol reduce oxidative/nitrosative stress generated by cigarette smoke extracts and IL-17A in human bronchial epithelial cells. Eur J Pharmacol 718: 418–427, 2013. [DOI] [PubMed] [Google Scholar]
- 18.Palmberg L, Larssson BM, Malmberg P, Larsson K. Airway responses of healthy farmers and nonfarmers to exposure in a swine confinement building. Scand J Work Environ Health 28: 256–263, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Poole JA, Dooley GP, Saito R, Burrell AM, Bailey KL, Romberger DJ, Mehaffy J, Reynolds SJ. Muramic acid, endotoxin, 3-hydroxy fatty acids, and ergosterol content explain monocyte and epithelial cell inflammatory responses to agricultural dusts. J Toxicol Environ Health 73: 684–700, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poole JA, Romberger DJ. Immunological and inflammatory responses to organic dust in agriculture. Curr Opin Allergy Clin Immunol 12: 126–132, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poole JA, Romberger DJ, Bauer C, Gleason AM, Sisson JH, Oldenburg PJ, West WW, Wyatt TA. Protein kinase Cε is important in modulating organic-dust-induced airway inflammation. Exp Lung Res 38: 383–395, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poole JA, Wyatt TA, Kielian T, Oldenburg P, Gleason AM, Bauer A, Golden G, West WW, Sisson JH, Romberger DJ. Toll-like receptor 2 regulates organic dust-induced airway inflammation. Am J Respir Cell Mol Biol 45: 711–719, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poole JA, Wyatt TA, Oldenburg PJ, Elliott MK, West WW, Sisson JH, Von Essen SG, Romberger DJ. Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am J Physiol Lung Cell Mol Physiol 296: L1085–L1095, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romberger DJ, Bodlak V, Von Essen SG, Mathisen T, Wyatt TA. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J Appl Physiol 93: 289–296, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Romberger DJ, Heires AJ, Nordgren TM, Souder CP, West W, Liu XD, Poole JA, Toews ML, Wyatt TA. Proteases in agricultural dust induce lung inflammation through PAR-1 and PAR-2 activation. Am J Physiol Lung Cell Mol Physiol 309: L388–L399, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spina D. Current and novel bronchodilators in respiratory disease. Curr Opin Pulm Med 20: 73–86, 2014. [DOI] [PubMed] [Google Scholar]
- 27.Spurzem JR, Gupta J, Veys T, Kneifl KR, Rennard SI, Wyatt TA. Activation of protein kinase A accelerates bovine bronchial epithelial cell migration. Am J Physiol Lung Cell Mol Physiol 282: L1108–L1116, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Takizawa H. Airway epithelial cells as regulators of airway inflammation. Int J Mol Med 1: 367–378, 1998. [DOI] [PubMed] [Google Scholar]
- 29.Theron AJ, Steel HC, Tintinger GR, Feldman C, Anderson R. Can the anti-inflammatory activities of β2-agonists be harnessed in the clinical setting? Drug Des Dev Ther 7: 1387–1398, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tosi MF, Stark JM, Smith CW, Hamedani A, Gruenert DC, Infeld MD. Induction of ICAM-1 expression on human airway epithelial cells by inflammatory cytokines: effects on neutrophil-epithelial cell adhesion. Am J Respir Cell Mol Biol 7: 214–221, 1992. [DOI] [PubMed] [Google Scholar]
- 31.Von Essen S, Donham K. Illness and injury in animal confinement workers. Occup Med 14: 337–350, 1999. [PubMed] [Google Scholar]
- 32.Von Essen S, Romberger D. The respiratory inflammatory response to the swine confinement building environment: the adaptation to respiratory exposures in the chronically exposed worker. J Agric Saf Health 9: 185–196, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z, Larsson K, Palmberg L, Malmberg P, Larsson P, Larsson L. Inhalation of swine dust induces cytokine release in the upper and lower airways. Eur Respir J 10: 381–387, 1997. [DOI] [PubMed] [Google Scholar]
- 34.Wyatt TA, Poole JA, Nordgren TM, DeVasure JM, Heires AJ, Bailey KL, Romberger DJ. cAMP-dependent protein kinase activation decreases cytokine release in bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 307: L643–L651, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wyatt TA, Sisson JH. Chronic ethanol downregulates PKA activation and ciliary beating in bovine bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 281: L575–L581, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Wyatt TA, Sisson JH, Von Essen SG, Poole JA, Romberger DJ. Exposure to hog barn dust alters airway epithelial ciliary beating. Eur Respir J 31: 1249–1255, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wyatt TA, Slager RE, Heires AJ, Devasure JM, Vonessen SG, Poole JA, Romberger DJ. Sequential activation of protein kinase C isoforms by organic dust is mediated by tumor necrosis factor. Am J Respir Cell Mol Biol 42: 706–715, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]