

Lymphatic endothelial cells, exposed to chronically elevated pulmonary lymph flow in a model of congenital heart disease with increased pulmonary blood flow, demonstrate disrupted nitric oxide (NO) signaling. Specifically, they have altered NO synthase expression and activity that result in increased accumulation of reactive oxygen species and decreased bioavailable NO.
Keywords: nitric oxide signaling, nitric oxide synthase
Abstract
Associated abnormalities of the lymphatic circulation are well described in congenital heart disease. However, their mechanisms remain poorly elucidated. Using a clinically relevant ovine model of a congenital cardiac defect with chronically increased pulmonary blood flow (shunt), we previously demonstrated that exposure to chronically elevated pulmonary lymph flow is associated with: 1) decreased bioavailable nitric oxide (NO) in pulmonary lymph; and 2) attenuated endothelium-dependent relaxation of thoracic duct rings, suggesting disrupted lymphatic endothelial NO signaling in shunt lambs. To further elucidate the mechanisms responsible for this altered NO signaling, primary lymphatic endothelial cells (LECs) were isolated from the efferent lymphatic of the caudal mediastinal node in 4-wk-old control and shunt lambs. We found that shunt LECs (n = 3) had decreased bioavailable NO and decreased endothelial nitric oxide synthase (eNOS) mRNA and protein expression compared with control LECs (n = 3). eNOS activity was also low in shunt LECs, but, interestingly, inducible nitric oxide synthase (iNOS) expression and activity were increased in shunt LECs, as were total cellular nitration, including eNOS-specific nitration, and accumulation of reactive oxygen species (ROS). Pharmacological inhibition of iNOS reduced ROS in shunt LECs to levels measured in control LECs. These data support the conclusion that NOS signaling is disrupted in the lymphatic endothelium of lambs exposed to chronically increased pulmonary blood and lymph flow and may contribute to decreased pulmonary lymphatic bioavailable NO.
NEW & NOTEWORTHY
Lymphatic endothelial cells, exposed to chronically elevated pulmonary lymph flow in a model of congenital heart disease with increased pulmonary blood flow, demonstrate disrupted nitric oxide (NO) signaling. Specifically, they have altered NO synthase expression and activity that result in increased accumulation of reactive oxygen species and decreased bioavailable NO.
the association between congenital heart disease (CHD) and congenital and acquired abnormalities of the lymphatic circulation has been well described (11, 19, 30, 39, 44, 53, 62, 65). In particular, patients with common congenital cardiac defects, like ventricular septal defects, have increased pulmonary blood flow (PBF) that leads to altered respiratory mechanics and respiratory distress, and lymphatic abnormalities have been implicated in these respiratory aberrations (65). Despite these observations, investigations into the mechanisms that underlie lymphatic aberrations in CHD are sparse. Using our established clinically relevant ovine model of a congenital cardiac defect with increased PBF (shunt), we have previously demonstrated that chronically increased PBF disrupts the normal postnatal development and function of the pulmonary lymphatic system (12, 13). Specifically, we found that chronically increased PBF and the resulting exposure to chronically elevated pulmonary lymph flow are associated with: 1) prolonged transit time through the pulmonary lymphatics, with decreased bioavailable nitric oxide (NOx) in pulmonary lymph effluent; 2) aberrations in lymphatic architecture, including alteration in the expression of proteins associated with lymphatic growth, such as vascular endothelial growth factor-c (VEGF-c); and 3) increased baseline tone and induced contractility, with attenuated endothelium-dependent relaxation of thoracic duct rings. Furthermore, we found that exogenously administered inhaled nitric oxide (NO) preserved pulmonary lymphatic flow in this setting (12, 13).
A small but expanding body of literature indicates that lymphatic vessel capacitance and pumping primarily dictate lymphatic function under normal physiological conditions (15, 16, 23–28, 55, 68), and that endothelial NO signaling is an important modulator of lymphatic pump activity and lymph flow (7, 25, 28, 32, 61). Based on our previous findings, we hypothesized that nitric oxide synthase (NOS) signaling is disrupted in the lymphatic endothelium of lambs exposed to chronically increased pulmonary blood and lymph flow (12, 13).
The objective of the present study was to test this hypothesis and potential mechanisms of disrupted NO signaling. To this end, late-gestation fetal lambs underwent in utero placement of an aortopulmonary vascular graft. Four weeks after birth, primary lymphatic endothelial cells (LECs) were isolated from the efferent lymphatic of the caudal mediastinal lymph node of both shunt and normal (control) lambs. We measured and compared NOx, NOS isoform expression and activity, levels of cellular nitration, and accumulation of reactive oxygen species (ROS). A better understanding of these mechanisms may lead to improved treatment and prevention strategies for congenital and acquired lymphatic abnormalities in the setting of CHD.
METHODS
Chronic model of increased PBF and pulmonary lymph flow.
As previously described in detail (51), an 8.0-mm Gore-Tex vascular graft, ∼2 mm length (W. L. Gore and Associates, Milpitas, CA), was anastomosed between the ascending aorta and main pulmonary artery in anesthetized late-gestation fetuses (137–141 days gestation, term = 145 days) from five mixed-breed Western ewes. Four weeks after spontaneous delivery, shunt lambs (n = 5) and normal age-matched control (n = 5) were anesthetized, mechanically ventilated, and instrumented to continuously measure hemodynamics and harvest tissue (51). Animals' vital signs, including core temperature, were monitored throughout the study, and they were given intravenous fluids and prophylactic antibiotics per protocol (51). For all shunt lambs, the ratio of pulmonary to systemic blood flow (Qp/Qs) was calculated using the Fick principle. For control lambs, the Qp/Qs was assumed to be 1:1.
At the end of each protocol, all lambs were killed with a lethal injection of pentobarbital sodium followed by bilateral thoracotomy as described in the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The Committees on Animal Research of the University of California, San Francisco, and the University of California, Davis, approved all protocols and procedures.
Isolation and culture of lymphatic endothelial cells.
After completion of baseline hemodynamic measurements (13) and following instillation of additional local anesthesia, a right thoracotomy was made in the sixth intercostal space, and a segment of the efferent vessel of the caudal mediastinal lymph node was visualized (12, 60) and harvested. Dissected lymphatic vessels were flushed with Ca2+-free Hanks's solution, cut into smaller segments, and filleted. They were placed endothelium layer face down on type IV collagen-coated plates (BioCoat; BD Biosciences 354453, Bedford MA). Tissue explants were maintained in BioCoat Endothelial Cell Growth Media (BD Biosciences) with 20% fetal bovine serum and a penicillin-streptomycin solution in a humidified chamber with 5% CO2 with room air. Cell growth was noticeable by 24 h, and the tissue explants were removed by 72 h. At 4–6 days, cells that exhibit cobblestone-like appearance were cloned by a cloning cylinder and were passaged at ∼70% confluence. LECs between passages 3 and 6 were used for subsequent experiments; all lines were confirmed positive for LYVE-1 and Prox1 expression (see below). In total, independently derived cell lines were established from five control and five shunt lambs; for all experiments, LECs from three control and three shunt lambs were used.
Immunofluorescence on LECs.
When cultured LECs reached 70% confluence, they were fixed with 4% paraformaldehyde and washed briefly in PBS/Tris buffer. Immunofluorescence was performed as described previously (12): cells were washed in TBS + Tween (0.03%) (TBSTw) 3 × 5 min, blocked with Dako Antibody Diluent (Dako, Carpentaria, CA), and exposed to primary antibody, goat anti-Prox1 (R&D Systems), diluted in blocking serum overnight at 4°C. Cells were then washed 3 × 5 min in TBSTw and exposed to an appropriate secondary antibody that was conjugated with Alexa Fluor (Invitrogen, Life Technologies, Carlsbad, CA) or Dylight (Thermo Fisher Scientific, Rockford, IL) in blocking serum for 60 min. Cells were washed in TBS for 4 × 5 min. Cells were mounted in Vectashield (Vector, Burlingame, CA) containing 4′,6-diamidino-2-phenylindole (DAPI) and were covered with a cover slip.
Images were taken with a Hamamatsu c10600 ORCA-R2 Digital Camera on a Zeiss Axio Imager Z2 using DIC objectives, the X-cite 120 Mercury/Halide System, and analyzed using ZEN pro 2012 software (Carl Zeiss Microimaging, Thornwood, NY). All images were subsequently processed using Adobe Photoshop CS5 software (Adobe, San Jose, CA).
Preparation of LEC protein extracts and Western blot analysis.
Preparation of protein from LECs for Western blot analysis was performed as previously described (5, 42, 64). Cell lysates from third- to sixth-passage LECs derived from control and shunt lambs were used. Protein concentration in each sample was quantified using a Nano Drop Spectrophotometer (ND-1000; Thermo Fisher Scientific, Waltham, MA). For Western blot analysis, 20 μg total protein were separated by 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Millipore). The membranes were then blocked with 5% nonfat dried milk in 130 mM NaCl and 25 mM Tris (TBS, pH 7.5) for 1 h at room temperature. Membranes were blocked and subsequently exposed to primary antibodies against endothelial nitric oxide synthase (eNOS; Santa Cruz), inducible nitric oxide synthase (iNOS; Santa Cruz), neuronal nitric oxide synthase (nNOS; Santa Cruz), LYVE-1 (Abcam), Prox1 (EMD Millipore), 90-kDa heat shock protein (HSP90) (BD Transduction Laboratories), caveolin-1 (Santa Cruz), calmodulin (Santa Cruz), phospho-eNOS-serine-1177 (Cell Signaling), or nitrotyrosine (CalBiochem), as well as β-actin (Abcam), which served as a loading control. Following incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies, chemiluminescence was then used to detect bands (SuperSignal West Pico Chemiluminescent Substrate kit; Pierce Biotechnology, Rockford, IL). Densitometry was performed using a public domain Java image-processing program, Image J (NIH Image).
Flow cytometry.
Passaged LEC lines were grown to 70% confluency. Before analysis by flow cytometry, cells were trypsinized (0.25%) for 5 min, fixed with 4% paraformaldehyde for 15 min, and washed briefly in PBS. Immunofluorescence was performed as described above and previously (12), except that washes were performed with PBS. Cells were exposed to primary antibody, either mouse anti-Prox1 (EMD Millipore), rabbit anti-LYVE-1 (Abcam), or the corresponding mouse (eBioscience) or rabbit (Sigma) isotype control, and then to the appropriate AlexaFuor 488 (Thermo Fisher Scientific)- or 647 (Jackson ImmunoResearch)-conjugated secondary antibody. Before analysis on a BD LSR II flow cytometer using FACSDiva software, cells were washed in PBS for 4 × 5 min, with the third wash containing DAPI. The dyes were excited using a 488-nm blue laser or a 640-nm red laser, and emissions were measured using a 530- or 670-nm filter, respectively. Data were analyzed using FlowJo version 9.3.2 software. Cells were gated for DAPI (405-nm violet laser with 450-nm filter) to exclude acellular debris and isolate nucleated cells.
Measurement of NOx.
To quantify bioavailable NO, the concentration of NO and its metabolites was determined in cell lysates from control and shunt LECs. Passaged LECs were harvested, rinsed in PBS, snap-frozen in liquid nitrogen, and stored at −80°C. Cells were subsequently homogenized in ×10 (weight to volume) 6% trichloroacetic acid. Samples were spun down (3,000 revolution/min, 4°C for 15 min), and supernatant was recovered for direct measurement. In solution, NO reacts with molecular oxygen to form nitrite, and with oxyhemoglobin and superoxide anion to form nitrate. Nitrite and nitrate are reduced using vanadium (III) and hydrochloric acid at 90°C. NO is purged from solution, resulting in a peak of NO for subsequent detection in micromoles per liter by chemiluminescence (NOA 280; Sievers Instruments, Boulder, CO), as previously described (6, 43, 69). The sensitivity is 1 × 10−12 moles, with a concentration range of 1 × 10−9 to 1 × 10−3 molar of nitrate.
Isolation of mRNA and quantitative real-time PCR.
Total RNA was isolated from control and shunt LECs using the Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA) per the manufacturer's instructions. For each sample, reverse transcription was performed with 1 μg of total RNA using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Quantitative Real-Time PCR amplification was carried out (in triplicate) using the iQ SYBR Green Supermix (Bio-Rad) on an Applied Biosystems 7300 Real-Time PCR System (Foster City, CA). The following eNOS primers were used: forward, gaggggctgtcattccacta; reverse, aggggtcttccagatggact, and were designed using public OligoPerfect Designer software (Life Technologies). Relative gene expression was analyzed. Differences in cycle threshold number (CT) were calculated by normalizing the sample cycle threshold of the targeted gene with that of the internal control reference gene GAPDH using primers to mouse GAPDH (PrimePCR SYBR Green Assay; Bio-Rad). The ΔΔCT [calculated as CT(target) − CT(reference)] method was used to determine relative abundance of expression, as described previously (38).
Immunoprecipitation.
Total protein was isolated from control and shunt LECs as above. Based on the manufacturer's instructions, specific antibodies were cross-linked with Protein A/G Magnetic Beads for 1 h at room temperature using the Pierce Crosslink Immunoprecipitation Kit (Pierce Biotechnology). Protein extracts were incubated with immunoprecipitation (IP) antibody rabbit anti-eNOS/protein A/G magnetic beads overnight 4°C. The antigen-antibody-beads were next washed two times with IP lysis/wash buffer. Finally, the antigen-antibody complexes were eluted and used for Western blot analysis.
NOS activity assay.
LECs derived from control and shunt lambs were homogenized, and NOS activity was determined using the conversion of l-[3H]arginine to l-[3H]citrulline, according to the manufacturer's instructions in the NOS Activity Assay Kit (Cayman Chemical, Ann Arbor, MI). All activities were normalized to the amount of protein in each lysate. To determine the contribution of iNOS to total NOS activity, assays were repeated without Ca2+ supplementation. As a control for specificity, assays were repeated in the presence of NG-nitro-l-arginine (l-NNA), a nonspecific NOS inhibitor. All assays were performed in triplicate.
Measurement of ROS.
Cultured control and shunt LECs were incubated with CellROX Deep Red (Life Technologies) at a final concentration of 5 μM for 30 min, after a subset of both control and shunt LECs was incubated with 20 μM N-{[3-(aminomethyl)phenyl]methyl}ethanimidamide dihydrochloride (1400W) (Sigma Aldrich, St. Louis, MO) for 6 h or 100 μM 2-(2-aminoethyl)isothiourea dihydrobromide (AET) (Sigma Aldrich) for 24 h. Some plates were used for immunofluorescence imaging. Otherwise, culture medium was removed, and cells were washed in PBS, fixed in 3.7% paraformaldehyde for 15 min, washed again in PBS, and trypsinized (0.25%) for 4 min. This reaction was stopped with the addition of 10% FBS DMEM and centrifuged at 500 g for 10 min at 4°C. Cell pellets were resuspended in PBS and counted using a Moxi Z miniautomated cell counter (ORFLO, Ketchum, ID); 5,000 cells/sample were placed (in triplicate) on a Costar 96-well plate and analyzed on a Molecular Devices Spectra Max M2 following the manufacturer's instructions.
Statistical analysis.
For Western blot analysis and measurement of NOx and NOS activity, means ± SD were calculated. Results for quantitative PCR are shown as means ± SE. ΔΔCT was calculated with GAPDH as reference and normalized to compare relative mRNA expression between control and shunt LECs. Comparisons between control and shunt samples were made with the unpaired t-test. Comparisons between untreated and treated (e.g., 1400W) samples were made with the paired t-test. A P value of <0.05 was considered statistically significant.
RESULTS
Isolation and identification of LECs.
We isolated primary LECs from the efferent lymphatic of the caudal mediastinal lymph node from 4-wk-old control and shunt lambs. This is the same vessel that we have previously cannulated to collect pulmonary lymph flow (12). We confirmed that each individual isolate of LECs expressed the lymphatic endothelial cell-specific markers Prox1 and LYVE-1 (Fig. 1, C–F). By flow cytometric analysis, 98.8% of nucleated cells in LEC isolates were positive for LYVE-1, and 99.9% were positive for Prox1 (Fig. 1, F and G). All shunt lambs had evidence of increased pulmonary blood flow, with a Qp/Qs, calculated using the Fick principle, of 2.4 ± 0.8. For all experiments that follow, except where noted, three control and three shunt lines were used.
Fig. 1.

Lymphatic endothelial cells (LECs) isolated from the efferent lymphatic vessel of the caudal mediastinal lymph node of control and shunt lambs. A–D: representative micrographs of immunofluorescence staining for nuclei with 4′,6-diamidino-2-phenylindole (DAPI, blue) (A and B) or the lymphatic-specific marker Prox1 (red) (C and D). E: Western blot demonstrates that LECs, but not pulmonary artery smooth muscle cells (SMC) isolated from control (n = 2, 2) and shunt (n = 2, 2) lambs each express the lymphatic-specific markers Prox1 and LYVE-1. F and G: flow cytometric analysis of a representative LEC isolate, gated on nucleated cells (DAPI positive). LECs stained with the appropriate isotype control (gray) are compared with LECs stained with LYVE-1 (green) or Prox1 (red). Greater than 98% of nucleated cells are LYVE-1 and Prox1 positive.
Decreased NOx and expression of NOS isoforms.
Similar to what we observed in the pulmonary lymph effluent (8), NOx levels were 6.7-fold lower in LECs derived from shunt lambs than control LECs, P < 0.05 (Fig. 2A). In addition, both eNOS mRNA and protein were decreased in shunt LECs (Fig. 2, B and C); eNOS mRNA was 4-fold lower, and eNOS protein expression was 3.2-fold lower than in control LECs, P < 0.05. As expected, nNOS was not detected by Western blot in control or shunt LECs (Fig. 2E), but, somewhat surprisingly, iNOS expression was 1.8-fold higher in shunt LECs than in control LECs, P < 0.01 (Fig. 2D).
Fig. 2.

Bioavailable NO (NOx) and endothelial nitric oxide synthase (eNOS) expression is decreased in shunt LECs, but inducible nitric oxide synthase (iNOS) expression is increased. A: NOx levels in control (40.9 ± 6.3 μM) and shunt (6.1 ± 2.7 μM) LECs, *P < 0.001. B: eNOS mRNA expression is decreased 4-fold in shunt LECs, *P < 0.05. Relative RNA expression quantified by quantitative real-time PCR (qPCR) and normalized to control. Data are shown as means ± SE. C: eNOS protein expression is decreased 3-fold in shunt LECs, *P < 0.05. D: iNOS protein expression is increased 1.8-fold in shunt LECs, *P < 0.01. E: neuronal nitric oxide synthase (nNOS) is not expressed in control or shunt LECs; +, positive control (tissue homogenate from right ventricle) for nNOS. Protein levels in control and shunt LECs quantified by Western blot. For presentation graphically, densitometry in each lane has been normalized to β-actin and to control. For all experiments, n = 3 control, 3 shunt.
NOS activity.
We performed an in vitro measure of total NOS activity and found that NOS activity per unit protein in shunt LECs under maximum enzyme reaction rate conditions was increased 2.3-fold compared with control LECs, P < 0.05 (Fig. 3A). Ca2+-independent activity, which is due to iNOS alone, was similarly increased in shunt LECs (Fig. 3B). In fact, the increase in total NOS activity in shunt LECs was due entirely to the increased iNOS activity. As a control, all NOS activity was abolished in the presence of l-NNA, a nonspecific NOS inhibitor (Fig. 3).
Fig. 3.
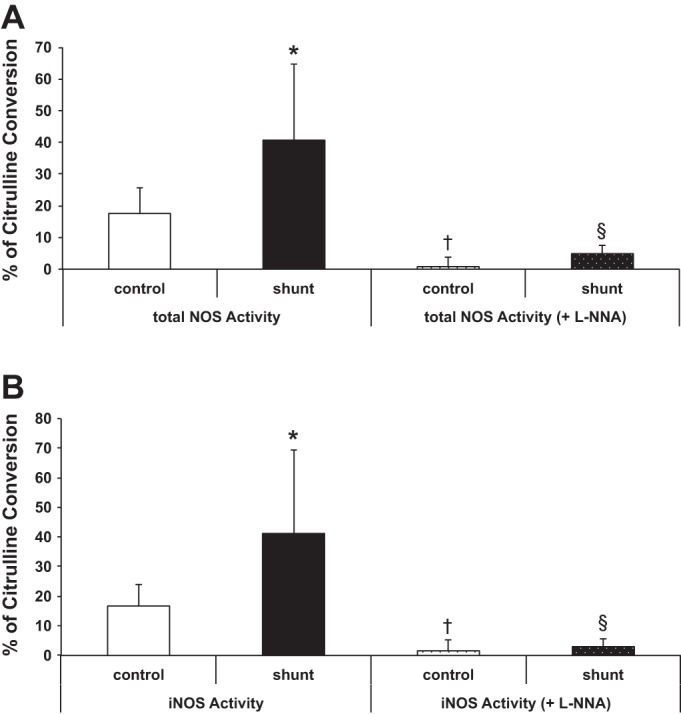
Nitric oxide synthase (NOS) activity in LECs, as a percentage of arginine converted to citrulline. A: total NOS activity (in the presence of Ca2+) is increased in shunt LECs, 40.7 ± 24% vs. 17.6 ± 8% in control LECs, as measured by an in vitro maximum enzyme reaction rate assay. NOS activity is abrogated in both control (0.8 ± 3%) and shunt (4.9 ± 2.7%) LECs in the presence of the nonspecific inhibitor NG-nitro-l-arginine (l-NNA). B: iNOS activity (NOS activity in the absence of Ca2+) is similarly increased in shunt LECs, 41.2 ± 28% vs. 16.7 ± 7% in control LECs, and l-NNA also inhibits iNOS activity in control (1.5 ± 3.7%) and shunt (2.9 ± 2.6%) LECs. For NOS activity assays, n = 3 control and 3 shunt and were tested in triplicate, *P < 0.05. NOS activity assays with l-NNA were tested in duplicate, control LECs vs. control LECs + inhibitor (†) and shunt LECs vs. shunt LECs + inhibitor (§), P < 0.05 for each.
Posttranslational regulation of eNOS.
The contribution of eNOS to the total NOS activity in control and shunt LECs, in the in vitro assay, was minimal (Fig. 3). We decided to examine more closely the protein-protein interactions that can regulate eNOS activity. An important activating phosphorylation event at Ser1177(18) was decreased 2.3-fold in shunt LECs, P < 0.05 (Fig. 4). Interestingly and perhaps paradoxically, the amount of eNOS associated with caveolin-1, a trafficking protein that inhibits eNOS activation (18), was decreased 2.1-fold in shunt LECs compared with control LECs, P < 0.05 (Fig. 4). Likewise, HSP90, a molecular chaperone known to stimulate eNOS activity, and calmodulin, whose binding to eNOS is required for activity (18), were each increased by 1.5-fold in shunt LECs, P < 0.05 (Fig. 4).
Fig. 4.
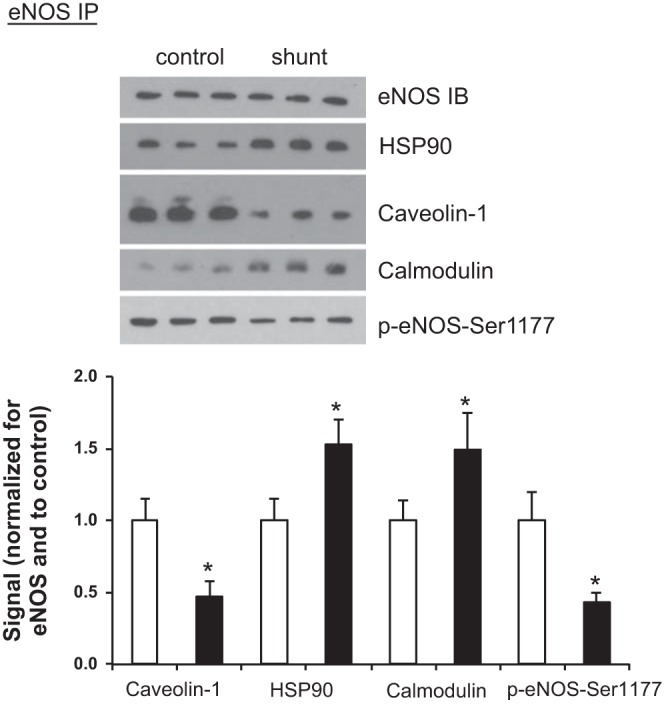
eNOS protein-protein interactions and Ser1177 phosphorylation in LECs. Following eNOS immunoprecipitation (IP) in control and shunt LECs, Western blots were performed for eNOS [immunoblot (IB)], 90-kDa heat shock protein (HSP90), caveolin-1, calmodulin, and phospho-eNOS-serine-1177. Normalized to control LECs, the steric inhibitor caveolin-1 associated with eNOS is decreased in shunt LECs (0.47 ± 0.11); conversely, the relative amount of the chaperone HSP90 associated with eNOS increases in shunt LECs (1.53 ± 0.17), and the relative amount of calmodulin associated with eNOS increases in shunt LECs to 1.49 ± 0.25. The relative amount of phosphorylated Ser1177 decreases in shunt LECs (0.43 ± 0.07). Data are shown as mean ratios [caveolin-1, HSP90, calmodulin, or phospho (p)-eNOS-Ser1177/eNOS] ± SD and normalized to control. For all experiments, n = 3 control, 3 shunt, with *P < 0.05.
Nitration, ROS, and the effect of iNOS inhibition.
With the increased iNOS activity and the decrease in bioavailable NO, we hypothesized that generation of ROS might scavenge bioavailable NO, leading to production of peroxynitrite and increased nitration. We used 3-nitrotyrosine to measure overall nitration and as a marker of peroxynitrite formation in control and shunt LECs. Total nitration in shunt LECs was increased 2.4-fold compared with control LECs, P < 0.05 (Fig. 5A), and we found a 1.4-fold increase in nitration of eNOS protein, P < 0.05 (Fig. 5B). We next used the fluorogenic probe CellROX Deep Red to measure accumulation of ROS. Compared with control LECs, we observed increased fluorescence signal in shunt LECs (Fig. 6B vs. 6A); when quantified using a microplate reader, shunt LECs had a 2.6-fold increase in ROS compared with control LECs, P < 0.05 (Fig. 6C).
Fig. 5.
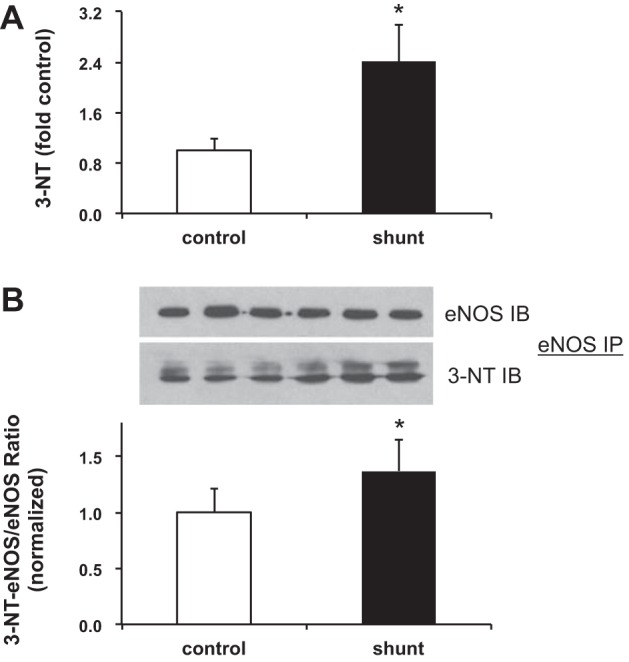
Nitration level in LECs. A: overall nitration is increased 2.4-fold in shunt LECs compared with control LECs, *P < 0.05. Nitrotyrosine (3-NT) levels in control and shunt LEC lysates, quantified by Western. For presentation graphically, densitometry in each lane has been normalized to β-actin and to control. B: nitration of eNOS is increased 1.4-fold in shunt LECs compared with control LECs, *P < 0.05. Following IP with an antibody against eNOS, the membrane was probed by IB for eNOS and 3-NT. Data are shown as a mean ratio of 3-NT-eNOS/eNOS ± SD. For all experiments, n = 3 control, 3 shunt.
Fig. 6.

Measurement of reactive oxygen species (ROS) in LECs. Micrographs (×10) of control (A) and shunt (B) LECs. Nuclei are stained with DAPI (blue), and activated (CellROX) fluorophore (red), marks ROS. C: ROS accumulation is increased 2.6-fold in shunt LECs (+CellROX) compared with control LECs (+CellROX), *P < 0.05. ROS was quantified using a fluorescence microplate reader; for each assay, n = 3 control and 3 shunt and were tested in duplicate; data are presented as mean signal intensities ± SD normalized to control. Note that, in the absence of CellROX, the signal is negligible in both control and shunt LECs (−CellROX). D: pharmacological inhibition of iNOS with the selective inhibitor N-{[3-(aminomethyl)phenyl]methyl}ethanimidamide dihydrochloride (1400W) results in a 2.5-fold reduction in ROS levels in shunt LECs (§P < 0.05) and is equivalent to ROS levels measured in control LECs. Note that iNOS inhibition does not affect ROS levels in control LECs; n = 3 control and 3 shunt and were tested in quadruplicate in the absence or presence of 20 μM 1400W for 6 h before addition of CellROX. E: pharmacological inhibition using the nonselective NOS inhibitor 2-(2-aminoethyl)isothiourea dihydrobromide (AET) results in 1.6-fold decreased accumulation of ROS in shunt LECs (§P < 0.05) and 2.9-fold decreased ROS in control LECs (†P < 0.05). For this assay, n = 2 control and 2 shunt and were tested in duplicate in the absence or presence of 100 μM AET for 24 h before addition of CellROX. All data are presented as mean signal intensities ± SD normalized to control.
To confirm that the additional accumulation of ROS was due to the increased iNOS activity in shunt LECs, we measured ROS in shunt LECs in the absence or presence of 1400W, a potent and specific inhibitor of iNOS. We found that, following treatment with 1400W, ROS levels decreased by 60% in shunt LECs, P < 0.05, and were equivalent to those measured in control LECs, with or without inhibitor (Fig. 6D). Interestingly, the eNOS that is present in LECs may also contribute to the accumulation of ROS. We measured ROS in control and shunt LECs that were treated with AET, an inhibitor of eNOS and iNOS. In the presence of AET, ROS levels decreased significantly in both control (66%) and shunt (36%) LECs (Fig. 6E). These data suggested that, whatever eNOS activity there is, may, in part, be uncoupled.
DISCUSSION
The regulation of endothelial gene expression through biomechanical forces is appreciated as a vital mechanism of normal and abnormal vascular tone and growth. Under normal conditions, the principal physiological stimulus of eNOS is laminar shear stress; activation of eNOS occurs within minutes of exposure and constantly adjusts the production of NO according to metabolic needs (4). However, chronic alterations in mechanical forces can disrupt these normal responses and result in endothelial dysfunction, with decreased bioavailable NO (4). In fact, decreased bioavailable NO is known to be central to the pathobiology of a wide array of vascular disorders and is often caused in large part by an increase in oxidant stress (8). Previously, we demonstrated decreased NOx levels in the pulmonary lymph effluent of shunt lambs. These shunt lambs are chronically exposed to two times the normal pulmonary lymph flow as control lambs, the associated doubling of the hydrostatic driving force in the pulmonary capillaries, and likely altered mechanical forces within the lymphatic circulation such as increased shear stress (12). Recently, others have shown that shear stress is also important for normal lymphatic development, remodeling, and function (47, 48, 54). In the current study, using LECs cultured from both control and shunt lambs, we elucidated potential mechanisms for the decreased NOx levels and impaired endothelium-dependent relaxation of the thoracic duct rings demonstrated in shunt lambs (12, 13): decreased eNOS expression and activity, increased iNOS expression and activity, and increased ROS and peroxynitrite production.
eNOS transcriptional regulation is a balance of a number of competing mechanisms (56). For example, as in other vascular endothelium, in cultured human lymphatic endothelial cells, even brief exposure to laminar shear stress (1–4 h) causes increased expression of both eNOS mRNA and protein (35), whereas hypoxia and thrombin can promote transcriptional repression of eNOS (56). Interestingly, shunt LECs are exposed to elevated pulmonary lymph flow in vivo for weeks, yet eNOS mRNA was decreased 4-fold and eNOS protein was decreased 3.2-fold compared with control LECs (Fig. 2, B and C). The duration of exposure to biomechanical forces may help explain this difference. For example, the rate of eNOS transcription returns to baseline with prolonged exposure to shear (14). The exact mechanisms by which chronic exposure to mechanical forces results in decreased eNOS transcription in vivo are unclear. Potential mechanisms include epigenetic changes that alter eNOS transcriptional stability, through decreased 3′-polyadenylation, antisense or micro-RNAs (52, 70), or changes in the efficiency of eNOS transcription, secondary to alterations to DNA methylation or modification of chromatin structure (9, 20). Of note, shunt lambs are not hypoxic; in fact, the aortopulmonary graft creates a persistent left to right shunt with increased oxygen content in the pulmonary vasculature (51). Interestingly, we have observed increased thrombin expression in the pulmonary vasculature of shunt lambs (unpublished observations), which may be a potential mechanism of eNOS transcriptional suppression in shunt LECs. Last, VEGF-c/vascular endothelial growth factor receptor 3 (VEGFR3) signaling has been implicated in promoting eNOS expression (66). It is interesting to note that VEGF-c expression is decreased in peripheral lung homogenate from shunt lambs (12), but the role of VEGF-c/VEGFR3 signaling and other potential mechanisms for the decrease in eNOS expression in shunt LECs warrant further study.
An increasing number of studies implicate oxidative stress in the pathogenesis of cardiovascular disease and the development of endothelial dysfunction (8, 34). The endothelium is a source of oxygen-derived free radical production, especially the superoxide anion (40, 41). NADPH oxidase and eNOS are two major sources of ROS, specifically superoxide and peroxynitrite (1, 50, 63, 71). In the current study, we demonstrate an accumulation of ROS in shunt LECs (2.6-fold higher than in control LECs; Fig. 6C); however, this is despite decreased eNOS expression and activity (Figs. 2B, 2C, and 3). Instead, nearly all of the increase in NOS activity and a majority of the ROS generation is due to an induction of iNOS (Figs. 2D, 3A, and 6C); indeed, pharmacological inhibition of iNOS decreased ROS accumulation to baseline levels observed in control LECs (Fig. 6D).
Unlike eNOS and nNOS, activation of iNOS does not require increases in Ca2+ to bind calmodulin (31). It is predominantly under transcriptional regulation by, among others, NF-κB, activator protein-1 (AP-1), and JAK-signal transducer and activator of transcription (STAT) signaling (2, 31, 33), often as part of an inflammatory response to infection. Of note, shunt lambs do not seem to show increased signs of inflammation or infection (Fineman, unpublished observations). It is interesting to note the relationship between iNOS and the peroxisome proliferator-activated receptor (PPAR) family of transcription factors. PPARs can antagonize AP-1, STAT, and NF-κB (10) and thus suppress iNOS expression and activity (58). We have previously shown that endothelial dysfunction in the pulmonary vasculature is associated with downregulation of PPARγ in the lungs of shunt lambs, and that, in this setting, a PPARγ agonist can preserve vascular function, in part by decreasing oxidative stress (45). Whether PPARγ plays a role in regulating iNOS expression and the accumulation of ROS and peroxynitrite in shunt LECs warrants further study.
Once expressed, iNOS is much more active than eNOS or nNOS, and thus can generate a large amount of NO that more often leads to the production of peroxynitrite, a very potent ROS (2). Peroxynitrite-mediated tyrosine nitration can also accelerate protein degradation (59). In the current study we have shown increased tyrosine nitration of eNOS in shunt LECs (Fig. 5B), which could further contribute to decreased eNOS protein levels in shunt LECs (Fig. 2C).
A number of factors such as intracellular location, protein-protein interactions, posttranslational modification, like nitration, and cofactor availability can all dynamically regulate eNOS activity (3, 18, 21, 22, 29, 46, 49, 57). For example, binding of eNOS by caveolin-1 inhibits eNOS activity by physically blocking the binding site for calmodulin, which is necessary for eNOS activity. HSP90 is a chaperone protein that promotes eNOS activity by supporting activating phosphorylation events and enhancing eNOS-calmodulin binding. In shunt LECs, we found that eNOS association with caveolin-1 decreased while eNOS association with HSP90 and calmodulin increased (Fig. 4), suggesting that there might be an effort to adapt to the decrease in eNOS mRNA and protein. However, a phosphorylation event at Ser1177, important for eNOS activation, was decreased in shunt LECs (Fig. 4); thus, on balance, despite some favorable protein-protein interactions with HSP90 and calmodulin, the decrease in eNOS expression and activity likely contributes to the measured decrease in bioavailable pulmonary lymphatic NO in shunt lambs (12).
Furthermore, eNOS can become uncoupled and itself produce superoxide and peroxynitrite if, for example, the stoichiometry becomes unbalanced (17), such as if there is insufficient cofactor (tetrahydrobiopterin) or substrate (l-arginine) (36), or if the balance of eNOS (de)phosphorylation shifts (37). These and other conditions can lead to increased superoxide production, superoxide-mediated scavenging of NO, and formation of peroxynitrite. Peroxynitrite can itself uncouple eNOS by disrupting its zinc-thiolate cluster, creating more superoxide (71). The exact mechanisms for eNOS uncoupling in shunt LECs are unclear, requiring further studies. Interestingly, eNOS-dependent superoxide production requires calmodulin binding (67), which was increased in shunt LECs (Fig. 4B).
Taken together, our data suggest the following model that leads to dysfunction of lymphatic endothelium in shunt lambs: chronically elevated PBF leads to increased hydrostatic forces and increased pulmonary lymph flow. Lymphatic endothelial cells exposed to this higher pulmonary lymph flow experience altered biomechanical forces that result in disrupted NO signaling. This is marked by 1) downregulation of eNOS transcript, protein, and activity and 2) upregulation of iNOS expression and activity that contributes to accumulation of ROS and further scavenging of NO, with peroxynitrite production and increased eNOS nitration. Both mechanisms serve to decrease bioavailable NO in the lymphatic vasculature, leading to impaired pulmonary lymphatic pumping and flow. Pulmonary lymphatic dysfunction in patients with congenital cardiac defects associated with increased PBF may persist for quite some time even after undergoing complete surgical repair. Therefore, therapies that target and augment lymphatic endothelial NO signaling may have benefit for these patients in the immediate postsurgical period and during outpatient convalescence, warranting further study.
GRANTS
This research was supported in part by grants from the National Institutes of Health (NIH) (K08-HL-116763 to S. A. Datar and HL-61284 to J. R. Fineman) and from the American Heart Association (12BGIA11540021 to S. A. Datar). Flow cytometry analysis was supported in part by the DRC Center Grant, NIH P30-DK-063720.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.A.D. and J.R.F. conception and design of research; S.A.D., W.G., Y.H., M.J.J., R.J.K., G.W.R., and P.E.O. performed experiments; S.A.D., W.G., and Y.H. analyzed data; S.A.D. and E.M. interpreted results of experiments; S.A.D. prepared figures; S.A.D. drafted manuscript; S.A.D., W.G., R.J.K., E.M., P.E.O., and J.R.F. edited and revised manuscript; S.A.D., W.G., Y.H., M.J.J., R.J.K., G.W.R., E.M., P.E.O., and J.R.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful to Linda Talken for care of the animals, Christine Sun for technical support, Ninnia Lescano for assistance with flow cytometry, Martina Steurer-Müller for statistical advice, and Jason Boehme for thoughtful discussions.
REFERENCES
- 1.Aggarwal S, Gross CM, Sharma S, Fineman JR, Black SM. Reactive oxygen species in pulmonary vascular remodeling. Comp Physiol 3: 1011–1034, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci 75: 639–653, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res 43: 521–531, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Balligand JL, Feron O, Dessy C. eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev 89: 481–534, 2009. [DOI] [PubMed] [Google Scholar]
- 5.Black SM, Bekker JM, Johengen MJ, Parry AJ, Soifer SJ, Fineman JR. Altered regulation of the ET-1 cascade in lambs with increased pulmonary blood flow and pulmonary hypertension. Pediatr Res 47: 97–106, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Black SM, Heidersbach RS, McMullan DM, Bekker JM, Johengen MJ, Fineman JR. Inhaled nitric oxide inhibits NOS activity in lambs: potential mechanism for rebound pulmonary hypertension. Am J Physiol Heart Circ Physiol 277: H1849–H1856, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Bohlen HG, Wang W, Gashev A, Gasheva O, Zawieja D. Phasic contractions of rat mesenteric lymphatics increase basal and phasic nitric oxide generation in vivo. Am J Physiol Heart Circ Physiol 297: H1319–H1328, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Chan Y, Fish JE, D'Abreo C, Lin S, Robb GB, Teichert AM, Karantzoulis-Fegaras F, Keightley A, Steer BM, Marsden PA. The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J Biol Chem 279: 35087–35100, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Chinetti G, Griglio S, Antonucci M, Torra IP, Delerive P, Majd Z, Fruchart JC, Chapman J, Najib J, Staels B. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J Biol Chem 273: 25573–25580, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Corbett CR, Dale RF, Coltart DJ, Kinmonth JB. Congenital heart disease in patients with primary lymphedemas. Lymphology 15: 85–90, 1982. [PubMed] [Google Scholar]
- 12.Datar SA, Johnson EG, Oishi PE, Johengen M, Tang E, Aramburo A, Barton J, Kuo HC, Bennett S, Xoinis K, Reel B, Kalkan G, Sajti E, Osorio O, Raff GW, Matthay MA, Fineman JR. Altered lymphatics in an ovine model of congenital heart disease with increased pulmonary blood flow. Am J Physiol Lung Cell Mol Physiol 302: L530–L540, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Datar SA, Oishi PE, Gong W, Bennett SH, Sun CE, Johengen M, Maki J, Johnson RC, Raff GW, Fineman JR. Altered reactivity and nitric oxide signaling in the isolated thoracic duct from an ovine model of congenital heart disease with increased pulmonary blood flow. Am J Physiol Heart Circ Physiol 306: H954–H962, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis ME, Cai H, Drummond GR, Harrison DG. Shear stress regulates endothelial nitric oxide synthase expression through c-Src by divergent signaling pathways. Circ Res 89: 1073–1080, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Davis MJ, Davis AM, Ku CW, Gashev AA. Myogenic constriction and dilation of isolated lymphatic vessels. Am J Physiol Heart Circ Physiol 296: H293–H302, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis MJ, Scallan JP, Wolpers JH, Muthuchamy M, Gashev AA, Zawieja DC. Intrinsic increase in lymphangion muscle contractility in response to elevated afterload. Am J Physiol Heart Circ Physiol 303: H795–H808, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol 46: 235–276, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res 75: 247–260, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dutka DP, Cousins C, Manhire AR. Lymphatic abnormalities in Alagille's syndrome. Br Heart J 65: 168–170, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fish JE, Matouk CC, Rachlis A, Lin S, Tai SC, D'Abreo C, Marsden PA. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem 280: 24824–24838, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Fleming I. Signal transduction of eNOS activation. Cardiovasc Res 43: 532–541, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn't calcium/calmodulin enough? J Pharmacol Exp Ther 299: 818–824, 2001. [PubMed] [Google Scholar]
- 23.Gashev AA. Lymphatic vessels: pressure- and flow-dependent regulatory reactions. Ann NY Acad Sci 1131: 100–109, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Gashev AA, Davis MJ, Delp MD, Zawieja DC. Regional variations of contractile activity in isolated rat lymphatics. Microcirculation 11: 477–492, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Gashev AA, Davis MJ, Zawieja DC. Inhibition of the active lymph pump by flow in rat mesenteric lymphatics and thoracic duct. J Physiol (Lond) 540: 1023–1037, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gashev AA, Delp MD, Zawieja DC. Inhibition of active lymph pump by simulated microgravity in rats. Am J Physiol Heart Circ Physiol 290: H2295–H2308, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Gasheva OY, Gashev AA, Zawieja DC. cGMP/PKG-mediated regulation of lymphatic contractility in rat thoracic duct. J Physiol In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gasheva OY, Zawieja DC, Gashev AA. Contraction-initiated NO-dependent lymphatic relaxation: a self-regulatory mechanism in rat thoracic duct. J Physiol (Lond) 575: 821–832, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Govers R, Rabelink TJ. Cellular regulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol 280: F193–F206, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Graziano JN, Heidelberger KP, Ensing GJ, Gomez CA, Ludomirsky A. The influence of a restrictive atrial septal defect on pulmonary vascular morphology in patients with hypoplastic left heart syndrome. Pediatr Cardiol 23: 146–151, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J Physiol Pharmacol 54: 469–487, 2003. [PubMed] [Google Scholar]
- 32.Hagendoorn J, Padera TP, Kashiwagi S, Isaka N, Noda F, Lin MI, Huang PL, Sessa WC, Fukumura D, Jain RK. Endothelial nitric oxide synthase regulates microlymphatic flow via collecting lymphatics. Circ Res 95: 204–209, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Hecker M, Cattaruzza M, Wagner AH. Regulation of inducible nitric oxide synthase gene expression in vascular smooth muscle cells. Gen Pharmacol 32: 9–16, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A. eNOS uncoupling in cardiovascular diseases–the role of oxidative stress and inflammation. Curr Pharm Des 20: 3579–3594, 2014. [DOI] [PubMed] [Google Scholar]
- 35.Kawai Y, Yokoyama Y, Kaidoh M, Ohhashi T. Shear stress-induced ATP-mediated endothelial constitutive nitric oxide synthase expression in human lymphatic endothelial cells. Am J Physiol Cell Physiol 298: C647–C655, 2010. [DOI] [PubMed] [Google Scholar]
- 36.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111: 1201–1209, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin MI, Fulton D, Babbitt R, Fleming I, Busse R, Pritchard KA Jr, Sessa WC. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem 278: 44719–44726, 2003. [DOI] [PubMed] [Google Scholar]
- 38.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Loscalzo ML, Van PL, Ho VB, Bakalov VK, Rosing DR, Malone CA, Dietz HC, Bondy CA. Association between fetal lymphedema and congenital cardiovascular defects in Turner syndrome. Pediatrics 115: 732–735, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Matsubara T, Ziff M. Increased superoxide anion release from human endothelial cells in response to cytokines. J Immunol 137: 3295–3298, 1986. [PubMed] [Google Scholar]
- 41.Matsubara T, Ziff M. Superoxide anion release by human endothelial cells: synergism between a phorbol ester and a calcium ionophore. J Cell Physiol 127: 207–210, 1986. [DOI] [PubMed] [Google Scholar]
- 42.McMullan DM, Bekker JM, Johengen MJ, Hendricks-Munoz K, Gerrets R, Black SM, Fineman JR. Inhaled nitric oxide-induced rebound pulmonary hypertension: role for endothelin-1. Am J Physiol Heart Circ Physiol 280: H777–H785, 2001. [DOI] [PubMed] [Google Scholar]
- 43.McMullan DM, Bekker JM, Parry AJ, Johengen MJ, Kon A, Heidersbach RS, Black SM, Fineman JR. Alterations in endogenous nitric oxide production after cardiopulmonary bypass in lambs with normal and increased pulmonary blood flow. Circulation 102: III172–III178, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Noonan JA, Walters LR, Reeves JT. Congenital pulmonary lymphangiectasis. Am J Dis Child 120: 314–319, 1970. [DOI] [PubMed] [Google Scholar]
- 45.Oishi PE, Sharma S, Datar SA, Kumar S, Aggarwal S, Lu Q, Raff G, Azakie A, Hsu JH, Sajti E, Fratz S, Black SM, Fineman JR. Rosiglitazone preserves pulmonary vascular function in lambs with increased pulmonary blood flow. Pediatr Res 73: 54–61, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Papapetropoulos A, Rudic RD, Sessa WC. Molecular control of nitric oxide synthases in the cardiovascular system. Cardiovasc Res 43: 509–520, 1999. [DOI] [PubMed] [Google Scholar]
- 47.Planas-Paz L, Lammert E. Mechanical forces in lymphatic vascular development and disease. Cell Mol Life Sci 70: 4341–4354, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Planas-Paz L, Strilic B, Goedecke A, Breier G, Fassler R, Lammert E. Mechanoinduction of lymph vessel expansion. EMBO J 31: 788–804, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pritchard K, Stepp D, Konduri G. Inhibition of heatshock protein 90 activity impairs vasorelaxation by increasing superoxide anion generation by endothelial nitric oxide synthase (eNOS) (Abstract). Circulation 500, 2001. [Google Scholar]
- 50.Pritchard KA, Groszek L, Smalley DM, Sessa WC, Wu M, Villalon P, Wolin MS, Stemerman MB. Native low-density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ Res 77: 510–518, 1995. [DOI] [PubMed] [Google Scholar]
- 51.Reddy VM, Meyrick B, Wong J, Khoor A, Liddicoat JR, Hanley FL, Fineman JR. In utero placement of aortopulmonary shunts. A model of postnatal pulmonary hypertension with increased pulmonary blood flow in lambs. Circulation 92: 606–613, 1995. [DOI] [PubMed] [Google Scholar]
- 52.Robb GB, Carson AR, Tai SC, Fish JE, Singh S, Yamada T, Scherer SW, Nakabayashi K, Marsden PA. Post-transcriptional regulation of endothelial nitric-oxide synthase by an overlapping antisense mRNA transcript. J Biol Chem 279: 37982–37996, 2004. [DOI] [PubMed] [Google Scholar]
- 53.Rychik J, Rome JJ, Collins MH, DeCampli WM, Spray TL. The hypoplastic left heart syndrome with intact atrial septum: atrial morphology, pulmonary vascular histopathology and outcome. J Am Coll Cardiol 34: 554–560, 1999. [DOI] [PubMed] [Google Scholar]
- 54.Sabine A, Agalarov Y, Maby-El Hajjami H, Jaquet M, Hagerling R, Pollmann C, Bebber D, Pfenniger A, Miura N, Dormond O, Calmes JM, Adams RH, Makinen T, Kiefer F, Kwak BR, Petrova TV. Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev Cell 22: 430–445, 2012. [DOI] [PubMed] [Google Scholar]
- 55.Scallan JP, Wolpers JH, Muthuchamy M, Zawieja DC, Gashev AA, Davis MJ. Independent and interactive effects of preload and afterload on the pump function of the isolated lymphangion. Am J Physiol Heart Circ Physiol 303: H809–H824, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Searles CD. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am J Physiol Cell Physiol 291: C803–C816, 2006. [DOI] [PubMed] [Google Scholar]
- 57.Shaul PW. Regulation of endothelial nitric oxide synthase: location, location, location. Annu Rev Physiol 64: 749–774, 2002. [DOI] [PubMed] [Google Scholar]
- 58.Slomiany BL, Slomiany A. Activation of peroxisome proliferator-activated receptor gamma suppresses inducible cyclooxygenase and nitric oxide synthase during oral mucosal ulcer healing. J Physiol Pharmacol 53: 159–169, 2002. [PubMed] [Google Scholar]
- 59.Souza JM, Choi I, Chen Q, Weisse M, Daikhin E, Yudkoff M, Obin M, Ara J, Horwitz J, Ischiropoulos H. Proteolytic degradation of tyrosine nitrated proteins. Arch Biochem Biophys 380: 360–366, 2000. [DOI] [PubMed] [Google Scholar]
- 60.Staub NC, Bland RD, Brigham KL, Demling R, Erdmann AJ 3rd, Woolverton WC. Preparation of chronic lung lymph fistulas in sheep. J Surg Res 19: 315–320, 1975. [DOI] [PubMed] [Google Scholar]
- 61.Tsunemoto H, Ikomi F, Ohhashi T. Flow-mediated release of nitric oxide from lymphatic endothelial cells of pressurized canine thoracic duct. Jpn J Physiol 53: 157–163, 2003. [DOI] [PubMed] [Google Scholar]
- 62.Vasquez JC, Rabah R, Delius RE, Walters HL. Hypoplastic left heart syndrome with intact atrial septum associated with deletion of the short arm of chromosome 18. Cardiovasc Pathol 12: 102–104, 2003. [DOI] [PubMed] [Google Scholar]
- 63.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA Jr.. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA 95: 9220–9225, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wedgwood S, Bekker JM, Black SM. Shear stress regulation of endothelial NOS in fetal pulmonary arterial endothelial cells involves PKC. Am J Physiol Lung Cell Mol Physiol 281: L490–L498, 2001. [DOI] [PubMed] [Google Scholar]
- 65.Wert SE. Normal and Abnormal Structural Development of the Lung. In: Fetal and Neonatal Physiology, edited by Polin RA, Fox WW, and Abman SH. Philadelphia, PA: Elsevier/Saunders, 2011, p. 874. [Google Scholar]
- 66.Wiig H, Schroder A, Neuhofer W, Jantsch J, Kopp C, Karlsen TV, Boschmann M, Goss J, Bry M, Rakova N, Dahlmann A, Brenner S, Tenstad O, Nurmi H, Mervaala E, Wagner H, Beck FX, Muller DN, Kerjaschki D, Luft FC, Harrison DG, Alitalo K, Titze J. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest 123: 2803–2815, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem 273: 25804–25808, 1998. [DOI] [PubMed] [Google Scholar]
- 68.Zawieja D. Contractile physiology of lymphatics. Lymph Res Biol 7: 87–96, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeballos GA, Bernstein RD, Thompson CI, Forfia PR, Seyedi N, Shen W, Kaminiski PM, Wolin MS, Hintze TH. Pharmacodynamics of plasma nitrate/nitrite as an indication of nitric oxide formation in conscious dogs. Circulation 91: 2982–2988, 1995. [DOI] [PubMed] [Google Scholar]
- 70.Zhang MX, Ou H, Shen YH, Wang J, Wang J, Coselli J, Wang XL. Regulation of endothelial nitric oxide synthase by small RNA. Proc Natl Acad Sci USA 102: 16967–16972, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest 109: 817–826, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]