

Abstract
Following myocardial infarction (MI), the left ventricle (LV) undergoes a series of cardiac wound healing responses that involve both the stimulation of robust inflammation to clear necrotic myocytes and tissue debris and the induction of extracellular matrix (ECM) protein synthesis to generate an infarct scar. The collective changes in myocardial structure and function are termed LV remodeling, and matrix metalloproteinase-9 (MMP-9) is a key instigator of post-MI LV remodeling. Through direct molecular effects on ECM and inflammatory protein turnover as well as indirect effects on major cell types that coordinate cardiac wound healing, namely the infiltrating leukocytes and the cardiac fibroblasts, MMP-9 coordinates multiple aspects of LV remodeling. In this review, we will discuss recent research that has expanded our understanding of post-MI LV remodeling, including recent proteomic advances focused on the ECM compartment to provide novel functional and translational insights. This overview will summarize how our understanding of MMP-9 has evolved over the last decade and will provide insight into future directions that will drive our understanding of MMP-9-directed cardiac ECM turnover in the post-MI LV.
Keywords: matrix metalloproteinases-9, signaling, transcription factors, extracellular matrix, proteomics
this article is part of a collection on Annual Meeting of the International Academy of Cardiovascular Sciences: North American Section. Other articles appearing in this collection, as well as a full archive of all collections, can be found online at http://ajpheart.physiology.org/.
The matrix metalloproteinase (MMP) family is comprised of zinc-dependent endopeptidase enzymes that share similar sequence homology and structure, and all MMPs by definition can cleave at least one extracellular matrix (ECM) protein. Based on domain structures and substrate preferences, MMPs can loosely be catalogued into one of six classes: 1) collagenases MMP-1, -8, and -13; 2) stromelysins MMP-3 and MMP-10; 3) gelatinases MMP-2 and MMP-9; 4) matrilysins MMP-7 and MMP-26; 5) six membrane-type MMPs; and 6) other types (e.g., MMP-28). Of note, this classification is an imprecise catalogue, as MT1-MMP (MMP-14), for example, fits into both the collagenase and MT-MMP groups (72). MMPs are expressed by a broad array of cell types, including the cardiac-relevant neutrophils, macrophages, fibroblasts, cardiomyocytes, endothelial cells, and lymphocytes (6, 95). Of the MMPs that have been evaluated in the setting of cardiovascular disease, MMP-9 plays a pivotal role in atherosclerosis, hypertension, myocardial infarction (MI), and heart failure. Under physiological conditions, MMP-9 expression at the gene and protein levels is low; MMP-9 is robustly elevated under pathological conditions including multiple cardiovascular diseases.
This review article will provide an overview summary of MMP-9 signaling at the molecular, cellular, and tissue levels to provide insight into MMP-9 pathogenic roles in the post-MI left ventricle (LV; Fig. 1). Delineating modulation at each level provides a foundation on which to form the global network of MMP-9 actions and can serve as a template for the examination of other MMPs in the system.
Fig. 1.
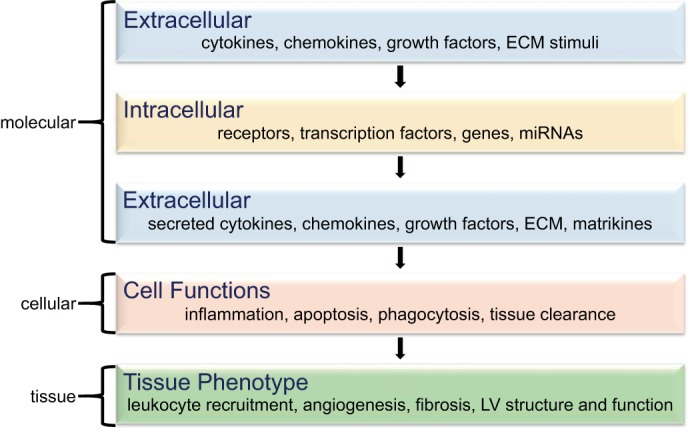
Simplified matrix metalloproteinase-9 (MMP-9) signaling network, with molecular inputs in layers 1, 2, and 3 and cellular and tissue level phenotype outputs in layers 4 and 5. ECM, extracellular matrix; LV, Left ventricular; miRNA, microRNAs.
Molecular Level MMP-9 Signaling
Figure 2 depicts the signaling cascade from stimulation of MMP-9 synthesis to its activation and proteolytic processing of a wide range of MI-relevant substrates. By in vitro cleavage assay or in silico evaluation, the MMP-9 substrate list includes ECM proteins (e.g., collagen, fibronectin, laminin, thrombospondin and tenascin-C), non-ECM substrates including a variety of cytokines and chemokines [e.g., interleukin (IL)-1β, CXC motif ligand (CXCL)1, CXCL4, CXCL5, CXCL7 and CXCL12], and novel substrates (e.g., CD36 and citrate synthase) (24, 25, 43, 47, 61, 62, 74, 106).
Fig. 2.
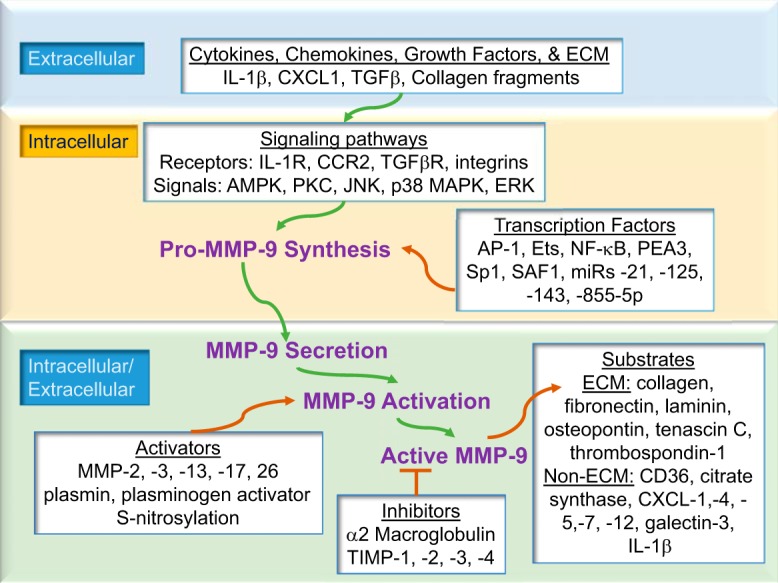
MMP-9 signaling begins at the extracellular-cell membrane interface, where cytokines, chemokines, growth factors, or ECM proteins engage cell surface receptors to transmit signaling cascades that culminate in MMP-9 release, activation, and proteolysis of substrates. The green arrows depict the signaling pathway, and the orange arrows depict modifiers of the pathway.
Stimuli that upregulate MMP-9 synthesis.
There are a number of well-known stimuli that elevate MMP-9 gene levels, including cytokines, chemokines, growth factors, and ECM proteins (10, 32, 33, 91, 103, 107). Below, we provide one example for a cytokine and one for a growth factor that demonstrate bidirectional signaling with MMP-9.
Tumor necrosis factor-α (TNF-α) is a classical proinflammatory cytokine synthesized as a membrane-bound protein that is subsequently cleaved to release a soluble form. Soluble TNF-α circulates as a stable homotrimer (10). Soluble TNF-α is an endogenous mediator of inflammatory processes leading to multiple cellular responses, including activation of genes involved in inflammatory and immunoregulatory responses, cell proliferation, and cell death (9). Elevated TNF-α levels in the LV leads to the local induction of myocardial MMP-9 and MMP-13 but not MMP-2 (10). Loss of TNF-α reduces post-MI leukocyte infiltration, a major MMP-9 source, and attenuates collagen degradation to decrease the incidence of LV rupture in mice (73). Partial inactivation of phosphatase and tensin homolog (PTEN) drives down post-MI TNF-α and MMP-9 expression, indicating that PTEN serves as a positive upstream modifier (73).
Transforming growth factor (TGF)-β1 is a pleiotropic factor with divergent roles in the post-MI setting (13). TGF-β1 has a bidirectional regulatory loop with MMP-9, such that their functional interplay is due to a heterotypic reciprocal interaction between the two factors. TGF-β1 needs to be proteolytically triggered by MMPs to exert its cellular functions, and activated TGF-β1 modulates the balance of ECM remodeling by controlling the expression of MMPs and their tissue inhibitors of metalloproteinases (TIMPs), particularly MMP-9 and TIMP-1 (50). MMP-9 initiates TGF-β1 signaling to maintain ECM integrity and stability (13). MMP-2 and MT1-MMP are also capable of activating TGF-β1 to maintain matrix integrity and stability (39, 48). In the mdx mouse model of muscular dystrophy, inhibition of MMP-9 by administration of a nuclear factor-κB (NF-κB) inhibitory peptide lowers the levels of active TGF-β1 concentrations and decreases cardiac fibrosis (55). Vaday and colleagues (32, 91, 107) showed that TGF-β1 acts as a potent suppressor of TNF-α-induced monocyte MMP-9 synthesis via a prostaglandin E2- and cAMP-dependent mechanism, indicating that TNF-α and TGF-β1 intersignal and that MMP-9 is both upstream and downstream of these signaling mechanisms. To delineate the precise role of MMP-9, it will be important to decipher the overall signaling network and the cross talk among the individual components.
Intracellular signaling cascades that stimulate MMP-9 expression.
Regulatory elements that are present on the MMP-9 gene include binding sites for NF-κB, activator protein-1 (AP-1), specificity protein 1 (Sp-1), serum amyloid A-activating factor (SAF)-1, E-twenty six (Ets) transcription factors, and polyomavirus enhancer A-binding protein-3 (PEA3) transcription factors (101). While we know these transcription factors control MMP-9 expression, whether these transcription factors have overlapping or distinct temporal and cellular expression profiles in the post-MI setting has not been entirely elucidated. For example, NF-κB and AP-1 have relevance in driving MMP-9 expression in neutrophils, macrophages, fibroblasts, and cardiomyocytes (17, 68, 87, 93); Sp-1 has been shown in neutrophils, macrophages, and myocytes (11, 53, 90), and SAF-1 is expressed in macrophages while Ets is found in fibroblasts (79, 85). Understanding the entire complement of regulatory elements of the MMP-9 gene within the different post-MI cell types over the time continuum of LV remodeling is still needed.
MMP promoter regions can be categorized into one of three groups based on the cis-element composition: 1) MMPs that contain a TATA box, an AP-1-binding site and an upstream PEA3-binding site to increase transcription by cytokines (e.g., MMP-9); 2) MMPs with a TATA box but no AP-1-binding site (e.g., MMP-8, -11, and -21); and 3) MMPs with multiple other transcription sites such as Sp-1 that binds to the GC box (e.g., MMP-2, -9, -14, and -28). (63) Note that MMP-9 falls in both the first and third groups due to binding sites for AP-1 and Sp-1 promoters (63).
The transcription factor NF-κB is a dimer protein that consists of five members: p50, p52, RelA/p65, c-Rel, and RelB (71). NF-κB activation and recruitment into the nucleus can be initiated by proinflammatory cytokines and growth factors. In vascular smooth muscle cells, IL-1α triggers NF-κB to strongly enhance MMP-9 expression (7, 21). The global absence of the NF-κB subunit p50 improves post-MI remodeling, in part by decreasing MMP-9 expression and lowering collagen content to improve survival and attenuate LV dilation (30).
In human carotid atherosclerotic plaques, CD147, a member of the immunoglobulin superfamily, mediates MMP-9 induction through extracellular signal-regulated kinase (ERK) and the nuclear translocation of NF-κB (46). In primary mouse cardiac fibroblasts, MMP-9 induction is mediated through multiple different transcription factors such as IκB kinase (IKK)/NF-κB, jun kinase (JNK)/AP-1, and specificity protein-1 (Sp1)-mediated reversion-inducing-cysteine-rich protein with kazal motifs (RECK) suppression (82). These studies provide strong evidence that NF-κB is a crucial transcription factor controlling MMP-9 synthesis. Depending on the cell type and stimulus used, the downstream mechanisms that follow NF-κB activation to enhance MMP-9 synthesis vary.
Overexpression of the ETS-related transcription factors EIA-F and PEA3 induce the MMP-9 promoter to increase MMP-9 synthesis (98). The fine tuning of MMP-9 expression is modified by the functional interplay among NF-κB, AP-1, and ETS factors (94), and Sp-1 inhibition can downregulate MMP-9 expression (70). SAF-1 is an inflammatory transcription factor that induces MMP-9 transcription through AP-1, and mutation of either SAF-1 or AP-1 compromises the transactivation potential and weakens cytokine responsiveness of the MMP-9 promoter (78).
MMP-9 can also be transcriptionally tuned by epigenetic mechanisms, including DNA methylation, histone acetylation, or chromatin remodeling (19). DNA methylation of cytosines within CpG islands in the promoter region inhibits MMP-9 gene expression, whereas chromatin remodeling promotes MMP-9 expression (19). Since LV remodeling involves both acute and chronic inflammation, the possibility that epigenetic mechanisms modify MMP-9 concentrations cannot be ruled out. Further studies are needed to understand the underlying epigenetic pathways that control MMP-9 gene expression.
MMP-9 expression can by posttranslationally fine-tuned by miRNAs. The ectopic expression of miR-885-5p or miR-491-5p reduces MMP-9 and inhibits cellular invasion in glioma cells (102). In multiple different types of cancer cells, miR-125, miR-21, miR-143, and miR-181 augment MMP-9 (56). Each of these miRNAs is also expressed in cardiac myocytes (18, 64, 77). This may provide another layer for MMP-9 release to be coordinated in a cell and time-dependent manner.
While we know that promoters among the structurally and functionally related MMP-2 and MMP-9 are distinct, MMP-9 transcription is not a well explored area in the setting of MI. This is due, in part, to the importance that has been placed on the activation step as the major point of regulation. Understanding how signal transduction cascades allow strict tuning of amplification, feedback, cross talk, and branching of the initial signals triggered among the different cell types to result in precise MMP-9 localization needs to be evaluated. Further studies are required to understand the full transcriptional control of MMP- 9 at the genomic level.
MMP-9 activation.
MMP-9 can be activated by a variety of endogenous and exogenous factors, including plasmin, heparin, chymase, and other MMPs such as MMP-3 (43). Historically, priority has been placed experimentally on measuring MMP activity as a marker of function. We now know that this may not be the perfect indicator, as zymograms evaluating activity in tissue samples do not take into consideration the presence of tissue inhibitors of metalloproteinases (TIMPs) and other endogenous inhibitors within the sample, and pro-MMP-9 can have activity when presented with substrate through a mechanism that does not require removal of the prodomain (43). While measuring pro- and active-MMP-9 amounts can give you an idea of recruitability and current activity potential, monitoring the actual generation of substrate cleavage products is a final confirmation of MMP activity.
MMP-9 inhibition.
Specific and selective MMP-9 inhibition has been difficult to achieve, for a variety of reasons that are well-reviewed elsewhere and briefly include the following: 1) promoter polymorphisms; 2) involvement of multiple MMPs in cardiovascular diseases; and 3) lack of knowledge of complete temporal and spatial profiles for each MMP and members of other protease families (26, 34, 43, 101, 105). Fields laboratory (52) has recently constructed an inhibitor with a Ki of 0.98 ± 0.09 nM for MMP-9; at low dose, this inhibitor is selective for MMP-9 over MMP-2, which has a Ki of 2.24 ± 0.11 nM. Studies evaluating the effects of this MMP-9 inhibitor on post-MI remodeling are warranted.
Cellular Sources of MMP-9
In the post-MI LV, neutrophils and macrophages are predominant early sources of MMP-9. In addition, fibroblasts, cardiomyocytes, and endothelial cells are relevant sources. Signaling initiated during the post-MI inflammatory response strongly stimulates MMP-9 in a variety of cell types (23, 67).
Neutrophils.
The post-MI inflammatory response includes robust leukocyte infiltration, and the neutrophils are an early leukocyte type to home to the infarct. Within 15 min of reperfusion, neutrophils are found in the infarcted LV and provide a first source of MMP-9 (58). Activated neutrophils degranulate to quickly release preformed MMP-9 (16, 20). The inactive pro-form of MMP-9 is stored in neutrophil gelatinase granules, indicating that MMP-9 derived from neutrophils is primarily posttranslationally controlled. MMP-9 release from neutrophils is stimulated by the phorbol ester formyl-Met-Leu-Phe, TNF-α, and IL-8 (15). In the extracellular space, pro-MMP-9 is activated by serine elastase and other proteinases (57, 58). Neutrophils isolated from human blood and stimulated with IL-8 showed robust MMP-9 release that was mediated through CXCR2 induction of two downstream signaling pathways, the first involving protein kinase C and ERK1/2 and the second involving Src-family kinases (15).
In addition to being released from neutrophils to degrade collagen, fibronectin, and other ECM components necessary for clearance of necrotic myocardium, MMP-9 can degrade intracellular proteins, including actin, tubulin, annexin 1, and high mobility group box 1 (HMGB1) protein, indicating that active MMP-9 may serve a protection feature to limit the injury induced by damage-associated molecular patterns (DAMPs) released from ischemic cells (14, 49). At the same time, MMP-9 signals back to the neutrophil to prevent apoptosis, which prolongs inflammation (25). The in vitro stimulation of isolated neutrophils with MMP-9 lowers caspase-9 expression, indicating that MMP-9 can directly control neutrophil apoptosis (25). While the above studies provide evidence that MMP-9 is involved in multiple neutrophil functions, the precise signaling pathways are not yet known.
Monocytes and macrophages.
Macrophages in the post-MI LV originate from circulating blood monocytes, and the conversion of monocytes to macrophages is accompanied by the induction of several cytokines, chemokines, and proteases. For example TNF-α, TGF-β, IL-1, MMPs -1, -2, -3, -7, -8, -9, -12, -14, and -28 as well as TIMPs -1, -2, -3, and -4 are markers of differentiated macrophages (100). A principal function of the macrophage post-MI is to enable wound healing and scar formation by phagocytosis of necrotic or apoptotic cells and by secretion of angiogenic molecules and growth factors (100).
While Mac-3-positive macrophages infiltrate into the infarct beginning at day 3 post-MI and peak at day 5 post-MI, Nahrendorf and colleagues (27, 31, 36, 38, 54, 59, 92) have reported an earlier pool that infiltrates beginning at the onset of ischemia. This indicates that the marker(s) used to identify the macrophage is important. In mice, MMPs in general and MMP-9 specifically are markers of proinflammatory M1 macrophages (8). Peripheral blood mononuclear cells from acute MI patients that were differentiated in vitro to macrophages produce high levels of MMP-9 compared with cells from healthy controls, indicating that macrophages are a significant cellular source of MMP-9 in humans as well (34).
Macrophage-specific transgenic overexpression of MMP-9 has been shown to improve post-MI cardiac function by blunting the inflammatory response and limiting ECM synthesis (105). On the other hand, targeted deletion of the MMP-9 gene also improves cardiac remodeling and survival post-MI albeit through different mechanisms. MMP-9 deletion stimulates the resolution of inflammation by increasing neutrophil apoptosis and macrophage phagocytosis and promoting neovascularization (25, 26, 37, 59). The fact that deletion and overexpression generate overall beneficial cardiac phenotypes indicates that LV remodeling can proceed through different pathways to end up at the same place.
Cardiac fibroblasts.
In addition to directly modulating macrophage function through multiple mechanisms, MMP-9 indirectly regulates fibroblast functions. MMP-9 secreted by M1 proinflammatory macrophages activates TGF-β to stimulate fibroblast proliferation (51). While fibroblasts are not the major source of MMP-9 in the post-MI LV, fibroblasts can elevate their MMP-9 in vitro expression in response to hypoxia or ischemia and oxidative stress (33, 83). The increase in MMP-9 expression in cardiac fibroblasts associates with a decrease in collagen synthesis rates. MMP-9 is also actively involved in cardiac fibroblast migration (97). MMP-9 levels increase in cardiac fibroblasts after stimulation with IL-1β or TNF-α through ERK1/2 and NF-κB signaling pathways (12). A common signaling pathway for MMP-9 synthesis in cardiac fibroblasts involves Notch 3 upregulation (107). Zhang et al. (107) showed that in post-MI mice, Notch3 siRNA augments MMP-9 expression levels to reduce cardiac fibrosis post-MI. Furthermore, Notch3 siRNA-dependent MMP-9 upregulation inhibited TGF-β1-induced fibroblast-myofibroblast transition, indicating that MMP-9 and downstream TGF-β1/Smad3 signaling are controlled by Notch 3. Notch 3 is activated by G protein-coupled receptors and the extracellular signal-regulated kinase pathway, which suggests that these signaling pathways may cross talk to trigger the downstream effects of MMP-9 (88).
Cardiac myocytes.
MI results in the dramatic loss of cardiac myocytes (74), and prolonged ischemia can cause myocyte vacuolization, often termed myocytolysis. Myocytolysis is characterized by cell swelling, myofibril lysis, and phagocytosis of necrotic myocytes (74). Ischemic cardiac myocytes are smaller in size with concomitant elevations in intracellular edema (74). Since myocardial ECM provides the structural integrity surrounding the myocyte, limiting MMP activity to the site of necrotic myocyte removal and keeping it at sufficient amounts are important (74). An imbalance between MMPs and TIMPs provides a major mechanism for the extension of infarction into the remote area (74). MMP-9 is expressed in infarcted myocytes post-MI (80). MMP-9 expression is induced in cultured rat cardiac myocytes exposed to a hypoxic environment (80). Aldosterone induces CaMKII, which can also increase the expression of MMP-9 in cardiomyocytes, indicating that MMP-9 production by myocytes occurs via several different mechanisms (35). Likewise, PPARβ/δ activation in cardiac myocytes inhibits reactive oxygen/nitrogen species generation and drives down MMP-9 gene expression (2). The PPARβ/δ agonist GW0742 markedly suppresses IL-1β-induced MMP-9 expression in vascular smooth muscle cells. PPAR signaling controls both MMP-2 and MMP-9, whereas TNF-α signaling controls only MMP-9 and not MMP-2 (2, 10). This stimulus-dependent selective induction of MMP-9 illustrates the fine tuning of a highly orchestrated process of MMP synthesis across a myriad of cellular sources.
Other cell types.
MMP-9 is also expressed in endothelial cells and lymphocytes (1). Endothelial cells are involved in the complex process of angiogenesis to form new blood vessels (81). Angiogensis includes multiple interactions between endothelial cells, surrounding smooth muscle cells, ECM, and angiogenic cytokines and growth factors (81). MMPs in general and MMP-9 specifically regulate the clearance of the ECM surrounding endothelial cells post-MI, which allows for cell proliferation and migration (4). VEGF initiates the release of pro-MMP-9 and induces migration of human CD34 endothelial progenitor and stem cells (33). This suggests that VEGF controls downstream MMP-9 signaling under hypoxic conditions. MMP-9 accelerates VEGF interaction with its receptor to trigger the angiogenic switch (4, 99).
In a chronic heart failure model, MMP-9 was shown to induce endothelial cell apoptosis and endothelial-myocyte uncoupling (74). At the same time, MMP-9 is required for adequate angiogenic revascularization of ischemic tissues and macrophage secreted MMP-9 is involved in capillary branching (45). In an indirect role, MMP-9 cleaves type IV collagen in the basement membrane to expose a cryptic regulatory peptide sequence that induces endothelial cell growth and migration (4.) MMP-9, therefore, is prominently involved in both inducing and limiting angiogenesis under pathophysiological conditions. This demonstrates the dual negative and positive functions of MMP-9.
Ras/mitogen-activated protein kinase (MAPK) signaling pathways may act as inhibitory signals for MMP-9 expression in T lymphocytes (29). Inhibition of phosphatidylinositide 3-kinases and MAPKs increase fibronectin-induced MMP-9 expression, revealing that inhibitory signals are transduced through Ras/Raf-1/MAPK pathways. Furthermore, upstream inhibition of the MAPK cascade by the G-protein inhibitor pertussis toxin enhances MMP-9 production (29).
Overall, a number of cell types contribute to the release of MMP-9 both in the LV infarct and remote regions. The cell source of MMP-9 is relevant, because MMP-9 function is dictated by the presence of substrates available for proteolytic processing. MMP, cell, and substrate presence are all managed in time and location. During the temporal evolution of remodeling, the orchestration among cells that both augment MMP-9 and provide a shift in substrates available merges to provide the net overall effect. For example, MMP-9 released from the neutrophil is high at day 1 post-MI, while MMP-9 from the macrophage is high at day 5 post-MI (58, 59). Likewise, IL-1β and other cytokines are elevated at day 1 post-MI, while TGF-β1 and other growth factors are elevated at day 5 post-MI (26, 75). Stimulation of neutrophil-derived MMP-9, therefore, would exert a greater effect on substrates present at day 1 and generate a different cell and tissue level phenotype compared with stimulation of macrophage-derived MMP-9.
MMP-9 inhibitors have been used to test effects at different stages of MI (42). The administration of the broad-spectrum MMP inhibitor CP-471,474 immediately post-MI decreased LV dilation 4 days post-MI in mice (74). CP-471,474 inhibits MMP-1, -2, -3, -9, and - 13. Administration of PD166793 in pigs 5 days after MI led to decrease in MI size and expansion rate by 2 wk post-MI (69). PD166793 inhibits MMP-1, -2, -3, -7, -9, -13, and -14. The MMP inhibitor PGE-530742 blocks MMP-2, -3, -9 and -13 reduces dilation after MI (104). The MMP inhibitor 2R-2-{5-[4-(ethyl-methylamino)phenyl] thiophene-2-sulfonylamino}-3-methylbutyric acid (TISAM) also showed beneficial changes to remodeling (66). In addition to MMP-2, TISAM also inhibits MMP-9, and -14. Doxycycline regulates coronary artery disease, in part by inhibiting MMP-2, -8, -9, and -13 (3, 42). The cannabinoid receptor antagonist rimonabant decreased MMP-9 activity and TGF-β1 expression in rats, leading to reduced collagen content and attenuation of fibrosis at 6 wk post-MI (84). Rimonabant regulated cardiac remodeling by indirect modulation of MMP expression and activity (84). Salvianolic acid A, a competitive inhibitor of MMP-9 prevented LV remodeling post-MI by preventing fibroblast proliferation and myofibroblast transdifferentiation (44). Even though these broad spectrum inhibitors showed a beneficial effect post-MI, clinical trials to date have not shown efficacy. This may be attributed to the lack of complete knowledge of protease roles in specific cell types and at specific times point post-MI. Cell-based targeting studies are needed to examine MMP-9 levels under a variety of conditions to provide information regarding the best time to promote or inhibit MMP-9.
Tissue Level Effects of MMP-9 on LV Function Post-MI
MI is an acute event that develops after prolonged disruption of blood supply leading to irreversible myocardial tissue injury (101). Acute MI is followed by a progressive wound healing process that can evolve to prolonged pathological remodeling of the LV. MMP-9 directs many aspects of the inflammatory and proliferative stages of acute MI (26). MMP-9 levels increase very early post-MI and remain high for the first week in both animal models of MI and in human patients with MI (25). The early increases in MMP-9 levels post-MI correlate with elevated leukocyte numbers and LV dimensions, as well as with impaired LV function (25, 101). Targeted deletion of MMP-9 reduces the number of macrophages post-MI leading to attenuated enlargement of the LV, prevents collagen accumulation, promotes neovascularization, and improves LV remodeling (34). Interestingly, transgenic overexpression of MMP-9 only in macrophages unexpectedly also showed improved cardiac function by stimulating inflammation resolution (105). These studies reveal that timing and cellular source of MMP-9 determine net function at the tissue level (Table 1).
Table 1.
A summary of MMP-9 effects in the left ventricle following myocardial infarction
| Model | Time of Post-MI Evaluation, days | Cellular Source | MMP-9 Expression | Post-MI Effects |
|---|---|---|---|---|
| MMP-9 null mice (25, 59, 76) | Up to 28 | Global | ↓ | Deletion: |
| ↑Neutrophil apoptosis, macrophage phagocytic potential, and neovascularization | ||||
| ↓Macrophage infiltration, collagen accumulation and LV enlargement and dysfunction | ||||
| Transgenic overexpression of MMP-9 in macrophages (105) | 5 | Macrophage | ↑ | Overexpression in macrophages: |
| ↓Inflammation, macrophage polarization, ECM synthesis, and LV dysfunction | ||||
| NF-κB p50 null mice (30) | 56 | Global deletion | ↓ | Deletion: |
| ↑Early survival | ||||
| ↓LV dilation | ||||
| Diabetic ischemia/reperfusion (5) | 30-min ischemia/2-h reperfusion | Global | ↑ | Diabetes: |
| ↑Vascular remodeling and cardiovascular complications |
MMP-9, matrix metalloproteinase-9; MI, myocardial infarction; LV, left ventricular; ECM, extracellular matrix.
Future Directions and Concluding Remarks
MMP-9 has both negative and positive effects in the post-MI LV. Thus any kind of therapeutic intervention targeting MMP-9 must be carefully evaluated. There are several areas where additional in vivo and in vitro studies are required to provide a more complete understanding of MMP-9 functions. The interplay among temporal and spatial effects on MMP-9 expression remains incompletely understood. MMP-9 interactions with other MMPs remains to be examined in terms of MMP competition for a particular substrate. Likewise, whether MMP-9 exhibits substrate preference when a variety of substrates are present has not been evaluated by competition assay. A more complete understanding of MMP-9 substrates is required to know which substrates are preferentially cleaved in the post-MI setting and what is the biological function of the MMP-9 generated substrate peptides (61).
There is a close association between MMP-9 expression and diabetes (41). Diabetes enhances vascular MMP-9 activity, presumably by increasing local oxidative stress (89). In animal MI models, MMP-9 expression is linked with diabetic microvascular complications. At 6 wk after induction of diabetes, increases in MMP-2 and MMP-9 were enhanced in the infarct region of diabetic rats following 30 min of ischemia and 2 h of reperfusion (5). Increased MMP-9 levels in ischemia/reperfusion-injured rats lead to vascular remodeling and cardiovascular complications (5). In a porcine model with streptozotocin-induced diabetes, MMP-2 and -9 levels decreased in serum and activities decreased in the LV myocardium compared with the control group (65). Diabetic patients have increased risk of developing heart failure post-MI (28). Human diabetic plaques showed increased MMP-9 levels and decreased collagen content, which could lead to vulnerable plaque formation (22). Hyperglycemia associates with impaired microvascular function and cardiac wound healing after MI (40). While the association between MMP-9 and diabetic complications is strong, whether MMP-9 could serve as a diagnostic marker for the progression of diabetic complications remains to be tested.
MMP-9 signaling is another area open for future investigation. The majority of studies to date use MMP-9 as an output measurement rather than an input signal (60). Experiments where MMP-9 itself is used as the input stimuli are needed to then evaluate downstream mechanisms. The MEROPS database assimilates information of MMPs and their substrates, including known cleavage sites (see http://merops.sanger.ac.uk) (105). By evaluating known cleavage sites in a variety of substrates, an MMP-9 consensus sequence has been generated (Fig. 3). A more complete understanding of MMP-9 and its substrate interactions will likely identify specific targets that could be inhibited or overexpressed to provide therapeutic means to improve LV remodeling or to serve as diagnostic early indicators of MI.
Fig. 3.
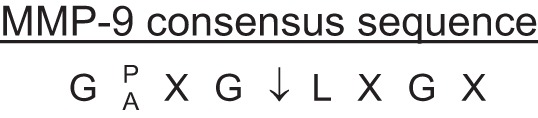
MMP-9 Substrate Consensus Sequences derived from analysis of known cleavage sites. Abbreviations for the amino acids are given, with X indicating any amino acid. Derived from http://merops.sanger.ac.uk.
In conclusion, MMP-9 is a complex protein that plays a key role in several molecular mechanisms operational across multiple cell types during the post-MI LV remodeling continuum (Fig. 4). MMP-9 targeted studies for selective inhibition (preferably within a specific cellular source) are warranted at both the in vitro and in vivo levels to elucidate the signaling mechanisms and provide new insights to improve outcomes for the post-MI patient.
Fig. 4.
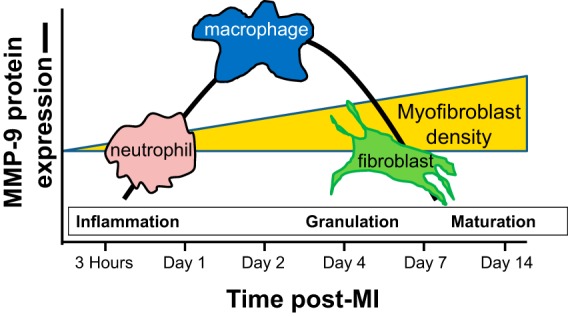
Diagram showing the temporal rise of MMP-9 post-myocardial infarction (MI) as it relates to the influx of neutrophils and macrophages and the activation of cardiac fibroblasts (34, 80, 86, 96).
GRANTS
This work was supported by American Heart Association Postdoctoral Grant 14POST18770012; National Institutes of Health Grants HL-075360, HL-129823, GM-114833, HL-051971, and GM-104357; and the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award 5I01BX000505.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
R.P.I., M.J., and M.L.L. edited and revised manuscript; R.P.I., M.J., and M.L.L. approved final version of manuscript; M.L.L. prepared figures; M.L.L. drafted manuscript.
REFERENCES
- 1.Baram D, Vaday GG, Salamon P, Drucker I, Hershkoviz R, Mekori YA. Human mast cells release metalloproteinase-9 on contact with activated T cells: juxtacrine regulation by TNF-alpha. J Immunol 167: 4008–4016, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Barlaka E, Gorbe A, Gaspar R, Paloczi J, Ferdinandy P, Lazou A. Activation of PPARbeta/delta protects cardiac myocytes from oxidative stress-induced apoptosis by suppressing generation of reactive oxygen/nitrogen species and expression of matrix metalloproteinases. Pharmacol Res 95–96: 102–110, 2015. [DOI] [PubMed] [Google Scholar]
- 3.Bench TJ, Jeremias A, Brown DL. Matrix metalloproteinase inhibition with tetracyclines for the treatment of coronary artery disease. Pharmacol Res 64: 561–566, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Bendeck MP. Macrophage matrix metalloproteinase-9 regulates angiogenesis in ischemic muscle. Circ Res 94: 138–139, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Bhatt LK, Veeranjaneyulu A. Enhancement of matrix metalloproteinase 2 and 9 inhibitory action of minocycline by aspirin: an approach to attenuate outcome of acute myocardial infarction in diabetes. Arch Med Res 45: 203–209, 2014. [DOI] [PubMed] [Google Scholar]
- 6.Bildyug NB, Voronkina IV, Smagina LV, Yudintseva NM, Pinaev GP. Matrix metalloproteinases in primary culture of cardiomyocytes. Biochemistry (Mosc) 80: 1318–1326, 2015. [DOI] [PubMed] [Google Scholar]
- 7.Bond M, Chase AJ, Baker AH, Newby AC. Inhibition of transcription factor NF-kappaB reduces matrix metalloproteinase-1, -3 and -9 production by vascular smooth muscle cells. Cardiovasc Res 50: 556–565, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Boorsma CE, Draijer C, Melgert BN. Macrophage heterogeneity in respiratory diseases. Mediators Inflamm 2013: 769214, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradham WS, Bozkurt B, Gunasinghe H, Mann D, Spinale FG. Tumor necrosis factor-alpha and myocardial remodeling in progression of heart failure: a current perspective. Cardiovasc Res 53: 822–830, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Bradham WS, Moe G, Wendt KA, Scott AA, Konig A, Romanova M, Naik G, Spinale FG. TNF-α and myocardial matrix metalloproteinases in heart failure: relationship to LV remodeling. Am J Physiol Heart Circ Physiol 282: H1288–H1295, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Brightbill HD, Plevy SE, Modlin RL, Smale ST. A prominent role for Sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J Immunol 164: 1940–1951, 2000. [DOI] [PubMed] [Google Scholar]
- 12.Brown RD, Jones GM, Laird RE, Hudson P, Long CS. Cytokines regulate matrix metalloproteinases and migration in cardiac fibroblasts. Biochem Biophys Res Commun 362: 200–205, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 74: 184–195, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cauwe B, Martens E, Proost P, Opdenakker G. Multidimensional degradomics identifies systemic autoantigens and intracellular matrix proteins as novel gelatinase B/MMP-9 substrates. Integr Biol (Camb) 1: 404–426, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Chakrabarti S, Patel KD. Regulation of matrix metalloproteinase-9 release from IL-8-stimulated human neutrophils. J Leukoc Biol 78: 279–288, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Chakrabarti S, Zee JM, Patel KD. Regulation of matrix metalloproteinase-9 (MMP-9) in TNF-stimulated neutrophils: novel pathways for tertiary granule release. J Leukoc Biol 79: 214–222, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Chandrasekar B, Freeman GL. Induction of nuclear factor kappaB and activation protein 1 in postischemic myocardium. FEBS Lett 401: 30–34, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Cheng Y, Zhang C. MicroRNA-21 in cardiovascular disease. J Cardiovasc Transl Res 3: 251–255, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chernov AV, Strongin AY. Epigenetic regulation of matrix metalloproteinases and their collagen substrates in cancer. Biomol Concepts 2: 135–147, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi KS, Grab DJ, Dumler JS. Anaplasma phagocytophilum infection induces protracted neutrophil degranulation. Infect Immun 72: 3680–3683, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou YC, Sheu JR, Chung CL, Chen CY, Lin FL, Hsu MJ, Kuo YH, Hsiao G. Nuclear-targeted inhibition of NF-kappaB on MMP-9 production by N-2-(4-bromophenyl) ethyl caffeamide in human monocytic cells. Chem Biol Interact 184: 403–412, 2010. [DOI] [PubMed] [Google Scholar]
- 22.Cipollone F, Iezzi A, Fazia M, Zucchelli M, Pini B, Cuccurullo C, De Cesare D, De Blasis G, Muraro R, Bei R, Chiarelli F, Schmidt AM, Cuccurullo F, Mezzetti A. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: role of glycemic control. Circulation 108: 1070–1077, 2003. [DOI] [PubMed] [Google Scholar]
- 23.Creemers EE, Cleutjens JP, Smits JF, Daemen MJ. Matrix metalloproteinase inhibition after myocardial infarction: a new approach to prevent heart failure? Circ Res 89: 201–210, 2001. [DOI] [PubMed] [Google Scholar]
- 24.de Castro Bras LE, Cates CA, DeLeon-Pennell KY, Ma Y, Iyer RP, Halade GV, Yabluchanskiy A, Fields GB, Weintraub ST, Lindsey ML. Citrate synthase is a novel in vivo matrix metalloproteinase-9 substrate that regulates mitochondrial function in the postmyocardial infarction left ventricle. Antioxid Redox Signal 21: 1974–1985, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeLeon-Pennell KY, Tian Y, Zhang B, Cates CA, Padmanabhan Iyer R, Cannon P, Shah P, Aiyetan P, Halade GV, Ma Y, Flynn E, Zhang Z, Jin YF, Zhang H, Lindsey ML. CD36 is a matrix metalloproteinase-9 substrate that stimulates neutrophil apoptosis and removal during cardiac remodeling. Circ Cardiovasc Genet 9: 14–25, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deten A, Volz HC, Briest W, Zimmer HG. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Cardiovasc Res 55: 329–340, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Dutta P, Nahrendorf M. Monocytes in myocardial infarction. Arterioscler Thromb Vasc Biol 35: 1066–1070, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res 66: 22–32, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Esparza J, Vilardell C, Calvo J, Juan M, Vives J, Urbano-Marquez A, Yague J, Cid MC. Fibronectin upregulates gelatinase B (MMP-9) and induces coordinated expression of gelatinase A (MMP-2) and its activator MT1-MMP (MMP-14) by human T lymphocyte cell lines. A process repressed through RAS/MAP kinase signaling pathways. Blood 94: 2754–2766, 1999. [PubMed] [Google Scholar]
- 30.Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, Bauersachs J. Absence of NF-kappaB subunit p50 improves heart failure after myocardial infarction. FASEB J 20: 1918–1920, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Frantz S, Nahrendorf M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res 102: 240–248, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao F, Chambon P, Offermanns S, Tellides G, Kong W, Zhang X, Li W. Disruption of TGF-beta signaling in smooth muscle cell prevents elastase-induced abdominal aortic aneurysm. Biochem Biophys Res Commun 454: 137–143, 2014. [DOI] [PubMed] [Google Scholar]
- 33.Gao Q, Guo M, Zeng W, Wang Y, Yang L, Pang X, Li H, Suo Y, Jiang X, Yu C. Matrix metalloproteinase 9 secreted by hypoxia cardiac fibroblasts triggers cardiac stem cell migration in vitro. Stem Cells Int 2015: 836390, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halade GV, Jin YF, Lindsey ML. Matrix metalloproteinase (MMP)-9: a proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol Ther 139: 32–40, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He BJ, Joiner ML, Singh MV, Luczak ED, Swaminathan PD, Koval OM, Kutschke W, Allamargot C, Yang J, Guan X, Zimmerman K, Grumbach IM, Weiss RM, Spitz DR, Sigmund CD, Blankesteijn WM, Heymans S, Mohler PJ, Anderson ME. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med 17: 1610–1618, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, van der Laan AM, Swirski FK, Weissleder R, Nahrendorf M. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res 115: 284–295, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 5: 1135–1142, 1999. [DOI] [PubMed] [Google Scholar]
- 38.Hulsmans M, Sam F, Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. J Mol Cell Cardiol 93: 149–155, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem 261: 4337–4345, 1986. [PubMed] [Google Scholar]
- 40.Iwakura K, Ito H, Ikushima M, Kawano S, Okamura A, Asano K, Kuroda T, Tanaka K, Masuyama T, Hori M, Fujii K. Association between hyperglycemia and the no-reflow phenomenon in patients with acute myocardial infarction. J Am Coll Cardiol 41: 1–7, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Iwasaka T, Takahashi N, Nakamura S, Sugiura T, Tarumi N, Kimura Y, Okubo N, Taniguchi H, Matsui Y, Inada M. Residual left ventricular pump function after acute myocardial infarction in NIDDM patients. Diabetes Care 15: 1522–1526, 1992. [DOI] [PubMed] [Google Scholar]
- 42.Iyer RP, de Castro Bras LE, Jin YF, Lindsey ML. Translating Koch's postulates to identify matrix metalloproteinase roles in postmyocardial infarction remodeling: cardiac metalloproteinase actions (CarMA) postulates. Circ Res 114: 860–871, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iyer RP, Patterson NL, Fields GB, Lindsey ML. The history of matrix metalloproteinases: milestones, myths, and misperceptions. Am J Physiol Heart Circ Physiol 303: H919–H930, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang B, Chen J, Xu L, Gao Z, Deng Y, Wang Y, Xu F, Shen X, Guo DA. Salvianolic acid B functioned as a competitive inhibitor of matrix metalloproteinase-9 and efficiently prevented cardiac remodeling. BMC Pharmacol 10: 10, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson C, Sung HJ, Lessner SM, Fini ME, Galis ZS. Matrix metalloproteinase-9 is required for adequate angiogenic revascularization of ischemic tissues: potential role in capillary branching. Circ Res 94: 262–268, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim JY, Kim WJ, Kim H, Suk K, Lee WH. The stimulation of CD147 induces MMP-9 expression through ERK and NF-kappaB in macrophages: implication for atherosclerosis. Immune Netw 9: 90–97, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kirk J. Thrombospondins in the transition from myocardial infarction to heart failure. J Mol Cell Cardiol 90: 102–110, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koenig GC, Rowe RG, Day SM, Sabeh F, Atkinson JJ, Cooke KR, Weiss SJ. MT1-MMP-dependent remodeling of cardiac extracellular matrix structure and function following myocardial infarction. Am J Pathol 180: 1863–1878, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13: 159–175, 2013. [DOI] [PubMed] [Google Scholar]
- 50.Krstic J, Santibanez JF. Transforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cells. ScientificWorldJournal 2014: 521754, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol 130: 147–158, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lauer-Fields JL, Whitehead JK, Li S, Hammer RP, Brew K, Fields GB. Selective modulation of matrix metalloproteinase 9 (MMP-9) functions via exosite inhibition. J Biol Chem 283: 20087–20095, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee BS, Oh J, Kang SK, Park S, Lee SH, Choi D, Chung JH, Chung YW, Kang SM. Insulin protects cardiac myocytes from doxorubicin toxicity by sp1-mediated transactivation of survivin. PLoS One 10: e0135438, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, Sena B, Chudnovskiy A, Panizzi P, Keliher E, Higgins JM, Libby P, Moskowitz MA, Pittet MJ, Swirski FK, Weissleder R, Nahrendorf M. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med 209: 123–137, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet 18: 2584–2598, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L, Li H. Role of microRNA-mediated MMP regulation in the treatment and diagnosis of malignant tumors. Cancer Biol Ther 14: 796–805, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin TC, Li CY, Tsai CS, Ku CH, Wu CT, Wong CS, Ho ST. Neutrophil-mediated secretion and activation of matrix metalloproteinase-9 during cardiac surgery with cardiopulmonary bypass. Anesth Analg 100: 1554–1560, 2005. [DOI] [PubMed] [Google Scholar]
- 58.Lindsey M, Wedin K, Brown MD, Keller C, Evans AJ, Smolen J, Burns AR, Rossen RD, Michael L, Entman M. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation 103: 2181–2187, 2001. [DOI] [PubMed] [Google Scholar]
- 59.Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, McClister DM Jr, Su H, Gannon J, MacGillivray C, Lee RT, Sinusas AJ, andSpinale FG. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol 290: H232–H239, 2006. [DOI] [PubMed] [Google Scholar]
- 60.Lindsey ML, Iyer RP, Jung M, DeLeon-Pennell KY, Ma Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J Mol Cell Cardiol 91: 134–140, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lindsey ML, Iyer RP, Zamilpa R, Yabluchanskiy A, DeLeon-Pennell KY, Hall ME, Kaplan A, Zouein FA, Bratton D, Flynn ER, Cannon PL, Tian Y, Jin YF, Lange RA, Tokmina-Roszyk D, Fields GB, de Castro Bras LE. A novel collagen matricryptin reduces left ventricular dilation post-myocardial infarction by promoting scar formation and angiogenesis. J Am Coll Cardiol 66: 1364–1374, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther 30: 31–41, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Loffek S, Schilling O, Franzke CW. Series “matrix metalloproteinases in lung health and disease”: Biological role of matrix metalloproteinases: a critical balance. Eur Respir J 38: 191–208, 2011. [DOI] [PubMed] [Google Scholar]
- 64.Lozano-Velasco E, Galiano-Torres J, Jodar-Garcia A, Aranega AE, Franco D. miR-27 and miR-125 distinctly regulate muscle-enriched transcription factors in cardiac and skeletal myocytes. Biomed Res Int 2015: 391306, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu L, Zhang Q, Pu LJ, Peng WH, Yan XX, Wang LJ, Chen QJ, Zhu ZB, Michel JB, Shen WF. Dysregulation of matrix metalloproteinases and their tissue inhibitors is related to abnormality of left ventricular geometry and function in streptozotocin-induced diabetic minipigs. Int J Exp Pathol 89: 125–137, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest 115: 599–609, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mauviel A. Cytokine regulation of metalloproteinase gene expression. J Cell Biochem 53: 288–295, 1993. [DOI] [PubMed] [Google Scholar]
- 68.McDonald PP, Bald A, Cassatella MA. Activation of the NF-kappaB pathway by inflammatory stimuli in human neutrophils. Blood 89: 3421–3433, 1997. [PubMed] [Google Scholar]
- 69.Mukherjee R, Brinsa TA, Dowdy KB, Scott AA, Baskin JM, Deschamps AM, Lowry AS, Escobar GP, Lucas DG, Yarbrough WM, Zile MR, Spinale FG. Myocardial infarct expansion and matrix metalloproteinase inhibition. Circulation 107: 618–625, 2003. [DOI] [PubMed] [Google Scholar]
- 70.Murthy S, Ryan AJ, Carter AB. SP-1 regulation of MMP-9 expression requires Ser586 in the PEST domain. Biochem J 445: 229–236, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1: a000034, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ohuchi E, Imai K, Fujii Y, Sato H, Seiki M, Okada Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J Biol Chem 272: 2446–2451, 1997. [DOI] [PubMed] [Google Scholar]
- 73.Parajuli N, Yuan Y, Zheng X, Bedja D, Cai ZP. Phosphatase PTEN is critically involved in post-myocardial infarction remodeling through the Akt/interleukin-10 signaling pathway. Basic Res Cardiol 107: 248, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Phatharajaree W, Phrommintikul A, Chattipakorn N. Matrix metalloproteinases and myocardial infarction. Can J Cardiol 23: 727–733, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD, Kass DA. Cardiomyocyte-specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ Res 114: 1246–1257, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramirez TA, Iyer RP, Ghasemi O, Lopez EF, Levin DB, Zhang J, Zamilpa R, Chou YM, Jin YF, Lindsey ML. Aliskiren and valsartan mediate left ventricular remodeling post-myocardial infarction in mice through MMP-9 effects. J Mol Cell Cardiol 72: 326–335, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rangrez AY, Massy ZA, Metzinger-Le Meuth V, Metzinger L. miR-143 and miR-145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet 4: 197–205, 2011. [DOI] [PubMed] [Google Scholar]
- 78.Ray A, Bal BS, Ray BK. Transcriptional induction of matrix metalloproteinase-9 in the chondrocyte and synoviocyte cells is regulated via a novel mechanism: evidence for functional cooperation between serum amyloid A-activating factor-1 and AP-1. J Immunol 175: 4039–4048, 2005. [DOI] [PubMed] [Google Scholar]
- 79.Ray BK, Shakya A, Turk JR, Apte SS, Ray A. Induction of the MMP-14 gene in macrophages of the atherosclerotic plaque: role of SAF-1 in the induction process. Circ Res 95: 1082–1090, 2004. [DOI] [PubMed] [Google Scholar]
- 80.Romanic AM, Burns-Kurtis CL, Gout B, Berrebi-Bertrand I, Ohlstein EH. Matrix metalloproteinase expression in cardiac myocytes following myocardial infarction in the rabbit. Life Sci 68: 799–814, 2001. [DOI] [PubMed] [Google Scholar]
- 81.Rundhaug JE. Matrix metalloproteinases, angiogenesis, and cancer: commentary re: A. C. Lockhart et al., Reduction of wound angiogenesis in patients treated with BMS-275291, a broad spectrum matrix metalloproteinase inhibitor. Clin. Cancer Res., 9: 00–00, 2003. Clin Cancer Res 9: 551–554, 2003. [PubMed] [Google Scholar]
- 82.Siddesha JM, Valente AJ, Sakamuri SS, Gardner JD, Delafontaine P, Noda M, Chandrasekar B. Acetylsalicylic acid inhibits IL-18-induced cardiac fibroblast migration through the induction of RECK. J Cell Physiol 229: 845–855, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol 280: C53–C60, 2001. [DOI] [PubMed] [Google Scholar]
- 84.Slavic S, Lauer D, Sommerfeld M, Kemnitz UR, Grzesiak A, Trappiel M, Thone-Reineke C, Baulmann J, Paulis L, Kappert K, Kintscher U, Unger T, Kaschina E. Cannabinoid receptor 1 inhibition improves cardiac function and remodelling after myocardial infarction and in experimental metabolic syndrome. J Mol Med (Berl) 91: 811–823, 2013. [DOI] [PubMed] [Google Scholar]
- 85.Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ. Origin of cardiac fibroblasts and the role of periostin. Circ Res 105: 934–947, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thompson MM, Squire IB. Matrix metalloproteinase-9 expression after myocardial infarction: physiological or pathological? Cardiovasc Res 54: 495–498, 2002. [DOI] [PubMed] [Google Scholar]
- 87.Thorp EB. Contrasting inflammation resolution during atherosclerosis and post myocardial infarction at the level of monocyte/macrophage phagocytic clearance. Front Immunol 3: 39, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tremblay I, Pare E, Arsenault D, Douziech M, Boucher MJ. The MEK/ERK pathway promotes NOTCH signalling in pancreatic cancer cells. PLoS One 8: e85502, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Uemura S, Matsushita H, Li W, Glassford AJ, Asagami T, Lee KH, Harrison DG, Tsao PS. Diabetes mellitus enhances vascular matrix metalloproteinase activity: role of oxidative stress. Circ Res 88: 1291–1298, 2001. [DOI] [PubMed] [Google Scholar]
- 90.Ueno-Shuto K, Kato K, Tasaki Y, Sato M, Sato K, Uchida Y, Sakai H, Ono T, Suico MA, Mitsutake K, Tokutomi N, Kai H, Shuto T. Lipopolysaccharide decreases single immunoglobulin interleukin-1 receptor-related molecule (SIGIRR) expression by suppressing specificity protein 1 (Sp1) via the Toll-like receptor 4 (TLR4)-p38 pathway in monocytes and neutrophils. J Biol Chem 289: 18097–18109, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vaday GG, Schor H, Rahat MA, Lahat N, Lider O. Transforming growth factor-beta suppresses tumor necrosis factor alpha-induced matrix metalloproteinase-9 expression in monocytes. J Leukoc Biol 69: 613–621, 2001. [PubMed] [Google Scholar]
- 92.van der Laan AM, Ter Horst EN, Delewi R, Begieneman MP, Krijnen PA, Hirsch A, Lavaei M, Nahrendorf M, Horrevoets AJ, Niessen HW, Piek JJ. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur Heart J 35: 376–385, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Van Linthout S, Miteva K, Tschope C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res 102: 258–269, 2014. [DOI] [PubMed] [Google Scholar]
- 94.Vellaichamy E, Khurana ML, Fink J, Pandey KN. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J Biol Chem 280: 19230–19242, 2005. [DOI] [PubMed] [Google Scholar]
- 95.Verma RP, Hansch C. Matrix metalloproteinases (MMPs): chemical-biological functions and (Q)SARs. Bioorg Med Chem 15: 2223–2268, 2007. [DOI] [PubMed] [Google Scholar]
- 96.Wagner DR, Delagardelle C, Ernens I, Rouy D, Vaillant M, Beissel J. Matrix metalloproteinase-9 is a marker of heart failure after acute myocardial infarction. J Card Fail 12: 66–72, 2006. [DOI] [PubMed] [Google Scholar]
- 97.Wang L, Ma YT, Xie X, Yang YN, Fu ZY, Liu F, Li XM, Chen BD. [Association of MMP9 gene -1562 C/T polymorphism with myocardial infarction in Uighur population of Xinjiang]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 28: 180–184, 2011. [DOI] [PubMed] [Google Scholar]
- 98.Westermarck J, Kahari VM. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J 13: 781–792, 1999. [PubMed] [Google Scholar]
- 99.Xu J, Rodriguez D, Petitclerc E, Kim JJ, Hangai M, Moon YS, Davis GE, Brooks PC. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J Cell Biol 154: 1069–1079, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yabluchanskiy A, Li Y, Chilton RJ, Lindsey ML. Matrix metalloproteinases: drug targets for myocardial infarction. Curr Drug Targets 14: 276–286, 2013. [PMC free article] [PubMed] [Google Scholar]
- 101.Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology (Bethesda) 28: 391–403, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yan W, Zhang W, Sun L, Liu Y, You G, Wang Y, Kang C, You Y, Jiang T. Identification of MMP-9 specific microRNA expression profile as potential targets of anti-invasion therapy in glioblastoma multiforme. Brain Res 1411: 108–115, 2011. [DOI] [PubMed] [Google Scholar]
- 103.Yang CQ, Li W, Li SQ, Li J, Li YW, Kong SX, Liu RM, Wang SM, Lv WM. MCP-1 stimulates MMP-9 expression via ERK 1/2 and p38 MAPK signaling pathways in human aortic smooth muscle cells. Cell Physiol Biochem 34: 266–276, 2014. [DOI] [PubMed] [Google Scholar]
- 104.Yarbrough WM, Mukherjee R, Escobar GP, Mingoia JT, Sample JA, Hendrick JW, Dowdy KB, McLean JE, Lowry AS, O'Neill TP, Spinale FG. Selective targeting and timing of matrix metalloproteinase inhibition in post-myocardial infarction remodeling. Circulation 108: 1753–1759, 2003. [DOI] [PubMed] [Google Scholar]
- 105.Zamilpa R, Ibarra J, de Castro Bras LE, Ramirez TA, Nguyen N, Halade GV, Zhang J, Dai Q, Dayah T, Chiao YA, Lowell W, Ahuja SS, D'Armiento J, Jin YF, Lindsey ML. Transgenic overexpression of matrix metalloproteinase-9 in macrophages attenuates the inflammatory response and improves left ventricular function post-myocardial infarction. J Mol Cell Cardiol 53: 599–608, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zamilpa R, Lopez EF, Chiao YA, Dai Q, Escobar GP, Hakala K, Weintraub ST, Lindsey ML. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics 10: 2214–2223, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang M, Pan X, Zou Q, Xia Y, Chen J, Hao Q, Wang H, Sun D. Notch3 ameliorates cardiac fibrosis after myocardial infarction by inhibiting the TGF-beta1/Smad3 pathway. Cardiovasc Toxicol 2015. October 20 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]