

Abstract
The objective of the present study was to examine the genetically determined differences in the natriuretic peptide receptor-A (NPRA) gene (Npr1) copies affecting the expression of cardiac hypertrophic markers, proinflammatory mediators, and matrix metalloproteinases (MMPs) in a gene-dose-dependent manner. We determined whether stimulation of Npr1 by all-trans retinoic acid (RA) and histone deacetylase (HDAC) inhibitor sodium butyric acid (SB) suppress the expression of cardiac disease markers. In the present study, we utilized Npr1 gene-disrupted heterozygous (Npr1+/−, 1-copy), wild-type (Npr1+/+, 2-copy), gene-duplicated (Npr1++/+, 3-copy) mice, which were treated intraperitoneally with RA, SB, and a combination of RA/SB, a hybrid drug (HB) for 2 wk. Untreated 1-copy mice showed significantly increased heart weight-body weight (HW/BW) ratio, blood pressure, hypertrophic markers, including beta-myosin heavy chain (β-MHC) and proto-oncogenes (c-fos and c-jun), proinflammatory mediator nuclear factor kappa B (NF-κB), and MMPs (MMP-2, MMP-9) compared with 2-copy and 3-copy mice. The heterozygous (haplotype) 1-copy mice treated with RA, SB, or HB, exhibited significant reduction in the expression of β-MHC, c-fos, c-jun, NF-κB, MMP-2, and MMP-9. In drug-treated animals, the activity and expression levels of HDAC were significantly reduced and histone acetyltransferase activity and expression levels were increased. The drug treatments significantly increased the fractional shortening and reduced the systolic and diastolic parameters of the Npr1+/− mice hearts. Together, the present results demonstrate that a decreased Npr1 copy number enhanced the expression of hypertrophic markers, proinflammatory mediators, and MMPs, whereas an increased Npr1 repressed the cardiac disease markers in a gene-dose-dependent manner.
Keywords: natriuretic peptide receptor-A, gene-disruption, fibrosis, hypertrophy, histone deacetylase, histone acetyltransferase
atrial and brain natriuretic peptides (ANP and BNP) activate guanylyl cyclase-A/natriuretic peptide receptor-A (GC-A/NPRA), which produces the intracellular second messenger cGMP. The ANP/NPRA system elicits natriuretic, diuretic, vasorelaxant, and antiproliferative responses, all of which contribute largely to the reduction of blood pressure and blood volume (40, 49, 64). Previous studies have suggested that NPRA signaling is involved not only in maintaining the arterial pressure but also in antagonizing the cardiac growth responses to hypertrophic stimuli (6, 14, 34). It has been shown that both external and internal stimuli regulate Npr1 (coding for GC-A/NPRA) expression and receptor signaling, including angiotensin II (ANG II), all-trans retinoic acid (RA), vitamin D, and extracellular osmolality (1, 8, 16, 21). In spite of the hallmark functional significance of NPRA in renal, vascular, and cardiac physiology, the precise mechanisms of receptor activation, regulation, and function at the molecular and genetic levels still remain elusive. Mice carrying the Npr1 gene disruption exhibit hypertension and congestive heart failure (45, 60, 68). On the other hand, Npr1 gene duplication reduces blood pressure, attenuates hypertrophic stimuli-induced cardiac myocyte growth, and lowers cardiac ANG II and aldosterone levels (46, 76, 77). A significant positive association between Npr1 variants and left ventricular mass index was found in patients with essential hypertension, suggesting that the ANP/NPRA system seems to contribute to cardiac remodeling in response to chronic high blood pressure (12, 24, 44, 55, 71). However, the cellular mechanisms by which the ANP/NPRA system blocks hypertrophic growth and prevents cardiac dysfunction are not well understood.
Several lines of evidence indicate that the activation of hypertrophic markers, matrix metalloproteinases (MMPs), and proinflammatory cytokines play a central pathophysiological roles in the development of cardiac hypertrophy, fibrosis, and heart failure in the experimental animal models and humans (11, 52, 58, 66). Previous studies have suggested that ANP/NPRA signaling acts as a negative regulator of inflammation and hypertrophic growth (14, 65, 67, 69). However, the studies on the role of ANP/NPRA in regulating the expression of hypertrophic and proinflammatory markers in vivo are limited. In the present study we have utilized Npr1 gene-disrupted heterozygous (haplotype, Npr1+/−), wild-type (Npr1+/+), and gene-duplicated (Npr1++/+) mice to examine whether Npr1 gene copy numbers affect the differential expression levels of cardiac hypertrophic markers, proinflammatory cytokines, and matrix enzymes in a gene-dose-dependent manner.
The findings of our previous studies have indicated that RA and sodium butyrate (SB) stimulate Npr1 gene transcription by inhibiting the histone deacetylases (HDACs) and activating the histone acetyltransferases (HATs) in cultured cells in vitro and intact animals in vivo (29–31). Inhibition of HDACs by RA and SB has also been shown to improve cardiovascular and renal disease conditions (3, 5, 36). Moreover, mixed esters of butyric acid and retinoic acid hybrid drug (HB) have also been reported to enhance cardiac repair of infarcted hearts in the experimental animals (61, 70). In the present study, we examined the effect of RA-, SB-, and HB-mediated effects on the cardiac hypertrophic and fibrotic markers, nuclear factor kappa B (NF-κB), and MMPs in haplotype Npr1+/− animals and compared the results with those of wild-type Npr1+/+ and heterozygous gene-duplicated Npr1++/+ mice. The current findings provide evidence that a decreased Npr1 copy number leads to cardiac remodeling and activation of hypertrophic markers, MMPs, and profibrotic cytokines, while enhanced expression of Npr1 by RA, SB, and HB as well as increased Npr1 gene copies suppressed the expression levels of cardiac disease markers.
MATERIALS AND METHODS
Materials.
RNeasy mini-kit for total RNA isolation, RT2 First Strand cDNA kit, and RT2 SYBR Green/ROX master mix were obtained from Qiagen (Valencia, CA). Sequence-specific oligonucleotides were purchased from Eurofins MWG (Operon). cGMP assay kit was obtained from Enzo Life Sciences (Farmingdale, NY). HDAC and HAT activity assay kits were purchased from Epigentek (Brooklyn, NY). RA, SB, and HB, primary antibodies for β-myosin heavy chain (β-MHC), α-skeletal actin (α-SK), c-fos, c-jun, MMP-2, MMP-9, tissue inhibitors of MMPs (TIMP-1, TIMP-2), transforming growth factor-β (TGF-β1), NF-κB, inhibitory kappa Bα (IκBα), HDAC1, HDAC2, p300, p300/cAMP-binding protein (CBP)-associated factor (PCAF), and TATA-binding protein (TBP) were obtained from Santa Cruz Biotechnology (San Diego, CA). Primary antibody for NPRA was produced as previously described (31, 39). Western blot detection reagent kit was obtained from Thermo Fisher Scientific (Rockford, IL). Texas red anti-rabbit IgG (H+L) and 4′,6-diamidino-2-phenylindole (DAPI) were obtained from Vector Laboratories (Burlingame, CA). All other reagents used were analytical grade.
Generation and genotyping of mice.
Npr1 gene-disrupted and gene-duplicated mice were produced by homologous recombination in embryonic stem cells (45, 46, 50). These mice were bred and maintained at the animal facility of the Tulane University Health Sciences Center. Animals were handled according to protocols approved by the Institutional Animal Care and Use Committee. The mouse colonies were housed under 12 h light/12 h dark cycles at 25°C and fed regular chow (Purina Laboratories, Framingham, MA); tap water was available ad libitum. All animals were littermate progeny of C57/BL6 genetic background and were designated as Npr1 gene-disrupted heterozygous (Npr1+/−, 1-copy), wild-type (Npr1+/+, 2-copy), and Npr1 gene-duplicated heterozygous (Npr1++/+, 3-copy) mice. In the present study, adult male mice were utilized. The animals were genotyped by PCR analyses of DNA isolated from tail biopsies using primer A (5′-GCTCTCTTGTCGCCGAATCT-3′), corresponding to the 5′-sequence of the mouse Npr1 gene common to both alleles (Npr1+/+) 2-copy: primer B (5′-TGTCACCATGGTCTGATCGC-3′), corresponding to the exon 1 sequence present only in the intact allele (Npr1+/−) 1-copy; and primer C (5′-CCTCTAGATGCATACATGTGCC-3′), corresponding to a sequence for heterozygous gene duplicated (Npr1++/+) 3-copy mice. PCR was done by our standard method as previously reported (45, 59, 60).
Treatment of mice with RA, SB, and HB drugs.
Adult male 1-copy (n = 24), 2-copy (n = 24), and 3-copy (n = 24) mice were divided into four experimental treatment groups: group I, vehicle (control); group II, RA (0.5 mg/kg/day); group III, SB (0.5 mg/kg/day); group IV, HB (1 mg/kg/day). Stock solutions RA and SB drugs were prepared each at 20 mg/ml, and HB was made at 10 mg/ml concentrations, respectively, in DMSO and stored at −80°C. On the day of injection, the drugs were thawed, diluted with olive oil to appropriate concentrations, and vortexed for 2 min at room temperature. Animals were injected intraperitoneally for 14 days, and the bioavailability of drugs was based on earlier published work, namely, for RA (20) and SB (63). The HB drug has also been previously described (18), and dose and method of drug delivery were based on our earlier in vivo study (30). Control groups were injected with vehicle (DMSO and olive oil). At the end of the experiment, animals were killed under CO2 anesthesia, plasma was collected, and the hearts were removed from mice for histological, biochemical and analytical analyses.
Assessment of blood pressure and cardiac function.
Systolic blood pressures (SBP) and heart rates were measured by a noninvasive computerized tail-cuff method using VisiTech 2000 as previously described (59). All mice were trained for 7 days for acclimatization, and the actual blood pressure was measured from the 10th day to 14th day of treatment period. Blood pressures and heart rates were calculated as the average of six to seven sessions/day for 5 consecutive days. The cardiac functions of control and drug-treated mice were analyzed using two-dimensional echocardiography as described previously (68).
Blood and tissue collection, cGMP assay.
Blood was collected by cardiac puncture from mice under CO2 anesthesia into chilled tubes containing 10 μl of heparin (1,000 USP units/ml). Plasma was separated by centrifuging the blood samples at 3,000 rpm for 10 min at 4°C and then stored at −80°C until use. Heart tissues were dissected, frozen in liquid nitrogen, and stored at −80°C. A portion of heart tissue was sliced, kept in 10% buffered formalin overnight, and processed for histological studies. The plasma samples were used for cGMP assay, using a direct enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer's protocol. The results are expressed as picomoles/ml of plasma.
Measurement of cardiac hypertrophy and fibrosis.
At the end of experiments, body weights of mice were measured. The hearts were dissected out, blotted, and weighed. The ratio of heart weight to body weight (HW/BW) was calculated as an index of cardiac hypertrophy. Paraffin-embedded tissue sections (5 μm) were stained with Masson's trichrome to disclose the presence of accumulated interstitial and perivascular collagen fiber as a marker of cardiac fibrosis. The ratio of interstitial fibrosis and perivascular collagen to the total left ventricular area was calculated from 20 randomly selected microscopic fields in five sections per heart, using ImagePro Plus image analysis software (Media Cybernetics, Silver Spring, MD).
Real-time RT-PCR analysis.
Total RNA was extracted using RNeasy plus mini-kit. First-strand cDNA was synthesized from 1 μg of total RNA in a final volume of 20 μl using RT2 First Strand kit. Real-time quantitative RT-PCR (qPCR) was done in Mx3000P real-time PCR system. Data were analyzed with MxPro QPCR software (Stratagene, La Jolla, CA). The primers used for MHC were 5′-TCCGCAAGGTGCAGCACGAG-3′ (sense) and 5′-CACGGGCACCCTTGGAGCTG-3′ (antisense); α-skeletal actin (SK) were 5′-ACTTCCTACCCTCGGCACCCA-3′ (sense) and 5′-CACCAGGCCAGAGCCGTTGT-3′ (antisense); c-FOS were 5′-CCACCGACCTGCCTGCAAGA-3′ (sense) and 5′-TGGCTTGGGCTCAGGGTCGT-3′ (antisense); c-JUN were 5′-TGCGCACAGCCCAGGCTAAC-3′ (sense) and 5′-CGGCTTCAGACTGCCCACCG-3′ (antisense); MMP-2 were 5′-CCGGCCACATCTGGCGTCTG-3′ (sense) and 5′-ACGGGGTCCCACGTCCCAAT-3′ (antisense); MMP-9 were 5′-GGCCGCTCGGATGGTTACCG-3′ (sense) and 5′-GCTCTCCTGCCGAGTTGCCC-3′ (antisense); TIMP-1 were 5′-GCCAACTCCGCCCTTCGCAT-3′ (sense) and 5′-GGGGGCCATCATGGTATCTCTGGT-3′ (antisense); TIMP-2 were 5′-TGCACCCGCAACAGGCGTTT-3′ (sense) and 5′-TGCTGAAGAGGGGGCCGTGT-3′ (antisense); and GAPDH were 5′-TCCCTCAAGATTGTCAGCAA-3′ (sense) and 5′-AGATCCACAAACGGATACATT-3′ (antisense). PCR amplifications (triplicates) were carried out in a 20-μl reaction volume using RT2 real-time SYBR Green/ROX PCR Master Mix. The reaction conditions were 95°C for 10 min, followed by 45 cycles at 95°C for 15 s and 60°C for 1 min; this was followed by 1 cycle at 95°C for 1 min, 55°C for 30 s, and 95°C for 30 s for the dissociation curve. The reaction mixture without template cDNA was used as a negative control. Threshold cycle numbers (CT) were determined with MxPro QPCR Software and transformed using the ΔCT comparative method. The mRNA expression was normalized to expression values of GAPDH (endogenous control) within each sample and relative to positive and negative controls. The level of gene expression was determined by the comparative CT method (ΔΔCT). After PCR amplification, a melting curve of each amplicon was determined to verify its accuracy.
Cytosolic and nuclear extract preparation.
The heart tissues were homogenized in an ice-cold 10 mM Tris·HCl buffer (pH 8.0) containing 0.32 M sucrose, 3 mM calcium chloride (CaCl2), 2 mM magnesium acetate (MgOAc), 0.1 mM ethylenediaminetetraacetic acid (EDTA), 0.5% Nonidet P-40 (NP-40), 1 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM orthovanadate, 30 mM sodium fluoride (NaF), and 10 μg/ml each of leupeptin, aprotinin, and pepstatin. The homogenate was centrifuged at 8,000 g at 4°C for 20 min. The supernatant was separated and stored as cytosolic fraction. The pellet was washed three times in wash buffer by resuspending it with a 20-gauge needle and centrifuging at 6,000 g. The pellet was resuspended in a low-salt buffer (20 mM HEPES, pH 7.9; 1.5 mM MgCl2; 20 mM KCl; 0.2 mM EDTA; 25% glycerol; 0.5 mM DTT; and 0.5 mM PMSF), incubated on ice for 5 min, and mixed with an equal volume of high-salt buffer (20 mM HEPES, pH 7.9; 1.5 mM MgCl2; 800 mM KCl; 0.2 mM EDTA; 1% NP-40; 25% glycerol; 0.5 mM DTT; 0.5 mM PMSF; 1 mM orthovanadate; 30 mM NaF; and 10 μg/ml each of leupeptin, aprotinin, and pepstatin). The mixture was incubated on ice for 30 min and centrifuged at 14,000 g for 20 min. The supernatant was separated and stored as nuclear fraction at −80°C.
Western blot analysis.
Tissue homogenate (30 μg of proteins) was mixed with an equal volume of sample loading buffer and separated under reducing conditions by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were transferred at 100 volts to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 1× Tris-buffered saline/Tween 20 (TBST) (pH 7.5) containing 5% nonfat milk powder for 1 h at room temperature, then incubated overnight at 4°C with specific antibodies to β-MHC, α-skeletal actin, c-fos, c-jun, MMP-2, MMP-9, TGF-β1, TIMP-1, TIMP-2, pNF-κB, pIκB, HDAC1, HDAC2, p300, PCAF, NPRA, and TBP at a dilution of 1:500 in TBST containing 3% nonfat milk powder. After three washes with TBST for 5 min each, the membrane was incubated for 1 h in horseradish peroxidase-conjugated corresponding secondary antibodies at a dilution of 1:5,000, washed three times with TBST, and developed using the West Femto Chemiluminescent substrate detection reagent kit. The luminescent signal was detected using the Alpha Innotech imaging system (San Loreno, CA).
HDAC and HAT activity assay.
Total HDAC and HAT activities were measured in nuclear extracts prepared from the heart tissues. HAT and HDAC activities were determined with a colorimetric ELISA assay kit (EpiQuik HDAC or HAT activity/inhibition kit). Absorbance was read at 450 nm, and activities were calculated using a standard curve for HDAC (ng·min−1·mg protein−1) and HAT (ng·h−1·mg protein−1) as per manufacturer's instructions.
Immunofluorescence staining and confocal microscopy image analysis.
For immunofluorescence studies, paraffin-embedded heart tissue sections (5 μm) were deparaffinized in xylenes (three times for 5 min each), and then slides were washed with phosphate-buffered saline (PBS) for 5 min. The slides were rehydrated serially with 100, 90, and 70% ethanol for 5 min each at room temperature. For the antigen retrieval, the slides were rinsed in distilled water and incubated for 20 min in preheated sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) and the slides were allowed to cool down for 20 min then washed (three times for 5 min each) in distilled water. Slides were incubated with 5% goat serum and 1% BSA in PBS for 1 h to block nonspecific protein interactions. Blocking buffer was gently tipped off and slides were incubated with anti-NF-κB antibody diluted (1:200) in 10% goat serum in PBS, overnight at 4°C. After being washed in PBS (three times for 5 min each), slides were incubated with the secondary antibody anti-rabbit IgG (1:2,000 dilutions) conjugated Texas red for 2 h at room temperature in the dark. The slides were washed three times for 15 min each in PBS, then counterstained with DAPI, and sealed with Fixogum rubber cement. Stained tissue sections were examined, and images were acquired using a TCS SP2 confocal laser scanning microscope (Leica Microsystems, Heidelberg, Germany). In all experiments, tissue sections were visualized using the same confocal microscope settings (i.e., sequential scans with wavelengths set as follows: blue, 358–461; red, 594–615), using a ×40 Apo-oil immersion objective using the LEICA Scan TCS-SP2 software. The fluorescence intensity was measured using the MetaMorph software (Molecular Devices, Downingtown, PA). NF-κB immunofluorescence intensity was quantified by measuring the fluorescence intensity in arbitrary units.
Statistical analysis.
The results are expressed as means ± SE. Statistical significance was evaluated by two-way analysis of variance, followed by Dunnett's multiple comparison tests using GraphPad Prism Software (GraphPad Software, San Diego, CA). A P value of <0.05 was considered significant.
RESULTS
The results demonstrate that treatment with RA, SB, or combination drug, HB, showed a significant reduction in HW/BW ratio (RA, 5.1 ± 0.2; SB, 5.2 ± 0.3; HB, 4.7 ± 0.2; vehicle, 6.5 ± 0.2) and restored SBP (RA, 111.40 ± 3; SB, 110.10 ± 3; HB, 108.6 ± 3; vehicle, 121.5 ± 4 mmHg) in 1-copy (Npr1+/−) mice compared with untreated control groups (Table 1). Moreover, 3-copy mice showed a further decrease in HW/BW ratio (RA, 4.6 ± 0.2; SB, 4.4 ± 0.3; HB, 4.4 ± 0.2) and SBP (RA, 87.10 ± 2; SB, 87.90 ± 2; and HB, 86.36 ± 3 mmHg) and slightly increased heart rate compared with vehicle-treated mice (Table 1). Echocardiographic analysis showed significantly reduced left ventricular end systolic dimension and left ventricular end diastolic dimension (P < 0.05), as well as increased fractional shortening in drug-treated Npr1+/− mice compared with vehicle-treated control groups (Table 2). Masson's trichrome staining of heart tissue sections indicates the intensity of collagen accumulation as reflected by blue staining (Fig. 1, A and B). A significant increase in interstitial fibrosis (>20%; P < 0.01) occurred in vehicle-treated 1-copy mice compared with age-matched 2-copy (7%) and 3-copy (3%) mice. In contrast, 1-copy mice treated with RA or SB alone or combination drug HB showed the attenuation of fibrosis by 42, 40, and 50% in mice treated with RA, SB, and HB, respectively, compared with vehicle-treated 1-copy control mice. Any abnormal accumulation of collagen was not observed in drugs-treated 2- or 3-copy mice.
Table 1.
HW/BW ratios, SBP, and HR of wild-type (2-copy), Npr1 gene-disrupted (1-copy), and Npr1 gene-duplicated (3-copy) control and drug-treated mice
| Groups/Indexes | HW/BW, mg/g | SBP, mmHg | HR, beats/min |
|---|---|---|---|
| 1-Copy vehicle | 6.5 ± 0.2‡ | 121.50 ± 4† | 446.5 ± 11† |
| 1-Copy + RA | 5.1 ± 0.2* | 111.40 ± 3* | 460.6 ± 10* |
| 1-Copy + SB | 5.2 ± 0.3* | 110.10 ± 3* | 457.6 ± 11* |
| 1-Copy + HB | 4.7 ± 0.2* | 108.60 ± 3* | 462.8 ± 10* |
| 2-Copy vehicle | 5.4 ± 0.2 | 100.9 ± 3 | 468.8 ± 10 |
| 2-Copy + RA | 4.9 ± 0.2 | 97.5 ± 2 | 462.3 ± 16 |
| 2-Copy + SB | 4.8 ± 0.1 | 98.1 ± 3 | 467.6 ± 14 |
| 2-Copy + HB | 4.5 ± 0.2* | 90.3 ± 3* | 479.1 ± 12* |
| 3-Copy vehicle | 4.7 ± 0.1‡ | 81.4 ± 2‡ | 487.5 ± 14‡ |
| 3-Copy + RA | 4.6 ± 0.2 | 87.1 ± 2 | 484.2 ± 10 |
| 3-Copy + SB | 4.4 ± 0.3 | 87.9 ± 2 | 483.9 ± 16 |
| 3-Copy + HB | 4.4 ± 0.2* | 86.3 ± 3* | 485.2 ± 12* |
Values are expressed as means ± SE (n = 8). Heart weight-to-body weight (HW/BW) ratio, systolic blood pressure (SBP), and heart rate (HR) of control, retinoic acid (RA), sodium butyrate (SB) and hybrid (HB) drug-treated mice. Statistical significance expressed as
P < 0.05,
P < 0.01 compared with 2-copy control;
P < 0.05,
P < 0.01 compared with vehicle-treated controls within the group.
Table 2.
Echocardiographic analysis of wild-type (2-copy), Npr1 gene-disrupted (1-copy), and Npr1 gene-duplicated (3-copy) control and drug-treated mice
| Groups/Indexes | LVEDS, mm | LVEDD, mm | IVSTD, mm | FS, % |
|---|---|---|---|---|
| 1-Copy vehicle | 2.6 ± 0.02‡ | 4.4 ± 0.01‡ | 0.88 ± 0.02‡ | 38.91 ± 1.6‡ |
| 1-Copy + RA | 2.5 ± 0.02 | 4.0 ± 0.02* | 0.87 ± 0.01 | 39.07 ± 2.7* |
| 1-Copy + SB | 2.5 ± 0.01 | 4.0 ± 0.01* | 0.87 ± 0.01 | 39.17 ± 3.8* |
| 1-Copy + HB | 2.1 ± 0.02§ | 3.7 ± 0.01§ | 0.67 ± 0.01§ | 46.35 ± 4.2§ |
| 2-Copy vehicle | 2.2 ± 0.01 | 3.9 ± 0.01 | 0.68 ± 0.01 | 45.82 ± 2.0 |
| 2-Copy + RA | 2.0 ± 0.01* | 3.8 ± 0.01 | 0.67 ± 0.01 | 46.52 ± 2.0 |
| 2-Copy + SB | 2.1 ± 0.01 | 3.7 ± 0.01* | 0.67 ± 0.01 | 46.88 ± 2.7 |
| 2-Copy + HB | 2.0 ± 0.01* | 3.6 ± 0.01* | 0.65 ± 0.01* | 47.07 ± 2.5* |
| 3-Copy vehicle | 1.9 ± 0.01‡ | 3.7 ± 0.01‡ | 0.66 ± 0.01‡ | 47.22 ± 1.9‡ |
| 3-Copy + RA | 1.8 ± 0.01 | 3.6 ± 0.01 | 0.65 ± 0.01 | 49.23 ± 2.0* |
| 3-Copy + SB | 1.8 ± 0.01 | 3.6 ± 0.01 | 0.64 ± 0.01* | 48.02 ± 2.4 |
| 3-Copy + HB | 1.7 ± 0.01* | 3.4 ± 0.01* | 0.63 ± 0.01* | 51.18 ± 1.7* |
Values are expressed as means ± SE (n = 8). Left ventricular end dimension (systolic) (LVEDS); left ventricular end dimension (diastolic) (LVEDD); interventricular end dimension (diastolic) (IVSTD); fractional shortening (FS) of control, retinoic acid (RA), sodium butyrate (SB), and hybrid (HB) drug-treated mice. Statistical significance expressed as
P < 0.05,
P < 0.01 compared with 2-copy control;
P < 0.05,
P < 0.01 compared with vehicle-treated controls within the group.
Fig. 1.

Analysis of interstitial collagen in control and drug-treated Npr1 gene-targeted mice hearts. Representative heart tissue sections (A) and the fibrosis analysis (B) of collagen from control, retinoic acid (RA), sodium butyrate (SB), and hybrid (HB) drug-treated mice stained with Masson's trichrome. The color blue is indicative of fibrosis (×40 magnification). Values are expressed as means ± SE (n = 6 animals in each group). Statistical significance expressed as $P < 0.05, $$P < 0.01 compared with 2-copy control; *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle-treated controls.
Heart tissue of 1-copy mice exhibited increased mRNA expression of β-MHC (2.45-fold, P < 0.01), α-SKA (2.25-fold, P < 0.01), and proto-oncogenes activator protein (AP-1) such as c-fos (3.15-fold, P < 0.01) and c-jun (3.0-fold, P < 0.01) compared with 2-copy and 3-copy mice (Fig. 2, A–D). In contrast, 1-copy mice treated with RA and SB either alone or with HB exhibited significantly attenuated (P < 0.05) protein levels of β-MHC (RA, 25%; SB, 27%, HB, 55%), α-SKA (RA, 22%, SB, 20%, HB, 42%), c-fos (RA, 22%; SB, 19%, HB, 35%), and c-jun (RA, 21%; SB, 22%, HB, 40%), respectively. The expression of MMP-2 and MMP-9 mRNA levels was significantly (P < 0.05) upregulated in the heart tissues of 1-copy mice compared with 2-copy control mice (Fig. 2, E and F). In contrast, expression of TIMP-1 and TIMP-2 mRNA transcripts was downregulated by 36 and 40%, respectively, in 1-copy cardiac tissue (Fig. 2, G and H). Protein levels of the hypertrophic markers, β-MHC (2.08-fold), α-SKA (2.30-fold), c-fos (2.83-fold), and c-jun (2.23-fold) were also markedly increased in 1-copy mice compared with 2-copy mice (Fig. 3). Increased expression of MMP-2 (2.59-fold; P < 0.01), MMP-9 (1.93-fold; P < 0.01), and TGF-β1 (2.42-fold; P < 0.01) proteins was also found in heart tissues of 1-copy mice with decreased expression of TIMP-1 and TIMP-2 proteins compared with 2- and 3-copy mice (Fig. 4). While the heart tissue of 1-copy mice treated with RA and SB alone or HB exhibited attenuated expression of MMP-2 (P < 0.01), MMP-9 (P < 0.01), and TGF-β1 (P < 0.01), as well as upregulated expression of TIMP-1 (HB, 51%, P < 0.05) and TIMP-2 (HB, 40%, P < 0.05) proteins. Furthermore, treatment with RA and SB alone or in combination potently attenuated (P < 0.05) the expression of fibrotic markers and activated the expression levels of TIMPs in age-matched 2- and 3-copy cardiac tissues (Fig. 4).
Fig. 2.

Expression analysis of hypertrophic and fibrotic marker genes in the hearts of control and drug-treated Npr1 gene-targeted mice. mRNA expression of β-myosin heavy chain (β-MHC; A), α-skeletal actin (α-SKA; B), c-fos (C), c-jun (D), MMP-2 (E), MMP-9 (F), TIMP-1 (G), and TIMP-2 (H) in the heart tissue of control, retinoic acid (RA), sodium butyrate (SB) and hybrid (HB) drug-treated mice, normalized to GAPDH expression. Values are expressed as means ± SE (n = 6 animals in each group). Statistical significance expressed as $P < 0.05, $$P < 0.01 compared with 2-copy control; *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle-treated controls within the group.
Fig. 3.

Analysis of hypertrophic and proto-oncogenes protein levels in the heart of control, drug-treated Npr1 gene-targeted mice: A–F: representative Western blots showing the expression of β-myosin heavy chain (β-MHC), α-skeletal actin (α-SKA), c-fos, and c-jun in the heart tissue of control, retinoic acid (RA), sodium butyrate (SB), and hybrid (HB) drug-treated mice, respectively. G–J: densitometry analysis of proteins normalized to β-actin expression. Values are expressed as means ± SE (n = 6 animals in each group). Statistical significance expressed as $P < 0.05, $$P < 0.01 compared with 2-copy control; * P < 0.05, **P < 0.01 compared with vehicle-treated controls within the group.
Fig. 4.

Analysis of matrix metallo proteinase (MMP) and tissue inhibitors of MMPs (TIMP) expression levels in the heart tissues of controls and drug-treated Npr1 gene-targeted mice. A–F: representative Western blots showing the expression of MMP-2, MMP-9, TGF-β1, TIMP-1, and TIMP-2 proteins in the heart tissue of control, retinoic acid (RA), sodium butyrate (SB), and hybrid (HB) drug-treated mice, respectively. G–K: densitometry analysis of proteins normalized to β-actin expression. Values are expressed as means ± SE (n = 6 animals in each group). Statistical significance expressed as $P < 0.05, $$P < 0.01 compared with 2-copy control; *P < 0.05, **P < 0.01 compared with vehicle-treated controls within the group.
The expression of phosphorylated-NF-κB p65 (p-NF-κB/p65) subunit and phosphorylated-IκBα (p-IκBα) protein levels in heart tissue from 1-, 2-, and 3-copy control and drug-treated mice was quantitated by Western blot analysis. Significant increases in the expression of p-p65 (3.0-fold; P < 0.01) and p-IκBα (2.6-fold; P < 0.01) protein levels were evident in vehicle-treated 1-copy mice. After treatment with RA and SB alone or HB, 1-copy mice exhibited significantly reduced expression of p-p65 (P < 0.01) and p-IκBα (P < 0.01) proteins in the heart tissues. No abnormal upregulation in the expression of p65 was found in age-matched 2-copy (wild-type) or 3-copy (gene-duplicated) mice hearts (Fig. 5, A–E). The immunofluorescence intensity of NF-κB analyzed by confocal microscopy, displayed 82.8, 29.13, and 19.86% expression in vehicle-treated 1-, 2-, and 3-copy Npr1 mice, respectively. The fluorescent intensity of NF-κB was increased by 2.8-fold in 1-copy compared with 2-copy mice (Fig. 5F). On the other hand, HB treatment significantly (P < 0.01) decreased the fluorescence intensity of NF-κB by 64% in 1-copy, 38% in 2-copy, and 41% in 3-copy mice compared with vehicle-treated control animals (Fig. 5, F and G).
Fig. 5.

Analysis of p65, IκB phosphorylation in the heart tissues of Npr1 gene-disrupted and Npr1 gene-duplicated control and drug-treated mice. A–C: representative Western blots showing the expression of p-NF-κB (p65) and p-IκB proteins in the heart tissue of control, retinoic acid (RA), sodium butyrate (SB), and hybrid (HB) drug-treated mice, respectively. D and E: densitometry analysis of proteins normalized to β-actin expression. F: immunofluorescence of NF-κB in vehicle-treated and HB-treated Npr1 gene-targeted mice hearts. Red fluorescence indicates NF-κB expression, and blue fluorescence shows the nucleus stained with DAPI. G: quantitative fluorescence intensity analysis of NF-κB expression quantified by intracellular fluorescence intensity. Values are expressed as means ± SE (n = 8 animals in each group). Statistical significance expressed as $P < 0.05, $$P < 0.01 compared with 2-copy control; *P < 0.05; **P < 0.01; ***P < 0.001 compared with vehicle-treated controls within the group. Scale bar = 50 μm.
One-copy mice exhibited significantly higher HDAC activity (4.7 ± 0.5 ng·min−1·mg protein−1, P < 0.01) than 2-copy (2.8 ± 0.3 ng·min−1·mg protein−1) and 3-copy mice (1.3 ± 0.4 ng·min−1·mg protein−1, P < 0.05) (Fig. 6A). In contrast, HDAC activity was significantly reduced in 1-copy mice with treatment of RA (3.3 ± 0.5 ng·min−1·mg protein−1, P < 0.01), SB (3.1 ± 0.2 ng·min−1·mg protein−1, P < 0.01), and HB (2.35 ± 0.5 ng·min−1·mg protein−1, P < 0.01) compared with vehicle-treated 1-copy mice. Similarly, there was significant reduction in HDAC activity in 2-copy mice (RA, 2.1 ± 0.15 ng·min−1·mg protein−1, P < 0.05; SB, 2.2 ± 0.22 ng·min−1·mg protein−1, P < 0.05; and HB, 1.6 ± 0.2 ng·min−1·mg protein−1, P < 0.05) and 3-copy mice (RA, 0.9 ± 0.23 ng·min−1·mg protein−1, SB, 0.84 ± 0.18 ng·min−1·mg protein−1, P < 0.05; and HB, 0.81 ± 0.3 ng·min−1·mg protein−1, P < 0.01) (Fig. 6A). The protein expression of HDAC1 and HDAC2 was also significantly (P < 0.05) upregulated in the heart tissues of 1-copy mice compared with 2-copy control mice; however, the expression of HDAC2 protein level was significantly decreased (P < 0.05) in HB-treated 1-copy mice (Fig. 6B). On the other hand, HAT activity was significantly reduced in 1-copy mice (14.1 ± 1.2 ng·h−1·mg protein−1, P < 0.01) but increased in 3-copy mice (34.2 ± 2.7) compared with 2-copy mice (23.2 ± 2.8 ng·h−1·mg protein−1) (Fig. 6C). A significant increase in HAT activity was observed in 1-copy mice treated with RA (18.0 ± 2.0 ng·h−1·mg protein−1, P < 0.05), SB (20.0 ± 2.0 ng·h−1·mg protein−1, P < 0.05), and HB (23.0 ± 1.60 ng·h−1·mg protein−1, P < 0.01). Similarly, there was a notable increase in HAT activity in HB-treated 2-copy (42.0 ± 4 ng·h−1·mg protein−1, P < 0.01) and 3-copy (52.0 ± 5.0 ng·h−1·mg protein−1, P < 0.01) mice compared with vehicle-treated controls (Fig. 6C). The protein expression of HATs, namely p300 and PCAF, was significantly (P < 0.05) downregulated in the cardiac tissues of 1-copy mice compared with 2-copy controls (Fig. 6D). Treatment with HB markedly induced protein expression of p300 in 1-copy mice and that of PCAF in 1-copy, 2-copy, and 3-copy mice compared with untreated control groups.
Fig. 6.

Assay of histone deacetylase (HDAC) and histone acetyltransferase (HAT) activity in the heart tissues of Npr1 gene-targeted mice. A: quantitative analysis of HDAC activity in the heart tissues of Npr1 gene-targeted mice. B: representative Western blot showing expression of HDAC1 and HDAC2 in the heart tissue of control (C) and hybrid (HB) drug-treated mice. Densitometry analysis was done by AlphaInnotech phosphoimager software. C: HAT activity in heart tissues of Npr1 gene-targeted mice treated with HB. Values are expressed as means ± SE (n = 8 animals in each group). Statistical significance expressed as $P < 0.05, $$P < 0.01 compared with 2-copy control; *P < 0.05, **P < 0.01 compared with vehicle-treated controls within the group.
The protein expression of NPRA in the heart tissues from 1-, 2-, and 3-copy controls and HB-treated mice was quantitated by Western blot analysis. Heart tissues of 1-copy mice exhibited significantly (P < 0.01) attenuated NPRA expression compared with 2-copy mice (Fig. 7A). In contrast, treatment with HB markedly upregulated the expression of NPRA in both 1-copy and 2-copy mice compared with vehicle-treated control groups. Plasma cGMP levels were significantly lower in vehicle-treated 1-copy mice (8.12 ± 2.35 pg/ml) compared with 2-copy (18.28 ± 5.38 pg/ml) and 3-copy (30.58 ± 5.93 pg/ml) mice (Fig. 7B), whereas treatments with RA, SB, and HB led to significantly (P < 0.01) increased levels of cGMP in the plasma of 1-copy mice (RA, 17.74 ± 3.01 pg/ml; SB, 16.88 ± 4.24 pg/ml; HB, 26.76 ± 5.67 pg/ml), which was further enhanced in 2-copy (RA, 31.05 ± 6.33 pg/ml; SB, 39.42 ± 6.71 pg/ml; HB, 47.51 ± 7.04 pg/ml), and 3-copy mice (RA, 45.56 ± 6.21 pg/ml; SB, 50.28 ± 7.27 pg/ml; HB, 67.25 ± 7.96 pg/ml).
Fig. 7.
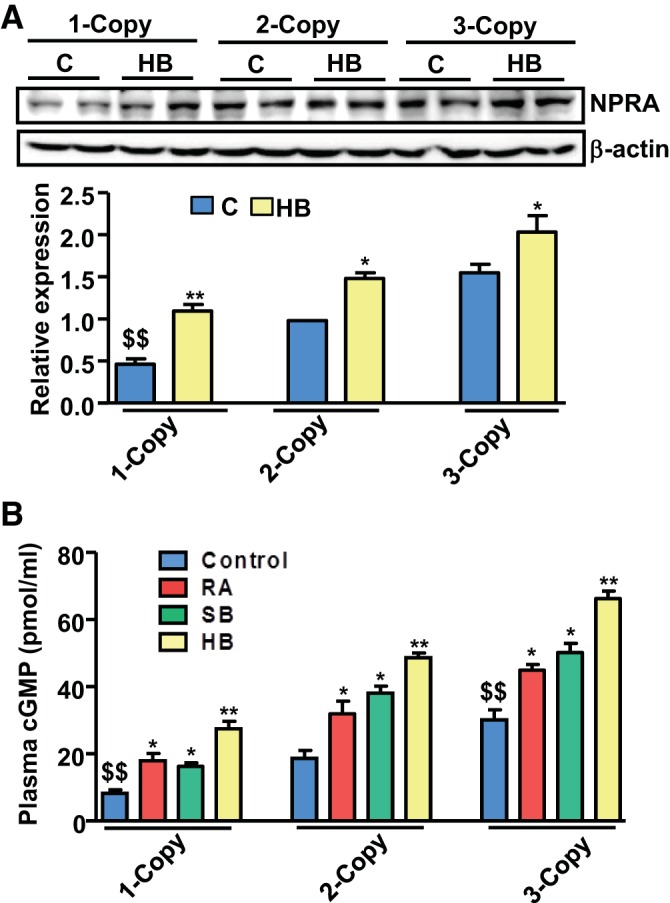
Cardiac protein expression of NPRA in control and drug-treated Npr1 gene-targeted mice. A: representative Western blot showing expression of NPRA in the heart tissue of control (C) and hybrid (HB) drug-treated mice. Densitometry analysis was done by AlphaInnotech phosphoimager software. B: plasma cGMP levels in control and drug-treated mice. Values are expressed as means ± SE of 3 independent experiments. *P < 0.05; **P < 0.01 (vehicle-treated vs. drug-treated same group); $$P < 0.01 (1-copy or 3-copy vs. 2-copy); n = 8 mice per group.
DISCUSSION
The results of the present study demonstrate that treatment with RA and SB either alone or in combination markedly suppresses the expression of hypertrophic markers, proinflammatory mediators, and MMPs in haplotype Npr1+/− heart tissues and also the basal levels in 2-copy mice hearts. The drug treatments suppressed the development of cardiac remodeling and fibrosis in Npr1+/− mice. The increase in HW/BW ratio in Npr1+/− mice indicates that cardiac hypertrophic growth occurred in these animals. Echocardiographic measurements show that treatment with RA and SB restored cardiac functions that were compromised in untreated haplotype Npr1+/− mice. Our findings are consistent with previous reports that an RA-mediated effect suppresses the hypertrophy of myocardial cells (48, 74). Chronic RA treatment has been shown to prevent ventricular fibrosis and improve impaired systolic heart function in rodent models of myocardial infarction (37, 47). Furthermore, the treatments with RA and SB protected the heart against fibrosis (15, 35, 62). The present results are consistent with the previous findings that RA exerts a beneficial effect in preventing cardiac remodeling, including the extracellular matrix (ECM), and fibrosis (9, 10). Cardiac hypertrophy has been shown to be associated with increased expression of β-MHC and α-SKA (53, 54). The present results suggest that an increased expression of hypertrophic marker genes such as β-MHC and α-SKA in the heart of Npr1+/− mice contributed to the development of cardiac hypertrophy in these animals. Expression levels of AP-1 molecules (c-fos and c-jun), which are functionally relevant molecular markers of ventricular hypertrophy, were also significantly increased in cardiac tissue of Npr1+/− mice. The attenuated expression of these hypertrophic markers and proto-oncogenes in RA- and SB-treated tissues suggests that these drugs exert their antihypertrophic effects by interacting with these key hypertrophic genes. A change in blood pressure could significantly affect the HW in a Npr1 gene-dose-dependent manner. Previous studies have indicated that high blood pressure is associated with an increase in the HW/BW ratios in vitamin D receptor gene-knockout mice (75). An association between increased HW/BW ratio and blood pressure has also been previously demonstrated (72). Our previous studies and the present results also suggest that Npr1 gene disruption causes the alteration in the gene expression of hypertrophic markers, proinflammatory mediators, and cytokines, which indicates that an increase in the HW/BW ratio that might be independent of blood pressure in these animals (67, 68). Thus, both the blood pressure and early gene activation might trigger cardiac hypertrophy by two independent pathways.
Cardiac remodeling is a dynamic process and excessive ECM deposition in the heart can lead to end-organ damage (7). MMPs and their natural inhibitors, TIMPs, are considered critical in cardiac tissue remodeling (43). The treatments with RA or SB, alone or in combination prevented the expression of MMPs and activated the expression of TIMPs in heart tissue of Npr1+/− mice. Moreover, in vivo studies have shown that RA not only prevents ventricular fibrosis and remodeling during the development of hypertension but also activates RA signaling after myocardial infarction (4, 37). Those previous findings support our hypothesis regarding the protective mechanisms of RA and SB in 1-copy haplotype Npr1 mice. TGF-β1 is a widely accepted profibrotic cytokine, which is attributed to the ECM remodeling and cardiovascular pathology that typically occurs in patients with chronic progressive cardiomyopathy (57). TGF-β1 exerts pleiotropic effects in the induction of tissue fibrosis, collagen synthesis, cytokine production, and myofibroblast trans-differentiation (23, 33, 56). The increase in the expression of TGF-β1 along with MMPs suggests a direct involvement of TGF-β1 in the cardiac remodeling process in Npr1+/− (1-copy) mice. In contrast, treating Npr1+/− mice with RA and SB alone or in combination prevented the expression of matrix synthesis in the cardiac tissue, suggesting that these drugs exert protective effects over the matrix remodeling process. NF-κB is a redox-sensitive transcription factor that directly regulates the expression of immediate- to early-response genes, cytokines, and inflammatory molecules (38, 73). It has been shown that NF-κB activity is significantly increased in vitamin A-deficient mice, while RA treatment attenuates its increased activation (2). In the present study, treatment with SB and RA or combination drug HB decreased the activation of p-NF-κB in Npr1+/− mice.
The findings of our current studies are consistent with previous reports demonstrating the inhibitory effect of HDACs on cardiac hypertrophy (26). Our results provide evidence that RA and SB prevent the cardiac remodeling and their cooperative effects in blocking the cardiac disorders by inhibiting HDAC 1 and HDAC 2 in Npr1+/− mice. Thus, the pharmacological inhibition of HDAC by RA and SB markedly repressed the expression of hypertrophic markers, matrix proteins, and proinflammatory mediators in the hearts of Npr1+/− mice. Evidence suggests that RA and SB induce epigenetic modifications at the promoter levels, as a consequence of acetylation, opened chromatin structure, and activation of transcriptional machinery to enhance gene expression (25, 29, 30, 32). The balance between HAT and HDAC is an essential regulatory switch, which can be achieved through the inhibition of the deacetylation process (17, 51). Our previous studies have also demonstrated that Npr1 gene contains various cis-acting elements and both RA and SB enhance its gene transcription and expression (30). Treatment with HB significantly induced cardiac NPRA expression in Npr1+/− mice. Moreover, reduced HDAC activity with stimulated HAT activity was observed in the heart tissues of RA- and SB-treated 1-copy mice, indicating that RA and SB exert a direct epigenetic modulatory effect on the cardiac-specific expression of Npr1. There was significant increase in protein levels of p300 and PCAF in HB-treated mice hearts. Earlier studies have shown that RA treatment was able to enhance p300 and PCAF protein expression and their binding to gene promoters (13, 17, 27). In the present study, we found that HDAC 1 and 2 proteins were overexpressed and treatment with HB significantly reduced HDAC 2 expression in Npr1+/− mice hearts. In general, HDACs control diverse processes, including hypertrophy and fibrosis (22, 26, 28), contractility (19), and energy metabolism (42). In vivo studies support the cardiac protective activity of HDAC inhibitors in the heart tissues (41). The present results demonstrate that RA and SB exert transcriptional regulatory effects on Npr1 gene expression and function in the cardiac tissues of haplotype 1-copy mice. Diagrammatic representation indicates the pathways involved in promoting the cardiac fibrosis and hypertrophic remodeling with decreased Npr1 expression in 1-copy mice; however, RA and SB treatments attenuated the abnormal fibrotic and hypertrophic remodeling in the hearts of Npr1+/− mice (Fig. 8).
Fig. 8.

Schematic representation of the proposed mechanisms by which RA and SB treatments inhibit the development of cardiac remodeling in Npr1+/− mice. Disruption of Npr1 gene leads to an unbalanced activation of NF-κB and AP-1 cascades that trigger the expression of MMPs, profibrotic cytokines, hypertrophic markers, NF-κB, AP-1, and HDACs, thereby activate specific molecular and structural changes leading to fibrosis and hypertrophic remodeling in 1-copy (Npr1+/−) mice hearts. Treatment with retinoic acid and sodium butyrate increases the production of cGMP thus inhibits the expression of MMPs, hypertrophic markers, NF-κB, AP-1, and HDACs, which protect against fibrosis and hypertrophic remodeling in Npr1+/− mice hearts. NF-κB, nuclear factor-kappa B; AP-1, activating protein-1; HDACs, histone deacetylases; HAT, histone acetyltransferase; MMPs, matrix metalloproteinases.
In summary, the present results provide direct evidence that RA, SB, and HB treatments synergistically inhibit cardiac hypertrophy and fibrosis, attenuating cardiac injury and dysfunction in Npr1+/− mice. Overall, the present findings provide important insights into the expression and function of Npr1, which might be critical for the physiological roles of GC-A/NPRA. The induced expression of Npr1 in the heart plays critical roles by inhibiting the expression levels of cardiac hypertrophic markers, proinflammatory, and profibrotic mediators, as well as diastolic disorders, thus providing possible molecular therapeutic targets in the prevention of cardiac remodeling and dysfunction.
GRANTS
This work was supported by National Institutes of Health Grants HL-057531 and HL-062147 and Core Facilities of IDeA Program.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
U.S. and K.N.P. conception and design of research; U.S., P.K., I.M., D.C., R.P., G.R., and K.N.P. performed experiments; U.S., P.K., I.M., and K.N.P. analyzed data; U.S., I.K., and K.N.P. interpreted results of experiments; U.S. and K.N.P. prepared figures; U.S. and K.N.P. drafted manuscript; U.S., P.K., and K.N.P. edited and revised manuscript; U.S., P.K., I.M., D.C., I.K., R.P., G.R., and K.N.P. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Meaghan Bloodworth and Whitney Nolan for excellent technical help and Kamala Pandey for assistance in the preparation of this manuscript. The authors are indebted to Professor Oliver Smithies for providing with the initial breeding pairs of Npr1 gene-targeted mice colonies.
REFERENCES
- 1.Arise KK, Pandey KN. Inhibition and down-regulation of gene transcription and guanylyl cyclase activity of NPRA by angiotensin II involving protein kinase C. Biochem Biophys Res Commun 349: 131–135, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Austenaa LM, Carlsen H, Ertesvag A, Alexander G, Blomhoff HK, Blomhoff R. Vitamin A status significantly alters nuclear factor-kappaB activity assessed by in vivo imaging. FASEB J 18: 1255–1257, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nature Struct Mol Biol 14: 1008–1016, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Bilbija D, Haugen F, Sagave J, Baysa A, Bastani N, Levy FO, Sirsjo A, Blomhoff R, Valen G. Retinoic acid signalling is activated in the postischemic heart and may influence remodelling. PLoS One 7: e44740, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush EW, McKinsey TA. Protein acetylation in the cardiorenal axis: the promise of histone deacetylase inhibitors. Circ Res 106: 272–284, 2010. [DOI] [PubMed] [Google Scholar]
- 6.Calderone A, Thaik CM, Takahashi N, Chang DL, Colucci WS. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J Clin Invest 101: 812–818, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camp TM, Smiley LM, Hayden MR, Tyagi SC. Mechanism of matrix accumulation and glomerulosclerosis in spontaneously hypertensive rats. J Hypertens 21: 1719–1727, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Chen S, Olsen K, Grigsby C, Gardner DG. Vitamin D activates type A natriuretic peptide receptor gene transcription in inner medullary collecting duct cells. Kidney Int 72: 300–306, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Choudhary R, Baker KM, Pan J. All-trans retinoic acid prevents angiotensin II- and mechanical stretch-induced reactive oxygen species generation and cardiomyocyte apoptosis. J Cell Physiol 215: 172–181, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Choudhary R, Palm-Leis A, Scott RC 3rd, Guleria RS, Rachut E, Baker KM, Pan J. All-trans retinoic acid prevents development of cardiac remodeling in aortic banded rats by inhibiting the renin-angiotensin system. Am J Physiol Heart Circ Physiol 294: H633–H644, 2008. [DOI] [PubMed] [Google Scholar]
- 11.de Jong S, van Veen TA, de Bakker JM, Vos MA, van Rijen HV. Biomarkers of myocardial fibrosis. J Cardiovasc Pharmacol 57: 522–535, 2011. [DOI] [PubMed] [Google Scholar]
- 12.Del Greco MF, Pattaro C, Luchner A, Pichler I, Winkler T, Hicks AA, Fuchsberger C, Franke A, Melville SA, Peters A, Wichmann HE, Schreiber S, Heid IM, Krawczak M, Minelli C, Wiedermann CJ, Pramstaller PP. Genome-wide association analysis and fine mapping of NT-proBNP level provide novel insight into the role of the MTHFR-CLCN6-NPPA-NPPB gene cluster. Hum Mol Genet 20: 1660–1671, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dietze EC, Caldwell LE, Marcom K, Collins SJ, Yee L, Swisshelm K, Hobbs KB, Bean GR, Seewaldt VL. Retinoids and retinoic acid receptors regulate growth arrest and apoptosis in human mammary epithelial cells and modulate expression of CBP/p300. Microsc Res Tech 59: 23–40, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Ellmers LJ, Scott NJ, Piuhola J, Maeda N, Smithies O, Frampton CM, Richards AM, Cameron VA. Npr1-regulated gene pathways contributing to cardiac hypertrophy and fibrosis. J Mol Endocrinol 38: 245–257, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 5: 15, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garg R, Pandey KN. Angiotensin II-mediated negative regulation of Npr1 promoter activity and gene transcription. Hypertension 41: 730–736, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Gaub P, Tedeschi A, Puttagunta R, Nguyen T, Schmandke A, Di Giovanni S. HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Different 17: 1392–1408, 2010. [DOI] [PubMed] [Google Scholar]
- 18.Gediya LK, Khandelwal A, Patel J, Belosay A, Sabnis G, Mehta J, Purushottamachar P, Njar VC. Design, synthesis, and evaluation of novel mutual prodrugs (hybrid drugs) of all-trans-retinoic acid and histone deacetylase inhibitors with enhanced anticancer activities in breast and prostate cancer cells in vitro. J Med Chem 51: 3895–3904, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Gupta MP, Samant SA, Smith SH, Shroff SG. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem 283: 10135–10146, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hisamori S, Tabata C, Kadokawa Y, Okoshi K, Tabata R, Mori A, Nagayama S, Watanabe G, Kubo H, Sakai Y. All-trans-retinoic acid ameliorates carbon tetrachloride-induced liver fibrosis in mice through modulating cytokine production. Liver Int 28: 1217–1225, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Hum D, Besnard S, Sanchez R, Devost D, Gossard F, Hamet P, Tremblay J. Characterization of a cGMP-response element in the guanylyl cyclase/natriuretic peptide receptor A gene promoter. Hypertension 43: 1270–1278, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Iyer A, Fenning A, Lim J, Le GT, Reid RC, Halili MA, Fairlie DP, Brown L. Antifibrotic activity of an inhibitor of histone deacetylases in DOCA-salt hypertensive rats. Br J Pharmacol 159: 1408–1417, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapur NK. Transforming growth factor-beta: governing the transition from inflammation to fibrosis in heart failure with preserved left ventricular function. Circ Heart Fail 4: 5–7, 2011. [DOI] [PubMed] [Google Scholar]
- 24.Karayannis G, Tsezou A, Giannatou E, Papanikolaou V, Giamouzis G, Triposkiadis F. Polymorphisms of renin-angiotensin system and natriuretic peptide receptor A genes in patients of Greek origin with a history of myocardial infarction. Angiology 61: 737–743, 2010. [DOI] [PubMed] [Google Scholar]
- 25.Kashyap V, Gudas LJ. Epigenetic regulatory mechanisms distinguish retinoic acid-mediated transcriptional responses in stem cells and fibroblasts. J Biol Chem 285: 14534–14548, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, Kim JK, Kim KK, Epstein JA, Kook H. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 113: 51–59, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Kim SH, Kang HJ, Na H, Lee MO. Trichostatin A enhances acetylation as well as protein stability of ERalpha through induction of p300 protein. Breast Cancer Res 12: R22, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, Hill JA. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 113: 2579–2588, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar P, Garg R, Bolden G, Pandey KN. Interactive roles of Ets-1, Sp1, and acetylated histones in the retinoic acid-dependent activation of guanylyl cyclase/atrial natriuretic peptide receptor-A gene transcription. J Biol Chem 285: 37521–37530, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar P, Periyasamy R, Das S, Neerukonda S, Mani I, Pandey KN. All-trans retinoic acid and sodium butyrate enhance natriuretic peptide receptor a gene transcription: role of histone modification. Mol Pharmacol 85: 946–957, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar P, Tripathi S, Pandey KN. Histone deacetylase inhibitors modulate the transcriptional regulation of guanylyl cyclase/natriuretic peptide receptor-a gene: interactive roles of modified histones, histone acetyltransferase, p300, AND Sp1. J Biol Chem 289: 6991–7002, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwon HS, Huang B, Ho Jeoung N, Wu P, Steussy CN, Harris RA. Retinoic acids and trichostatin A (TSA), a histone deacetylase inhibitor, induce human pyruvate dehydrogenase kinase 4 (PDK4) gene expression. Biochim Biophys Acta 1759: 141–151, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res 74: 207–212, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, Novak L, Renfrow MB, Chen YF. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res 102: 185–192, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Li YY, McTiernan CF, Feldman AM. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc Res 46: 214–224, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, Lu L, Tao BB, Zhu YC. All-trans retinoic acid inhibits the increases in fibronectin and PAI-1 induced by TGF-beta1 and Ang II in rat mesangial cells. Acta Pharmacol Sin 29: 1035–1041, 2008. [DOI] [PubMed] [Google Scholar]
- 37.Lu L, Yao T, Zhu YZ, Huang GY, Cao YX, Zhu YC. Chronic all-trans retinoic acid treatment prevents medial thickening of intramyocardial and intrarenal arteries in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 285: H1370–H1377, 2003. [DOI] [PubMed] [Google Scholar]
- 38.Machado RA, Constantino Lde S, Tomasi CD, Rojas HA, Vuolo FS, Vitto MF, Cesconetto PA, de Souza CT, Ritter C, Dal-Pizzol F. Sodium butyrate decreases the activation of NF-kappaB reducing inflammation and oxidative damage in the kidney of rats subjected to contrast-induced nephropathy. Nephrol Dialysis Transplant 27: 3136–3140, 2012. [DOI] [PubMed] [Google Scholar]
- 39.Mani I, Garg R, Tripathi S, Pandey KN. Subcellular trafficking of guanylyl cyclase/natriuretic peptide receptor-A with concurrent generation of intracellular cGMP. Biosci Rep 35: e00260, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGrath MF, de Bold AJ. Determinants of natriuretic peptide gene expression. Peptides 26: 933–943, 2005. [DOI] [PubMed] [Google Scholar]
- 41.McKinsey TA. The biology and therapeutic implications of HDACs in the heart. Hand Exp Pharmacol 206: 57–78, 2011. [DOI] [PubMed] [Google Scholar]
- 42.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev 21: 1790–1802, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 69: 562–573, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Newton-Cheh C, Larson MG, Vasan RS, Levy D, Bloch KD, Surti A, Guiducci C, Kathiresan S, Benjamin EJ, Struck J, Morgenthaler NG, Bergmann A, Blankenberg S, Kee F, Nilsson P, Yin X, Peltonen L, Vartiainen E, Salomaa V, Hirschhorn JN, Melander O, Wang TJ. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat Genet 41: 348–353, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, Pandey KN, Milgram SL, Smithies O, Maeda N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci USA 94: 14730–14735, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliver PM, John SW, Purdy KE, Kim R, Maeda N, Goy MF, Smithies O. Natriuretic peptide receptor 1 expression influences blood pressures of mice in a dose-dependent manner. Proc Natl Acad Sci USA 95: 2547–2551, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paiva SA, Matsubara LS, Matsubara BB, Minicucci MF, Azevedo PS, Campana AO, Zornoff LA. Retinoic acid supplementation attenuates ventricular remodeling after myocardial infarction in rats. J Nutr 135: 2326–2328, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Palm-Leis A, Singh US, Herbelin BS, Olsovsky GD, Baker KM, Pan J. Mitogen-activated protein kinases and mitogen-activated protein kinase phosphatases mediate the inhibitory effects of all-trans retinoic acid on the hypertrophic growth of cardiomyocytes. J Biol Chem 279: 54905–54917, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Pandey KN. Biology of natriuretic peptides and their receptors. Peptides 26: 901–932, 2005. [DOI] [PubMed] [Google Scholar]
- 50.Pandey KN, Oliver PM, Maeda N, Smithies O. Hypertension associated with decreased testosterone levels in natriuretic peptide receptor-A gene-knockout and gene-duplicated mutant mouse models. Endocrinology 140: 5112–5119, 1999. [DOI] [PubMed] [Google Scholar]
- 51.Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol 2011: 371832, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Polyakova V, Loeffler I, Hein S, Miyagawa S, Piotrowska I, Dammer S, Risteli J, Schaper J, Kostin S. Fibrosis in endstage human heart failure: severe changes in collagen metabolism and MMP/TIMP profiles. Int J Cardiol 151: 18–33, 2011. [DOI] [PubMed] [Google Scholar]
- 53.Ramamurthy B, Hook P, Jones AD, Larsson L. Changes in myosin structure and function in response to glycation. FASEB J 15: 2415–2422, 2001. [DOI] [PubMed] [Google Scholar]
- 54.Reggiani C, Bottinelli R, Stienen GJ. Sarcomeric myosin isoforms: fine tuning of a molecular motor. News Physiol Sci 15: 26–33, 2000. [DOI] [PubMed] [Google Scholar]
- 55.Rubattu S, Bigatti G, Evangelista A, Lanzani C, Stanzione R, Zagato L, Manunta P, Marchitti S, Venturelli V, Bianchi G, Volpe M, Stella P. Association of atrial natriuretic peptide and type a natriuretic peptide receptor gene polymorphisms with left ventricular mass in human essential hypertension. J Am Coll Cardiol 48: 499–505, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Ruiz-Ortega M, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J. TGF-beta signaling in vascular fibrosis. Cardiovasc Res 74: 196–206, 2007. [DOI] [PubMed] [Google Scholar]
- 57.Sanderson JE, Lai KB, Shum IO, Wei S, Chow LT. Transforming growth factor-beta(1) expression in dilated cardiomyopathy. Heart 86: 701–708, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sangaralingham SJ, Burnett JC Jr, McKie PM, Schirger JA, Chen HH. Rationale and design of a randomized, double-blind, placebo-controlled clinical trial to evaluate the efficacy of B-type natriuretic Peptide for the preservation of left ventricular function after anterior myocardial infarction. J Card Fail 19: 533–539, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi SJ, Nguyen HT, Sharma GD, Navar LG, Pandey KN. Genetic disruption of atrial natriuretic peptide receptor-A alters renin and angiotensin II levels. Am J Physiol Renal Physiol 281: F665–F673, 2001. [DOI] [PubMed] [Google Scholar]
- 60.Shi SJ, Vellaichamy E, Chin SY, Smithies O, Navar LG, Pandey KN. Natriuretic peptide receptor A mediates renal sodium excretory responses to blood volume expansion. Am J Physiol Renal Physiol 285: F694–F702, 2003. [DOI] [PubMed] [Google Scholar]
- 61.Simioniuc A, Campan M, Lionetti V, Marinelli M, Aquaro GD, Cavallini C, Valente S, Di Silvestre D, Cantoni S, Bernini F, Simi C, Pardini S, Mauri P, Neglia D, Ventura C, Pasquinelli G, Recchia FA. Placental stem cells pretreated with a hyaluronan mixed ester of butyric and retinoic acid to cure infarcted pig hearts: a multimodal study. Cardiovasc Res 90: 546–556, 2011. [DOI] [PubMed] [Google Scholar]
- 62.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 87: 1285–1342, 2007. [DOI] [PubMed] [Google Scholar]
- 63.Sun X, Zhang B, Hong X, Zhang X, Kong X. Histone deacetylase inhibitor, sodium butyrate, attenuates gentamicin-induced nephrotoxicity by increasing prohibitin protein expression in rats. Eur J Pharmacol 707: 147–154, 2013. [DOI] [PubMed] [Google Scholar]
- 64.Tremblay J, Hum DH, Sanchez R, Dumas P, Pravenec M, Krenova D, Kren V, Kunes J, Pausova Z, Gossard F, Hamet P. TA repeat variation, Npr1 expression, and blood pressure: impact of the Ace locus. Hypertension 41: 16–24, 2003. [DOI] [PubMed] [Google Scholar]
- 65.Tsukagoshi H, Shimizu Y, Kawata T, Hisada T, Shimizu Y, Iwamae S, Ishizuka T, Iizuka K, Dobashi K, Mori M. Atrial natriuretic peptide inhibits tumor necrosis factor-alpha production by interferon-gamma-activated macrophages via suppression of p38 mitogen-activated protein kinase and nuclear factor-kappa B activation. Regul Pept 99: 21–29, 2001. [DOI] [PubMed] [Google Scholar]
- 66.Vanderheyden M, Paulus WJ, Voss M, Knuefermann P, Sivasubramanian N, Mann D, Baumgarten G. Myocardial cytokine gene expression is higher in aortic stenosis than in idiopathic dilated cardiomyopathy. Heart 91: 926–931, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vellaichamy E, Das S, Subramanian U, Maeda N, Pandey KN. Genetically altered mutant mouse models of guanylyl cyclase/natriuretic peptide receptor-A exhibit the cardiac expression of proinflammatory mediators in a gene-dose-dependent manner. Endocrinology 155: 1045–1056, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vellaichamy E, Khurana ML, Fink J, Pandey KN. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J Biol Chem 280: 19230–19242, 2005. [DOI] [PubMed] [Google Scholar]
- 69.Vellaichamy E, Zhao D, Somanna N, Pandey KN. Genetic disruption of guanylyl cyclase/natriuretic peptide receptor-A upregulates ACE and AT1 receptor gene expression and signaling: role in cardiac hypertrophy. Physiol Genomics 31: 193–202, 2007. [DOI] [PubMed] [Google Scholar]
- 70.Ventura C, Cantoni S, Bianchi F, Lionetti V, Cavallini C, Scarlata I, Foroni L, Maioli M, Bonsi L, Alviano F, Fossati V, Bagnara GP, Pasquinelli G, Recchia FA, Perbellini A. Hyaluronan mixed esters of butyric and retinoic Acid drive cardiac and endothelial fate in term placenta human mesenchymal stem cells and enhance cardiac repair in infarcted rat hearts. J Biol Chem 282: 14243–14252, 2007. [DOI] [PubMed] [Google Scholar]
- 71.Volpe M, Rubattu S, Burnett J Jr. Natriuretic peptides in cardiovascular diseases: current use and perspectives. Eur Heart J 35: 419–425, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wallen WJ, Cserti C, Belanger MP, Wittnich C. Gender-differences in myocardial adaptation to afterload in normotensive and hypertensive rats. Hypertension 36: 774–779, 2000. [DOI] [PubMed] [Google Scholar]
- 73.Wan F, Lenardo MJ. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harbor Perspect Biol 1: a000067, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang HJ, Zhu YC, Yao T. Effects of all-trans retinoic acid on angiotensin II-induced myocyte hypertrophy. J Appl Physiol 92: 2162–2168, 2002. [DOI] [PubMed] [Google Scholar]
- 75.Xiang W, Kong J, Chen S, Cao LP, Qiao G, Zheng W, Liu W, Li X, Gardner DG, Li YC. Cardiac hypertrophy in vitamin D receptor knockout mice: role of the systemic and cardiac renin-angiotensin systems. Am J Physiol Endocrinol Metab 288: E125–E132, 2005. [DOI] [PubMed] [Google Scholar]
- 76.Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF. Expression of constitutively active guanylate cyclase in cardiomyocytes inhibits the hypertrophic effects of isoproterenol and aortic constriction on mouse hearts. J Biol Chem 278: 47694–47699, 2003. [DOI] [PubMed] [Google Scholar]
- 77.Zhao D, Das S, Pandey KN. Interactive roles of NPR1 gene-dosage and salt diets on cardiac angiotensin II, aldosterone and pro-inflammatory cytokines levels in mutant mice. J Hypertens 31: 134–144, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]