

Abstract
Hepatic glucose production (HGP) normally begins just prior to birth. Prolonged fetal hypoglycemia, intrauterine growth restriction, and acute hypoxemia produce an early activation of fetal HGP. To test the hypothesis that prolonged hypoxemia increases factors which regulate HGP, studies were performed in fetuses that were bled to anemic conditions (anemic: n = 11) for 8.9 ± 0.4 days and compared with control fetuses (n = 7). Fetal arterial hematocrit and oxygen content were 32% and 50% lower, respectively, in anemic vs. controls (P < 0.005). Arterial plasma glucose was 15% higher in the anemic group (P < 0.05). Hepatic mRNA expression of phosphonenolpyruvate carboxykinase (PCK1) was twofold higher in the anemic group (P < 0.05). Arterial plasma glucagon concentrations were 70% higher in anemic fetuses compared with controls (P < 0.05), and they were positively associated with hepatic PCK1 mRNA expression (P < 0.05). Arterial plasma cortisol concentrations increased 90% in the anemic fetuses (P < 0.05), but fetal cortisol concentrations were not correlated with hepatic PCK1 mRNA expression. Hepatic glycogen content was 30% lower in anemic vs. control fetuses (P < 0.05) and was inversely correlated with fetal arterial plasma glucagon concentrations. In isolated primary fetal sheep hepatocytes, incubation in low oxygen (3%) increased PCK1 mRNA threefold compared with incubation in normal oxygen (21%). Together, these results demonstrate that glucagon and PCK1 may potentiate fetal HGP during chronic fetal anemic hypoxemia.
Keywords: glucose, oxygen, PEPCK, glycogen, hepatocyte
glucose is the principal energy substrate for the fetus and is essential for normal fetal metabolism and growth. The transport of glucose from the mother to the fetus occurs by facilitated diffusion across the placenta. Therefore, the rate of fetal glucose uptake depends largely on the maternal arterial plasma glucose concentration (19). Normally, transported maternal arterial glucose is the sole source of fetal glucose, and there is no fetal hepatic glucose production (HGP) (20).
As part of the transition to extrauterine life, fetal HGP is activated just prior to delivery (13). At birth, infants have a period of reduced glucose intake when they are removed from their placental glucose supply and their mother's milk is being established. Thus, to avoid hypoglycemia, the neonate must rely on endogenous HGP. Fetal sheep at 0.97 gestation have increased endogenous HGP, as well as higher hepatic levels of the gluconeogenic enzymes phosphoenolpyruvate carboxykinase (PEPCK, encoded by PCK1) and glucose-6-phosphatase (G6Pase, encoded by G6PC) compared with 0.95 gestation (11, 13). PEPCK catalyzes the rate-limiting step, and G6Pase catalyzes the final step in gluconeogenesis. Increased perinatal transcription of PCK1 is activated by rising plasma concentrations of glucagon and glucocorticoids (13, 17, 36). There also is activation of glycogenolysis by increased secretion and plasma concentrations of glucagon and catecholamines around the time of birth (17, 36). Increased gluconeogenesis and glycogenolysis maintain glucose homeostasis in the neonate by matching glucose utilization with increased HGP (36).
Normal fetuses have little, if any, HGP prior to 0.97 gestation (11, 13). Animal studies, however, have shown that certain conditions can increase factors that regulate fetal HGP. One of these conditions is decreased fetal glucose supply and fetal plasma glucose concentrations produced during maternal fasting and maternal hypoglycemia (11, 26, 50). Other conditions that also increase these factors and fetal HGP include acute maternal hypoxia, bilateral uterine artery ligation, umbilical cord compression, and intrauterine growth restriction from placental insufficiency (PI-IUGR) (5, 15, 25, 34, 38, 40, 48). A characteristic that is common to these conditions is fetal hypoxemia or low fetal blood oxygen concentration. Studies specifically testing the impact of low blood oxygen concentration and/or low Po2 on factors regulating fetal HGP are limited to acute hypoxemia, intermittent hypoxemia, or hypoxemia coupled with hypoglycemia such as PI-IUGR (5, 15, 40, 44, 48, 50). The effect of chronic sustained fetal hypoxemia without hypoglycemia on the factors that regulate HGP has not been studied.
The goal of our study was to test the impact of experimental chronic anemic hypoxemia on factors that regulate fetal HGP, such as fetal arterial glucose, lactate, and hormone concentrations, as well as hepatic mRNA expression of key gluconeogenic enzymes and hepatic glycogen content. We hypothesized that chronic fetal hypoxemia would increase plasma arterial glucagon, cortisol, and hepatic PCK1 mRNA expression and decrease hepatic glycogen content. To test this hypothesis, we induced prolonged experimental fetal anemic hypoxemia by reducing the fetal blood oxygen carrying capacity through daily bleeding of the fetus.
MATERIALS AND METHODS
Fetal sheep model of chronic anemic hypoxemia.
Studies were conducted on pregnant Columbia-Rambouillet sheep carrying a singleton fetus at the Perinatal Research Center in Aurora, Colorado, with approval of the Institutional Animal Care and Use Committee, University of Colorado School of Medicine. This center is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) and is compliant with the Animal Welfare Act and Public Health Service Policy. At 120 ± 0.3 days gestational age (dGA; term ∼148 dGA), fetal catheters were surgically placed into the descending aorta and femoral vein via pedal incisions and into the common umbilical vein via one of the umbilical veins. Maternal catheters were placed into the femoral artery and vein via a groin incision. Surgery was performed with appropriate anesthesia, and pain was controlled, as previously described (47).
Beginning at least 5 days after surgery, fetal sheep were bled (anemic, n = 11) with isovolumetric replacement by 0.9% NaCl to anemic conditions for an average of 8.9 ± 0.4 days before umbilical blood flow and fetal glucose, lactate, and oxygen uptake measurements were performed. Anemic fetuses were compared with control fetuses who were not bled, but were otherwise treated the same as the anemic fetuses (CON; n = 7). The goal for fetal arterial oxygen content in the anemic group was 2.0 mmol/l, as this is the arterial oxygen content measured in PI-IUGR sheep fetuses (7, 28, 47). The amount of blood removed daily was determined with a previously established formula taking into account fetal hematocrit and the goal arterial oxygen content (22).
Biochemical analysis.
Fetal and maternal arterial blood samples were obtained daily. Arterial plasma glucose and lactate concentrations were measured using Yellow Springs Instrument 2700 (Yellow Springs Instruments, Yellow Springs, OH) and blood hematocrit, pH, partial pressure of oxygen, partial pressure of carbon dioxide (CO2), and oxygen-hemoglobin saturation were measured with the blood gas analyzer ABL 520 (Radiometer, Copenhagen, Denmark). Oxygen content of the blood was calculated by the ABL 520 analyzer. Fetal arterial plasma insulin was measured by enzyme-linked immunosorbent assay (ELISA) (Alpco, Windham, NH; intra-assay and interassay coefficients of variation = 5.6% and 4.7%, respectively; sensitivity = 0.14 ng/ml) (37) and norepinephrine by HPLC (model 2475 Waters, Milford, MA; intra- and interassay coefficients of variation = 9.2% and 9.0%, respectively; sensitivity = 170 pg/ml) (47) throughout the study. Fetal arterial plasma cortisol was measured by ELISA (Alpco Immunoassays; intra- and interassay coefficients of variation = 4.6% and 5.8%, respectively; sensitivity = 1.0 ng/ml) and glucagon by radioimmunoassay (Millipore, Billerica, MA; intra- and interassay CVs: 4.8% and 11.7%; sensitivity = 18.5 pg/ml) (37) on day 0 (baseline) and on the final day of the study before in vivo metabolic studies were performed.
Umbilical blood flow and nutrient uptake studies.
Studies were performed on the final day of the experimental period to determine umbilical blood flow and glucose, lactate, and oxygen uptake. Because of catheter failure, complete metabolic studies were completed on seven animals in each study group. One anemic fetus did not survive to necropsy following the metabolic study, but there were no other unplanned fetal deaths. Fetal umbilical blood flow was calculated using an ethanol tracer and the transplacental diffusion technique (32). Glucose, lactate, oxygen, and ethanol were measured from four blood draws obtained from the fetal umbilical vein and descending aorta after the ethanol tracer reached a steady state. During the study period, fetal blood removed was replaced with a fetal transfusion of heparinized maternal whole blood diluted with 0.9% NaCl to match the hematocrit of the fetal blood (47). Nutrient uptake by the fetus was calculated using the Fick principle (32). Fetal oxygen extraction efficiency was calculated with the equation: Oxygen extraction (%) = ([O2]v − [O2]a)/[O2]v × 100, where [O2]a is the fetal arterial oxygen content and [O2]v is the umbilical venous oxygen content (4).
Fetal and fetal liver weight.
The day after the metabolic studies, the mother and fetus were euthanized (37). Fetal organs were collected and weighed. A portion of the right lobe of the fetal liver was snap frozen in liquid nitrogen and then stored at −80°C. One anemic fetus did not survive to necropsy following the metabolic study. Other than not surviving to necropsy, we did not detect anything unusual about this fetus and have included it in the in vivo analysis.
Glycogen content.
Glycogen content in the liver was determined as previously described (29). Results are expressed as milligrams of glycogen per grams of tissue (wet weight).
Real-time quantitative PCR.
RNA was extracted from pulverized liver tissue and reverse transcribed to cDNA. Quantitative PCR was performed as previously described (50). Previously validated quantitative PCR primers and assays were used for PCK1: G6PC, peroxisome proliferator-activated receptor-γ coactivator (PGC1A), glucose-regulated protein, 78 kDa (GRP78; heat shock 70-kDa protein 5 [HSPA5]), DNA-damage inducible transcript 3 (DDIT3), pyruvate dehydrogenase kinase 4 (PDK4), lactate dehydrogenase A (LDHA), phosphofructokinase 1 (PFK1), and endothelin 1 (EDN1) (6, 16, 39, 48, 50). Additional primers were developed and validated for vascular endothelial growth factor A (VEGFA; forward: TTGCCTTGCTGCTCTACCTT; reverse: GGGCACACACTCCAGACTTT), erythropoietin (EPO; forward: CCCAGACACCAAGGTTAACT; reverse: GAAAGATAGCTTCTGAGAGCA), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 1(PFKB1), and the glucagon receptor (GCGR; forward: ATGCTGTTCGTCATCCCCTG; reverse: AAGCGCAGAATCCACCAGAA). The housekeeping genes GAPDH and RPS15 were not different between groups and were used to normalize results for the genes of interest (14).
Western blot analysis.
Tissue lysates were prepared from pulverized frozen liver tissue, and Western immunoblotting was performed as previously described (50). PEPCK, G6Pase, hepatocyte nuclear factor-4α (HNF4α), actin, protein kinase B (AKT), phosphorylated AKT (S473/T308), AMPK, phosphorylated AMPK (T172), cyclic AMP response element binding-protein (CREB), and phosphorylated CREB (S133) were used as previously described (47, 50). The membrane for glucose-regulated protein 78 kDa (GRP78) was blocked for 1 h in TBS with 0.01% Tween 20 (TBST; Bio-Rad, Hercules, CA) and 5% wt/vol nonfat dried milk. A monoclonal rat IgG anti-GRP78 antibody (Santa Cruz Biotechnology; Santa Cruz, CA) was diluted to 1:500 in TBST with 5% BSA and placed on the membrane overnight. Quantification of bands was performed by densitometry (Image J) or Image Studio (LI-COR) and was normalized to actin. For phosphorylated proteins, a ratio to the densitometry of the respective total protein was calculated.
Primary fetal hepatocytes.
Primary fetal hepatocytes were isolated from four additional late-gestation fetal sheep, as previously described (47). Briefly, a piece of the right lobe was flushed with perfusion buffer and then with digestion buffer containing collagenase. The digested tissue was filtered and spun, and isolated hepatocytes were washed, plated, and allowed to attach for 4 h in DMEM (1.1 mM glucose, 2 mM lactate, 2 mM glutamine, 1 mM pyruvate, 1× nonessential amino acids, 1× penicillin-streptomycin) supplemented with 0.1 nM insulin, 10 mM dexamethasone, and 10% FBS. After attachment, the cells were washed and incubated in nonsupplemented DMEM. The cells were then incubated in two different oxygen concentrations (21% vs. 3% oxygen) and 5% CO2 (balanced with nitrogen) with or without 100 μmol/l cAMP and 500 nmol/l dexamethasone for 24 h (47). RNA was then extracted from the hepatocytes, reverse transcribed, and quantitative PCR performed for PCK1, PGC1A, and RPS15.
Statistical analysis.
Statistical analysis was performed using SAS version 9.1 or GraphPad Prism 6.0. Results are expressed as means ± SE. A mixed-model ANOVA was performed to determine effects of treatment group (control or anemic), time (days of treatment), and treatment-time interactions for in vivo measurements made more than once. Repeated measures made in the same animal or hepatocytes isolated from the same animals also were accounted for. If P ≤ 0.05 in the ANOVA, individual means were compared by Fischer's least significant difference test. Data obtained at only one time point were compared using Student's t-test or Mann-Whitney U-test. Statistical significance was declared at P ≤ 0.05.
RESULTS
Maternal and fetal biochemistry and necropsy data.
Daily bleeding resulted in significant reduction in both the fetal arterial hematocrit and oxygen content beginning on day 1 in the anemic group (P < 0.001; Fig. 1, A and B). At the end of the study period, when umbilical blood flow and fetal nutrient uptakes were measured, the blood hematocrit and oxygen content were 32% and 50% lower, respectively, in the anemic vs. control group. There were no differences in pH, partial pressure of CO2, or arterial plasma lactate on any study day between groups (Table 1). There was a decrease in the partial pressure of oxygen and hemoglobin-oxygen saturation in both groups over time, but the groups were not different from each other (Fig. 1, C and D). By the end of the study, plasma glucose concentrations were 15% higher in the anemic vs. control group (P < 0.05; Fig. 2A).
Fig. 1.

Effect of chronic fetal anemic hypoxemia on fetal biochemistry. A: fetal arterial hematocrit was significantly lower in the anemic (□; n = 11) fetuses compared with controls (●; n = 7) (*P < 0.001). B: fetal arterial oxygen content was significantly lower in the anemic compared with the control fetuses (*P < 0.001). The partial pressure of arterial oxygen (Po2; C) and the hemoglobin-oxygen arterial saturation (So2; D) decreased in both groups compared with their baselines (#P < 0.01), but the groups were not different from each other. For all panels, the mean and SE are plotted, statistical analysis was performed by using mixed-model ANOVA.
Table 1.
Maternal and fetal biochemistry, umbilical blood flow, and lactate uptake at the end of the study
| Control | Anemic | |
|---|---|---|
| Maternal data | ||
| Blood pH | 7.45 ± 0.01 | 7.42 ± 0.02 |
| Blood Pco2, mmHg | 34.4 ± 1.0 | 37.7 ± 2.1 |
| Blood Po2, mmHg | 96.0 ± 2.3 | 88.3 ± 1.5 |
| Blood So2, % | 96.4 ± 0.7 | 96.2 ± 1.0 |
| Blood O2 Content, mmol/l | 5.28 ± 0.25 | 5.61 ± 0.27 |
| Blood hematocrit, % | 27.8 ± 1.3 | 30.3 ± 1.5 |
| Plasma glucose, mmol/l | 4.04 ± 0.34 | 3.99 ± 0.19 |
| Plasma lactate, mmol/l | 0.94 ± 0.15 | 1.05 ± 0.19 |
| Fetal data | ||
| Blood pH | 7.34 ± 0.01 | 7.34 ± 0.00 |
| Blood Pco2, mmHg | 51.7 ± 1.0 | 52.6 ± 0.9 |
| Plasma lactate, mmol/l | 2.28 ± 0.21 | 2.46 ± 0.17 |
| Umbilical blood flow, ml·min−1·kg−1 | 156 ± 14 | 139 ± 11 |
| Net lactate uptake, μmol·kg−1·min−1 | 28.0 ± 2.4 | 23.5 ± 2.9 |
| Maternal-fetal arterial glucose difference, mmol/l | 2.87 ± 0.28 | 2.93 ± 0.15 |
Values are expressed as means ± SE; n = 7 for control and n = 11 for anemic except for umbilical blood flow and lactate uptake (anemic: n = 7).
Pco2, partial pressure of carbon dioxide; Po2, partial pressure of oxygen; So2, oxygen saturation.
Fig. 2.

Effect of chronic fetal anemic hypoxemia on fetal glucose. A: fetal arterial plasma glucose concentrations were measured throughout the study. Glucose concentrations were significantly higher in the anemic fetuses (□; n = 11) at the end of the study period compared with controls (■; n = 7; P < 0.05 by mixed-model ANOVA). B: there was no difference in fetal glucose uptake between groups when measured at the end of the study. Plotted values are means ± SE. *P < 0.05 control vs. anemic.
There were no differences in maternal pH, partial pressure of oxygen, partial pressure of CO2, oxygen-hemoglobin saturation (So2), oxygen content, hematocrit, glucose, or lactate concentration on any day of study (Table 1).
Fetal necropsy data are shown in Table 2. The anemic fetuses had heavier right ventricular weight compared with controls (P < 0.05). There were no other differences between the groups.
Table 2.
Fetal necropsy data
| Control | Anemic | |
|---|---|---|
| Gestational age, days | 134.3 ± 0.5 | 134.6 ± 0.4 |
| Fetal weight, g | 2911 ± 159 | 3043 ± 113 |
| Fetal sex, % female | 100% | 55% |
| Crown rump length, cm | 48.1 ± 1.6 | 47.5 ± 1.6 |
| Pancreas, g | 2.96 ± 0.25 | 3.08 ± 0.20 |
| Liver, g | 89.9 ± 7.2 | 108.2 ± 8.9 |
| Kidneys, g | 18.75 ± 0.84 | 18.78 ± 0.72 |
| Perirenal adipose tissue, g | 9.43 ± 0.72 | 10.98 ± 0.93 |
| Spleen, g | 6.26 ± 0.41 | 5.63 ± 0.40 |
| Adrenal glands, g | 0.44 ± 0.06 | 0.41 ± 0.02 |
| Lungs, g | 108.0 ± 3.4 | 103.3 ± 3.2 |
| Heart (total), g | 20.27 ± 1.92 | 22.53 ± 0.93 |
| Left ventricle + septum, g | 8.25 ± 0.50 | 8.45 ± 0.56 |
| Right ventricle, g | 3.69 ± 0.31 | 5.44 ± 0.47* |
| Total no. placentomes | 74.3 ± 4.0 | 83.9 ± 5.1 |
| Total placental weight, g | 325.9 ± 35.5 | 347.8 ± 24.4 |
Values are expressed as means ± SE; n = 7 for control and n = 11 for anemic for gestational age, weight, and sex. n = 10 for anemic for fetal measurements.
Statistical significance (P ≤ 0.05).
Fetal hormones.
Fetal arterial plasma norepinephrine and insulin concentrations were not statistically different between the two groups (Fig. 3, A and B). Fetal arterial cortisol concentrations were increased 90% on the final day in the anemic group compared with baseline (P < 0.05; Fig. 3C), but they were not different from the controls on the final day (Fig. 3C). By the final day of the study, arterial glucagon concentrations in anemic fetuses were significantly increased compared with baseline and 70% higher than controls (P < 0.05; Fig. 3D).
Fig. 3.

Effect of chronic fetal anemic hypoxemia on fetal hormone concentrations. Fetal arterial plasma levels of norepinephrine (A) and insulin (B) were not significantly different between control (solid bars; n = 7) and anemic (open bars; n = 11) fetuses. C: cortisol increased over the course of the study, such that in the anemic fetuses, it was significantly higher at the end compared with their baseline (P < 0.05). D: glucagon increased over the course of the study such that in the anemic fetuses, it was significantly higher at the end compared with their baseline, as well as on the final day being higher than controls (P < 0.05). Mean ± SEs are plotted. Statistical analysis was done through mixed-model ANOVA. *P < 0.05 between indicated values.
Fetal metabolic studies.
Umbilical blood flow, fetal glucose and lactate uptake rates, and the maternal-to-fetal arterial glucose concentration difference were not different between groups (Table 1, Fig. 2B). The anemic fetuses had significantly lower whole body oxygen utilization despite having a higher oxygen extraction compared with controls (P < 0.05; Fig. 4, A and B). There was a significant correlation between fetal arterial oxygen content and fetal oxygen extraction, such that at low oxygen contents, there was increased fetal oxygen extraction (P < 0.001; Fig. 4C).
Fig. 4.
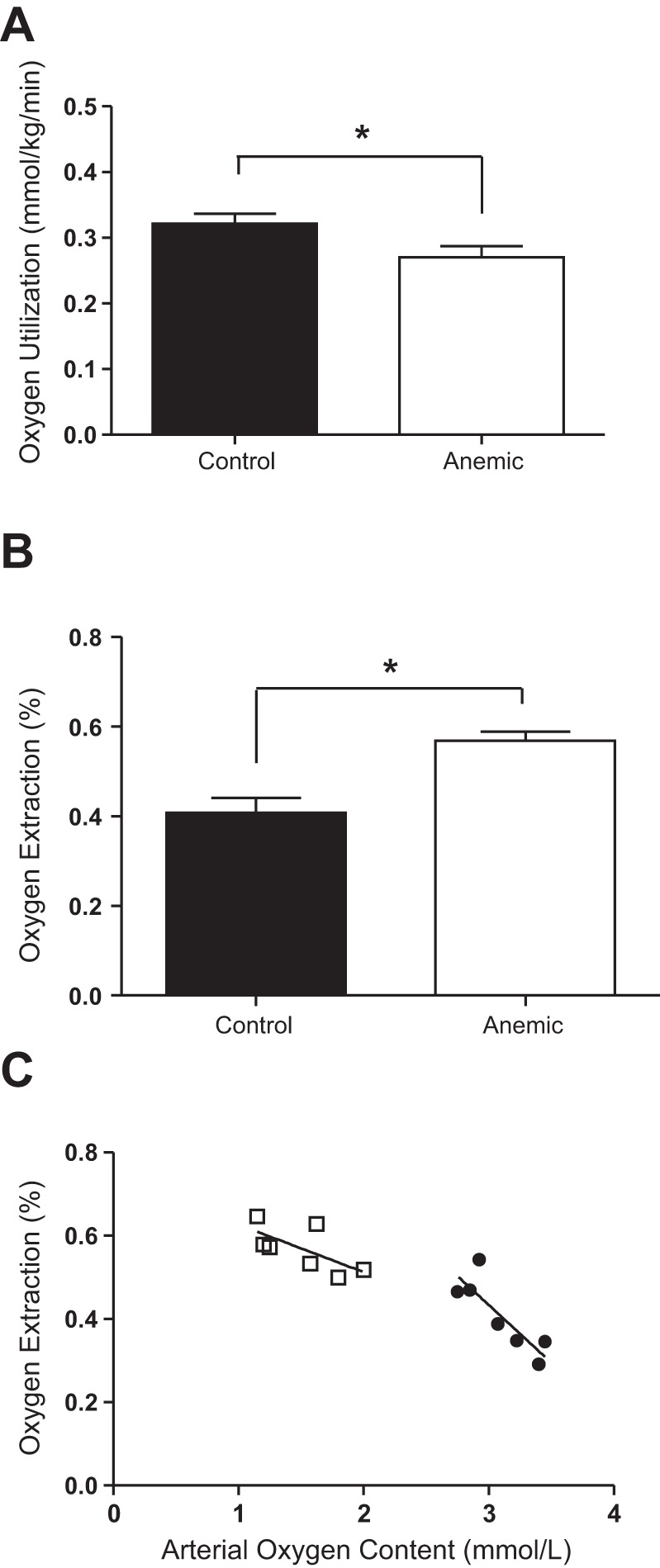
Effect of chronic fetal anemic hypoxemia on fetal oxygen utilization and extraction. A: whole body oxygen utilization was significantly less in anemic (n = 7) fetuses than controls (n = 7; *P < 0.05). B: fetal oxygen extraction was significantly greater in anemic fetuses (n = 7) than controls (n = 7; *P < 0.01). C: in both anemic (□) and control (●) fetuses, oxygen extraction was correlated with arterial oxygen content such that at lower levels of arterial oxygen, there was higher placental oxygen extraction. Groups were compared by Student's t-test. Means ± SEs are plotted.
Hepatic PCK1, G6PC, and glycogen.
Hepatic PCK1 mRNA was twofold greater in the anemic group compared with controls (P < 0.05; Fig. 5A). The tendency for higher levels of hepatic G6PC mRNA in anemic fetuses was not statistically significant (Fig. 5D). Fetal arterial glucagon concentrations were significantly correlated with both hepatic PCK1 and G6PC mRNA levels (P < 0.05; Fig. 5, B and E). Fetal arterial cortisol concentrations did not correlate with either hepatic PCK1 or G6PC mRNA (Fig. 5, C and F). The hepatic protein expressions of PEPCK and glucose-6-phosphatase were not different between the groups (Table 3). Hepatic glycogen concentration was significantly lower in the anemic fetuses compared with controls (P < 0.05; Fig. 5G) and was inversely correlated to fetal arterial plasma glucagon concentrations (P < 0.05), but not fetal cortisol concentrations (Fig. 5, H and I).
Fig. 5.

Effect of chronic fetal anemic hypoxemia on hepatic PCK1 mRNA, G6PC mRNA, and glycogen. A: hepatic PCK1 mRNA levels were higher in anemic fetuses (n = 10) vs. controls (n = 7) (P < 0.05). There was a significant correlation between final day anemic (□) and control (●) fetal arterial plasma glucagon and hepatic PCK1 mRNA (B; r2 = 0.52, P < 0.05), but not with fetal arterial plasma cortisol concentrations (C). D: G6PC mRNA levels were not statistically different between groups. There was a significant correlation between final day anemic (□) and control (●) fetal arterial plasma glucagon and G6PC mRNA (E; r2 = 0.29, P < 0.05), but not with fetal arterial plasma cortisol concentrations (F). G: hepatic glycogen levels were greater in anemic vs. CON fetuses. There was an inverse correlation between final day anemic (□) and control (●) fetal arterial plasma glucagon and hepatic glucagon (H; r2 = 0.52, P < 0.05), but not with fetal arterial plasma cortisol concentrations (I). Median and interquartile ranges are plotted for PCK1 and G6PC mRNA, and mean ± SE are plotted for glycogen. *P < 0.05 vs. controls.
Table 3.
Expression of genes and proteins in the fetal liver
| Control | Anemic | |
|---|---|---|
| Hepatic mRNA Normalized to GAPDH and RPS15 | ||
| Peroxisome proliferator-activated receptor-γ | 1.00 ± 0.27 | 0.90 ± 0.34 |
| Coactivator 1α (PGC1A) | ||
| Activating transcription factor 2 (ATF2) | 1.00 ± 0.29 | 0.68 ± 0.20 |
| Hypoxia inducible factor 1α (HIF1A) | 1.00 ± 0.34 | 0.61 ± 0.23 |
| Vascular endothelial growth factor A (VEGFA) | 1.00 ± 0.17 | 0.82 ± 0.20 |
| Erythropoietin (EPO) | 1.00 ± 0.36 | 1.54 ± 0.95 |
| Endothelin 1 (EDN1) | 1.00 ± 0.17 | 0.81 ± 0.06 |
| Phosphofructokinase 1 (PFK1) | 1.00 ± 0.27 | 1.59 ± 0.21 |
| 6-phospho-2-kinase/fructose 2,6 bisphosphatase 1 | 1.00 ± 0.27 | 0.39 ± 0.07* |
| (PFKFB1) | ||
| Lactate dehydrogenase A (LDHA) | 1.00 ± 0.14 | 1.26 ± 0.42 |
| Pyruvate dehydrogenase kinase 4 (PDK4) | 1.00 ± 0.31 | 0.84 ± 0.41 |
| DNA damage inducible transcript 3 (DDIT3) | 1.00 ± 0.11 | 0.85 ± 0.08 |
| Heat shock 70-kDa protein 5 (HSPA5) | 1.00 ± 0.10 | 0.73 ± 0.07* |
| Glucagon receptor (GCGR) | 1.00 ± 0.11 | 0.77 ± 0.06* |
| Hepatic Proteins | ||
| PEPCK (to Actin) | 1.00 ± 0.10 | 1.12 ± 0.10 |
| Glucose-6-phosphatase (to Actin) | 1.00 ± 0.27 | 0.88 ± 0.30 |
| Total AMPK (to Actin) | 1.00 ± 0.30 | 0.55 ± 0.14 |
| T172 phosphorylated AMPK (to total) | 1.00 ± 0.19 | 0.95 ± 0.13 |
| Total CREB (to Actin) | 1.00 ± 0.31 | 1.23 ± 0.24 |
| S133 phosphorylated CREB (to total) | 1.00 ± 0.28 | 1.17 ± 0.27 |
| Total AKT (to Actin) | 1.00 ± 0.09 | 1.02 ± 0.09 |
| S473/T308 phosphorylated AKT (to total) | 1.00 ± 0.10 | 0.98 ± 0.19 |
| HNF4α (to Actin) | 1.00 ± 0.12 | 1.05 ± 0.20 |
| GRP78 (to Actin) | 1.00 ± 0.37 | 0.70 ± 0.22 |
Values are expressed as means ± SE; n = 7 for control and n = 10 for anemic.
Statistical significance (P ≤ 0.05).
Cellular nutrient sensors and intracellular signaling.
Hepatic levels of mRNA and protein of genes associated with cellular nutrient sensing, selected for their responsiveness to acute oxygen deprivation (including HIF1α target genes: VEGFA, EPO, EDN1, PFK1, PFKFB1, PDK4, and LDHA), were largely unchanged by chronic anemic hypoxemia (Table 3). Similarly unchanged were intracellular signaling proteins AMPK, CREB, and AKT. Chronic anemic hypoxemia reduced hepatic mRNA expression of the glucagon receptor (GCCR) (P < 0.05; Table 3).
Fetal hepatocytes.
The roles of oxygen and hormone stimulation on expression of genes involved in regulation of HGP were tested in primary fetal sheep hepatocytes. In response to 3% oxygen, mRNA expression of both PCK1 and PGC1A increased twofold to threefold compared with 21% oxygen (P < 0.001; Fig. 6). Expression of PCK1 and PGC1A also increased in response to cAMP + dexamethasone treatment (P < 0.005; Fig. 6).
Fig. 6.
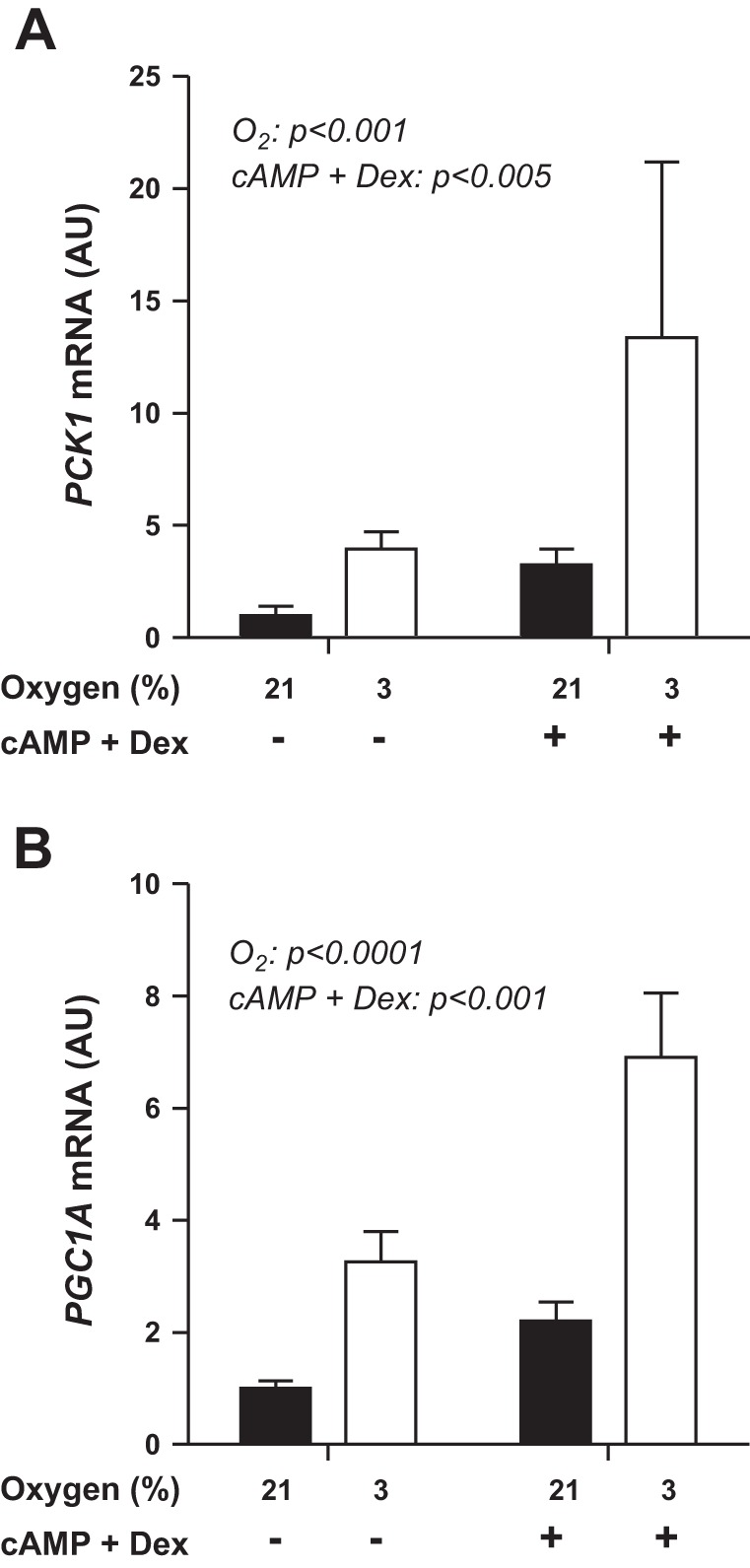
PCK1 and PGC1A mRNA levels are increased in fetal hepatocytes incubated in 3% oxygen compared with 21% oxygen. 3% vs. 21% oxygen and the presence of cAMP + dexamethasone (Dex) both significantly increased mRNA expression of PCK1 (A) and PGC1A (B). Statistical analysis was performed by mixed-model ANOVA.
DISCUSSION
In this study, we determined that low fetal blood oxygen concentrations as a result of chronic fetal anemia increases circulating glucagon concentrations, which were associated with increased hepatic PCK1 mRNA and decreased hepatic glycogen content. These results show that fetal anemic hypoxemia is sufficient to stimulate and sustain increases in factors that are associated with activation of both gluconeogenesis and glycogenolysis. Furthermore, these results demonstrate that fetal hypoglycemia (a hallmark of PI-IUGR) is not necessary for this response, even though it can, by itself, lead to fetal HGP (21). Supporting this conclusion is our finding that exposure to either 3% oxygen or glucogenic activators (cAMP + dexamethasone) increases expression of PCK1 and PGC1A in isolated fetal hepatocytes. This is the first study to test the impact of chronic (>24 h) fetal anemic hypoxemia independent of placental insufficiency and fetal hypoglycemia on fetal metabolic and endocrine responses, thereby identifying novel mechanisms by which fetal oxygen concentrations can regulate fetal glucose metabolism.
To determine the fetal endocrine response to anemic hypoxemia, we measured the concentrations of several hormones throughout the study. Fetal glucagon concentrations increased in the anemic group and correlated with both PCK1 and G6PC mRNA. Glucagon has been previously shown to activate fetal HGP, but the concentrations of glucagon required were greater than 1,000 pg/ml, suggesting fetal hepatic resistance to glucagon relative to adults (10, 45). In our study, physiological concentrations of glucagon, 20–80 pg/ml, correlated with higher PCK1 and G6PC mRNA levels. It may be that fetal anemic hypoxemia augments the physiological hyperglucagonemia leading to the transcriptional activation of PCK1, while in fetal normoxemia, much higher concentrations of glucagon are required to stimulate fetal gluconeogenesis. Lower hepatic glucagon receptor mRNA levels in anemic fetuses is evidence of ongoing hepatic glucagon signaling, consistent with a dose-dependent decrease in GCCR mRNA in primary hepatocytes following incubation with glucagon (1).
Fetal cortisol concentrations increased in the anemic fetuses, but also started lower than controls. By the end of the study, cortisol concentrations were not different between the two groups. These mixed results make it difficult to speculate about the role of cortisol in regulating hepatic PCK1 in this set of animals, despite the well-described role for cortisol in activating gluconeogenesis (12, 13, 38). Whether this represents another pathway partially responsible for the hepatic phenotype in anemic fetuses is not clear. To demonstrate the capacity of hormone signaling, as well as oxygen to increase PCK1 and G6PC mRNA, we performed in vitro studies with isolated fetal hepatocytes. cAMP + dexamethasone treatment increased PCK1 mRNA levels in these cells. Interestingly, although we did not detect increased hepatic PGC1A mRNA in the anemic fetuses, both 3% oxygen and cAMP + dexamethasone treatment in isolated hepatocytes increased PGC1A expression. Thus, oxygen clearly has the capacity to increase hepatocyte PGC1A expression, but in vivo fetal hypoxemia does not.
Since physiological increases in glucagon were associated with increased hepatic expression of gluconeogenic genes, we analyzed components of the cAMP-dependent glucagon-signaling pathway. PI-IUGR fetuses have activation of PCK1 associated with increased glucagon, PGC1α (PGC1A), and phosphorylated CREB (29, 48). However, we found no change in PGC1A or phosphorylated CREB in the livers of anemic fetuses on the terminal day of the study. This may be due to three factors. First, levels may have been increased earlier in the study representing an acute fetal response to hypoxemia (2, 41). Second, there may be fundamental differences in the fetal response to chronic hypoxemia and PI-IUGR. Fetuses affected by PI-IUGR experience global nutrient restriction, including reduced oxygen supply and blood concentrations (47, 48). Further, this oxygen deprivation occurs throughout most of the latter half of gestation, in contrast to the nine days of anemic hypoxemia tested in the current model. Factors other than fetal hypoxemia, to which the fetus is exposed for longer than 9 days, may be responsible for increased PGC1A and phosphorylated CREB in PI-IUGR. Third, and likely related to the duration and global nutrient restriction noted above, PI-IUGR fetuses have a much more robust increase in PCK1 and G6PC mRNA levels compared with the anemic fetuses (29, 30, 47, 48). The modest increase in PCK1 and G6PC mRNA in the anemic livers compared with models of placental insufficiency is consistent with the similar level of hepatic protein expression of PEPCK. It is notable that while both PI-IUGR and anemic fetuses have increased hepatic PCK1, substantial differences in hepatic signaling are apparent, likely due to different hepatic nutrient (glucose and oxygen) supplies.
We did not find any differences in hepatic AMPK phosphorylation or GRP78, two proteins normally activated by acute nutrient deprivation and/or hypoxemia (Table 3) (18, 24). Three reasons may account for this lack of activation. First, although AMPK and GRP78 are activated by hypoxemia, the anemic animals did not have a significant decrease in their Po2 compared with controls, and hepatocytes might not have experienced tissue hypoxia, which may be required to activate AMPK and GRP78 (18, 24, 48). Supporting the conclusion that the liver did not experience hypoxia is the lack of change noted in any of the hypoxia-responsive genes, such as VEGFA, EPO, EDN1, PFK1, PFKBP1, LDHA, PDK4, DDIT3, and HSPA5 (8, 9, 43). Second, although anemic fetuses had low oxygen content, they were not deprived of glucose or lactate, which also are important for activating AMPK and GRP78 (48). Lastly, most studies demonstrating activation of AMPK and GRP78 focus on acute oxygen deprivation, often in vitro (18, 24).
Fetal oxygen utilization was significantly decreased in the anemic group compared with controls. This is despite higher oxygen extraction in the anemic fetuses. In previous reports (4, 52), fetuses were shown to maintain normal oxygen utilization during graded reductions in oxygen supply produced experimentally by partial occlusion of the maternal terminal aorta, causing uterine blood flow restriction. This increased oxygen extraction enabled normal fetal oxygen utilization during graded hypoxemia in this model. In contrast, the anemic fetuses in the present study were unable to maintain oxygen utilization despite increased fetal oxygen extraction. In the uterine blood flow restriction paradigm, the effect of acute hypoxemia on fetal oxygen consumption was tested (4, 52). In the current study, we tested the effects over a more prolonged period (9 days). Indeed, in chronic hypoxemia due to placental insufficiency, fetal sheep also tend to have slightly lower oxygen consumption rates than normal (47).
We also have shown in the current studies that chronic fetal anemic hypoxemia increased fetal glucose concentrations without a change in fetal insulin concentrations. This was not due to an increase in the rate of placental glucose transfer as a possible cause of elevated fetal glucose in anemia. Elevated fetal glucose concentrations in anemic fetuses were likely mediated by high fetal glucagon and cortisol concentrations. Even the slight, though not statistically significant, increase in mean fetal norepinephrine concentrations may have played a role (27, 30, 31). As the focus of the current study was on the factors that regulate fetal HGP, we did not determine the relative contribution of HGP and decreased fetal glucose utilization for the increase in fetal glucose concentrations.
The model of anemia, or reduced oxygen carrying capacity, for hypoxemia was chosen because it did not allow fetuses to compensate by increasing total hemoglobin, and because we could isolate the effects of hypoxemia from hypoglycemia. Kitanaka et al. (23) showed that fetuses exposed to 28 days of hypoxemia via high altitude responded by increasing their hemoglobin and hematocrit over time to normalize arterial oxygen contents; these fetuses grew at normal rates, indicative of normal fetal nutrient metabolism. In contrast, our model allowed us to chronically match the mean oxygen content of PI-IUGR fetuses (2.0 mmol/l) (7, 28, 47). However, comparison of sheep models of human PI-IUGR fetuses exposes limitations of this study, including length and timing of hypoxemia. Studies done to model chronic PI-IUGR in sheep result in fetal hypoxemia beginning as early as day 90 in gestation (33). In the current study, fetal hypoxemia occurred on average for 9 days at 0.85 gestation. In contrast, the chronically anemic fetal sheep is an appropriate model for human chronic fetal anemia, such as occurs in chronic fetomaternal hemorrhage and subacute placental abruption, and also may help model aspects of fetal hypoxemia observed in cases of twin-twin transfusion syndrome, diabetic pregnancy, and some cases of post-date pregnancies (3, 42, 46). Future studies to induce experimental anemia earlier in gestation are planned. The studies also are limited by the random allocation of all female fetuses into the control group. We chose to include all animals in the anemic group, as we have never detected sex differences in outcomes reported (29, 38, 47, 48, 50). Furthermore, for the most important outcomes, PCK1 mRNA, G6PC mRNA, and glucagon concentrations, the two highest values in the anemic group were females, and the mean concentrations within the anemic group were higher for females than for males.
Perspectives and Significance
In conclusion, we speculate that in pregnancies complicated by fetal hypoxemia, such as intrauterine growth restriction, low oxygen concentrations in conjunction with elevated glucagon play a role in activating factors associated with fetal hepatic gluconeogenesis and glycogenolysis resulting in HGP. These are important findings, as complications of pregnancy that lead to premature activation of fetal HGP predispose to later-life development of hepatic insulin resistance, hyperglycemia, and ultimately Type 2 diabetes mellitus by unknown mechanisms (35, 49, 51).
GRANTS
P. J. Rozance was supported by National Institutes of Health (NIH) Grants R01 DK-088139 and K08 HD-060688 (to P. J. Rozance, principle investigator). L. D. Brown was supported by NIH Grants K12 HD-057022 Building Interdisciplinary Careers in Women's Health Scholar Award, and R01 HD-079404-01A1. S. R. Wesolowski was supported by K01 DK-090199 and R03 DK-102972. W. W. Hay Jr. was supported by NIH Grants T32007186-32 (principle investigator and program director), K12HD068372 (PD), and NIH-NCATS UL1TR001082, TL1TR001081, and KL2TR001080 (codirector), and a Grand Challenges Exploration Grant from the Bill and Melinda Gates Foundation (OPP1061082).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.C., S.R.W., J.B., S.S.J., W.W.H.J., and P.J.R. conception and design of research; C.C., S.R.W., J.B., J.L.B., L.D.B., R.B.W., W.W.H.J., and P.J.R. performed experiments; C.C., S.R.W., J.L.B., and P.J.R. analyzed data; C.C., S.R.W., J.L.B., L.D.B., S.S.J., R.B.W., W.W.H.J., and P.J.R. interpreted results of experiments; C.C., S.R.W., and P.J.R. prepared figures; C.C. drafted manuscript; C.C., S.R.W., J.B., J.L.B., L.D.B., S.S.J., R.B.W., W.W.H.J., and P.J.R. edited and revised manuscript; C.C., S.R.W., J.B., J.L.B., L.D.B., S.S.J., R.B.W., W.W.H.J., and P.J.R. approved final version of manuscript.
ACKNOWLEDGMENTS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institute of Child Health and Human Development.
REFERENCES
- 1.Abrahamsen N, Lundgren K, Nishimura E. Regulation of glucagon receptor mRNA in cultured primary rat hepatocytes by glucose and cAMP. J Biol Chem 270: 15,853–15,857, 1995. [DOI] [PubMed] [Google Scholar]
- 2.Beitner-Johnson D, Millhorn DE. Hypoxia induces phosphorylation of the cyclic AMP response element-binding protein by a novel signaling mechanism. J Biol Chem 273: 19,834–19,839, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Berry SM, Puder KS, Bottoms SF, Uckele JE, Romero R, Cotton DB. Comparison of intrauterine hematologic and biochemical values between twin pairs with and without stuck twin syndrome. Am J Obstet Gynecol 172: 1403–1410, 1995. [DOI] [PubMed] [Google Scholar]
- 4.Boyle DW, Hirst K, Zerbe GO, Meschia G, Wilkening RB. Fetal hind limb oxygen consumption and blood flow during acute graded hypoxia. Pediatr Res 28: 94–100, 1990. [DOI] [PubMed] [Google Scholar]
- 5.Bristow J, Rudolph AM, Itskovitz J, Barnes R. Hepatic oxygen and glucose metabolism in the fetal lamb. Response to hypoxia. J Clin Invest 71: 1047–1061, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown LD, Rozance PJ, Bruce JL, Friedman JE, Hay WWJ, Wesolowski SR. Limited capacity for glucose oxidation in fetal sheep with intrauterine growth restriction. Am J Physiol Regul Integr Comp Physiol 309: R920–R928, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown LD, Rozance PJ, Thorn SR, Friedman JE, Hay WWJ. Acute supplementation of amino acids increases net protein accretion in IUGR fetal sheep. Am J Physiol Endocrinol Metab 303: E352–E364, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dengler VL, Galbraith MD, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol 49: 1–15, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8: 705–713, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Devaskar SU, Ganguli S, Styer D, Devaskar UP, Sperling MA. Glucagon and glucose dynamics in sheep: evidence for glucagon resistance in the fetus. Am J Physiol Endocrinol Metab 246: E256–E265, 1984. [DOI] [PubMed] [Google Scholar]
- 11.Fowden AL, Forhead AJ. Adrenal glands are essential for activation of glucogenesis during undernutrition in fetal sheep near term. Am J Physiol Endocrinol Metab 300: E94–E102, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fowden AL, Mijovic J, Silver M. The effects of cortisol on hepatic and renal gluconeogenic enzyme activities in the sheep fetus during late gestation. J Endocrinol 137: 213–222, 1993. [DOI] [PubMed] [Google Scholar]
- 13.Fowden AL, Mundy L, Silver M. Developmental regulation of glucogenesis in the sheep fetus during late gestation. J Physiol 508: 937–947, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gadhia MM, Maliszewski AM, O'Meara MC, Thorn SR, Lavezzi JR, Limesand SW, Hay WWJ, Brown LD, Rozance PJ. Increased amino acid supply potentiates glucose-stimulated insulin secretion but does not increase β-cell mass in fetal sheep. Am J Physiol Endocrinol Metab 304: E352–E362, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gentili S, Morrison JL, McMillen IC. Intrauterine growth restriction and differential patterns of hepatic growth and expression of IGF1, PCK2, and HSDL1 mRNA in the sheep fetus in late gestation. Biol Reprod 80: 1121–1127, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gien J, Tseng N, Seedorf G, Roe G, Abman SH. Endothelin-1 impairs angiogenesis in vitro through Rho-kinase activation after chronic intrauterine pulmonary hypertension in fetal sheep. Pediatr Res 73: 252–262, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Girard J. Metabolic adaptations to change of nutrition at birth. Biol Neonate 58 Suppl 1: 3–15, 1990. [DOI] [PubMed] [Google Scholar]
- 18.Hardie DG. Sensing of energy and nutrients by AMP-activated protein kinase. Am J Clin Nutr 93: 891S–896S, 2011. [DOI] [PubMed] [Google Scholar]
- 19.Hay WWJ, Molina RA, DiGiacomo JE, Meschia G. Model of placental glucose consumption and glucose transfer. Am J Physiol Regul Integr Comp Physiol 258: R569–R577, 1990. [DOI] [PubMed] [Google Scholar]
- 20.Hay WWJ, Sparks JW, Quissell BJ, Battaglia FC, Meschia G. Simultaneous measurements of umbilical uptake, fetal utilization rate, and fetal turnover rate of glucose. Am J Physiol Endocrinol Metab 240: E662–E668, 1981. [DOI] [PubMed] [Google Scholar]
- 21.Houin SS, Rozance PJ, Brown LD, Hay WWJ, Wilkening RB, Thorn SR. Coordinated changes in hepatic amino acid metabolism and endocrine signals support hepatic glucose production during fetal hypoglycemia. Am J Physiol Endocrinol Metab 308: E306–E314, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jonker SS, Scholz TD, Segar JL. Transfusion effects on cardiomyocyte growth and proliferation in fetal sheep after chronic anemia. Pediatr Res 69: 485–490, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitanaka T, Alonso JG, Gilbert RD, Siu BL, Clemons GK, Longo LD. Fetal responses to long-term hypoxemia in sheep. Am J Physiol Regul Integr Comp Physiol 256: R1348–R1354, 1989. [DOI] [PubMed] [Google Scholar]
- 24.Koong AC, Auger EA, Chen EY, Giaccia AJ. The regulation of GRP78 and messenger RNA levels by hypoxia is modulated by protein kinase C activators and inhibitors. Radiat Res 138: S60–S63, 1994. [PubMed] [Google Scholar]
- 25.Lane RH, Flozak AS, Ogata ES, Bell GI, Simmons RA. Altered hepatic gene expression of enzymes involved in energy metabolism in the growth-retarded fetal rat. Pediatr Res 39: 390–394, 1996. [DOI] [PubMed] [Google Scholar]
- 26.Lemons JA, Moorehead HC, Hage GP. Effects of fasting on gluconeogenic enzymes in the ovine fetus. Pediatr Res 20: 676–679, 1986. [DOI] [PubMed] [Google Scholar]
- 27.Leos RA, Anderson MJ, Chen X, Pugmire J, Anderson KA, Limesand SW. Chronic exposure to elevated norepinephrine suppresses insulin secretion in fetal sheep with placental insufficiency and intrauterine growth restriction. Am J Physiol Endocrinol Metab 298: E770–E778, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Limesand SW, Rozance PJ, Macko AR, Anderson MJ, Kelly AC, Hay WWJ. Reductions in insulin concentrations and beta-cell mass precede growth restriction in sheep fetuses with placental insufficiency. Am J Physiol Endocrinol Metab 304: E516–E523, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Limesand SW, Rozance PJ, Smith D, Hay WW. Increased insulin sensitivity and maintenance of glucose utilization rates in fetal sheep with placental insufficiency and intrauterine growth restriction. Am J Physiol Endocrinol Metab 293: E1716–E1725, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Limesand SW, Rozance PJ, Zerbe GO, Hutton JC, Hay WWJ. Attenuated insulin release and storage in fetal sheep pancreatic islets with intrauterine growth restriction. Endocrinology 147: 1488–1497, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Macko AR, Yates DT, Chen X, Green AS, Kelly AC, Brown LD, Limesand SW. Elevated plasma norepinephrine inhibits insulin secretion, but adrenergic blockade reveals enhanced beta-cell responsiveness in an ovine model of placental insufficiency at 0.7 of gestation. J Dev Orig Health Dis 4: 402–410, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maliszewski AM, Gadhia MM, O'Meara MC, Thorn SR, Rozance PJ, Brown LD. Prolonged infusion of amino acids increases leucine oxidation in fetal sheep. Am J Physiol Endocrinol Metab 302: E1483–E1492, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrison JL. Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin Exp Pharmacol Physiol 35: 730–743, 2008. [DOI] [PubMed] [Google Scholar]
- 34.Narkewicz MR, Carver TD, Hay WWJ. Induction of cytosolic phosphoenolpyruvate carboxykinase in the ovine fetal liver by chronic fetal hypoglycemia and hypoinsulinemia. Pediatr Res 33: 493–496, 1993. [DOI] [PubMed] [Google Scholar]
- 35.Peterside IE, Selak MA, Simmons RA. Impaired oxidative phosphorylation in hepatic mitochondria in growth-retarded rats. Am J Physiol Endocrinol Metab 285: E1258–E1266, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Rozance PJ, Hay WWJ. Early human development. Early Hum Dev 86: 275–280, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rozance PJ, Limesand SW, Barry J, Brown L, Hay W. Glucose replacement to euglycemia causes hypoxia, acidosis, and decreased insulin secretion in fetal sheep with intrauterine growth restriction. Pediatr Res 65: 72–78, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rozance PJ, Limesand SW, Barry JS, Brown LD, Thorn SR, LoTurco D, Regnault TRH, Friedman JE, Hay WWJ. Chronic late-gestation hypoglycemia upregulates hepatic PEPCK associated with increased PGC1α mRNA and phosphorylated CREB in fetal sheep. Am J Physiol Endocrinol Metab 294: E365–E370, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rozance PJ, Limesand SW, Hay WWJ. Decreased nutrient-stimulated insulin secretion in chronically hypoglycemic late-gestation fetal sheep is due to an intrinsic islet defect. Am J Physiol Endocrinol Metab 291: E404–E411, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Rudolph CD, Roman C, Rudolph AM. Effect of acute umbilical cord compression on hepatic carbohydrate metabolism in the fetal lamb. Pediatr Res 25: 228–233, 1989. [DOI] [PubMed] [Google Scholar]
- 41.Shoag J, Arany Z. Regulation of hypoxia-inducible genes by PGC-1α. Arterioscler Thromb Vasc Biol 30: 662–666, 2010. [DOI] [PubMed] [Google Scholar]
- 42.Stanek J. Comparison of placental pathology in preterm, late-preterm, near-term, and term births. Am J Obstet Gynecol 210: 234 e1–e6, 2014. [DOI] [PubMed] [Google Scholar]
- 43.Stow LR, Jacobs ME, Wingo CS, Cain BD. Endothelin-1 gene regulation. FASEB J 25: 16–28, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stratford LL, Hooper SB. Effect of hypoxemia on tissue glycogen content and glycolytic enzyme activities in fetal sheep. Am J Physiol Regul Integr Comp Physiol 272: R103–R110, 1997. [DOI] [PubMed] [Google Scholar]
- 45.Teng C, Battaglia FC, Meschia G, Narkewicz MR, Wilkening RB. Fetal hepatic and umbilical uptakes of glucogenic substrates during a glucagon-somatostatin infusion. Am J Physiol Endocrinol Metab 282: E542–E550, 2002. [DOI] [PubMed] [Google Scholar]
- 46.Teramo KA. Obstetric problems in diabetic pregnancy. The role of fetal hypoxia. Best Pract Res Clin Endocrinol Metab 24: 663–671, 2010. [DOI] [PubMed] [Google Scholar]
- 47.Thorn SR, Brown LD, Rozance PJ, Hay WWJ, Friedman JE. Increased hepatic glucose production in fetal sheep with intrauterine growth restriction is not suppressed by insulin. Diabetes 62: 65–73, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thorn SR, Regnault TRH, Brown LD, Rozance PJ, Keng J, Roper M, Wilkening RB, Hay WWJ, Friedman JE. Intrauterine growth restriction increases fetal hepatic gluconeogenic capacity and reduces messenger ribonucleic acid translation initiation and nutrient sensing in fetal liver and skeletal muscle. Endocrinology 150: 3021–3030, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thorn SR, Rozance PJ, Brown LD, Hay WWJ. The intrauterine growth restriction phenotype: fetal adaptations and potential implications for later life insulin resistance and diabetes. Semin Reprod Med 29: 225–236, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thorn SR, Sekar SM, Lavezzi JR, O'Meara MC, Brown LD, Hay WWJ, Rozance PJ. A physiological increase in insulin suppresses gluconeogenic gene activation in fetal sheep with sustained hypoglycemia. Am J Physiol Regul Integr Comp Physiol 303: R861–R869, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vuguin P, Raab E, Liu B, Barzilai N, Simmons R. Hepatic insulin resistance precedes the development of diabetes in a model of intrauterine growth retardation. Diabetes 53: 2617–2622, 2004. [DOI] [PubMed] [Google Scholar]
- 52.Wilkening RB, Meschia G. Fetal oxygen uptake, oxygenation, and acid-base balance as a function of uterine blood flow. Am J Physiol Heart Circ Physiol 244: H749–H755, 1983. [DOI] [PubMed] [Google Scholar]