

Abstract
Objective
To identify novel prognostic and therapeutic markers for PARP inhibitors in BRCA wild type ovarian cancer (OvCa).
Methods
BRCAness status was defined by analyzing whole-exome deep sequencing data from 220 BRCAwt OvCa cases in TCGA. Thirty-three DNA-repair genes were screened in an integrated manner for BRCA-independent mechanism of BRCAness using multiple-dimensional genomic data. Publicly available databases and siRNA knock-down were used for external validation and evaluation of drug response in OvCa cell lines.
Results
In 220 BRCAwt OvCa patients, tumors exhibiting the BRCAness signature have enhanced OS (HR [95% CI]=0.33 [0.15-0.69], P=0.004) and PFS (HR [95% CI]=0.51 [0.24-1.08], P=0.077), strongly suggesting a BRCA-independent mechanism of drug sensitivity in those patients. Systematic screening of driving molecular events of BRCAness revealed that RAD50 deletion is a marker of BRCAness. The RAD50 deletion occurred in 18% of BRCAwt OvCa patients. RAD50 deletion led to its decreased mRNA expression in tumors (Fold Change=0.63, P=3.56 × 10-13). In BRCAwt patients, RAD50 deletion was associated with significantly better OS (HR [95% CI]=0.44 [0.25-0.78], P=0.005) and PFS (HR [95% CI]=0.60 [0.37-0.99], P=0.044), adjusted by age and stage. Knockdown of RAD50 expression augmented OvCa cell's responses to cisplatin and olaparib. Among 19 OvCa cell lines, the RAD50 copy number deletion is significantly associated with better responses to two structurally distinct PARPis (i.e. olaparib and rucaparib).
Conclusion
Our study identified the copy number deletion of RAD50 as a candidate marker for survival and response to PARPis in BRCAwt OvCa tumors.
Keywords: High-grade serous ovarian cancer, PARP inhibitor, BRCAness, Homologous recombination
Introduction
Ovarian cancer (OvCa) is the most lethal gynecological cancer in the world. OvCa is clinically and pathologically heterogeneous with different subtypes and etiologies [1]. This clinical heterogeneity can be further observed among same pathological subtype, reflecting by distinct molecular signatures in mRNA [2], protein [3] and miRNA [4] expression profiles. Standard treatment strategies have largely ignored the heterogeneity and individuality of different molecular subtypes of OvCa. Current treatment of OvCa entails surgical resection of the tumor followed by adjuvant platinum-based chemotherapy with or without paclitaxel. Despite an acceptable initial response to chemotherapy, most women eventually develop chemotherapy-resistant disease and die of recurrent disease [5, 6]. Understanding the mechanism(s) that drive the heterogeneous chemotherapy responses is critical to prolonging OvCa patients' survival. Scientific studies (including our work) combined with clinical trials have demonstrated that homologous recombination (HR) deficiency might be the Achilles heel of this deadly disease. These studies have demonstrated that loss of HR DNA repair function results in elevated genome instability, which confers OvCa tumor cells' sensitivity to both frontline therapy (e.g., cisplatin) [7] and developing anti-cancer therapies such as poly (ADP-ribose) polymerase inhibitors (PARPi) [8]. Promoted by these studies, olaparib, one of the PARPi drugs, was recently approved by FDA to treat BRCA1 and BRCA2 germline-mutated OvCa patients. More PARPi drugs are undergoing active phase II and III investigation for OvCa patients according to clinical.trial.gov. However, the most established mechanism of HR deficiency, BRCA1 and 2 germline mutations, only account for approximately 15% of the OvCa patients [9]. For those patients, it has been demonstrated that PARPi might serve as a treatment. There are many remaining questions including (1) whether PARPi can also have a therapeutic effect for the 85% BRCA wild type (BRCAwt) patients and (2) how to stratify the BRCAwt patients for PARPi treatment. Previous studies have shown that BRCA-like phenotype (also known as BRCAness) can be present in roughly 50% of cases of high-grade disease [10]. However, the underlying mechanisms and clinically actionable markers for BRCAness in BRCAwt OvCa patients remain elusive.
In this study, we investigated whether BRCAness can be quantified in BRCAwt OvCa patients using multidimensional genomic and clinical data from The Cancer Genome Atlas (TCGA) project. We further performed integrative screening to identify the underlying mechanisms and potential mark for BRCAness and PARPi response in BRCAwt patients.
Methods
Patients
The copy number alteration, mRNA gene expression, and clinical data of three hundred sixteen high-grade serous OvCa patients were obtained from TCGA controlled-access portal with IRB and dbGap approval. Detailed patient information, including age at diagnosis, tumor stage and grade in different BRCAness statuses is listed in Table 1. The interval from the date of initial surgical removal to patient death or last contact (censored) was calculated as OS duration. The interval from the date of initial surgical removal to the date of progressed/recurred disease or last contact (censored) was calculated as PFS duration.
Table 1. Age and Tumor Characteristics of Patients With Different BRCAness Status.
| Characteristic | All Cases | BRCA Wild-Type | BRCA Mutation/Methylation | P Value | ||
|---|---|---|---|---|---|---|
| Hypomutation | Intermediate | Hypermutation | ||||
| No. of cases | 316 | 44 | 157 | 19 | 96 | |
| Age, mean [range], y | 60.6 [27-88] | 59.5 [35-82] | 62.3 [40-87] | 63.4 [40-83] | 57.5 [27-79] | 0.006 |
| Tumor stage | ||||||
| II | 14 (4) | 0 | 7 (5) | 2 (10) | 5 (5) | 0.33 |
| III | 248 (79) | 38 (86) | 118 (75) | 14 (74) | 78 (82) | |
| IV | 53 (17) | 6 (14) | 32 (20) | 3 (16) | 12 (13) | |
| Missing, No. | 1 | 0 | 0 | 0 | 1 | |
| Tumor grade | ||||||
| 2 | 28 (9) | 2 (5) | 16 (11) | 2 (11) | 8 (8) | 0.68 |
| 3 | 281 (91) | 41 (95) | 136 (89) | 17 (89) | 87 (92) | |
| Missing, No. | 7 | 1 | 5 | 0 | 1 | |
Systematic screening of driving molecular events of BRCAness in HR pathway genes
We first calculated the Pearson Correlation Coefficient between the copy number alteration and mRNA gene expression of the candidate genes, indicating the mRNA expression changed by the copy number variation. Then the correlation between the copy number alteration and the genome stability represented by the mutation frequency in each patient sample, is also calculated to show how the copy number variation in the HR pathways genes is associated with the genome stability. We selected genes with high correlation between their copy number variation and mRNA expression alteration, and whose copy number variation showed high association with genome stability for further study.
Analysis of Whole-Exome Mutation Data
In total, 19,359 mutations across 316 OvCa samples were previously characterized by TCGA working group [8]. To determine the hypermutation status of each OvCa patient group, we used an enrichment score (ES) [11] as we previously described. The maximum absolute ES was obtained if the N samples in the patient group were ranked the top (or bottom) N samples among all OvCa cases. The ES was measured for each patient group. We permuted the patients' labels 106 times to generate a background ES distribution and calculate the false-discovery rate (FDR).
Genomic and Chemotherapy response Analyses in Cell Lines
Almost 700 cancer cell lines across 16 cancer types and the response information (IC25, IC50, IC75, IC90, etc.) to 138 anti-cancer drugs were downloaded form Genomics of Drug Sensitivity in Cancer [12]. All cell lines were identity authenticated to exclude cross-contaminated or synonymous lines. Total copy number values were determined from Affymetrix SNP6.0 microarray data. And the copy number segments in each of the cell lines were further predicted using the ‘PICNIC’ algorithm. For RAD50 gene to be classified as deleted, the entire RAD50 coding sequence must be contained in one contiguous segment defined by PICNIC, and have a PICNIC score less than or equal to three. Gene expression was determined by using the Affymetrix HT-HGU122A microarray.
Cell Lines, Cell Culture, Reagents
Human ovarian cancer cell line HeyA8 was obtained from the American Type Culture Collection (ATCC, Manassas, VA). The cell line has been authenticated using the STR Short Tandem Repeat (STR) Method.
siRNA Transfection and MTT Assay
Twenty-four hours after transfection with 20 nm si-RAD50-1, si-RAD50-2, or si-Ctrl, cells were seeded onto 96-well plates (1 × 103 cells/well) and treated with a titration of cisplatin or olaparib. The medium and drug were replenished at day three for olaparib treatment. After incubation for five (cisplatin) or seven (olaparib) days, cell viability was estimated using the MTT reagent (Sigma Chemical, St. Louis, MO), and surviving fractions were calculated. Cell survival was calculated by normalizing the absorbance to that of untreated controls.
Statistical Analysis
Standard statistical tests were used to analyze the clinical and genomics data, including the chi-square test, Fisher's exact test, Wilcoxon rank-sum test, log-rank test, and Cox proportional hazard analysis were used to analyze the clinical and genomics data. Significance was defined as P<0.05. When multiple testing correction was required, Benjamini-Hochberg multiple testing and permutation-based FDR estimation correction [13] were used to estimate the false-estimated rate. Analyses were primarily performed using R.
Result
BRCAness and its association with survival in BRCAwt Ovarian Cancer
In our previous analysis using same cohort of 316 OvCa patients from TCGA, we have identified 37 cases and 29 cases with BRCA1 and BRCA2 non-synonymous mutations, respectively [7]. Among 37 BRCA1 mutated cases, 27 have germ-line mutations and 10 have somatic mutations. Among 29 BRCA2 mutated cases, 20 have germ-line mutations and nine have somatic mutations. Two BRCA2 germline mutations and one BRCA1 germline mutation were recently revealed to be SNPs rather than deleterious germline mutations [14]. Those three mutations, therefore, were not considered as BRCA mutations in the current study. We further identified 33 OvCa cases with BRCA1 epigenetically silenced in the tumor by integrating DNA methylation and gene expression data [7]. No promoter hypermethylation of BRCA2 was observed across 316 TCGA samples. These observations are consistent with the previous report that BRCA1/2 germline mutations account for ∼15% of OvCa cancer cases [15, 16] and epigenetic inactivation of BRCA1 gene accounts for another 10% of OvCa cancer cases [17]. The BRCA1 mutation, BRCA2 mutation and BRCA1 epigenetic silence are strictly mutually exclusive. No patient in the TCGA data set has two kinds of BRCA deficiencies. In total, there are 96 patients with either BRCA1/2 mutation or BRCA1 DNA methylation and 220 patients without any known BRCA deficiencies (Table 1). The 5-year survival rate of patients with BRCA deficiency was 44% (95% CI [confidence interval]=34%-58%, Figure S1), which is significantly better than BRCAwt cases, with 5-year survival rates of 25% (95% CI= [18%-34%], Figure S1) in the unadjusted (log-rank P=0.002) and adjusted (P=0.02, HR [hazard ratio]=0.67, 95% CI=0.48-0.94) models.
We used an enrichment score (ES) combined with tumor mutation rate to determine hypermutated and hypomutated tumors in OvCa [17]. Briefly, all 316 OvCa cases were decreasingly ordered on the basis of their mutation rates. The higher the mutation rate the more genome instability the tumor is. The hypermutated samples were defined as top 15% ranked patients, which is the turning point of the fitting curve. And bottom 15% ranked tumors are accordingly defined as hypomutated samples. As we expected, the hypermutated samples are significantly enriched with BRCA deficiency samples (Odds ratio=3.13, P=0.001). The other 19 samples without any BRCA deficiency were thus defined as BRCAness tumors.
Among the 220 BRCAwt patients, the 5-year overall survival rate of 19 patients with BRCAness signature was 55% (95% CI=32%-93%), which is significantly better than that of 44 Hypomutated (5-year rate=15%, 95% CI=7%-36%) and 156 Intermediate patients (5-year rate=34%, 95% CI=16%-35%) in the unadjusted (log-rank P=0.002, Table 2 and Figure 1C). The association of BRACness and the overall survival remained significant when we adjusted for age and tumor stage (P=0.004, HR=0.33, 95% CI=0.15-0.69, Table 2). Consistent with observations in OS, the different status of BRCAness in 220 OvCa wt patients could predict longer PFS in both unadjusted (P=0.02, Figure 1D) and adjusted (P=0.077, HR=0.51, 95% CI=0.24-1.08, Table 2) models. A direct comparison between patients with BRCAness signature and BRCA deficient patients revealed that patients with BRCAness signature had a comparable 3-year (75%, 95% CI=56%-100%) and 5-year (55%, 95% CI=32%-93%) overall survival rate in survival compared with BRCA deficient patients (3-year survival rate 71%, 95% CI=61%-82% and 5-year survival rate 44%, 95% CI=34%-58%). These observations suggest that tumors with BRCAness signature exhibit similar clinical characters, genome instability, and prognosis with BRCA deficient patients.
Table 2. Multivariable Models for Overall Survival and Progression-Free Survival of BRCA wild type OvCa patients with different BRCAness status.
| Overall Survival | Progression-Free Survival | |||||||
|---|---|---|---|---|---|---|---|---|
| 3-Year Rate, % (95% CI) | 5-Year Rate, % (95% CI) | HR (95% CI) | P Value | 3-Year Rate, % (95% CI) | 5-Year Rate, % (95% CI) | HR (95% CI) | P Value | |
| BRCAness | ||||||||
| Hypomutation | 40(26-60) | 15 (7-36) | 1 [Reference] | 8(2-27) | 0* | 1 [Reference] | ||
| Intermediate | 61(53-71) | 34(16-35) | 0.60(0.39-0.91) | 0.016 | 16(10-26) | 12(6-21) | 0.80(0.53-1.21) | 0.29 |
| BRCAness | 75(56-100) | 55(32-93) | 0.33(0.15-0.69) | 0.004 | 31(13-75) | 21(6-68) | 0.51(0.24-1.08) | 0.077 |
| Tumor Stage | ||||||||
| II | 89(71-100) | 67(36-100) | 1 [Reference] | 75(50-100) | 37(9-100) | 1 [Reference] | ||
| III | 59(51-68) | 21(14-31) | 2.34(0.73-7.43) | 0.15 | 13(8-21) | 8(4-16) | 4.06(1.27-12.92) | 0.018 |
| IV | 48(33-70) | 34(20-58) | 2.40(0.71-8.08) | 0.16 | 14(5-39) | 14 (5-39) | 4.22(1.25-14.22) | 0.020 |
| Age increase of 1.0 ye | 1.02(1.00-1.03) | 0.04 | 0.99(0.98-1.01) | 0.34 | ||||
all patients in the hypomutation group lived no more than 5-years
Figure 1. The BRCAness and its association with survival.
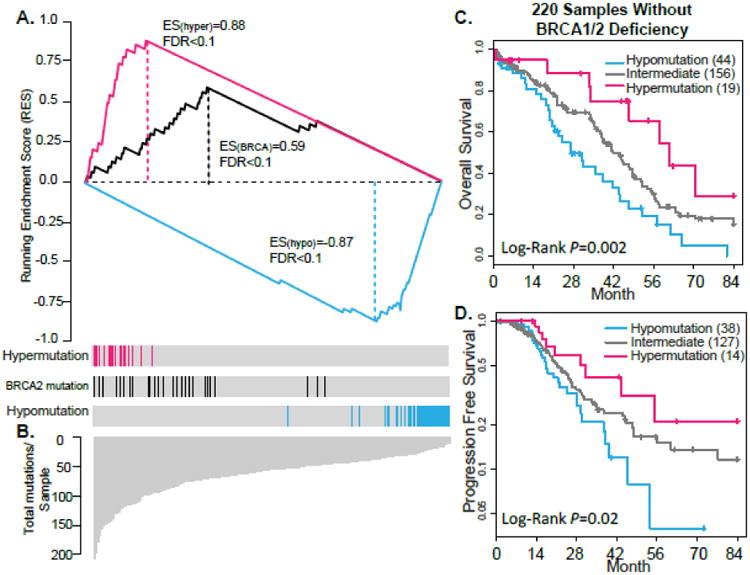
(A) Enrichment Score (ES) test of genome instability in BRCAness, Hypomutated and BRCA2-mutated cases (positive control). The bottom portion of the plot shows the total numbers of non-synonymous mutations of 316 decreasingly ranked OvCa cases. The height of each line indicates the number of non-synonymous mutations in each OvCa case. The middle portion of the plot shows where the samples with BRCAness (red bar) or BRCA2 mutations (black bar) and hypomutation (blue bar) appear in the ranked list of samples in the bottom portion. The top portion of the plot shows the running ES for the BRCA1- or BRCA2-mutated cases. (B & C) Kaplan-Meier survival curves for OS (B) and PFS (C) durations of BRCAness, Intermediate, and Hypomutated patients in 220 BRCAwt cases. The percent probability of survival is plotted versus time since diagnosis in months.
Systematic screening of driving molecular events of BRCAness
Because the tumors with BRCAness signature do not have any BRCA deficiency, we first hypothesized that deficiency in other DNA damage repair genes may contribute to this BRCA-independent mechanism(s) of genome instability and association with survival. To systematically screen of driving molecular events of BRCAness, we investigated the mutations of DNA damage repair genes using whole exome DNA seq data. The mutation analysis has identified scattered somatic mutations in 28 among 33 DNA damage repair genes from HR, Mismatch Repair, and Fanconi Anemia pathways (Figure S2A). Further survival analysis didn't link the candidate-mutated genes with patient survival or BRCAness (Figure S2B and S2C).
Because accumulated evidence [3, 18, 19] suggests that copy number alterations may play a dominant role in serous OvCa tumorigenesis and progression, we next investigated if the copy number alterations of the DNA damage repair genes are accountable for BRCA-independent mechanism of BRCAness. We conducted the screen by integrating copy number and gene expression data in 220 BRCAwt patients as described in Methods. Briefly, we performed correlation analysis between gene copy number alteration and mutation rate. For each gene, a Pearson correlation coefficient was calculated, reflecting the association of gene copy number and genomic instability. To ensure the copy number alteration will result in corresponding gene expression changes, we further calculated the correlation coefficient between same gene's copy number and mRNA expression data. The candidate genes that may be accountable for BRCAness can be prioritized based on two criteria: 1) each gene's copy number was significantly correlated with BRCAness and 2) the copy number alteration of the gene will cause corresponding changes in the mRNA expression level (Table S1).
RAD50 copy number deletion is enriched in tumors with BRCAness signature and significantly associated with better prognosis
The top ranked molecular event in hypermutated BRCAwt patients is RAD50 deletion (Figure 2A). RAD50 copy number deletion occurred in 19% (Figure 2C) of OvCa tumors. Compared to RAD50 diploid tumors, RAD50 expression was significantly lower in RAD50 deleted tumors (Fold change [FC]=0.63, two-sample T-test P=3.56 × 10-13). RAD50 deletion is significantly enriched with patients with BRCAness-positive tumors (ES=0.40, FDR<0.1) and correlated with higher mutation rate in BRCAwt patients (R=0.18, P=0.003, Table S1). The 5-year survival rate of RAD50 deletion BRCAwt patients was 41% (95% CI=24%-70%, Table 3), which is significantly better than that of rest of BRCAwt cases, with 5-year survival rates of 22% (15%-31%, Table 3) in both unadjusted (log-rank P=0.006, Figure 2D) and adjusted (P=0.005, HR [hazard ratio]=0.44, 95% CI=0.27-0.84, Table 3) models.
Figure 2. Identification of copy number altered DNA repair genes responsible for BRCAness and hypomutation in BRCAwt ovarian cancer HR pathways.
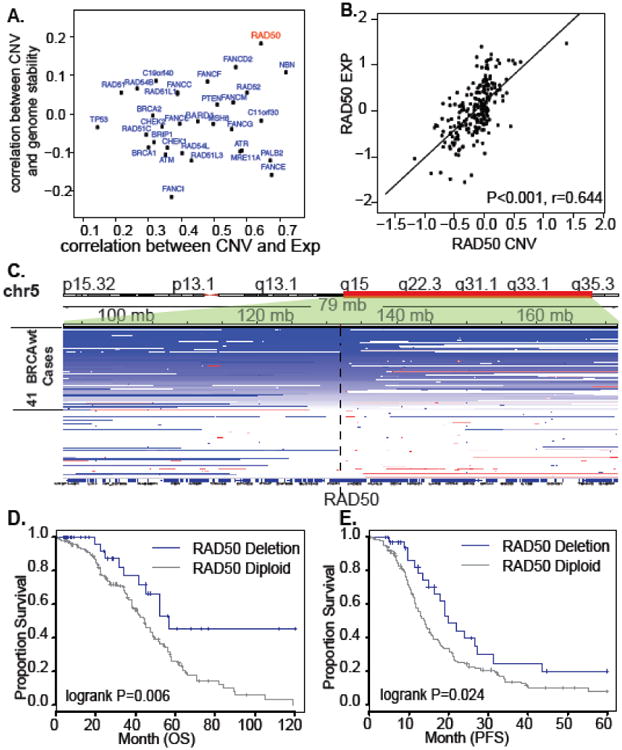
(A) Screening of copy number alterations for BRCAness. X-axis: correlation coefficient between gene copy number variation (CNV) and gene expression (EXP); Y-axis: correlation coefficient between gene copy number variation (CNV) and genome stability indicated by mutation rate. (B) Copy number deleted region covering RAD50 gene in 36 BRCAwt ovarian samples. Tumors are aligned in the rows. Deleted regions (blue) and the location of RAD50 are shown. (C) Association of expression and copy number status for RAD50 in 220 BRCAwt tumor samples. D & E. Kaplan-Meier survival curves for OS (D) and PFS (E) durations of RAD50 deletion and diploid tumors in 220 BRCAwt cases. The percent probability of survival is plotted versus time since diagnosis in months.
Table 3. Multivariable Models for Overall Survival and Progression-Free Survival of BRCA wild type OvCa patients with different RAD50 copy number status.
| Overall Survival | Progression-Free Survival | |||||||
|---|---|---|---|---|---|---|---|---|
| 3-Year Rate, % (95% CI) | 5-Year Rate, % (95% CI) | HR (95% CI) | P Value | 3-Year Rate, % (95% CI) | 5-Year Rate, % (95% CI) | HR (95% CI) | P Value | |
| RAD50 status | ||||||||
| RAD50 not deletion | 56 (48-64) | 22 (15-31) | 1 [Reference] | 13 (8-22) | 8 (4-17) | 1 [Reference] | ||
| RAD50 deletion | 69 (53-89) | 41 (24-70) | 0.44(0.25 -0.78) | 0.00 5 | 25 (12-52) | 20 (8-47) | 0.60(0.37-0.99) | 0.044 |
| Tumor Stage | ||||||||
| II | 89 (71-100) | 67 (36-100) | 1 [Reference] | 75 (50-100) | 38 (9-100) | 1 [Reference] | ||
| III | 59 (51-68) | 21 (14-31) | 2.63(0.83-8.30) | 0.100 | 13 (8-21) | 8 (4-16) | 4.15(1.31-13.12) | 0.016 |
| IV | 48 (33-70) | 34 (20-58) | 2.55(0.76-8.55) | 0.131 | 14 (5-39) | 14 (5-39) | 4.15(1.23-13.95) | 0.022 |
| Age increase of 1.0 ye | 1.02(1.00-1.04) | 0.011 | 0.99(0.98-1.01) | 0.523 | ||||
RAD50 copy number deletion predicts PARP inhibitors sensitivity in BRCAwt OvCa cell lines
To directly investigate the association of PARPi drugs and copy number status of RAD50 in ovarian cancer, we analyzed the cell line copy number data in 19 BRCAwt OvCa cell lines. Among the 19 OvCa cell lines, 9 were identified as RAD50 deletion, 10 were identified as diploid (see Methods and Table S2) [20]. Consistent with our observations in patient samples, the expression of RAD50 is significantly correlated with copy number alterations in OvCa cell lines [21] (Figure S3).
We next determined the association between copy number alterations of RAD50 and drug response in same 19 OvCa cell lines. The analysis revealed a significant association between the RAD50 deletion and sensitivity to rucaparib (PARPi, AG-014699) in OvCa cell lines (geometric mean IC50 for RAD50 deleted cells 20 uM versus 148 uM for RAD50 diploid cells, P<0.01, Figure 3A). We observed essentially same augmentation of drug sensitivity in RAD50 deleted OvCa cell lines treated by a structurally distinct PARP inhibitor, olaparib (AZD2281) (geometric mean IC50 for RAD50 deleted cells 99 uM versus 365 uM for diploid, P<0.05, Figure 3B).
Figure 3. Loss of RAD50 predicts drug sensitivity in BRCAwt OvCa cell lines.
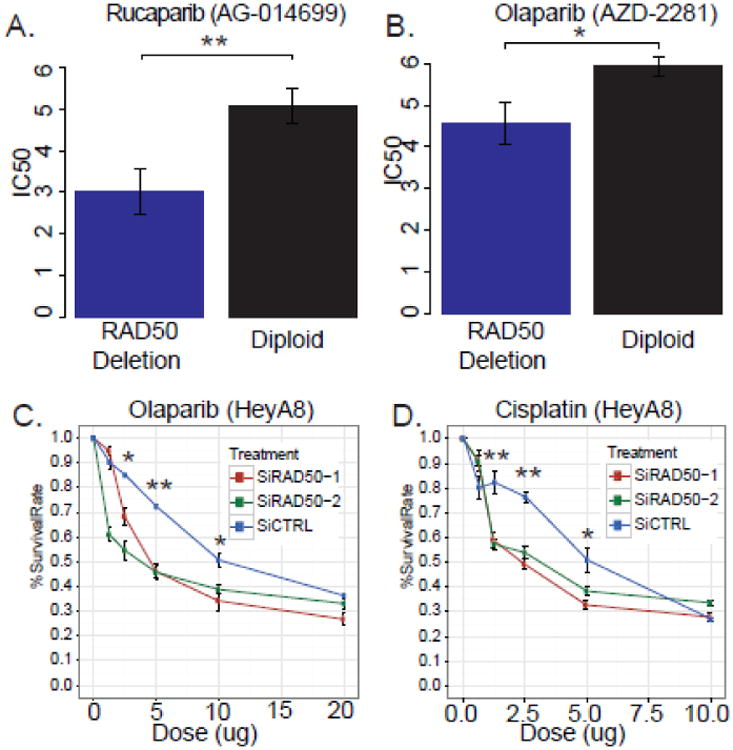
(A) Drug response of Rucaparib in ovarian cancer cells with different RAD50 copy number statuses (B) Drug response of Olaprib in ovarian cancer cells with different RAD50 copy number statuses (C & D) HeyA8 cells were transfected with SiCTRL, SiRAD50-1, SiRAD50-2 and then treated with Olaparib (C) or Cisplatin (D). Cell viability was assayed by MTT assay. Curves were generated from three independent experiments. * P<0.05; ** P<0.01, two-sided Student's t test. Data represent the mean±SD from three independent experiments.
We further tested whether knockdown of RAD50 can sensitize ovarian cancer cell olaparib treatment. Two RAD50 siRNAs (i.e. SiRAD50-1 and SiRAD50-2) were transferred in HeyA8 cells, which is RAD50 diploid. As expected, both RAD50-siRNAs led to higher sensitivity to olaparib than controls (survival percent for 1.25 μM olaparib treatment, SiCTRL vs SiRAD50-2: 90.168±2.662 vs 60.986±2.907, P=0.0007; survival percent for 2.5 μM olaparib treatment, SiCTRL vs SiRAD50-2: 85.020±0.481 vs 54.549±4.117, P=0.005) (Figure 3C).
The effects of RAD50 knockdown on response to cisplatin treatment in HeyA8 cell was also investigated using same siRNAs. Both RAD50-siRNAs led to higher sensitivity to cisplatin than controls (survival percent for 1.25 μM cisplatin treatment, SiCTRL vs SiRAD50-2: 82.302±4.872 vs 57.341±2.232, P=0.0085; 2.5 μM cisplatin treatment, SiCTRL vs SiRAD50-2: 76.395±2.187 vs 53.910±2.665, P=0.0007) (Figure 3D).
Discussion
In this study, we successfully identified the tumors with BRCAness signature using whole-exome deep sequencing data. Our data have demonstrated that BRCAness OvCa tumors resembled to BRCA deficient tumors in terms of hypermutation in whole-exome and enhanced patient survival. Further integrative screening of potential driver molecular events of BRCAness revealed that RAD50 deletion is a key marker of BRCAness and therefore may be predictive of OvCa drug responses and better prognosis. Following this lead, we demonstrated that loss of RAD50, either through copy number deletion or siRNA knock-down, indeed augmented OvCa cell's responses to Cisplatin and PARPi drugs.
The notion of BRCAness was prompted by a study showing that a substantial proportion of OvCa tumors harbor a molecular signature similar to BRCA1/2 germline mutated cancers [22-25]. These data indicate that sporadic ovarian cancers have a high prevalence of BRCA-like phenotypes, consistent with the known sensitivity of ovarian cancers to cisplatin. In accordance with this observation, clinical trial studies focusing on PARPi in OvCa have demonstrated that the patients without BRCA1/2 deficiency can be responsive to PARPi [9, 26]. These observations call for additional predictive and surrogate biomarkers for BRCAness, which will be remarkably helpful for better management of OvCa using PARPi. To this end, extensive efforts have been placed on identification of the marker or underlying mechanism of the BRCAness. Konstantinopoulos et al. analyzed expression profile from 131 patients and identified a 60-gene BRCA-like signature for BRCAness [27]. Abkevich et al. hypothesized that genomic patterns of loss of heterozygosity (LOH) can be representative to HR deficiency and established the Myriad HRD assay [28]. Popova et al. developed a method based on chromosomal copy number instability to predict BRCA1/2 inactivation in basal-like breast carcinomas [29]. Most recently, Liu et al. comprehensively investigated mutation data of 512 OvCa patients and successfully identified mutations in eight ADAMTS genes as potential markers of prognosis and drug sensitivity. They further revealed ADAMTS-mutated cases exhibited a distinct mutation spectrum and were significantly associated with hypermutated tumors [30]. These studies provide potential assays/markers of selecting patients for genetic testing or recruitment to clinical trials of PARPis. However, most of the studies based on sporadic OvCa patients, which are loosely defined BRCAwt patients without considering the BRCA somatic mutation and BRCA1 promoter hypermethylation. In this study, with the availability of the TCGA multiple-dimensional genomics and epigenetic data, we were able to specifically focus on strictly defined BRCAwt patients and cell lines. We have shown, in both BRCAwt patient cohort and cell line, that somatic copy number deletion of RAD50 may lead to BRCAness, manifested by higher mutation rate in genome-wide and consistently treatment sensitivity to olaparib and rucaparib. This provides biomarker for further stratifying the BRCAwt patients for response to PARPi, which can be translated to clinical trial and precision medicine in OvCa patient in the near future.
Consistent with previous studies, which mostly focused on gene mutation or gene expression signatures, we systematically screened for markers of BRCAness by investigating copy number alteration events. This is partially inspired by the recent observations that OvCa has a relatively simple mutational spectrum, while remarkably complicated copy number disarray across the genome. The frequency of copy number alterations in OvCa stands in striking contrast to that in glioblastoma, breast cancer and endometrial cancer, where there were far fewer chromosome arm-level or focal copy number alterations but more recurrently mutated genes [3, 18, 19]. This strategy has indeed revealed some copy number altered DNA repair genes may be attributable to BRCAness. In addition to RAD50, which is the top ranked gene in the screening, there are FANCD2 and NBN (also known as NBS1). Interestingly, RAD50 and NBN are two of the three subunits of Mre11-Rad50-Nbs1 (MRN) complex. The observation that the top genetic events associated with BRCAness are both from MRN complex strongly suggested MRN complex plays an important role in BRCA-independent mechanisms for regulation of OvCa BRCAness and drug response. Accumulating evidences have established the essential role that MRN complex plays in HR repair, meiotic recombination, cell cycle checkpoints, and maintenance of telomeres [4]. Homozygous knockouts of single member in MRN complex can lead to embryonic lethality in mouse [31-33]. In human, mutations in the members of MRN complex predispose the carrier to severe cancer susceptibility phenotypes [34, 35]. For example, Nijmegen breakage syndrome caused by mutation of NBN [36] is characterized by a high susceptibility to lymphoma. Further analysis revealed that RAD50 mutated human cells exhibit a cellular phenotype that includes sensitivity to ionizing radiation, a deficit in DNA DSB repair, and chromosomal instability (MIM 604391; MIM 251260) [37]. In the future, functional studies involving animal model will be needed to mechanistically determine MRN complex's role in regulation of OvCa BRCAness and drug response.
In summary, the discovery that OvCa tumors with RAD50 deletion exhibit BRCAness and better response to both cisplatin and PARPis will provides an important biomarker for further stratifying the BRCAwt patients for treatment of olaparib and rucaparib. This would expand our understanding of BRCAness in BRCAwt OvCa and help to better define the population of patients eligible for PARPi treatment and clinical trials in the near future.
Supplementary Material
Highlights.
Whole-exome analysis identifies BRCAness OvCa tumors
BRCAness tumors resembles BRCA deficient OvCa tumors by exhibiting similar genome instability and good prognosis
RAD50 deletion/knock-down predicts BRCAness and augments OvCa cell's responses to Cisplatin, Olaparib, and Rucaparib
Acknowledgments
This study was partially supported by a grant from the Elsa U. Pardee Foundation (Dr. Yang), the Career Development Award of RPCI-UPCI Ovarian Cancer SPORE (P50 CA159981to Drs. Yang and Edwards), Funding for the Genome Data Analysis Centers from the National Institutes of Health (U24 CA143835 to Dr. Zhang), funding for the Cancer Systems Informatics Center from the National Foundation for Cancer Research (Dr. Zhang), the MDACC Ovarian Cancer SPORE (P50 CA083639 to Dr. Sood) and U54 CA151668 (Dr. Sood).
Footnotes
Conflict of interest statement: No potential conflicts of interest were disclosed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kobel M, et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 2008;5(12):e232. doi: 10.1371/journal.pmed.0050232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tothill RW, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14(16):5198–208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 3.TCGA. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang D, et al. Integrated analyses identify a master microRNA regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer cell. 2013;23(2):186–99. doi: 10.1016/j.ccr.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jemal A, et al. Cancer statistics, 2004. CA Cancer J Clin. 2004;54(1):8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 6.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9(6):415–28. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang D, et al. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA. 2011;306(14):1557–65. doi: 10.1001/jama.2011.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu G, et al. Augmentation of response to chemotherapy by microRNA-506 through regulation of RAD51 in serous ovarian cancers. J Natl Cancer Inst. 2015;107(7) doi: 10.1093/jnci/djv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hennessy BT, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570–6. doi: 10.1200/JCO.2009.27.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan DS, et al. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol. 2008;26(34):5530–6. doi: 10.1200/JCO.2008.16.1703. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang W, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41(Database issue):D955–61. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Royal Statistical Society Series B. 1995;57(1):289. [Google Scholar]
- 14.Swisher E, Walsh T. BRCA1 and BRCA2 mutations in ovarian cancer. JAMA. 2012;307(4):359–60. doi: 10.1001/jama.2012.9. author reply 360-1. [DOI] [PubMed] [Google Scholar]
- 15.Berchuck A, et al. Frequency of germline and somatic BRCA1 mutations in ovarian cancer. Clin Cancer Res. 1998;4(10):2433–7. [PubMed] [Google Scholar]
- 16.Takahashi H, et al. Mutations of the BRCA2 gene in ovarian carcinomas. Cancer Res. 1996;56(12):2738–41. [PubMed] [Google Scholar]
- 17.Baldwin RL, et al. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res. 2000;60(19):5329–33. [PubMed] [Google Scholar]
- 18.Zack TI, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45(10):1134–40. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–50. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jazaeri AA, et al. Gene expression profiles of BRCA1-linked, BRCA2-linked, and sporadic ovarian cancers. J Natl Cancer Inst. 2002;94(13):990–1000. doi: 10.1093/jnci/94.13.990. [DOI] [PubMed] [Google Scholar]
- 23.Liu G, et al. Differing clinical impact of BRCA1 and BRCA2 mutations in serous ovarian cancer. Pharmacogenomics. 2012;13(13):1523–35. doi: 10.2217/pgs.12.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mezzanzanica D. Ovarian cancer: a molecularly insidious disease. Chin J Cancer. 2015;34(1):1–3. doi: 10.5732/cjc.014.10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun Y, et al. Key nodes of a microRNA network associated with the integrated mesenchymal subtype of high-grade serous ovarian cancer. Chin J Cancer. 2015;34(1):28–40. doi: 10.5732/cjc.014.10284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McNeish IA, et al. Results of ARIEL2: A Phase 2 trial to prospectively identify ovarian cancer patients likely to respond to rucaparib using tumor genetic analysis. 2015 ASCO Annual Meeting. 2015 [Google Scholar]
- 27.Konstantinopoulos PA, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–61. doi: 10.1200/JCO.2009.27.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abkevich V, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. J Cancer. 2012;107(10):1776–82. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Popova T, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72(21):5454–62. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, et al. Association of Somatic Mutations of ADAMTS Genes With Chemotherapy Sensitivity and Survival in High-Grade Serous Ovarian Carcinoma. JAMA Oncol. 2015;1(4):486–94. doi: 10.1001/jamaoncol.2015.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao Y, Weaver DT. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res. 1997;25(15):2985–91. doi: 10.1093/nar/25.15.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo G, et al. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci U S A. 1999;96(13):7376–81. doi: 10.1073/pnas.96.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu J, et al. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol. 2001;11(2):105–9. doi: 10.1016/s0960-9822(01)00019-7. [DOI] [PubMed] [Google Scholar]
- 34.Uchisaka N, et al. Two brothers with ataxia-telangiectasia-like disorder with lung adenocarcinoma. J Pediatr. 2009;155(3):435–8. doi: 10.1016/j.jpeds.2009.02.037. [DOI] [PubMed] [Google Scholar]
- 35.Regal JA, et al. Disease-associated MRE11 mutants impact ATM/ATR DNA damage signaling by distinct mechanisms. Hum Mol Genet. 2013;22(25):5146–59. doi: 10.1093/hmg/ddt368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Burgt I, et al. Nijmegen breakage syndrome. J Med Genet. 1996;33(2):153–6. doi: 10.1136/jmg.33.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waltes R, et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet. 2009;84(5):605–16. doi: 10.1016/j.ajhg.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.