

Abstract
Both Fras1 and Itga8 connect mesenchymal cells to epithelia by way of an extracellular ‘Fraser protein complex’ that functions in signaling and adhesion; these proteins are vital to the development of several vertebrate organs. We previously found that zebrafish fras1 mutants have craniofacial defects, specifically, shortened symplectic cartilages and cartilage fusions that spare joint elements. During a forward mutagenesis screen, we identified a new zebrafish mutation, b1161, that we show here disrupts itga8, as confirmed using CRISPR-generated itga8 alleles. fras1 and itga8 single mutants and double mutants have similar craniofacial phenotypes, a result expected if loss of either gene disrupts function of the Fraser protein complex. Unlike fras1 mutants or other Fraser-related mutants, itga8 mutants do not show blistered tail fins. Thus, the function of the Fraser complex differs in the craniofacial skeleton and the tail fin. Focusing on the face, we find that itga8 mutants consistently show defective outpocketing of a late-forming portion of the first pharyngeal pouch, and variably express skeletal defects, matching previously characterized fras1 mutant phenotypes. In itga8 and fras1 mutants, skeletal severity varies markedly between sides, indicating that both mutants have increased developmental instability. Whereas fras1 is expressed in epithelia, we show that itga8 is expressed complementarily in facial mesenchyme. Paired with the observed phenotypic similarity, this expression indicates that the genes function in epithelial-mesenchymal interactions. Similar interactions between Fras1 and Itga8 have previously been found in mouse kidney, where these genes both regulate Nephronectin (Npnt) protein abundance. We find that zebrafish facial tissues express both npnt and the Fraser gene fibrillin2b (fbn2b), but their transcript levels do not depend on fras1 or itga8 function. Using a revertible fras1 allele, we find that the critical window for fras1 function in the craniofacial skeleton is between 1.5 and 3 days post fertilization, which coincides with the onset of fras1-dependent and itga8-dependent morphogenesis. We propose a model wherein Fras1 and Itga8 interact during late pharyngeal pouch morphogenesis to sculpt pharyngeal arches through epithelial-mesenchymal interactions, thereby stabilizing the developing craniofacial skeleton.
Introduction
Epithelia are comprised of cohesive planar sheets of cells, whereas mesenchyme is comprised of loosely associated cells embedded in abundant matrix; interactions between these two tissue types are critical to animal development. The epithelially-expressed gene Fras1 (Gautier et al., 2008; McGregor et al., 2003; Vrontou et al., 2003) and the mesenchymally-expressed gene Integrin alpha8 (Itga8) (Benjamin et al., 2009; Schnapp et al., 1995a) are each thought to mediate epithelial-mesenchymal interactions (Smyth and Scambler, 2005). Mammalian Itga8 is a transmembrane protein involved in cell adhesion (Schnapp et al., 1995b) and signaling (Linton et al., 2007; Pitera et al., 2012). Mature Fras1 protein is secreted into the extracellular matrix, where it also has been implicated in cell adhesion and signaling (Carney et al., 2010; McGregor et al., 2003; Vrontou et al., 2003). Both Fras1 and Itga8 single mutant mice exhibit similar severe epithelial-mesenchymal adhesion defects (Benjamin et al., 2009; Petrou, 2005), kidney agenesis (Müller et al., 1997; Pitera et al., 2008; Vrontou et al., 2003) and lung defects (Benjamin et al., 2009; Petrou, 2005); suggesting a connection between these two genes (McGregor et al., 2003; Pitera et al., 2008). Furthermore, adhesion assays in cell culture reveal that mammalian Itga8 binds to Frem1, which functions in the core ‘Fraser protein complex’ (FPC) (Kiyozumi et al., 2006, 2005). The FPC is a ternary structure made of three enormous proteins, Fras1 and Fras-Related ECM (Frem) proteins Frem1 and Frem2 (Carney et al., 2010; Kiyozumi et al., 2006; Pavlakis et al., 2011). The FPC is found in the sub-lamina densa (Dalezios et al., 2007) of the epithelial basal lamina (Carney et al., 2010; Kiyozumi et al., 2006), which is the portion of the basal lamina directly apposed to embryonic mesenchyme. Other proteins associate with the core FPC, for example normal FPC function may sometimes require itga8. In cell culture, Itga8 binds directly to the Arg-Gly-Asp (RGD)-containing domain of Frem1, and RGD-binding is necessary for Frem1-mediated cell adhesion (Kiyozumi et al., 2005). However the Frem1 RGD domain is dispensable for mouse kidney, skin, and limb development (Kiyozumi et al., 2012). In mouse kidneys, Itga8 also binds directly to the RGD domain of Nephronectin (Npnt), another ECM glycoprotein (Brandenberger, 2001; Sato et al., 2009); Npnt binds to Frem1 (Kiyozumi et al., 2012), and Npnt is itself required for kidney formation (Linton et al., 2007), suggesting that Npnt may help to link Itga8 with the FPC. However, it was unclear from previous work how important Itga8 is to FPC-mediated epithelial mesenchymal interactions, and which tissues require Itga8-FPC interaction.
Defective interactions between epithelia and mesenchyme underlie many diseases, including Fraser syndrome and related diseases (Smyth and Scambler, 2005). Fraser syndrome is a human congenital disorder that affects many tissues including skin, kidneys, lungs, and craniofacial structures. Symptomatic severity varies between Fraser syndrome patients, and can even vary between the left and right sides of individual patients. The causes of Fraser syndrome variability are not yet fully understood, but variation is likely influenced by genetics, environment, and stochastic effects on development (Slavotinek et al., 2006; Talbot et al., 2012). Mutations in any of several genes cause Fraser syndrome or related Fraser-spectrum diseases. For example, truncating lesions in human FRAS1 (McGregor et al., 2003; Slavotinek et al., 2006; van Haelst et al., 2008), FREM2 (Shafeghati et al., 2008), and GRIP1 (Schanze et al., 2014; Vogel et al., 2012) each cause Fraser syndrome. Although several Fraser genes have been identified, lesions in these genes do not explain every case of Fraser syndrome, indicating that more Fraser-genes await discovery (van Haelst et al., 2008). Additionally, mutations in FREM1 can cause less severe Fraser-spectrum diseases (Alazami et al., 2009; Nathanson et al., 2013; Slavotinek et al., 2011). Hypomorphic lesions in FRAS1, FREM1, and other Fraser-genes also cause less-severe Fraser-spectrum diseases, including ‘isolated congenital anomalies of kidney and urinary tract’ (CAKUT) (Kohl et al., 2014). Recently, a homozygous ITGA8 lesion was identified in one CAKUT patient, implicating ITGA8 in this Fraser-spectrum disease (Kohl et al., 2014). Similarly, kidney formation is severely disrupted in mouse itga8 mutants (Müller et al., 1997), and in mutants for several FPC components (Jadeja et al., 2005; Smyth et al., 2004; Vrontou et al., 2003). In addition to mouse models, human Fraser syndrome has been modeled using zebrafish; in zebrafish, fras1 is vital to normal tail epithelial adhesion (Carney et al., 2010) and craniofacial development (Talbot et al., 2012). Prior to our current study, Itga8 function had not been investigated in zebrafish, and had not been implicated in craniofacial development in any organism.
In this study, we find that itga8 is necessary for normal craniofacial development in zebrafish, and that the faces of itga8 mutants closely phenocopy fras1 mutants, suggesting that the two genes function similarly, perhaps through interactions via the FPC. This inference is supported by our observation that fras1;itga8 double mutants have faces similar to the two single mutants. In contrast, itga8 appears to be dispensable for tail epithelial morphology, indicating that fras1 and itga8 are only co-required in some tissue contexts. In pharyngeal arches, we find itga8 mRNA expression in mesenchyme adjacent to epithelial fras1 expression, suggesting that Itga8 and the FPC may interact at epithelial-mesenchymal boundaries in the face. Itga8 may interact with the FPC through several mechanisms, such as protein-protein adhesion, the proteins regulating one another, and/or co-regulating downstream targets; some of these mechanisms have been confirmed previously in mouse kidneys (Kiyozumi et al., 2012; Pitera et al., 2008). However, a survey of several candidate genes expressed during zebrafish facial development revealed no evidence for signal transduction downstream of fras1 or itga8; instead, adhesive interactions between the two proteins may be of particular importance. We previously found that fras1 is required for a late forming portion of the first endodermal pouch, termed late-p1 (Talbot et al., 2012). Here we find that, like fras1, itga8 is also required for late-p1 formation. The itga8 defects in skeleton and epithelia begin to appear during the period of late-p1 formation; we demonstrate, using a conditional allele, that fras1 function is specifically required during this time period. We propose a model that Itga8 interacts with the FPC at the boundary between pharyngeal epithelia and mesenchyme; this interaction occurs during late-p1 morphogenesis, and sculpts both pharyngeal endoderm and the mesenchymally derived skeleton.
Materials and Methods
Fish maintenance, husbandry, strains, and genotyping
Fish were raised as described (Kimmel et al., 1995; Westerfield, 2007). Mutant lines were maintained on the AB background. The following lines have been previously described: sox9azc81tg (hereafter: sox9a:EGFP) (Bonkowsky and Chien, 2005; Eames et al., 2013), Tg(hsp70l:Cre)zdf13 (Feng et al., 2007), fras1te262d (Carney et al., 2010), and fras1mn0156Gt (Clark et al., 2011). We identified fras1b1048 mutants using previously identified fully penetrant tail fin blisters, or previously described PCR genotyping protocols (Carney et al., 2010). A screen of N-ethyl-N-nitrosourea (ENU) mutagenized Alcian blue and Alizarin red stained gynogenetic embryos (Beattie et al., 1999) identified b1161 mutants. Molecular cloning of the b1161 lesion (Fig. 1B) is described below. The PCR primers itga8IDF (CCCAGTTACATAACAAAGGTCCGAG) and itga8IDR (TAAGCCCAGTCAAGTTTTTGCC) produce a 510 bp band in wild type, a 431 bp band in b1161 mutants, and both sizes in heterozygous fish.
Figure 1.

Skeletal defects in b1161 mutants are caused by lesions in itga8. (A, B) Alcian and alizarin staining of (A) wild type and (B) itga8b1161 homozygotes shows skeletal defects caused by itga8 mutations. Homozygous itga8 mutants often show cartilage fusions in the first two pharyngeal arches and defects in symplectic length. (C) Linkage analysis reveals no recombinants (0/696 individuals) between b1161 and the itga8b1161, placing them at the same map position. Map distances (blue) of additional markers from b1161 are shown above in Mb and below in cM. (D) Sequence of the itga8b1161 lesion reveals a 7 bp insertion in exon 25 (green), followed by a 79 bp deletion (orange) (wild type: GenBank JN399198, b1161: GenBank JN399198). This lesion results in protein truncation after amino acid 845 of 1059 with the predicted addition of 21 aberrant amino acids (EFTHWSWRPRLFRTLQSYWAS). (E) Sequence of two CRISPR-induced itga8 lesions, itga8oz6 (11 bp deletion) and itga8oz7 (5 bp deletion), reveal that both introduce frameshifts after amino acid 79 of 1059; itga8oz6 causes an immediate stop codon after the frameshift, while itga8oz7 introduces 30 aberrant amino acids (HLSAGDCGGRSGVLLPLAGIRPRLLPPDPL) before terminating. (F) Itga8 protein diagram with locations of oz6, oz7, and b1161 mutations along with predicted protein motifs. Protein motifs are designated as follows: signal sequence (pink box), integrin beta domains (teal circles); integrin alpha domain (purple oval), transmembrane domain (peach box), and an intracellular integrin domain (red box). Cartilage abbreviations: Meckel's (Me), Retroarticular process (Ra), Palatoquadrate (Pq), Symplectic (Sy), Ceratohyal (Ch), Interhyal (Ih). Scale bar (100 μm) in B also applies to A.
CRISPR mutagenesis of itga8
The CRISPR target sequence (AAAGCGAACACCTCTCAGCC) was cloned into plasmid pDR274 (Hwang et al., 2013). Mutagenesis, mutant recovery, and subsequent outcrosses were performed as described (Talbot and Amacher, 2014). Mutants were initially identified by high resolution melt analysis (HRMA) (Dahlem et al., 2012), after PCR amplification using primers itga8_HRM_F (AGCATGTCGGTGTTGGTTG) and itga8_HRM_R (AGGAGTCTGGGTCTGATGC). itga8oz6 and itga8oz7 lesions (Fig. 1C) were identified using sequencing primers itga8_Seq_F (AGCACCACCAATATGGACCAAC) and itga8_Seq_R (GGAATTTATGCAGCCGAGTCTG). Both the oz6 and oz7 lesions destroy a DdeI (NEB R0175S) restriction enzyme site found in the wild-type allele, so in subsequent generations itga8 CRISPR mutations were genotyped by amplifying templates with itga8_Seq_F/itga8_Seq_R primers and then digesting the PCR products with DdeI.
Conditional induction of fras1
Animals heterozygous for the fras1te262d allele, which also carried a single copy of the heat-shock inducible Cre transgene Tg(hsp70l:Cre)zdf13, were pair-wise crossed to animals heterozygous for the fras1mn0156Gt revertible conditional allele (Clark et al., 2011). Heat-shock treatment (5 minutes at 40°C) at 24, 32, 48 or 72 hours post fertilization (hpf) induced Cre expression in animals that inherited the transgene; siblings lacking the Cre transgene served as controls. Animals were raised to 6 days post fertilization (dpf), then fixed and stained for cartilage and bone. Homozygous fras1te262d/mn0156Gt larvae were identified by the fully penetrant tail phenotype and hsp70l:Cre heterozygotes were identified by PCR with the primers creF (GCGGCATGGTGCAAGTTGAAT) and creR (CGTTCACCGGCATCAACGTTT). Heat shock, even as early as 24 hpf, did not alter the tail phenotype (not shown). Several replicates of each reversion time point were scored for head skeletal phenotypes as described (Talbot et al., 2012) to compare the penetrance of each phenotype in reverted (Cre+) animals to control (Cre−) animals.
Tissue labeling
Alcian and Alizarin staining performed as described (https://wiki.zfin.org/x/cwDI) (Walker and Kimmel, 2007). Skeletal defects were scored and statistically analyzed as described (Talbot et al., 2012). For antibody labeling and RNA in situ hybridization, melanogenesis was inhibited by raising embryos in 0.0015% PTU (Westerfield, 2007). RNA in situ hybridization followed by NBT/BCIP colorimetric labeling was performed as described (Rodriguez-Mari et al., 2005). Detailed protocols for whole mount fluorescent RNA in situ hybridization (https://wiki.zfin.org/x/0wHI) (Talbot et al., 2010), fluorescent RNA in situ hybridization on tissue sections (http://wiki.zfin.org/x/XQBrAQ), and antibody labeling on tissue sections (https://wiki.zfin.org/x/XACiAQ) (Talbot et al., 2012) are available online. Epithelia were labeled with Anti-P63 (4A4, SCBT). Epithelial nuclei were labeled with Anti-P63 (4A4, SCBT). Four antibodies to human ITGA8 (SCBT H-180, T-20, S-16, Sigma-Aldrich HPA003432) failed to show specific expression patterns when tested on zebrafish. To generate RNA probes for fbn2a, fbn2b, gdnfa, grip1, grip2b, itga8, and npnt transcripts, cDNA fragments were amplified using primer pairs shown in Table S1. Probes covering separate regions of the itga8 transcript reveal similar expression patterns (not shown). The resulting PCR fragments were cloned into pCR4-TOPO (Invitrogen), and clones from these fragments were used as templates to generate RNA probes. Other mRNA probes used were col2a1 (Yan et al., 1995), dlx2a (Akimenko et al., 1994), and fras1 (Carney et al., 2010). Images were processed with LSM, Volocity, ImageJ, and Metamorph software packages.
Quantification of phenotypic variation
Quantification of skeletal fusions, endoderm-ectoderm distances, and symplectic lengths were performed and statistically analyzed as previously described (Talbot et al., 2012). The present study includes previously reported wild type and fras1 mutant data that were collected at the same time as data for the itga8 mutant and published separately (Talbot et al., 2012). We reproduce these data here for the convenience of the reader, with permission from the Company of Biologists. The specific items are wild type and fras1 data shown in Fig. 3D-E, wild type and fras1 data shown in Fig. 6E, and fras1 mutant data shown in Fig. 7A.
Figure 3.

Phenotypic variation in fras1 and itga8 mutant phenotypes shows fluctuating asymmetry. (A-C’) Facial cartilage skeleton marked by sox9a:GFP expression at 7.5 dpf, region shown is boxed in red in A”. Compared to wild type (A), the itga8b1161 mutant skeleton is often asymmetric (B, C). For instance, the fish in (B) shows an extended symplectic cartilage fused to the ceratohyal cartilage on the right side and an unfused, severely shortened, symplectic phenotype on its left side (B’). In another example, both “Short Sy” and “Fused Sy-Ch” symplectic phenotypes (C) are found in a fish presenting only subtle defects on the opposite side (C’). (D, E) At 7.5 dpf, average symplectic length in itga8 mutants is shorter than wild type, but comparable to fras1 mutants. Symplectic cartilages in itga8 mutants show asymmetry similar to fras1 mutants, which is twice as high as wild-type asymmetry. (E) Plot of symplectic lengths measured on left and right sides, with grouped 95% density ellipses. Symplectic lengths for wild types were along the diagonal as expected for a high degree of left/right correlation, but for itga8b1161 and fras1b1048 mutants, symplectics were much shorter and do not correlate well between sides. Scale bar (A) is 100 μm, applicable to A-C’. Error bars (D) show 95% confidence intervals: 1.95 times standard error.
Figure 6.

Late-p1 is severely affected in itga8 and fras1 single and double mutants. (A-D) Tissue sections labeled with anti-P63 (epithelial nuclei) and sox9a:EGFP (cartilage) at 72 hpf, and oriented as shown in A'. The late-forming outpocketing of pouch-1 (late-p1) is indicated with a yellow dotted line in wild type. However, late-p1 is absent in (B) itga8b1161, (C) fras1b1048, and (D) fras1b1048;itga8b1161 double mutants, leaving a large gap (blue line) between endoderm and ectoderm. (E) Symplectic cartilage length and ectoderm-endoderm gap distance were measured on confocal stacks of whole-mounted embryos expressing sox9a:GFP and labeled with anti-P63. Each measurement pair is plotted as a dot, with 95% density ellipses shown for each genotype.
Figure 7.

fras1 and itga8 mutant defects appear between 36 and 72 hpf, during a critical window for fras1 function. (A) Symplectic length and endoderm-ectoderm gap distance, measured at 36 hpf and 72 hpf, as shown in (Talbot et al., 2012). For both symplectic length and endodermectoderm gap distance, mutants are significantly different (*, p<0.01) from wild type at 72 hpf, but the differences are not significant (n.s.) at 36 hpf. Symplectic cartilage was marked using sox9a:GFP and epithelial tissues were marked using anti-P63. Symplectic cartilage length and the endoderm-ectoderm gap were measured and recorded for each individual embryo. (B) Diagram of the fras1 revertible allele experiment. Embryos trans-heterozygous for fras1mn0156Gt and fras1te262d were heat-shocked at different developmental stages to induce Cre recombinase expression in the half of the clutch that inherited the hsp70l:Cre transgene. Cre activity removes the insertion trap transgene (which, when present, encodes an mRFP tag and a premature stop codon after exon 15), and restores fras1 function. Skeletal preparations of heat-shocked animals carrying the Cre transgene (Cre+, reverted) were compared to siblings that did not inherit the transgene (Cre−, control). (C-E) Larvae heat shocked at the indicated time were stained for cartilage and bone at 6 dpf, genotyped for hsp70l:Cre, and each fish was scored for the indicated skeletal trait. Graphs show the penetrance per fish, defined as the percent of fish with a given defect on at least one side of the embryo. Error bars in A are 95% confidence intervals: 1.95 times the standard error. Error bars in C-E are standard deviations; * indicates P<0.05, and ** denotes P<0.001.
Results
itga8 lesions cause facial skeletal defects
In a forward genetic screen for zebrafish craniofacial mutants we identified a mutant, b1161, showing specific cartilage defects that vary in severity from fish to fish (Fig. 1A, B). The b1161 facial cartilage defects are very similar to defects caused by fras1 mutation, suggesting that b1161 might either disrupt fras1, or a gene of similar function. Although facial defects are similar to fras1 mutants (Table 1), b1161 complements fras1b1048 (Table 2), eliminating the former possibility. Bulked segregant analysis using RAD-tagged SNPs (Floragenex) placed b1161 on a 10 Mb region of linkage group 16, a location later refined using traditional Z markers to a 4 Mb interval (Fig. 1C); this interval contains dozens of genes centered around itga8 (Fig. 1C), which is itself an excellent candidate. We sequenced itga8 genomic DNA and cDNA and found that both itga8b1161 cDNA and itga8b1161 genomic DNA contains a large indel in exon 25 that is never detected in wild-type siblings (Fig. 1D). To confirm that the b1161 lesion was indeed due to disruption of itga8, we used CRISPR-induced mutagenesis to generate two additional itga8 alleles (oz6 and oz7, Fig. 1E). Whereas the b1161 lesion frame-shifts the Itga8 protein midway through the integrin alpha domain, itga8oz6 and itga8oz7 both terminate the protein much earlier (Fig. 1F). Complementation tests between the b1161 ENU allele and the CRISPR-induced itga8 alleles confirm that the facial phenotypes in b1161 are caused by itga8 mutation (Table 1). Additionally, when homozygous, all three alleles show similar levels of overall phenotypic severity (Table 1), indicating that all three alleles cause equivalently severe loss of function. Homozygous itga8b1161 mutants sometimes survive to adulthood and are fertile, producing offspring that lack both maternal and zygotic gene function; these ‘maternal-zygotic’ embryos show penetrance indistinguishable from the offspring of heterozygous parents (Table 1), indicating that itga8 mutant phenotypic variation is not caused by maternally-deposited wild-type transcripts or protein. Hence, we conclude that severe zygotic loss of itga8 function results in variable craniofacial skeletal defects.
Table 1.
itga8 mutant fish show partially penetrant craniofacial defects
| Genotypea | nb | Me-PQ fusionc | Sy-Ch fusionc | Sy shortc |
|---|---|---|---|---|
| Siblings | 611 | 0% | 0% | 0% |
| itga8b1161 (zyg) | 129 | 26% | 64% | 42% |
| itga8b1161 (mat+zyg) | 54 | 26% | 55% | 43% |
| itga8oz6 | 41 | 9% | 34% | 73% |
| itga8oz7 | 18 | 28% | 44% | 67% |
| itga8oz6/oz7 | 16 | 38% | 31% | 78% |
| itga8oz6/b1161 | 30 | 5% | 38% | 53% |
| fras1te262 | 147 | 19% | 57% | 68% |
See Methods for details. “Siblings” includes both wild type and individuals heterozygous for b1161. itga8b1161 (zyg) fish are derived from incross of b1161 heterozygotes; itga8b1161 (mat+zyg) fish from an incross of b1161 homozygotes.
n, number of individuals for which both left and right sides of the face were scored for cartilage defects.
Fish were labeled with alcian/alizarin-stain to reveal cartilage and bone defects at 6 dpf. Individual defects are shown as the percentage of occurrence per side. Me-PQ fusion = fusion between Meckel's and palatoquadrate cartilages; Sy-Ch fusion = fusion between symplectic and ceratohyal cartilages; Sy short = shortened symplectic cartilage.
Table 2.
Penetrance of cartilage defects in fras1/itga8 single and double mutants.
| Genotypea | nb | Me-Pq fusionc | Sy-Ch fusionc | Sy shortc |
|---|---|---|---|---|
| Siblings | 225 | 2% | 0% | 0% |
| itga8b1161 | 78 | 36% | 18% | 51% |
| fras1b1048 | 63 | 60% | 50% | 72% |
| fras1b1048; itga8b1161 | 33 | 77% | 55% | 88% |
See methods for details. The “Siblings” category includes genetically wild type fish, siblings heterozygous for fras1, siblings heterozygous for itga8, and siblings trans-heterozygous for both genes; the heterozygous siblings appear phenotypically normal.
n, number of individuals for which both left and right sides of the face were scored for cartilage defects.
Fish scored live for skeletal defects at 7 dpf, using sox9a:EGFP expression to mark cartilages. Individual defects are shown as the percentage of occurrence per side. Me-PQ fusion = fusion between Meckel's and palatoquadrate cartilages; Sy-Ch fusion = fusion between symplectic and ceratohyal cartilages; Sy short = shortened symplectic cartilage.
Facial skeleton morphology suggests that itga8 and fras1 are co-required for craniofacial development
Craniofacial phenotypic similarities between fras1 and itga8 mutants suggest that these two genes may be required for similar processes. For instance, both fras1 and itga8 mutants display the same three cartilage defects – short symplectic, symplectic-ceratohyal fusion, and Meckel's-palatoquadrate fusion (Table 1, Fig. 2). For mutants in both genes, cartilage fusions characteristically spare joint elements (retroarticular process and interhyal cartilage; Fig. 2). To test the genetic relationship between fras1 and itga8, we constructed fras1b1048;itga8b1161 double mutants, and found that the double mutants have skeletal phenotypes similar to those of single mutants (Fig. 2A-E). In fras1 and itga8 single and double mutants, each skeletal defect shows partial penetrance (Table 2). Penetrance of the three cartilage defects are significantly higher in fras1 mutants compared to itga8 mutants (Tables 2, S2), and although the double mutant has higher penetrance than fras1 for some defects, these differences are not statistically significant (Tables 2, S2). In contrast to their craniofacial similarities, fras1 and itga8 mutant phenotypes differ in the tail; itga8b1161 mutants, as well as itga8oz6 and itga8oz7 mutants, have wild-type tail fin morphology whereas fras1 mutants and all other Fraser mutants found to date have blistered fins (Carney et al., 2010) (compare Fig. 2F, G with Fig. 2H, I; data not shown). The phenotypic differences between head and tail indicate that itga8 function is essential in some, but not all, tissues that also require fras1 function. Because all aspects of facial skeletal defects in the double mutant are comparable to either single mutant, we propose that fras1 and itga8 are both required for the same steps of craniofacial development, with both genes being necessary for facial FPC function.
Figure 2.

Comparison of skeletal, endodermal, and tail ectoderm phenotypes between wild type, itga8b1161 mutants, fras1b1048 mutants, and itga8b1161;fras1b1048 double mutants. (A) Illustration of a zebrafish larva, indicating regions shown in subsequent (B-I) panels. (B-E) Skeletal morphology is revealed using sox9a:EGFP expression (cartilage) and alizarin red staining (bone) at 7 dpf; itga8 and fras1 single and double mutants display similar cartilage defects, in particular, Meckel's-palatoquadrate joint fusion (arrowhead) and symplectic-ceratohyal fusions (asterisk). (F-I) Bright field images showing normal fin fold morphology (outlined blue) in (F) wild type and (G) itga8b1161 individuals versus the “blister phenotypes” in (H) fras1b1048 and (I) itga8b1161;fras1b1048 mutants. Scale bars (E, I) are 100 μm. Scale bar in E applies to B-E; Scale bar in I applies to F-I.
itga8 mutants show fluctuating asymmetry, similar to fras1 mutants
In fras1 mutants, skeletal defect severity varies approximately randomly between the left and right sides of embryos (Talbot et al., 2012). This type of variation, termed ‘fluctuating asymmetry’, may result from underlying developmental instability, i.e., the loss of buffering of stochastic noise (Graham et al., 2010). The hypothesis that loss of either fras1 or itga8 both disrupt FPC function predicts that itga8 and fras1 mutants both might show similar levels of phenotypic variation. Just like fras1 mutants, the left and right sides of individual itga8 mutant embryos are often asymmetric, differing dramatically in phenotypic severity (Fig. 3A-C). Rarely, fras1 and itga8 mutant faces appear almost normal, with only one skeletal defect found unilaterally; also rarely, in the most severe fish all three defects are seen bilaterally. Thus, itga8 mutants exhibit both asymmetry (Fig. 3D, E) and phenotypic variation (Fig. 3E) indistinguishable from variation in fras1 mutants (Talbot et al., 2012). For each defect there is a range of severity; for instance, symplectic lengths range continuously from 40-174 μm, overlapping the wild-type range 128-245 μm (Fig. 3E). When individual cartilages were measured twice, we found that measurement error is small compared to phenotypic severity (Fig. 3D). Phenotypes are not biased to the left or right sides, ruling out directional asymmetry (Fig. 3E). When variable phenotypes meet these conditions, the variation is thought to be caused by developmental instability (Dongen, 2006). These findings indicate that both fras1 and itga8 mutations reduce buffering of stochastic developmental variation during facial morphogenesis.
itga8 mRNA is expressed in pharyngeal arch mesenchyme surrounded by epithelia expressing fras1 mRNA
The phenotypic similarity we discovered between fras1 and itga8 mutants suggests that the products of these genes should be concurrently expressed either in the same tissue or in physically interacting tissues, such as facial epithelia and mesenchyme. Consistent with previous reports (Carney et al., 2010; Gautier et al., 2008; Talbot et al., 2012; Westcot et al., 2015), fras1 transcript is prominently expressed in ectoderm and endoderm lining pharyngeal arches (Fig. 4A, B). At 36 hpf and 72 hpf, itga8 mRNA is expressed in arch mesenchyme, but not in the ectoderm or endoderm that line the arches (Fig. 4C, D). Because itga8 and fras1 transcripts show no tissue overlap (Fig. 4A-G), we conclude that itga8 is not expressed in epithelia. Because itga8 transcript has little overlap with col2a1 transcript (Fig. 4F, G), a cartilage marker (Yan et al., 1995), we propose that itga8 likely down-regulates when mesenchyme differentiates into cartilage. Consistent with previous reports (Carney et al., 2010; Talbot et al., 2012), Fras1 protein is deposited at the basal surface of wild-type epithelia (Fig. 5D), potentially exposing it to adjacent mesenchyme. These expression analyses provide strong support for our hypothesis that the FPC interacts with Itga8 at the interface of pharyngeal arch epithelia and mesenchyme.
Figure 4.

fras1 and itga8 are expressed in adjacent facial tissues. (A-D) Colorimetric in situ hybridization, developed using NBT/BCIP on wild-type sections, showing fras1 expression in pharyngeal and ectodermal epithelia (A, B) and itga8 expression in mesenchyme (C, D) at 36 hpf (A, C) and 72 hpf (B,D). (E) Color-coded diagram of lateral and transverse sections from a wild-type 60 hpf embryo, showing the first two pharyngeal arches (green), ectoderm (orange), cartilage (blue), and endoderm (red). (F, G) Fluorescent RNA in situ for fras1, itga8, and col2a1 expression in a 60 hpf transverse section of wild-type embryos at (F) low and (G) higher magnification. All scale bars: 50 μm.
Figure 5.

fras1 and itga8 do not regulate one another, nor do they regulate candidate targets npnt or fbn2b. (A-C) RNA in situ hybridization for itga8 transcripts on transverse sections in 60 hpf wild-type and mutant embryos, developed colorimetrically, oriented as shown in C'. These transcripts appear similar in (A) wild type, (B) itga8 mutants, and (C) fras1 mutants. The black crescent in (B) is eye pigment. (D, E) 72 hpf tissue sections, labeled for Fras1 protein, epithelial nuclei (anti-P63) and cartilage (sox9a:GFP). Fras1 protein levels and localization appears similar in wild type (D) and itga8b1161 mutants (E). (F-Q) Triple in situ hybridization for fras1, npnt, and fbn2b transcripts on 60 hpf tissue sections, oriented as in Fig. 4E. (F-H) Merged overlays of all three probes, which are also shown separately for fras1 (I-K), npnt (L-N) , and fbn2b (O-Q). For all three genes, expression is similar between wild type, fras1 mutants, and itga8 mutants. All scale bars: 50 μm. Scale bar in A applies to A-C. Scale bar, in D applies D, E. Scale bar in F applies to F-Q.
fras1 and itga8 do not regulate one another, nor do they regulate npnt or fbn2b
Based on these data, we hypothesized that Fras1 and Itga8 shape facial morphology via a combination of tissue adhesive and/or regulatory interactions dependent upon the FPC, as has been indicated previously in mouse kidney (Kiyozumi et al., 2012; Pitera et al., 2008). To test this hypothesis, we first examined whether fras1 and/or itga8 regulate one another at the level of transcript expression. In itga8b1161 and fras1b1048 mutants, itga8 expression appears normal (Fig. 5A-C). Additionally, itga8b1161 mutants express and localize Fras1 protein normally (Fig. 5D, E), and also express normal fras1 transcripts (Fig. 5F-K). These results indicate that fras1 and itga8 do not regulate one another, at least at the transcript level. To test whether fras1 and itga8 regulate shared downstream transcriptional targets, we examined the expression of several candidate genes identified in previous studies (Carney et al., 2010; Kiyozumi et al., 2012; Linton et al., 2007; Pitera et al., 2008) including fbn2a, fnb2b, gdnfa, grip1, grip2b, and npnt (Fig. S1). Several candidates (fbn2a, gdnfa, grip1, and grip2b) have little or no expression in pharyngeal regions at 60 or 72 hpf, but two candidates are prominently expressed during facial development (Fig. S1): fbn2b is expressed primarily in facial mesenchyme, and npnt is expressed in endoderm and adjacent mesenchyme (Fig. S1, S2). However, expression of fbn2b (Fig. 5L-N) and npnt (Fig. 5O-Q) appears largely normal in fras1b1048 and itga8b1161 mutants. To summarize, we find no evidence that fras1 and itga8 regulate one another transcriptionally, nor that itga8 regulates Fras1 post-transcriptionally, nor that fras1 or itga8 control the transcriptional expression of candidate targets npnt or fbn2b.
itga8 is necessary for normal facial endodermal morphology
At the cellular level zebrafish fras1 functions in endoderm to produce late ventral out-pocketing of pharyngeal pouch-1 (late-p1) during facial development. Outpocketing fails in fras1 mutants, which in turn appears responsible for the skeletal defects in the mesenchyme (Talbot et al., 2012). To explain the similarities between fras1 and itga8 mutant skeletal defects, we hypothesize that mesenchymally-derived Itga8 is also required for FPC function during late-p1 formation. In support, every itga8 mutant examined showed late-p1 defects, indicated by an increased distance between endoderm and ectoderm at 72 hpf, and the severity of itga8b1161 mutant late-p1 defects (Fig. 6) is indistinguishable from late-p1 defects found in fras1b1048 mutants (Talbot et al., 2012). In both mutants, the pouch phenotype is fully penetrant, in contrast to the partially penetrant cartilage phenotypes. Serial sections of itga8 mutants confirm that the defect spans the entirety of late-p1 (Movie 1). Similar to skeletal defects, severity of late-p1 defects do not appear enhanced in fras1b1048;itga8b1161 double mutants when compared to the two single mutants (Fig. 6A-D). Although the late-p1 defect in mutants is both penetrant and severe, the degree of late-p1 severity does vary among individuals, as we could examine quantitatively in single mutants (Fig. 6E). Thus, our data show that like fras1, itga8 is essential for generating a discrete epithelial structure, late-p1, in the zebrafish face.
itga8 mutant endodermal and skeletal phenotypes arise concurrently with fras1 phenotypes
Interestingly, the late-p1 variation just described does not predict the severity of skeletal defect (short Sy) when both were measured on the same sides of individual fish at 72 hpf (Fig. 6E). Again, itga8b1161 mutants and fras1b1048 mutants are similar to one another in this respect. Given the overall similarity between fras1 and itga8 mutant phenotypes, we reasoned that defects might arise concurrently in the two mutants. In fras1 mutants, craniofacial defects become apparent between 36 and 72 hpf (Talbot et al., 2012), during a period of dramatic craniofacial morphogenesis that includes both endodermal outpocketing and skeletal morphogenesis. Indeed, as in fras1 mutants, we find that itga8 mutant facial morphology looks normal at 36 hpf, but is overtly abnormal by 72 hpf (Fig. 7A). These matching temporal correlations add further support to the hypothesis that both mutants are disrupting the same underlying process, that we suppose to be the functioning of the FPC in late-p1 morphogenesis.
The critical period for fras1 function is during late-p1 outpocketing
Craniofacial phenotypes in itga8b1161 and fras1b1048 mutants begin to diverge from wild-type during the time-window of late-p1 morphogenesis, perhaps suggesting that they are required during this morphogenesis. However, knowing the time of phenotypic onset does not necessarily reveal when fras1 and itga8 are required – for some genes, earlier misfunctioning can sometimes result in a later mutant phenotype. To directly test when fras1 is required for facial mutagenesis, that is, the ‘critical period’ for gene function, we used a recently described, Cre-revertable fras1 allele (mn0156Gt) (Clark et al., 2011). The gene-trapped (mutant) allelic form contains an exon that terminates fras1 transcript prematurely. When Cre recombinase is expressed, the gene trap cassette is excised, restoring wild-type function (Fig. 7B). Heat shock induction of Cre fully rescues craniofacial phenotypes in fras1mn0156Gt mutants only when heat shock is applied at or before 32 hpf (Fig. 7C-E). In contrast, reversion of fras1 to wild type at 72 hpf or later yields skeletal defects indistinguishable from other mutant alleles (Fig. 7C-E). At 48 hpf, part way through late-p1 development (Talbot et al., 2012), fras1 reversion provides an intermediate level of rescue (Fig. 7C-E). The same trend was found for all three fras1 mutant skeletal phenotypes, suggesting that a common underlying defect causes all three phenotypes. To ascertain whether late p1 was also restored, we examined whole-mounted animals with differential interference contrast (DIC) optics and found a nearly perfect correlation between reversion of the skeletal defects and reversion of pouch outpocketing in the Cre-expressing (N=31/32) and Cre-nonexpressing (N=20/20) heat-shocked animals (examples and quantitation in Figure S3). These experiments support the hypothesis that fras1 sculpts wild-type late-p1 and skeleton when late-p1 is outpocketing – the time when endodermal and skeletal phenotypes first appear in fras1 and itga8 mutants.
Discussion
itga8 and fras1 facilitate epithelial-mesenchymal interactions during facial development
Epithelial-mesenchymal interaction defects underlie some mammalian Fras1 mutant phenotypes (Smyth and Scambler, 2005). Here, we show that zebrafish itga8 mutants have craniofacial defects that strongly resemble fras1 mutants. Although fras1 and itga8 appear to function in different tissues, fras1;itga8 double mutants closely resemble the two single mutants. Since both mutations affect epithelial tissues (late-p1) and mesenchyme-derived tissues (cartilage), we infer that the two genes facilitate epithelial-mesenchymal interactions during craniofacial development. Our favorite physical model to explain these epithelial-mesenchymal interactions (Fig 8A) comes primarily from studies in mouse. In this model Fras1 and Itga8 proteins function in the ECM (Kiyozumi et al., 2006; Pavlakis et al., 2011), and Itga8 interacts with Fras1 by binding to the FPC core protein Frem1 (Kiyozumi et al., 2005) or other FPC-associated proteins like Npnt (Kiyozumi et al., 2012). We suspect that Itga8 interacts with the core FPC proteins, rather than being itself part of this core complex because, unlike core FPC components (Kiyozumi et al., 2006), itga8 is not essential for Fras1 localization. This model suggests a number of studies, biochemical or molecular in nature, that could provide direct evidence relevant to this issue. The present study reveals how important interaction is to the function of both genes. fras1 and itga8 single mutants show similar severe skeletal and endodermal phenotypes that share similar time of phenotype onset and similar phenotypic variation; these striking similarities suggest that most of Fras1 and Itga8 function is mediated by their interactions (indirectly at the molecular level, via the FPC). Consistent with that idea, double mutant embryos have phenotypes similar to both single mutants. However, the higher defect penetrance we observe in fras1 mutants, compared to itga8 mutants, may suggest that Fras1 protein accomplishes a small portion of its function independently of Itga8 protein. Together, our findings suggest that interactions between fras1 and itga8 products are vital to epithelial-mesenchymal interactions that sculpt zebrafish facial morphology.
Figure 8.
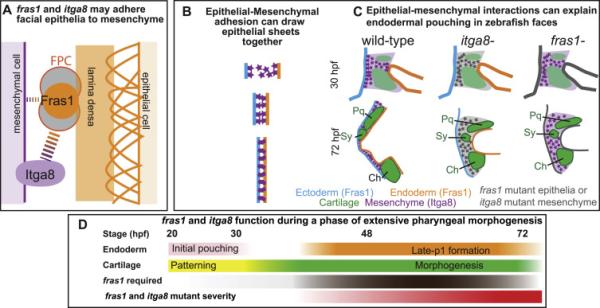
Epithelial-mesenchymal Fras1-Itga8 interactions sculpt zebrafish facial development. (A) Proposed structure of a Fras1-Itga8 interacting complex. Fras1 protein (orange circle) is part of the FPC (gray filled circles), that interacts with Itga8, directly or indirectly (dotted lines), to attach mesenchymal cells to the lamina densa (orange bar), which is itself attached (orange arches) to epithelial cells. Fras1 may be able to participate in weak epithelial/mesenchymal interactions independently of Itga8 (narrow dotted lines), but most of their function occurs via one another (broad dotted lines). (B) Diagram modeling how epithelial-mesenchymal adhesion might narrow the space between endoderm and ectoderm, e.g. during endodermal pouching. (C) Illustration of late-p1 formation, with mesenchyme and skeletal elements shown. In fras1 and itga8 mutants, epithelial-mesenchymal interactions are lost and late-p1 fails to form. (D) Timeline of fras1 and itga8 functions during different stages of facial development.. Rescue experiments indicate that fras1 is required (brown) during the cartilage morphogenesis phase of development (green), concurrent with late-p1 formation (orange). Fras1 is dispensable during early endodermal pouching (pink) and cartilage patterning (yellow). In both fras1 and itga8 mutants, phenotypes start to diverge from wild type by 36 hpf, and are severe by 72 hpf (red).
Interaction between Fras1 and Itga8 may utilize a combination of tissue adhesion and signaling
Both Fras1 and Itga8 are known to mediate cell adhesion and cell signaling (Short et al., 2007; Zargham, 2010). For instance, cultured Itga8 mutant cells from mouse lung and kidney are less adhesive and more migratory than wild-type counterparts (Benjamin et al., 2009; Bieritz et al., 2003; Short et al., 2007). In cell culture, Frem1-Itga8 binding increases cell adhesion (Kiyozumi et al., 2005), though their interaction may require additional proteins in vivo (Kiyozumi et al., 2012). In support of a tissue adhesion mechanism, adhesive defects could explain the observed epithelial (Fig. 8A-C) and skeletal (Talbot et al., 2012) defects found in zebrafish fras1 and itga8 mutants. Nonetheless, signaling functions may be key to understanding Fras1 and Itga8 activity. For instance, during mammalian kidney development, Itga8 and Fras1 are both necessary to activate transcription of Gdnf mRNA (Linton et al., 2007; Pitera et al., 2008). Gdnf protein in turn activates Fgf signaling, and fittingly, kidney development is rescued in both Itga8 and Fras1 mutants when the Fgf inhibitor Sprouty1 is also mutated (Linton et al., 2007; Pitera et al., 2012). During zebrafish craniofacial development Fgfs are essential for endodermal and skeletal morphogenesis (Crump, 2004; David et al., 2002; Larbuisson et al., 2013; Walshe and Mason, 2003), though phenotypes caused by Fgf loss do not resemble fras1 or itga8 mutant phenotypes. Although we did not detect gdnfa expression in any zebrafish craniofacial tissue, it is possible that Fras1 or Itga8 stimulate Fgf activity or another signaling activity, via a different mediator, during craniofacial development.
fras1-itga8 mediated epithelial-mesenchymal interactions sculpt craniofacial morphology
We propose that during zebrafish facial development, Fras1 and Itga8 help bind pharyngeal arch epithelia to arch mesenchyme, thereby generating late-p1 (Fig. 8B, C). We propose that itga8-expressing arch mesenchyme, when situated between two fras1-expressing epithelial sheets, draws them together (Fig. 8B) during late-p1 development (Fig. 8C). Consistent with this idea, we observe the strongest itga8 transcript expression in undifferentiated arch mesenchyme, and expression subsequently decreases as the tissue differentiates into skeleton. Since both mesenchyme and perichondrium contact the endoderm, Itga8 could mediate adhesion between either tissue and Fras1-expressing epithelia to drive late-p1 formation (Fig. 8C). We previously proposed that late-p1 defects account for the skeletal defects observed in fras1 mutants (Talbot et al., 2012); it follows that late-p1 defects also account for itga8 mutant skeletal defects. There are many precedents for our claim that endoderm influences skeletal morphogenesis; for example, in zebrafish itga5 mutants, loss of an early-forming portion of endodermal pouch 1 causes hyomandibular cartilage defects (Crump et al., 2004). Thus, while not ruling out the possibility of signaling functions, a simple adhesive model for Itga8 and Fras1 function can explain both skeletal and epithelial defects in fras1 and itga8 single and double mutants.
Variable facial phenotypes may be explained by late-p1 defects
In both itga8 and fras1 mutants, skeletal phenotypes are highly variable. We previously argued that strong loss of fras1 function leads to variable phenotypes, because fras1 mutants lose the developmental buffering provided by late-p1 (Talbot et al., 2012). Here, we bolster this argument by demonstrating that phenotypes are also variable in itga8 mutants, which lack the mesenchymal portion of the proposed Fras1/Itga8 containing complex. fras1 and itga8 are each required for late-p1 formation, though the degree of late-p1 phenotypic severity varies among mutant individuals. We know the presence of late-p1 is not absolutely required for seemingly normal cartilage morphology because the severities of particular phenotypes (e.g. symplectic cartilage length) can be mild or undetected in individual fras1 or itga8 mutants, which consistently display severe late-p1 defects. Therefore, we propose that the presence of the well-formed pouch stabilizes, or “buffers”, development by providing the correct environment for reliable skeletal morphogenesis. For instance, wild-type late-p1 may provide a physical barrier preventing skeletal fusion in wild-type embryos, but in mutants late-p1 absence allows skeletal elements to move more freely and sometimes fuse (Figure 8C). We investigated other potential sources of variation, but find these to be unlikely explanations. For example, the degree of skeletal variation is not influenced by maternal genotype. Phenotypic variation is similar for all three itga8 alleles, including alleles produced in different AB sub-lines (b1161 vs. oz6, oz7). Phenotypic variation is likely not due to hypomorphic residual function because all three itga8 alleles are putative null alleles that truncate the protein prior to critical domains. Matching what we show for zebrafish, ITGA8 may also be required to stabilize human development; a potentially hypomorphic human lesion may cause kidney disease, with severity ranging in different individuals from complete kidney agenesis (severe) to specific defects within kidneys (milder) (Humbert et al., 2014; Kohl et al., 2014). To our knowledge, the development of pharyngeal pouches has not yet been critically examined in mammalian Itga8 or Fras1 mutants. Perhaps ITGA8 helps sculpt developmental buffers during human development, possibly in the same way zebrafish Itga8 helps sculpt late-p1 during craniofacial development.
Fras1 and Itga8 sculpt facial shape during a critical phase of morphogenesis
Early pharyngeal arch development can be divided into two major phases: patterning and morphogenesis (Fig. 8D). Arch patterning occurs between 20 and 30-36 hpf, when pharyngeal arch mesenchyme is divided into major domains by signaling and transcription factors (Alexander et al., 2014; Miller, 2003; Miller et al., 2000; Nichols et al., 2013; Talbot et al., 2010; Zuniga et al., 2010). During this same phase, early endodermal pouch-1 forms (Schilling and Kimmel, 1994) and is a rich source of patterning signals (Choe and Crump, 2015). Then, between 30-36 and 72 hpf, pharyngeal arches undergo dramatic morphogenesis in response to earlier patterning events. During this same period, cartilages acquire their larval shapes (Knight and Schilling, 2006) and late-p1 forms (Talbot et al., 2012). We find that restoration of fras1 expression fully rescues both skeletal and endodermal development even when it is restored as late as 32 hpf, indicating that fras1 function is dispensable during the early facial patterning phase. However, there is only moderate rescue of craniofacial development when the fras1 mutation is reverted at 48 hpf, and no rescue at 72 hpf, indicating that fras1+ is necessary during the morphogenesis phase of cartilage and late-p1 development. We propose that, although Fras1 is expressed prior to craniofacial morphogenesis, its primary function in pharyngeal endoderm is to directly mold facial morphology. Similar to fras1 single mutants (Talbot et al., 2012), itga8 mutant defects also arise between 36 and 72 hpf; we propose that the reason why itga8 mutant phenotypes arise concurrently is because during this time period Itga8 protein functions (via the Fras1-containing FPC), to help drive this morphogenesis.
Fras1 and Itga8 may mediate epithelial-mesenchymal interactions in several, but not all, tissues
In this study, we primarily focused on the role of fras1 and itga8 in shaping facial morphologies, but it is worth contemplating how craniofacial fras1 and itga8 functions compare to their functions in other tissues. Itga8 does not necessarily interact with Fras1 in all fras1-expressing tissues; for instance we show that itga8 is dispensable for tail fin development, a process that requires fras1 and all other FPC-related genes investigated to date (Carney et al., 2010; Richardson et al., 2013). Although the composition of Fras1-containing complexes may differ between tissues, the co-requirement of fras1 and itga8 in the developing zebrafish face mirrors previous proposals that these two genes each mediate epithelial-mesenchymal interactions leading to mammalian kidney formation (Kiyozumi et al., 2012; Kohl et al., 2014; McGregor et al., 2003; Pitera et al., 2008). Mutation of mouse Fras1 (Petrou et al., 2005) or Itga8 (Benjamin et al., 2009) also results in similar discrete lung defects; specifically, both mutations disrupt epithelial-mesenchymal interactions, leading to fusion of medial and caudal lobes in the right lung. In mouse kidney, Npnt directly binds to Itga8 with high affinity, and also to Frem1, indicating that Npnt helps link Itga8 to the FPC (Kiyozumi et al., 2012). Consistent with this connection, zebrafish endoderm expresses high levels of npnt at sites of late-p1 formation. In mouse kidney, both Fras1 and Itga8 regulate downstream targets including Npnt protein (Kiyozumi et al., 2012; Pitera et al., 2008). However, regulation appears to occur post-transcriptionally, because Npnt transcript levels are normal in mouse Fras1 mutants (Kiyozumi et al., 2012). Similarly, we find that zebrafish npnt transcripts appear normal in fras1 and itga8, though our studies do not test Npnt regulation at the protein level. Therefore, while Fras1 and Itga8 may facilitate epithelial-mesenchymal interactions only in certain tissues, they may use similar mechanisms to interact in multiple tissues.
Conclusion
Previous research proposed that Fras1 and Itga8 physically interact in an adhesive complex. We propose that fras1 and itga8 are both vital to epithelial-mesenchymal interactions that sculpt facial morphology when late-p1 forms, during a phase of dramatic skeletal morphogenesis. A deeper understanding of how these two genes interact in different tissues, and the relative importance of adhesion versus signaling for their interactions, will provide further insights into normal embryonic development and Fraser-spectrum diseases.
Supplementary Material
Highlights.
itga8 is necessary for zebrafish facial endoderm and cartilage development.
itga8 is expressed in facial mesenchyme, surrounded by fras1-expressing epithelia.
Craniofacial defects in itga8 mutants phenocopy fras1 and fras1;itga8 mutants.
In faces, fras1 and itga8 function between 1.5 and 3 days post fertilization.
We propose that itga8 and fras1 interactions stabilize zebrafish facial development.
Acknowledgements
We thank the Amacher Lab fish facility, and colleagues at the Ohio State University for providing excellent fish care, helpful conversations and support during isolation of itga8oz6 and itga8oz7. We thank the University of Oregon fish facility and colleagues for all other fish care and support.
Funding
This work was supported by the National Institutes of Health (grants RO1 DE13834 to C.B.K.; DE020076 to J.H.P.; HD22486 to J.H.P, C.B.K. and J.C.T.; R01 GM088041 to S.L.A; NINDS T32 NS077984 to J.C.T.; K99/R00 DE024190 to J.T.N.) and by the Pelotonia Postdoctoral Fellowship Program (to J.C.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
J.C.T., J.H.P., and C.B.K. designed the experiments. J.C.T., J.T.N., Y.L.Y., I.F.L., C.B.K. and R.A.B. performed the experiments. C.B.K, J.H.P., and S.L.A. provided expertise, reagents, and infrastructure. J.C.T. wrote the paper, with additional writing contributions by J.T.N. and Y.L.Y.; all authors edited the paper.
References
- Akimenko M-A, Ekker M, Wegner J, Lin W, Westerfield M. Combinatorial Expression of Three Zebrafish Genes Related to Distal-Less: Part of a Homeobox Gene Code for the Head. J. Neurosci. 1994;14:3475–3486. doi: 10.1523/JNEUROSCI.14-06-03475.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alazami AM, Shaheen R, Alzahrani F, Snape K, Saggar A, Brinkmann B, Bavi P, Al-Gazali LI, Alkuraya FS. FREM1 Mutations Cause Bifid Nose, Renal Agenesis, and Anorectal Malformations Syndrome. Am. J. Hum. Genet. 2009;85:414–418. doi: 10.1016/j.ajhg.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander C, Piloto S, Le Pabic P, Schilling TF. Wnt Signaling Interacts with Bmp and Edn1 to Regulate Dorsal-Ventral Patterning and Growth of the Craniofacial Skeleton. PLoS Genet. 2014;10:e1004479. doi: 10.1371/journal.pgen.1004479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie CE, Raible DW, Henion PD, Eisen JS. Early pressure screens. Methods Cell Biol. 1999;60:71–86. [PubMed] [Google Scholar]
- Benjamin JT, Gaston DC, Halloran BA, Schnapp LM, Zent R, Prince LS. The role of integrin α8β1 in fetal lung morphogenesis and injury. Dev. Biol. 2009;335:407–417. doi: 10.1016/j.ydbio.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieritz B, Spessotto P, Colombatti A, Jahn A, Prols F, Hartner A. Role of alpha8 integrin in mesangial cell adhesion, migration, and proliferation. Kidney Int. 2003;64:119–127. doi: 10.1046/j.1523-1755.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- Bonkowsky JL, Chien C-B. Molecular cloning and developmental expression of foxP2 in zebrafish. Dev. Dyn. 2005;234:740–746. doi: 10.1002/dvdy.20504. [DOI] [PubMed] [Google Scholar]
- Brandenberger R. Identification and characterization of a novel extracellular matrix protein nephronectin that is associated with integrin alpha8beta1 in the embryonic kidney. J. Cell Biol. 2001;154:447–458. doi: 10.1083/jcb.200103069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney TJ, Feitosa NM, Sonntag C, Slanchev K, Kluger J, Kiyozumi D, Gebauer JM, Talbot JC, Kimmel CB, Sekiguchi K. Genetic analysis of fin development in zebrafish identifies furin and hemicentin1 as potential novel fraser syndrome disease genes. PLoS Genet. 2010;6:e1000907. doi: 10.1371/journal.pgen.1000907. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe CP, Crump JG. Dynamic epithelia of the developing vertebrate face. Curr. Opin. Genet. Dev. 2015;32:66–72. doi: 10.1016/j.gde.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KJ, Balciunas D, Pogoda H-M, Ding Y, Westcot SE, Bedell VM, Greenwood TM, Urban MD, Skuster KJ, Petzold AM, Ni J, Nielsen AL, Patowary A, Scaria V, Sivasubbu S, Xu X, Hammerschmidt M, Ekker SC. In vivo protein trapping produces a functional expression codex of the vertebrate proteome. Nat. Methods. 2011;8:506–512. doi: 10.1038/nmeth.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump JG. An essential role for Fgfs in endodermal pouch formation influences later craniofacial skeletal patterning. Development. 2004;131:5703–5716. doi: 10.1242/dev.01444. [DOI] [PubMed] [Google Scholar]
- Crump JG, Swartz ME, Kimmel CB. An Integrin-Dependent Role of Pouch Endoderm in Hyoid Cartilage Development. PLoS Biol. 2004;2:e244. doi: 10.1371/journal.pbio.0020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlem TJ, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, Locke AS, Weis AM, Voytas DF, Grunwald DJ. Simple Methods for Generating and Detecting Locus-Specific Mutations Induced with TALENs in the Zebrafish Genome. PLoS Genet. 2012;8:e1002861. doi: 10.1371/journal.pgen.1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalezios Y, Papasozomenos B, Petrou P, Chalepakis G. Ultrastructural localization of Fras1 in the sublamina densa of embryonic epithelial basement membranes. Arch. Dermatol. Res. 2007;299:337–343. doi: 10.1007/s00403-007-0763-8. [DOI] [PubMed] [Google Scholar]
- David NB, Saint-Etienne L, Tsang M, Schilling TF, Rosa FM. Requirement for endoderm and FGF3 in ventral head skeleton formation. Development. 2002;129:4457–4468. doi: 10.1242/dev.129.19.4457. [DOI] [PubMed] [Google Scholar]
- Dongen SV. Fluctuating asymmetry and developmental instability in evolutionary biology: past, present and future. J. Evol. Biol. 2006;19:1727–1743. doi: 10.1111/j.1420-9101.2006.01175.x. [DOI] [PubMed] [Google Scholar]
- Eames BF, DeLaurier A, Ullmann B, Huycke TR, Nichols JT, Dowd J, McFadden M, Sasaki MM, Kimmel CB. FishFace: interactive atlas of zebrafish craniofacial development at cellular resolution. BMC Dev. Biol. 2013;13:23. doi: 10.1186/1471-213X-13-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Langenau DM, Madge JA, Quinkertz A, Gutierrez A, Neuberg DS, Kanki JP, Thomas Look A. Heat-shock induction of T-cell lymphoma/leukaemia in conditional Cre/lox-regulated transgenic zebrafish. Br. J. Haematol. 2007;138:169–175. doi: 10.1111/j.1365-2141.2007.06625.x. [DOI] [PubMed] [Google Scholar]
- Gautier P, Naranjo-Golborne C, Taylor MS, Jackson IJ, Smyth I. Expression of the fras1/frem gene family during zebrafish development and fin morphogenesis. Dev. Dyn. 2008;237:3295–3304. doi: 10.1002/dvdy.21729. [DOI] [PubMed] [Google Scholar]
- Graham JH, Raz S, Hel-Or H, Nevo E. Fluctuating Asymmetry: Methods, Theory, and Applications. Symmetry. 2010;2:466–540. [Google Scholar]
- Humbert C, Silbermann F, Morar B, Parisot M, Zarhrate M, Masson C, Tores F, Blanchet P, Perez M-J, Petrov Y, Khau Van Kien P, Roume J, Leroy B, Gribouval O, Kalaydjieva L, Heidet L, Salomon R, Antignac C, Benmerah A, Saunier S, Jeanpierre C. Integrin Alpha 8 Recessive Mutations Are Responsible for Bilateral Renal Agenesis in Humans. Am. J. Hum. Genet. 2014;94:288–294. doi: 10.1016/j.ajhg.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh J-RJ, Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadeja S, Smyth I, Pitera JE, Taylor MS, van Haelst M, Bentley E, McGregor L, Hopkins J, Chalepakis G, Philip N, Perez Aytes A, Watt FM, Darling SM, Jackson I, Woolf AS, Scambler PJ. Identification of a new gene mutated in Fraser syndrome and mouse myelencephalic blebs. Nat. Genet. 2005;37:520–525. doi: 10.1038/ng1549. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kiyozumi D, Osada A, Sugimoto N, Weber CN, Ono Y, Imai T, Okada A, Sekiguchi K. Identification of a novel cell-adhesive protein spatiotemporally expressed in the basement membrane of mouse developing hair follicle. Exp. Cell Res. 2005;306:9–23. doi: 10.1016/j.yexcr.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Kiyozumi D, Sugimoto N, Sekiguchi K. Breakdown of the reciprocal stabilization of QBRICK/Frem1, Fras1, and Frem2 at the basement membrane provokes Fraser syndrome-like defects. Proc. Natl. Acad. Sci. 2006;103:11981–11986. doi: 10.1073/pnas.0601011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyozumi D, Takeichi M, Nakano I, Sato Y, Fukuda T, Sekiguchi K. Basement membrane assembly of the integrin 8 1 ligand nephronectin requires Fraser syndrome-associated proteins. J. Cell Biol. 2012;197:677–689. doi: 10.1083/jcb.201203065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight RD, Schilling TF. Neural Crest Induction and Differentiation. Springer; 2006. Cranial neural crest and development of the head skeleton; pp. 120–133. [DOI] [PubMed] [Google Scholar]
- Kohl S, Hwang D-Y, Dworschak GC, Hilger AC, Saisawat P, Vivante A, Stajic N, Bogdanovic R, Reutter HM, Kehinde EO, Tasic V, Hildebrandt F. Mild Recessive Mutations in Six Fraser Syndrome-Related Genes Cause Isolated Congenital Anomalies of the Kidney and Urinary Tract. J. Am. Soc. Nephrol. 2014;25:1917–1922. doi: 10.1681/ASN.2013101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larbuisson A, Dalcq J, Martial JA, Muller M. Fgf receptors Fgfr1a and Fgfr2 control the function of pharyngeal endoderm in late cranial cartilage development. Differentiation. 2013;86:192–206. doi: 10.1016/j.diff.2013.07.006. [DOI] [PubMed] [Google Scholar]
- Linton JM, Martin GR, Reichardt LF. The ECM protein nephronectin promotes kidney development via integrin 8 1-mediated stimulation of Gdnf expression. Development. 2007;134:2501–2509. doi: 10.1242/dev.005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor L, Makela V, Darling SM, Vrontou S, Chalepakis G, Roberts C, Smart N, Rutland P, Prescott N, Hopkins J. Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat. Genet. 2003;34:203–208. doi: 10.1038/ng1142. others. [DOI] [PubMed] [Google Scholar]
- Miller CT. Two endothelin 1 effectors, hand2 and bapx1, pattern ventral pharyngeal cartilage and the jaw joint. Development. 2003;130:1353–1365. doi: 10.1242/dev.00339. [DOI] [PubMed] [Google Scholar]
- Miller CT, Schilling TF, Lee K, Parker J, Kimmel CB. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development. 2000;127:3815–3828. doi: 10.1242/dev.127.17.3815. [DOI] [PubMed] [Google Scholar]
- Müller U, Wang D, Denda S, Meneses JJ, Pedersen RA, Reichardt LF. Integrin α8β1 is critically important for epithelial–mesenchymal interactions during kidney morphogenesis. Cell. 1997;88:603–613. doi: 10.1016/s0092-8674(00)81903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson J, Swarr DT, Singer A, Liu M, Chinn A, Jones W, Hurst J, Khalek N, Zackai E, Slavotinek A. Novel FREM1 mutations expand the phenotypic spectrum associated with manitoba-oculo-tricho-anal (MOTA) syndrome and bifid nose renal agenesis anorectal malformations (BNAR) syndrome. Am. J. Med. Genet. A. 2013;161:473–478. doi: 10.1002/ajmg.a.35736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols JT, Pan L, Moens CB, Kimmel CB. barx1 represses joints and promotes cartilage in the craniofacial skeleton. Development. 2013;140:2765–2775. doi: 10.1242/dev.090639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlakis E, Chiotaki R, Chalepakis G. The role of Fras1/Frem proteins in the structure and function of basement membrane. Int. J. Biochem. Cell Biol. 2011;43:487–495. doi: 10.1016/j.biocel.2010.12.016. [DOI] [PubMed] [Google Scholar]
- Petrou P. Basement Membrane Distortions Impair Lung Lobation and Capillary Organization in the Mouse Model for Fraser Syndrome. J. Biol. Chem. 2005;280:10350–10356. doi: 10.1074/jbc.M412368200. [DOI] [PubMed] [Google Scholar]
- Pitera JE, Scambler PJ, Woolf AS. Fras1, a basement membrane-associated protein mutated in Fraser syndrome, mediates both the initiation of the mammalian kidney and the integrity of renal glomeruli. Hum. Mol. Genet. 2008;17:3953–3964. doi: 10.1093/hmg/ddn297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitera JE, Woolf AS, Basson MA, Scambler PJ. Sprouty1 haploinsufficiency prevents renal agenesis in a model of Fraser syndrome. J. Am. Soc. Nephrol. 2012;23:1790–1796. doi: 10.1681/ASN.2012020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Gebauer JM, Zhang J-L, Kobbe B, Keene DR, Karlsen KR, Richetti S, Wohl AP, Sengle G, Neiss WF, Paulsson M, Hammerschmidt M, Wagener R. AMACO is a component of the basement membrane-associated Fraser complex. J. Invest. Dermatol. 2013;134:1313–1322. doi: 10.1038/jid.2013.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Marí A, Yan Y-L, BreMiller RA, Wilson C, Cañestro C, Postlethwait JH. Characterization and expression pattern of zebrafish anti-Müllerian hormone (amh) relative to sox9a, sox9b, and cyp19a1a, during gonad development Gene Expr. Patterns. 2005;5:655–667. doi: 10.1016/j.modgep.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Sato Y, Uemura T, Morimitsu K, Sato-Nishiuchi R, Manabe R. -i., Takagi J, Yamada M, Sekiguchi K. Molecular Basis of the Recognition of Nephronectin by Integrin 8 1. J. Biol. Chem. 2009;284:14524–14536. doi: 10.1074/jbc.M900200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanze D, Kayserili H, Satkın BN, Altunoglu U, Zenker M. Fraser syndrome due to mutations in GRIP1 -Clinical phenotype in two families and expansion of the mutation spectrum. Am. J. Med. Genet. A. 2014;164:837–840. doi: 10.1002/ajmg.a.36343. [DOI] [PubMed] [Google Scholar]
- Schilling TF, Kimmel CB. Segment and cell type lineage restrictions during pharyngeal arch development in the zebrafish embryo. Development. 1994;120:483–494. doi: 10.1242/dev.120.3.483. [DOI] [PubMed] [Google Scholar]
- Schnapp LM, Breuss JM, Ramos DM, Sheppard D, Pytela R. Sequence and tissue distribution of the human integrin alpha 8 subunit: a beta 1-associated alpha subunit expressed in smooth muscle cells. J. Cell Sci. 1995a;108:537–544. doi: 10.1242/jcs.108.2.537. [DOI] [PubMed] [Google Scholar]
- Schnapp LM, Hatch N, Ramos DM, Klimanskaya IV, Sheppard D, Pytela R. The human integrin α8β1 functions as a receptor for tenascin, fibronectin, and vitronectin. J. Biol. Chem. 1995b;270:23196–23202. doi: 10.1074/jbc.270.39.23196. [DOI] [PubMed] [Google Scholar]
- Shafeghati Y, Kneipert A, Vakili G, Zenker M. Fraser Syndrome Due to Homozygosity for a Splice Site Mutation of FREM2. Am. J. Med. Genet. A. 2008;146A:529–531. doi: 10.1002/ajmg.a.32091. [DOI] [PubMed] [Google Scholar]
- Short K, Wiradjaja F, Smyth I. Let's stick together: The role of the Fras1 and Frem proteins in epidermal adhesion. IUBMB Life. 2007;59:427–435. doi: 10.1080/15216540701510581. [DOI] [PubMed] [Google Scholar]
- Slavotinek A, Li C, Sherr EH, Chudley AE. Mutation analysis of theFRAS1 gene demonstrates new mutations in a propositus with Fraser syndrome. Am. J. Med. Genet. A. 2006;140A:1909–1914. doi: 10.1002/ajmg.a.31399. [DOI] [PubMed] [Google Scholar]
- Slavotinek AM, Baranzini SE, Schanze D, Labelle-Dumais C, Short KM, Chao R, Yahyavi M, Bijlsma EK, Chu C, Musone S, Wheatley A, Kwok P-Y, Marles S, Fryns J-P, Maga AM, Hassan MG, Gould DB, Madireddy L, Li C, Cox TC, Smyth I, Chudley AE, Zenker M. Manitoba-oculo-tricho-anal (MOTA) syndrome is caused by mutations in FREM1. J. Med. Genet. 2011;48:375–382. doi: 10.1136/jmg.2011.089631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth I, Du X, Taylor MS, Justice MJ, Beutler B, Jackson IJ. The extracellular matrix gene Frem1 is essential for the normal adhesion of the embryonic epidermis. Proc. Natl. Acad. Sci. U. S. A. 2004;101:13560–13565. doi: 10.1073/pnas.0402760101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth I, Scambler PJ. The genetics of Fraser syndrome and the blebs mouse mutants. Hum. Mol. Genet. 2005;14:R269–R274. doi: 10.1093/hmg/ddi262. [DOI] [PubMed] [Google Scholar]
- Talbot JC, Amacher SL. A streamlined CRISPR pipeline to reliably generate zebrafish frameshifting alleles. Zebrafish. 2014;11:583–585. doi: 10.1089/zeb.2014.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot JC, Johnson SL, Kimmel CB. hand2 and Dlx genes specify dorsal, intermediate and ventral domains within zebrafish pharyngeal arches. Development. 2010;137:2507–2517. doi: 10.1242/dev.049700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot JC, Walker MB, Carney TJ, Huycke TR, Yan Y-L, BreMiller RA, Gai L, DeLaurier A, Postlethwait JH, Hammerschmidt M, Kimmel CB. fras1 shapes endodermal pouch 1 and stabilizes zebrafish pharyngeal skeletal development. Development. 2012;139:2804–2813. doi: 10.1242/dev.074906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenaz P, Ruttimann UE, Unser M. A pyramid approach to subpixel registration based on intensity. Image Process. IEEE Trans. On. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- van Haelst MM, Maiburg M, Baujat G, Jadeja S, Monti E, Bland E, Pearce K, Fraser Syndrome Collaboration Group. Hennekam RC, Scambler PJ. Molecular study of 33 families with Fraser syndrome new data and mutation review. Am. J. Med. Genet. A. 2008;146A:2252–2257. doi: 10.1002/ajmg.a.32440. [DOI] [PubMed] [Google Scholar]
- Vogel MJ, van Zon P, Brueton L, Gijzen M, van Tuil MC, Cox P, Schanze D, Kariminejad A, Ghaderi-Sohi S, Blair E, Zenker M, Scambler PJ, Ploos van Amstel HK, van Haelst MM. Mutations in GRIP1 cause Fraser syndrome. J. Med. Genet. 2012;49:303–306. doi: 10.1136/jmedgenet-2011-100590. [DOI] [PubMed] [Google Scholar]
- Vrontou S, Petrou P, Meyer BI, Galanopoulos VK, Imai K, Yanagi M, Chowdhury K, Scambler PJ, Chalepakis G. Fras1 deficiency results in cryptophthalmos, renal agenesis and blebbed phenotype in mice. Nat. Genet. 2003;34:209–214. doi: 10.1038/ng1168. [DOI] [PubMed] [Google Scholar]
- Walshe J, Mason I. Fgf signalling is required for formation of cartilage in the head. Dev. Biol. 2003;264:522–536. doi: 10.1016/j.ydbio.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Westcot SE, Hatzold J, Urban MD, Richetti SK, Skuster KJ, Harm RM, Lopez Cervera R, Umemoto N, McNulty MS, Clark JJ, Hammerschmidt M, Ekker SC. Protein-trap insertional mutagenesis uncovers new genes involved in zebrafish skin development, including a Neuregulin 2a-based ErbB signaling pathway required during median fin fold morphogenesis. PLoS ONE. 2015;10:e0130688. doi: 10.1371/journal.pone.0130688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book: A guide for the laboratory use of zebrafish (Danio rerio) University of Oregon Press; Eugene: 2007. [Google Scholar]
- Yan Y-L, Hatta K, Riggleman B, Postlethwait JH. Expression of a type II collagen gene in the zebrafish embryonic axis. Dev. Dyn. 1995;203:363–376. doi: 10.1002/aja.1002030308. [DOI] [PubMed] [Google Scholar]
- Zargham R. Tensegrin in context: Dual role of α8 integrin in the migration of different cell types. Cell Adhes. Migr. 2010;4:485–490. doi: 10.4161/cam.4.4.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga E, Stellabotte F, Crump JG. Jagged-Notch signaling ensures dorsal skeletal identity in the vertebrate face. Development. 2010;137:1843–1852. doi: 10.1242/dev.049056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.