

Abstract
Objectives
To review all patients with SCO2 mutations and to describe a Brazilian patient with cardioencephalomyopathy carrying compound heterozygous mutations in SCO2, one being the known pathogenic p.E140K mutation and the other a novel 12–base pair (bp) deletion at nucleotides 1519 through 1530 (c.1519_1530del).
Design
Case report and literature review.
Setting
University hospital
Patient
Infant girl presenting with an encephalomyopathy, inspiratory stridor, ventilator failure, progressive hypotonia, and weakness, leading to death.
Main Outcome Measures
Clinical features, neuro-imaging findings, muscle biopsy with histochemical analysis, and genetic studies.
Results
This infant girl was the first child of healthy, nonconsanguineous parents. She developed progressive muscular hypotonia and ventilatory failure. At the end of the first month of life, she developed cardiomegaly and signs of cardiac failure. Routine blood tests showed lactic acidosis and mild elevation of the creatine kinase level. Brain magnetic resonance imaging showed increased T2 and fluid-attenuated inversion recovery signals in the putamen bilaterally. Nerve conduction studies showed severe axonal sensorimotor neuropathy. Muscle biopsy revealed a neurogenic pattern with mitochondrial proliferation and total absence of cytochrome-c oxidase histochemical stain. Sequencing of SCO2 showed that the patient had compound heterozygote SCO2 mutations: the previously described c.1541G A (p.E140K) mutation and a novel 12-bp deletion at nucleotides 1519 through 1530 (c.1519_1530del). The patient died at age 45 days.
Conclusions
Our findings and the literature review indicate that it is important to consider the diagnosis of mitochondrial disease in newborns with hypotonia and cardiomyopathy. In our case, the accurate diagnosis of SCO2 mutations is particularly important for genetic counseling.
HUMAN SCO2 PROTEIN IS essential for assembly of the catalytic core of cytochrome-c oxidase (COX), complex IV of the mitochondrial respiratory chain. Mutations in SCO2 (GenBank AF177385) cause an infantile cardioencephalomyopathy with severe deficiency of COX in heart, brain, and muscle.1,2
The main clinical manifestations are progressive muscular hypotonia, hypertrophic cardiomyopathy, and encephalopathy. The disease typically presents during the neonatal period, and the majority of the patients die in the first year of life. Cardiopulmonary failure is the most common cause of death.2
Almost all reported patients have had the p.E140K missense mutation, which can be homozygous or compound heterozygous. Patients with a homozygous p.E140K mutation show milder clinical phenotypes than those with a compound heterozygous mutation.2 Only about 50 cases have been described worldwide.1-15 Herein, we describe a Brazilian patient with a fatal cardioencephalomyopathy due to compound heterozygous SCO2 mutations, one being p.E140K and the other a 12–base pair (bp) deletion (c.1519_1530del).
REPORT OF A CASE
This infant girl is the first child of healthy, nonconsanguineous parents. After an uneventful pregnancy, she was born at 39 weeks’ gestation by vaginal delivery with fetal extraction difficulties. At birth, her weight was 2600 g, her length was 49.5 cm, and Apgar scores were 9 at 1 minute and 10 at 5 minutes; however, the newborn had inspiratory stridor, hypotonic lower limbs, and hypertonic upper limbs. She developed progressive muscular hypotonia and ventilatory failure, prompting transfer to the neonatal intensive care unit. At the end of the first month of life, she developed cardiomegaly (as shown on chest radiography) and signs of cardiac failure. Routine blood tests showed lactic acidosis and mild elevation of the creatine kinase level. Brain magnetic resonance imaging showed increased T2 and fluid-attenuated inversion recovery signals in the putamen bilaterally, and proton magnetic resonance spectroscopy showed an increased lactic acid doublet peak in the ventricular system and cerebral parenchyma (Figure 1). Nerve conduction studies and electromyography showed severe axonal sensorimotor neuropathy. Muscle biopsy revealed a neurogenic pattern with mitochondrial proliferation and total absence of COX histochemical stain (Figure 2). There was mild lipid and glycogen accumulation in muscle fibers. Sequencing of SCO2 showed that the patient had compound heterozygous SCO2 mutations, one being the previously described c.1541G A (p.E140K) mutation and the other a novel 12-bp deletion at nucleotides 1519 through 1530 (c.1519_1530del). Segregation analysis revealed that the father was a heterozygous carrier of the p.E140K mutation and the mother showed the deletion in heterozygosity (Figure 3). Both parents were clinically asymptomatic. The patient died at age 45 days.
Figure 1.

Brain magnetic resonance imaging (A) and proton magnetic resonance spectroscopy (B) showing a Leigh syndrome–like pattern with a lactate doublet peak in the brain tissue and ventricular system. NAA indicates N-acetylaspartate.
Figure 2.
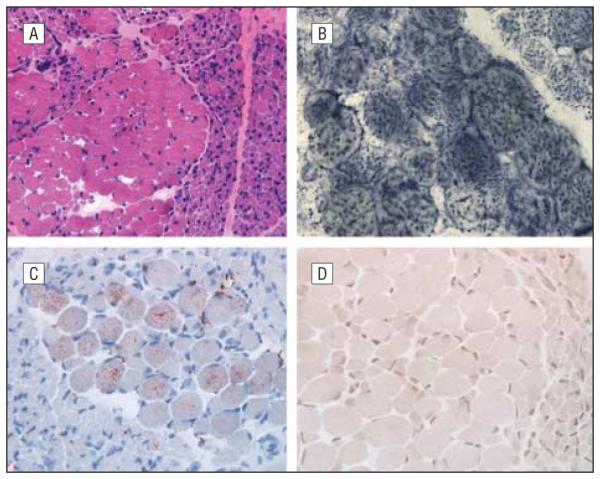
Muscle biopsy. A, Neurogenic pattern evident in hematoxylin-eosin staining (original magnification ×10). B, Mitochondrial proliferation revealed by succinate dehydrogenase histochemistry (original magnification ×40). C, Some hypertrophic fibers with mild lipid excess demonstrated by oil red 0 staining (original magnification ×20). D, Cytochrome-c oxidase histochemistry showing absence of detectable activity in all fibers (original magnification ×20).
Figure 3.
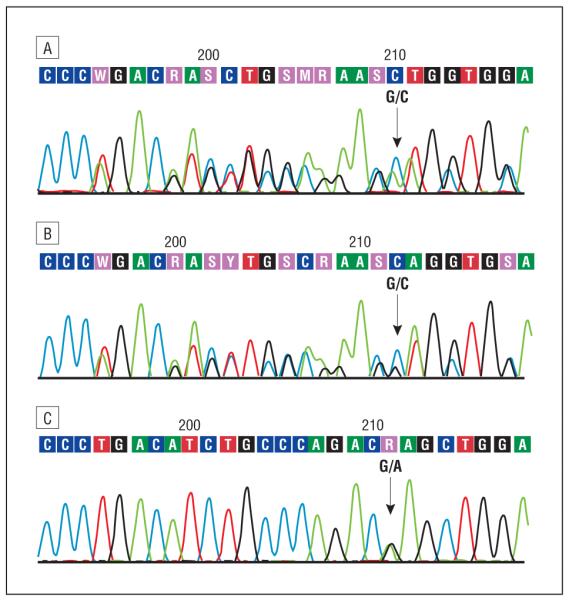
Sequencing analysis of SCO2. A, Representative electropherogram of the DNA (sense strand) from the patient showing the compound heterozygous mutations c.1541G A (p.E140K) and a 12–base pair (bp) deletion at c.1519-1530. B, Representative electropherogram of the DNA (sense strand) from the patient’s mother showing the deletion (12 bp at c.1519-1530) in heterozygosity, without the p.E140K mutation. C, Representative electropherogram of the DNA (sense strand) from the patient’s father showing the heterozygous p.E140K mutation.
COMMENT
The SCO2 protein is a metallochaperone that is essential for assembly of the catalytic core of COX.1 Numerous additional proteins are required for efficient COX assembly and maintenance. Mutations in different nuclear genes encoding these proteins have been described, including SURF1, SCO1, SCO2, COX10, and COX15. The phenotypes associated with these genetic defects are typically severe and fatal.6
To date, about 50 patients with mutations in SCO2 have been described.1-15 The p.E140K mutation was found in all patients either in heterozygosity or in association with a second mutation (compound heterozygosity), except one patient described in 2009 by Mobley et al13 who had a homozygous p.G193S mutation (Table). Patients harboring a homozygous p.E140K mutation have shown late onset and longer survival compared with patients with compound heterozygous SCO2 mutations.2,6,12,14,15 Our patient showed the common p.E140K mutation and a novel 12-bp deletion at nucleotides 1519 through 1530. The SCO proteins contain a conserved pair of cysteines separated by 3 residues, a motif that has been proposed to bind copper. Both genetic defects in our patient are adjacent to the SCO2 CXXXC copper-binding motif between Cys133 and Cys137, which is a critical region of SCO2.1 The clinical consequence of this compound heterozygous mutation was a severe phenotype and early death.
Table.
Clinical and Genetic Data of Published Cases With SCO2 Mutations
| Source | Phenotype | Muscle | Genotype |
|---|---|---|---|
| Papadopoulou et al,1 1999 |
Cardioencephalomyopathy, 3 patients | Absence of COX activity | p.Q53X/E140K in 2 patients, p.E140K/S225F in 1 patient |
| Jaksch et al,3 2000 | Cardioencephalomyopathy, 3 patients | Absence of COX activity | E140K/R171W in 1 patient, E140K/R90X in 2 patients (siblings) |
| Sue et al,4 2000 | Encephalopathy and hypertrophic cardiomyopathy, 3 patients |
Absence of COX activity | Compound heterozygosity, p.E140K/Q53X in 2 patients, p.E140K/S222F in 1 patient |
| Salviati et al,5
2002 |
Cardioencephalomyopathy and SMA phenotype, 1 patient |
Absence of COX activity, neurogenic pattern |
Compound heterozygosity, p.E140K and 10-bp duplication of nucleotides 1302-1311 |
| Sacconi et al,6 2003 | Leigh syndrome, 1 patient | Absence of COX activity | Compound heterozygosity, p.E140K/L151P |
| Foltopoulou et al,7
2004 |
Cardioencephalomyopathy, 3 patients | Absence of COX activity | E140K, S225F |
| Tarnopolsky et al,8
2004 |
Cardiomyopathy and SMA, 1 patient | Absence of COX activity, neurogenic pattern |
Compound heterozygosity, p.E140K and p.C133Y |
| Tay et al,9 2004 | Cardioencephalomyopathy, 1 patient | Absence of COX activity, neurogenic pattern |
Compound heterozygosity, p.E140K and p.Q53X |
| Vesela et al,10 2004 | Cardioencephalomyopathy, 6 patients | Absence of COX activity | p.E140K homozygosity in 5 patients, p.E140K/Q53X compound heterozygosity in 1 patient |
| Leary et al,11 2006 | Cardiomyopathy, 1 patient | Absence of COX activity | Homozygous p.E140K, hemizygous 16-bp intron deletion |
| Knuf et al,12 2007 | Fatal cardioencephalomyopathy, 1 patient | Absence of COX activity | Compound heterozygosity, E140K/V160G |
| Verdijk et al,2 2008 | Fatal cardioencephalomyopathy, 1 patient | Absence of COX activity | Compound heterozygosity, p.E140K/W36X |
| Mobley et al,13
2009 |
Fatal infantile cardioencephalomyopathy, 1 patient |
Homozygous p.G193S | |
| Pronicki et al,14
2010 |
Floppy infant with stridor and respiratory insufficiency, hypertrophic cardiomyopathy, 18 patients |
Neurogenic and other patterns, absence of COX activity |
p.E140K in all patients, p.Q53X in 1 patient, p.M177T in 1 patient |
| Joost et al,15 2010 | Leigh syndrome and cardioencephalomy- opathy, 1 patient (mother with mental retardation) |
Total absence of COX activity | Compound heterozygosity, c.418G>A (father), 19-bp insertion at position 17 (mother) |
| Present case | Cardioencephalomyopathy, stridor, neuropathy, 1 patient |
Total absence of COX activity | Compound heterozygosity, p.E140K and 12-bp deletion (c.1519_1530del) |
Abbreviations: bp, base pair; COX, cytochrome-c oxidase; SMA, spinal muscular atrophy.
In the initial description of SCO2 mutations, patients were noted to have abnormalities in the nervous system, heart, and skeletal muscle (cardioencephalomyopathy).1 Since then, other manifestations of SCO2 mutations have included Leigh syndrome, hypertrophic cardiomyopathy, lactic acidosis, stridor with ventilator insufficiency, and a spinal muscular atrophy–like phenotype.2-15 Our patient showed a complex phenotype characterized by encephalopathy, cardiomyopathy, peripheral neuropathy, stridor, ventilator insufficiency, and lactic acidosis. Brain magnetic resonance imaging showed increased T2 and fluid-attenuated inversion recovery signals in the putamen bilaterally, indicating Leigh syndrome. Proton magnetic resonance spectroscopy showed a lactic acid peak in the ventricular system and cerebral parenchyma as commonly observed in other mitochondrial diseases. The patient developed cardiomyopathy, which was ultimately fatal.
Nerve conduction studies and electromyography in our patient revealed a severe axonal sensorimotor neuropathy. In 1999, Papadopoulou et al1 described 3 patients with SCO2 mutations, but among them just 1 showed peripheral neuropathy. The electrophysiological findings have rarely been reported. This fact could be related to the difficulty of performing this examination in very young infants as well as the severity and early fatal outcome of the disease.
Among patients described with SCO2 mutations, muscle histopathological analysis has revealed varying abnormalities. In 2010, Pronicki et al14 described the pathological changes in muscles of 18 affected infants and noted 4 different patterns: type A, the spinal muscular atrophy pattern characterized by the presence of distinct atrophic and hypertrophic muscle fibers arranged in groups; type B, the spinal muscular atrophy–like pattern with a mixture of fibers of varying diameters generally not grouped; type C, dispersed and grouped neurogenic atrophy with small angulated fibers strongly reactive for oxidative enzymes; and type D, a nonspecific pattern with variability of muscle fiber size. Among the 18 patients, those with homozygous or compound heterozygous SCO2 mutations showed the type A or B pattern in muscle biopsy, while patients with uncertain or unconfirmed molecular background of the disease had the type C or D pattern. Our patient with the p.E140K and c.1519_1530del mutations showed fascicules with groups of atrophic or hypertrophic fibers compatible with the type A histopathological pattern (spinal muscular atrophy pattern).
The frequencies of causes of COX deficiency are unknown, but among children with COX deficiency, SCO2 mutations appear to be frequent genetic causes. In 2006, Böhm et al16 published a retrospective multicenter study of 180 children with COX deficiency. They identified pathogenic mutations in 75 patients and showed a striking prevalence of 2 nuclear gene mutations: c.845_846del_CT in SURF1 and a c.1541G A (p.E140K) transition in SCO2 in the Slavic population. In addition, Vesela et al10 studied 26 children with COX deficiency and found SCO2 mutations in 6 patients, leading them to conclude that SCO2 mutations are not rare in their population.
More recently, Honzik et al17 published a retrospective study that evaluated 461 patients with confirmed mitochondrial disease. The neonatal onset was reported in 28% of the patients, and a high incidence of neonatal cardiomyopathy was observed among them (40%). They proposed a diagnostic flowchart applicable to ill neonates suspicious for mitochondrial disorders. In this approach, the sequencing analysis of SCO1 and SCO2 was suggested if the neonate presented with a cardiomyopathy.
Our findings and the literature review indicate that it is important to consider the diagnosis of mitochondrial disease in newborns with hypotonia and cardiomyopathy. In our case, the accurate diagnosis of SCO2 mutations was important as it allows genetic counseling and enables pregestation or prenatal diagnosis in subsequent pregnancies of the parents.
Footnotes
Author Contributions: Study concept and design: Gurgel-Giannetti. Acquisition of data: Gurgel-Giannetti, Oliveira, and Martins. Analysis and interpretation of data: Gurgel-Giannetti, Oliveira, Brasileiro Filho, Vainzof, and Hirano. Drafting of the manuscript: Gurgel-Giannetti, Martins, and Hirano. Critical revision of the manuscript for important intellectual content: Gurgel-Giannetti, Oliveira, Brasileiro Filho, Vainzof, and Hirano. Obtained funding: Oliveira. Administrative, technical, and material support: Oliveira, Brasileiro Filho, Martins, and Hirano. Study supervision: Gurgel-Giannetti and Vainzof.
Conflict of Interest Disclosures: None reported.
REFERENCES
- 1.Papadopoulou LC, Sue CM, Davidson MM, et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. 1999;23(3):333–337. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- 2.Verdijk RM, de Krijger R, Schoonderwoerd K, et al. Phenotypic consequences of a novel SCO2 gene mutation. Am J Med Genet A. 2008;146A(21):2822–2827. doi: 10.1002/ajmg.a.32523. [DOI] [PubMed] [Google Scholar]
- 3.Jaksch M, Ogilvie I, Yao J, et al. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum Mol Genet. 2000;9(5):795–801. doi: 10.1093/hmg/9.5.795. [DOI] [PubMed] [Google Scholar]
- 4.Sue CM, Karadimas C, Checcarelli N, et al. Differential features of patients with mutations in two COX assembly genes, SURF-1 and SCO2. Ann Neurol. 2000;47(5):589–595. [PubMed] [Google Scholar]
- 5.Salviati L, Sacconi S, Rasalan MM, et al. Cytochrome c oxidase deficiency due to a novel SCO2 mutation mimics Werdnig-Hoffmann disease. Arch Neurol. 2002;59(5):862–865. doi: 10.1001/archneur.59.5.862. [DOI] [PubMed] [Google Scholar]
- 6.Sacconi S, Salviati L, Sue CM, et al. Mutation screening in patients with isolated cytochrome c oxidase deficiency. Pediatr Res. 2003;53(2):224–230. doi: 10.1203/01.PDR.0000048100.91730.6A. [DOI] [PubMed] [Google Scholar]
- 7.Foltopoulou PF, Zachariadis GA, Politou AS, Tsiftsoglou AS, Papadopoulou LC. Human recombinant mutated forms of the mitochondrial COX assembly Sco2 protein differ from wild-type in physical state and copper binding capacity. Mol Genet Metab. 2004;81(3):225–236. doi: 10.1016/j.ymgme.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Tarnopolsky MA, Bourgeois JM, Fu MH, et al. Novel SCO2 mutation (G1521A) presenting as a spinal muscular atrophy type I phenotype. Am J Med Genet A. 2004;125A(3):310–314. doi: 10.1002/ajmg.a.20466. [DOI] [PubMed] [Google Scholar]
- 9.Tay SK, Shanske S, Kaplan P, DiMauro S. Association of mutations in SCO2,a cytochrome c oxidase assembly gene, with early fetal lethality. Arch Neurol. 2004;61(6):950–952. doi: 10.1001/archneur.61.6.950. [DOI] [PubMed] [Google Scholar]
- 10.Vesela K, Hansikova H, Tesarova M, et al. Clinical, biochemical and molecular analyses of six patients with isolated cytochrome c oxidase deficiency due to mutations in the SCO2 gene. Acta Paediatr. 2004;93(10):1312–1317. doi: 10.1080/08035250410008761. [DOI] [PubMed] [Google Scholar]
- 11.Leary SC, Mattman A, Wai T, et al. A hemizygous SCO2 mutation in an early on-set rapidly progressive, fatal cardiomyopathy. Mol Genet Metab. 2006;89(1-2):129–133. doi: 10.1016/j.ymgme.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 12.Knuf M, Faber J, Huth RG, Freisinger P, Zepp F, Kampmann C. Identification of a novel compound heterozygote SCO2 mutation in cytochrome c oxidase deficient fatal infantile cardioencephalomyopathy. Acta Paediatr. 2007;96(1):130–132. doi: 10.1111/j.1651-2227.2007.00008.x. [DOI] [PubMed] [Google Scholar]
- 13.Mobley BC, Enns GM, Wong LJ, Vogel H. A novel homozygous SCO2 mutation, p.G193S, causing fatal infantile cardioencephalomyopathy. Clin Neuropathol. 2009;28(2):143–149. doi: 10.5414/npp28143. [DOI] [PubMed] [Google Scholar]
- 14.Pronicki M, Kowalski P, Piekutowska-Abramczuk D, et al. A homozygous mutation in the SCO2 gene causes a spinal muscular atrophy like presentation with stridor and respiratory insufficiency. Eur J Paediatr Neurol. 2010;14(3):253–260. doi: 10.1016/j.ejpn.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Joost K, Rodenburg R, Piirsoo A, van den Heuvel B, Zordania R, Ounap K. A novel mutation in the SCO2 gene in a neonate with early-onset cardioencephalomyopathy. Pediatr Neurol. 2010;42(3):227–230. doi: 10.1016/j.pediatrneurol.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 16.Böhm M, Pronicka E, Karczmarewicz E, et al. Retrospective, multicentric study of 180 children with cytochrome C oxidase deficiency. Pediatr Res. 2006;59(1):21–26. doi: 10.1203/01.pdr.0000190572.68191.13. [DOI] [PubMed] [Google Scholar]
- 17.Honzik T, Tesarova M, Magner M, et al. Neonatal onset of mitochondrial disorders in 129 patients: clinical and laboratory characteristics and a new approach to diagnosis. J Inherit Metab Dis. 2012;35(5):749–759. doi: 10.1007/s10545-011-9440-3. [DOI] [PubMed] [Google Scholar]