

Abstract
Myostatin (MSTN), a negative regulator of muscle growth and size, is increased after acute myocardial infarction (AMI) but timing of upregulation after injury is not known. In this study, we investigated the timing of the MSTN/AKT/p38 pathway activation in heart and skeletal muscle after AMI, as well as the potential effect of cardiac injury-related MSTN endocrine signaling on skeletal muscle and other circulating growth factors.
Methods
Coronary artery ligation was performed in C57BL/6 mice at age 8 weeks to induce AMI. Mice were sacrificed at different time points (10m, 1h, 2h, 6h, 12h, 24h, 1 week, 2 weeks, 1 months and 2 months) after surgery (n=3 per time point, n=18 total).
Results
Cardiac and circulating MSTN upregulation occurred as early as 10 minutes after AMI. Two months after AMI, increased cardiac MSTN/SMAD2,3 and p38 together with decreased IGF-1/AKT signaling suggest an anti-hypertrophic profile. In skeletal muscle, an absence of local MSTN increase was accompanied by increased MSTN-dependent SMAD2,3 signaling, suggestive of paracrine effects due to cardiac-derived MSTN. Protein degradation by the ubiquitin-proteasome system in the skeletal muscle was also evident. Serum from 24h post-MI mice effectively induced a MSTN-dependent increase in atrogin1 and MuRF1.
Conclusion
Our study shows that cardiac MTSN activation occurs rapidly after cardiac ischemia and may be involved in peripheral protein degradation in the skeletal muscle by activating atrogin1 and MuRF1.
Keywords: grow factors, myostatin, ischemia, heart failure, cardiac cachexia
Graphical abstract

1. Introduction
Myostatin (MSTN), also known as growth differentiation factor-8 (GDF8), is a signaling protein from the TGF-β superfamily whose best-known function is the inhibition of skeletal muscle growth. MSTN knockout mice, created in 1997 by McPherron et al. [1], show enlarged muscles due to both hypertrophy and hyperplasia. Since its discovery, the involvement of MSTN in cachexia, muscle atrophy, and dystrophy has been widely studied, and multiple efforts have been made to develop MSTN inhibiting drugs to treat patients with muscle disorders.
The role of MSTN in the heart was first suggested by Sharma et al. [2], who found expression of MSTN in fetal and adult hearts. Moreover, they found upregulated cardiac MSTN 12 hours after myocardial infarction. This finding has been confirmed in several later studies, which have described increased cardiac MSTN in rat models after one month of volume overload [3], and two months after myocardial infarction [4]. These findings have been corroborated in humans as well, as increased cardiac MSTN in patients with decompensated heart failure [5] and congenital heart disease [6] has been demonstrated. The significance of cardiac MSTN upregulation in heart failure is still under debate. It has been suggested that MSTN activation can be a physiological compensatory effect to curb pathological hypertrophy in heart failure, and a recent study has affirmed that genetic inactivation of MSTN in adult mice is enough to cause cardiac hypertrophy and heart failure [7]. However, different studies have suggested that MSTN blockade-mediated cardiac hypertrophy resembles to eccentric physiological hypertrophy found in athletes [8,9] making it clinically desirable in the setting of severe ventricular dysfunction.
Insulin-like growth factor 1 (IGF-1), through the stress sensor p38 MAP kinase, is the main regulator of cardiac MSTN [10]. In a feedback loop, MSTN inhibits IGF-1-dependent prohypertrophic serine/threonine kinase AKT [11], and inhibition of p38 by MSTN has also been described [11]. In this complicated regulatory network, it is likely that the balance between the cardiac pro-hypertrophic and anti-hypertrophic branches is what defines the resulting tropism and consequently functional and clinical outcome. In our study in congenital heart disease, we found that increased MSTN/IGF-1 ratio correlated with ventricular dysfunction [6].
Cardiac MSTN can also exert endocrine actions. Heineke et al. [12] affirmed that it is cardiac MSTN, and not local skeletal muscle MSTN, which mediates atrophy of the skeletal muscle in a murine model of pressure overload. However, other authors have reported increased local MSTN in the skeletal muscle of rats two months after myocardial infarction, which they postulate is circulating tumor necrosis factor (TNF)-α-dependent [13]. The regulation of the MSTN/AKT/p38 pathway in heart and skeletal muscle immediately following cardiac ischemia has not been analyzed to date. In this study, we investigated the timing of the MSTN/Akt/p38 pathway activation in heart and skeletal muscle after myocardial infarction, as well as the time course of circulating growth factors and the potential effect of MSTN endocrine signaling on skeletal muscle.
2. Materials and methods
2.1 In vivo experiment
This study was approved by the institutional Animal Care and Use Committee of Columbia University, which conforms to the “Guide for Care and Use of Laboratory Animals” published by the National Institutes of Health (NIH publication 85-23, revised 1996). Thirty-three eight-week old male C57BL/6 mice were randomly assigned to the different experimental groups. Mice underwent surgical ligation of the left anterior descending coronary artery (LAD) through a left thoracotomy incision, and were sacrificed at the following timepoints: 10 min, 1 hr, 2 hr, 6 hr, 12 hr, 24 hr, 1 week, 2 weeks, 1 month and 2 months after surgery (n=3-4/time point). Sham mice underwent thoracotomy incision, but no ligation. After the corresponding time, mice were sacrificed and heart, gastrocnemius muscle and blood were collected. Blood was allowed to clot, centrifuged and serum was collected. Tissue was stored at −80 C until analysis. Additional mice (n=4/group) were subjected to LAD ligation or sham operated and sacrificed after 24h to obtain serum for the in vitro studies.
2.2 Cell culture
To study the effect of circulating factors in mice subjected to LAD ligation, we used C2C12 mouse muscle cells (American Type Culture Collection, Manassas, VA, USA). C2C12 muscle cells were grown in high glucose Dulbeco's Modified Eagle's Medium (DMEM) supplemented with 10% non-heat inactivated fetal bovine serum (FBS, American Type Culture Collection, Manassas, VA, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin in 5% CO2 atmosphere at 37°C. Cells were used at low passage (between passages 2 and 4) for all experiments to maintain the differentiation potential of the cultures. Cells were grown in 10% FBS until they reached approximately 70% confluence, at which time the medium was replaced with DMEM containing 2% FBS for induction of differentiation into myotubes. When myotubes had formed, cytosine arabinoside (10 μM) was added to the culture medium for 48 h in order to remove any remaining dividing myoblasts. Myotubes were treated for 24 h 10% of pooled serum from mice 24h after LAD ligation, in the presence or absence of 1μM of the selective inhibitor of transforming growth factor-β type I receptor GW788388, which inhibits MSTN signaling (Tocris Bioscience, Ellisville, MI, USA) in 0.01% DMSO. Control myotubes were treated 10% serum from sham mice and 0.01% DMSO).
2.3. Preparation of protein lysates
Total cell and tissue lysates were prepared by homogenizing a portion of the tissue directly in RIPA buffer containing Protease Inhibitor Cocktail Tablets (Roche Applied Science, Indianapolis, IN, USA). Serum extracts were diluted in RIPA buffer with the protease inhibitor. Myotube extracts were briefly sonicated using a Model 100 Sonic Dismembrator (Fisher Scientific, Asheville, NC, USA). Cell, tissue and serum extracts were centrifuged at 14,000 g for 10 min at 4°C. Concentrations of lysates were determined by using the Bradford Protein Assay Kit (Fisher Scientific) with bovine serum albumin as standard.
2.4. Western blotting
Western blotting was performed to determine protein levels of circulating, cardiac and gastrocnemius MSTN, Follistatin (FST), IGF-1, activin A and GDF11; cardiac and gastrocnemius total and phosphorylated SMAD 2,3, total and phosphorylated AKT, total and phosphorylated p38 and gastrocnemius atrogin1 and MuRF1. Samples (50 μg) were denatured at 95°C for 10 minutes and loaded in 12% Tris-glycine polyacrylamide gels and SDS-PAGE was performed under reducing conditions. Electrophoresis (1h at 100V) was performed in a Miniprotean II cell (Bio-Rad Ltd, Hercules, CA, USA) followed by wet transfer of proteins (4°C for 1 h at 100 V) onto PVDF membranes in a mini trans-blot transfer cell (Bio-Rad Ltd) filled with transfer buffer (20% methanol). Blots were blocked for 1 hour in 5% non-fat milk diluted in Tris-buffered saline with 0.1% Tween-20 (TBST). For phosphorylated proteins, 5% BSA was used for blocking instead of milk. Blots were then incubated overnight at 4°C with primary antibody diluted 1:1000 in 5% milk/BSA TBST. The following primary antibodies and the HPR-linked appropriate secondary antibodies were used: a rabbit polyclonal anti-MSTN antibody (Abcam, Cambridge, MA, USA), a rabbit polyclonal anti-SMAD 2,3 antibody (Abcam), a rabbit polyclonal anti-P-SMAD2,3 antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), a rabbit polyclonal ani-IGF-1 (Abcam), a rabbit polyclonal anti-AKT (Cell Signaling Technology, Danvers, MA, USA), a rabbit polyclonal anti-P-AKT (Cell Signaling Technology), a rabbit polyclonal anti-p38 (Cell Signaling Technology), a rabbit polyclonal anti-P-p38 (Cell Signaling Technology), a rabbit polyclonal anti-atrogin-1 (Abcam), a rabbit polyclonal anti-MuRF1 (Abcam), a rabbit polyclonal anti-FST (Thermo Scientific, Waltham, MA, USA), and a rabbit polyclonal anti-GDF11 antibody (Abcam). A mouse monoclonal anti-GAPDH antibody (Santa Cruz) was used as a loading control. After washing in TBST, blots were incubated in the presence of a peroxidase-labeled secondary antibody diluted 1:5000 for 1 h at room temperature. Blots were washed again and incubated with HyGLO™ Quick Spray Chemiluminescent HRP Antibody Detection Reagent (Denville Scientific Inc., Denville, NJ, USA) for 1 min, followed by autoradiography. Immunoreactive bands were analyzed using the public domain Image J program (NIH, Frederick, MD, USA). The bands were quantified by densitometry and normalized to the appropriate loading controls.
2.5. ELISA
Serum concentrations of MSTN were determined by ELISA using a commercial kit from R&D Systems (Minneapolis, MN, USA), following manufacturer's directions.
2.6. Statistics
Results are reported as means ± SEM. Statistical analysis was performed by ANOVA followed by Tukey's post hoc test as appropriate. A p value less than 0.05 was considered statistically significant.
3. Results
3.1. Cardiac MSTN is upregulated rapidly after AMI
Cardiac protein changes after myocardial infarction (MI) are shown in Figure 1. Western blot analysis revealed significantly increased MSTN as early as 10 minutes after AMI (Figure 1A). The biggest increase occurred between 12 and 24 hours after MI, and MSTN levels continued to increase but plateaued by 1 week and slightly decreased at the 2-month time point. Cardiac active SMAD 2,3, which is the main MSTN signaling effector, was significantly increased at 2 hours post-MI and continued to increase even up to 2 months (Figure 1B), suggesting sustained MSTN cardiac signaling. Neither cardiac activin A, a similar protein to MSTN with comparable ant-hypertrophic action, nor MSTN natural inhibitor FST appeared significantly upregulated in the myocardium at any of the time points studied (Figure 1C). GDF11, another member the TGF-β superfamily closely related to MSTN, was not detected in the myocardium. Cardiac IGF-1 followed a trend very similar to MSTN, with an initial increase at 10 minutes and sustained activation after one week (Figure 1D). Cardiac active AKT was significantly increased 24 h after surgery. From two weeks after AMI onwards, however, activation of AKT decreased in the myocardium, to levels comparable to sham mice after 2 months, presumably due to inhibitory effects of MSTN. MAP kinase p38 activation was apparent 10 min post-AMI, and was consistently increased from 1 week after AMI onwards.. These results suggest a parallel activation of a pro-hypertrophic AKT-dependent response together with pro-atrophic MSTN/SMAD2,3 signaling in the myocardium following ischemia.
Figure 1.
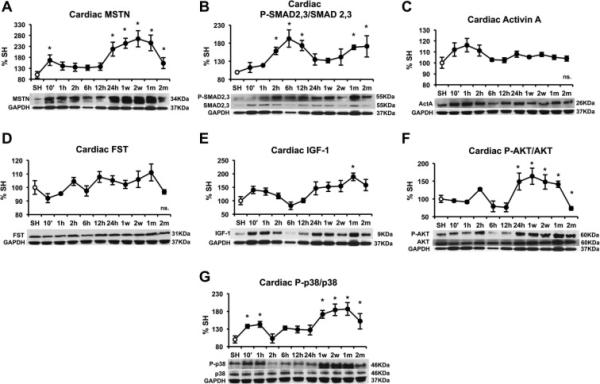
Time course of cardiac protein levels after myocardial infarction induced by left anterior descending coronary artery (LAD) ligation in male C57/B6 mice. (A) MSTN, (B) PSMAD2,3/SMAD2,3 (C) Activin A, (D) Follistatin (FST), (E) IGF-1, (F) P-AKT/AKT and (G) P-p38/p38. Protein levels were measured by western blot, normalized against GAPDH, and expressed as percentage of the Sham (SH) mice. Images show representative blots. Data represent mean± standard error. (n=3). *P<0.05 vs sham mice by one-way ANOVA.
3.2. Increased MSTN signaling and atrophy markers in the skeletal muscle after AMI are not mediated by local MSTN
In the gastrocnemius muscle MSTN remained unaltered after AMI (Figure 2A), while SMAD2,3 activation occurred 10 min post AMI and remained activated after 24 hours (Figure 2B). This increase in SMAD signaling did not appear to be mediated by local signaling factors, as local activin A and FST, like MSTN, remained unchanged (Figure 2C and 2D); this data suggests that SMAD2,3 activation may be exerted by circulating factors. Pro-hypertrophic signaling by IGF-1 was not modified in the gastrocnemius muscle (Figure 2E), while AKT signaling was initially increased after 24 hours but significantly decreased after 2 months (Figure 2F). p38 activation occurred after 1 week and remained increased at all the time points studied (Figure 2G). The E3 ubiquitin-ligases remained upregulated after 24 hours in the case of atrogin-1 and after 12 hours post-ischemia in the case of MuRF1, indicating the activation of a protein degradation program by the ubiquitin-proteasome system. These data suggest the activation of an atrophic program in the skeletal muscle, which could be mediated by SMAD2,3 signaling but without the involvement of local skeletal muscle-derived MSTN.
Figure 2.
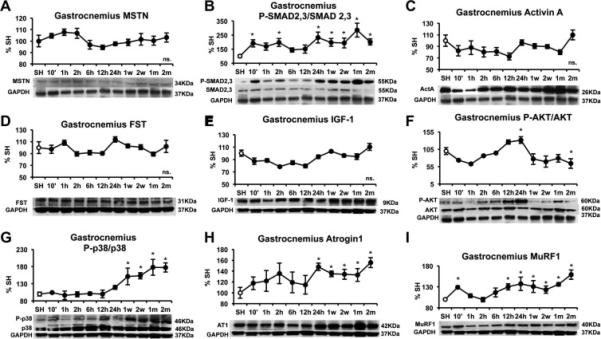
Time course of gastrocnemius protein levels after myocardial infarction induced by left anterior descending coronary artery (LAD) ligation in male C57/B6 mice. (A) MSTN, (B) PSMAD2,3/SMAD2,3 (C) Activin A, (D) Follistatin (FST), (E) IGF-1, (F) P-AKT/AKT, (G) P-p38/p38, (H) Atrogin-1 and (I) MuRF-1. Protein levels were measured by western blot, normalized against GAPDH, and expressed as percentage of the Sham (SH) mice. Images show representative blots. Data represent mean± standard error. (n=3). *P<0.05 vs sham mice by one-way ANOVA.
3.3 Circulating MSTN is consistently increased after AMI
The analysis of circulating factors after AMI revealed increased circulating MSTN 10 minutes after AMI as seen by ELISA (Figure 3A) and western blot (Figure 3B). The increase in serum activin A (Figure 3C) and GDF11 (Figure 3D) was not significant at any time point, while FST was significantly increased after 24 hours (Figure 3E). Circulating IGF-1 was not significantly increased at any time point. We postulated that increased circulating MSTN together with unchanged IGF-1 could result in potential peripheral anti-hypertrophic signaling
Figure 3.
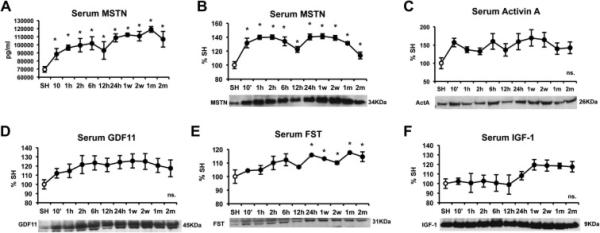
Time course of circulating protein levels after myocardial infarction induced by left anterior descending coronary artery (LAD) ligation in male C57/B6 mice. (A) MSTN levels as measured by ELISA, (B) MSTN levels as measured by western blot, (C) Activin A, (D) GDF11, (E) Follistatin (FST), and (F) IGF-1. Protein levels were measured by western blot, normalized total protein volume, and expressed as percentage of the Sham (SH) mice. Images show representative blots. Data represent mean± standard error. (n=3). *P<0.05 vs sham mice by one-way ANOVA.
3.4 Circulating factors after AMI can cause a SMAD 2,3-dependent increase in atrogin-1 and MuRF-1
Finally, we studied the response of serum from 24h post AMI mice on C2C12 murine myotubes in order to validate the hypothesis that circulating MSTN-related signaling proteins act in a paracrine fashion. At this time point increased circulating MSTN was accompanied by significantly high gastrocnemius atrogin1 and MuRF1 in vivo. By co-treatment with the MSTN signaling inhibitor GW788388 (Myo-I), we were able to discern whether the effects of the serum on myotubes were dependent on signaling by MSTN or other related factors. Treatment of cultured C2C12 cells with 5% of 24h post-AMI serum for 24h resulted in unchanged MSTN protein levels (Figure 4A) but increased in SMAD2,3 activation (Figure 4B), which was blunted by Myo-I, indicating that SMAD2,3 upregulation by 24h post-MI serum is specifically exerted by MSTN or some other member of the TGF-β superfamily. Surprisingly, 24h post-MI serum induced an increase in AKT activation (Figure 4C), which was also attenuated by Myo-I. P38 activation was still significant in cells treated by Myo-I, indicating that p38 activation in the muscle is only partially mediated by MSTN family (Figure 4D). 24h post–AMI serum significantly increased atrogin1 (Figure 4E) and MuRF1 (Figure 4F), and this effect was prevented by Myo-I in the case of atrogin1 and significantly attenuated in the case of MuRF-1. Taken together, these results suggest that increased circulating MSTN after AMI is involved in the activation of protein degradation by the ubiquitin proteasome system in the skeletal muscle.
Figure 4.
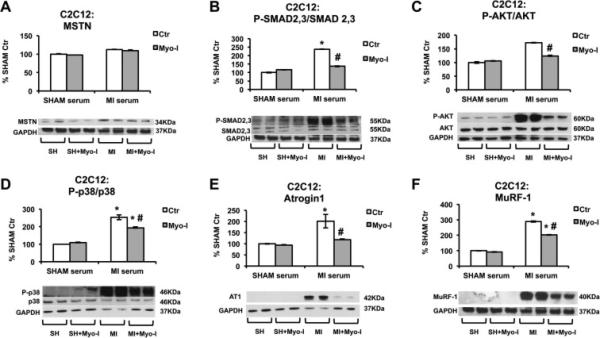
Effect of circulating serum from 24h post AMI mice on C2C12 skeletal muscle cells. (A) MSTN, (B) P-SMAD2,3/SMAD, (C) P-AKT/AKT, (D) P-p38/p38 2,3, (E) atrogin-1, and (F) MuRF1, protein levels in myotubes treated for 24h hours with 5% SHAM (SH) or 24h post AMI (24h) serum in the presence or absence of 1 μM Myo-I (GW788388). Proteins were measured by western blot, normalized against GAPDH, and expressed as percentage of the Control (Ctrl) + Sham (SH) serum myotubes. Images show representative blots. Data represent mean± standard error. (n=4). *P<0.05 vs. corresponding Ctr by two-way ANOVA.
4. Discussion
The present results show that cardiac MTSN activation occurs rapidly after ischemia and may also be involved in protein degradation in the skeletal muscle. The major findings of our study were as follows: (1) MSTN cardiac protein levels are activated rapidly after ischemia, (2) Activation of SMAD2,3 pathway in the skeletal muscle mediate activation of an atrophy program by the ubiquitin proteasome system, but this is not mediated by local MSTN. The in vitro data provide supportive evidence that circulating MSTN, likely from cardiac origin, mediates skeletal muscle effects.
The early upregulation of MSTN after ischemia has important implications for management of cardiac remodeling. In the myocardium, IGF-1 dependent pro-hypertrophic branch AKT, as well as the stress sensor p38 are activated after AMI We hypothesize that MSTN acts as a compensatory factor to prevent excessive cardiac hypertrophy after ischemia. After 2 months, however, the balance between hypertrophic IGF-1/AKT and pro-atrophic p38/MSTN/SMAD is shifted towards atrophy. This imbalance could mediate the cardiac wall thinning and dilation that occurs in end-stage heart failure. Echocardiogram analysis from our lab shows heart failure with decreased fractional shortening as soon as two weeks after LAD ligation, and further worsened function after 2 months (see supplementary data).
Our study supports early studies indicating that cardiac MSTN may mediate skeletal muscle atrophy [12] in heart failure. To our knowledge, this is the first study that reports a role of cardiac MSTN on skeletal muscle immediately after ischemia and the first study showing increased skeletal muscle atrogin1 and MurF1 by circulating endogenous MSTN. While local skeletal muscle MSTN remained unchanged after MI, increased SMAD 2,3 suggested a role of circulating MSTN on the muscle, which was confirmed by our studies on C2C12 myotubes. Increased local skeletal muscle MSTN has been reported in several models of cachexia, like certain types of cancer [14], AIDS [15] or COPD [16]. However, muscle MSTN remained unchanged or was decreased in other physiological situations accompanied by muscle atrophy such as sepsis [17], arthritis [18] or heart failure [19]. Increased circulating MSTN in heart failure is a well-established fact, and the study by Heineke et al. [12], together with our results show the importance of cardiac factors on skeletal muscle tropism.
McFarlane et al. [20] described the activation of the ubiquitin proteasome protein degradation by recombinant MSTN. In contrast to our study, they described decreased phosphorylation of AKT after MSTN treatment. Decreased AKT activation by MSTN has been previously demonstrated, so the increased AKT phosphorylation in response to 24h post-MI serum is likely a consequence of a different protein of the TGF-β superfamily. Indeed, TGF-β is capable of inducing AKT activation [21]. Other members of the TGF-β superfamily signal through SMAD2,3 and have pro-atrophic (or anti-hypertrophic) actions, and may partially mediate the effects that we report on myotubes. Activin A has redundant actions with MSTN and has reported to be stronger than MSTN promoting skeletal muscle atrophy [22]. GDF11 has also been recently identified as an important circulating factor mediating cardiac remodeling [23] and skeletal muscle atrophy [24]. The very similar structure of MSTN and GDF11 makes it difficult to differentiate the actions of both and it is possible that the antibodies used in this study offer some level of GDF11/GDF8 cross-reaction. The initial activation of MSTN after myocardial ischemia that we report may be exerted in different manners. Myocyte stretch and angiotensin II can activate MSTN via IGF-1/p38 [10,25], and our data show that MSTN increase is accompanied by p38 activation. TNF-α, which is activated upon ischemia, can also be triggering MSTN activation.
One limitation of our study that must be noted is that the nature of LAD ligation results in certain variability in the size of ischemia, so the specific dimension of protein upregulation/downregulation in each time point must be taken cautiously. Second, although variability among data within timepoints is low and overall trends of protein levels appear consistent, the small sample size may make interpreting individual time points difficult.
Our observations are important from a clinical standpoint regarding cardiac remodeling and cachexia management, as they support the hypothesis that inhibition of circulating MSTN may be an interesting tool to prevent muscle atrophy in patients with heart failure. The prevalence of cardiac cachexia has been estimated to be between 8-42% depending on the diagnosis criteria [26] and negatively affects the outcome of the disease. The early activation of MSTN, and the shown fact that as early as 24 hours following myocardial infarction circulating factors from the MSTN family can activate protein degradation pathways in the skeletal muscle, suggest that a treatment that blocks circulating MSTN should be administered early after myocardial ischemia to prevent muscle wasting. What may be the best for the skeletal muscle may be, however, counterproductive for the myocardium, as MSTN after 24h can act as a compensatory factor for increased pro-hypertrophic AKT signaling. While it has been previously reported that complete inactivation of MSTN is enough to cause hypertrophy and heart failure [7], it is possible that a timely blockade of MSTN after myocardial infarction, at a time when prohypertrophic signaling by AKT has remitted, could be beneficial to prevent cardiac dysfunction and wall thinning. Potential pharmacological MSTN blockade must take these different factors into account and more studies are needed to study the potential of MSTN inhibition in cardiac remodeling and muscle wasting in cardiac cachexia.
Supplementary Material
Highlights.
Cardiac MSTN activation occurs rapidly after cardiac ischemia
Skeletal muscle MSTN remains unchanged after cardiac ischemia
Skeletal muscle SMAD2,3, atrogin-1 and MuRF1 are increased after cardiac ischemia
Circulating MSTN is consistently increased after cardiac ischemia
Increased serum MSTN after ischemia can activate atrophy in the skeletal muscle
Acknowledgements
Source of Funding: National Center for Advancing Translational Sciences, NIH (UL1 TR000040), formerly the National Center for Research Resources (RR024156) (I.G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McPherron AC, Lawler AM AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 2.Sharma M, Kambadur R, Matthews KG, et al. Myostatin, a transforming growth factor-beta superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J. Cell. Physiol. 1999;180:1–9. doi: 10.1002/(SICI)1097-4652(199907)180:1<1::AID-JCP1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 3.Shyu KG, Lu MJ, Wang BW, et al. Myostatin expression in ventricular myocardium in a rat model of volume-overload heart failure. Eur. J. Clin. Invest. 2006;36:713–719. doi: 10.1111/j.1365-2362.2006.01718.x. [DOI] [PubMed] [Google Scholar]
- 4.Lenk K, Schur R, Linke A, et al. Impact of exercise training on myostatin expression in the myocardium and skeletal muscle in a chronic heart failure model. Eur. J. Heart Fail. 2009;11:342–348. doi: 10.1093/eurjhf/hfp020. [DOI] [PubMed] [Google Scholar]
- 5.George I, Bish LT, Kamalakkannan G, et al. Myostatin activation in patients with advanced heart failure and after mechanical unloading. Eur. J. Heart Fail. 2010;12:444–453. doi: 10.1093/eurjhf/hfq039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bish LT, George I, Maybaum S, et al. Myostatin is elevated in congenital heart disease and after mechanical unloading. PLoS One. 2011;6:e23818. doi: 10.1371/journal.pone.0023818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biesemann N, Mendler L, Wietelmann A, et al. Myostatin regulates energy homeostasis in the heart and prevents heart failure. Circ. Res. 2014;115:296–310. doi: 10.1161/CIRCRESAHA.115.304185. [DOI] [PubMed] [Google Scholar]
- 8.Jackson MF, Luong D, Vang DD, et al. The aging myostatin null phenotype: reduced adiposity, cardiac hypertrophy, enhanced cardiac stress response, and sexual dimorphism. J. Endocrinol. 2012;213:263–275. doi: 10.1530/JOE-11-0455. [DOI] [PubMed] [Google Scholar]
- 9.Rodgers BD, Interlichia JP, Garikipati DK, et al. Myostatin represses physiological hypertrophy of the heart and excitation-contraction coupling. J. Physiol. 2009;587:4873–4886. doi: 10.1113/jphysiol.2009.172544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shyu KG, Ko WH, Yang WS, et al. Insulin-like growth factor-1 mediates stretch-induced upregulation of myostatin expression in neonatal rat cardiomyocytes. Cardiovasc. Res. 2005;68:405–414. doi: 10.1016/j.cardiores.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 11.Morissette MR, Cook SA, Foo S, et al. Myostatin regulates cardiomyocyte growth through modulation of Akt signaling. Circ. Res. 2006;99:15–24. doi: 10.1161/01.RES.0000231290.45676.d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heineke J, Auger-Messier M, Xu J, et al. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation. 2010;121:419–425. doi: 10.1161/CIRCULATIONAHA.109.882068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lenk K, Schur R, Linke A A, et al. Impact of exercise training on myostatin expression in the myocardium and skeletal muscle in a chronic heart failure model. Eur. J. Heart Fail. 2009;11:342–348. doi: 10.1093/eurjhf/hfp020. [DOI] [PubMed] [Google Scholar]
- 14.Aversa Z, Bonetto A, Penna F, et al. Changes in myostatin signaling in non-weight-losing cancer patients. Ann. Surg. Oncol. 2012;19:1350–1356. doi: 10.1245/s10434-011-1720-5. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, et al. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc. Natl. Acad. Sci. U. S. A. 1998;95:14938–14943. doi: 10.1073/pnas.95.25.14938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plant PJ, Brooks D, Faughnan M, et al. Cellular markers of muscle atrophy in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2010;42:461–471. doi: 10.1165/rcmb.2008-0382OC. [DOI] [PubMed] [Google Scholar]
- 17.J Smith I, Aversa Z, Alamdari N, et al. Sepsis downregulates myostatin mRNA levels without altering myostatin protein levels in skeletal muscle. J. Cell. Biochem. 2010;111:1059–1073. doi: 10.1002/jcb.22796. [DOI] [PubMed] [Google Scholar]
- 18.Castillero E, Martín AI, López-Menduiña M, et al. IGF-I system, atrogenes and myogenic regulatory factors in arthritis induced muscle wasting. Mol. Cell. Endocrinol. 2009;309:8–16. doi: 10.1016/j.mce.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 19.Lima AR, Martinez PF, Okoshi K, et al. Myostatin and follistatin expression in skeletal muscles of rats with chronic heart failure. Int. J. Exp. Pathol. 2010;91:54–62. doi: 10.1111/j.1365-2613.2009.00683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McFarlane C, Plummer E, Thomas M, et al. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J. Cell. Physiol. 2006;209:501–514. doi: 10.1002/jcp.20757. [DOI] [PubMed] [Google Scholar]
- 21.Kato M, Putta S, Wang M, et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat. Cell. Biol. 2009;11:881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen JL, Walton KL, Winbanks CE, et al. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 2014;28:1711–1723. doi: 10.1096/fj.13-245894. [DOI] [PubMed] [Google Scholar]
- 23.Loffredo FS, Steinhauser ML, Jay SM, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153:828–839. doi: 10.1016/j.cell.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Souza TA, Chen X, Guo Y, et al. Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol. Endocrinol. 2008;22:2689–2702. doi: 10.1210/me.2008-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang BW, Chang H, Kuan P, et al. Angiotensin II activates myostatin expression in cultured rat neonatal cardiomyocytes via p38 MAP kinase and myocyte enhance factor 2 pathway. J. Endocrinol. 2008;197:85–93. doi: 10.1677/JOE-07-0596. [DOI] [PubMed] [Google Scholar]
- 26.Anker SD, Ponikowski P, Varney S, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. 1997;349:1050–1053. doi: 10.1016/S0140-6736(96)07015-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.