

Abstract
Isolated familial pseudohyperkalemia is a dominant red cell trait characterized by cold-induced ‘passive leak’ of red cell potassium ions into plasma. The causative gene of this condition is ABCB6, which encodes an erythrocyte membrane ABC transporter protein bearing the Langereis blood group antigen system. In this study analyzing three new families, we report the first functional characterization of ABCB6 mutants, including the homozygous mutation V454A, heterozygous mutation R276W, and compound heterozygous mutations R276W and R723Q (in trans). All these mutations are annotated in public databases, suggesting that familial pseudohyperkalemia could be common in the general population. Indeed, we identified variant R276W in one of 327 random blood donors (0.3%). Four weeks’ storage of heterozygous R276W blood cells resulted in massive loss of potassium compared to that from healthy control red blood cells. Moreover, measurement of cation flux demonstrated greater loss of potassium or rubidium ions from HEK-293 cells expressing ABCB6 mutants than from cells expressing wild-type ABCB6. The R276W/R723Q mutations elicited greater cellular potassium ion efflux than did the other mutants tested. In conclusion, ABCB6 missense mutations in red blood cells from subjects with familial pseudohyperkalemia show elevated potassium ion efflux. The prevalence of such individuals in the blood donor population is moderate. The fact that storage of blood from these subjects leads to significantly increased levels of potassium in the plasma could have serious clinical implications for neonates and infants receiving large-volume transfusions of whole blood. Genetic tests for familial pseudohyperkalemia could be added to blood donor pre-screening. Further study of ABCB6 function and trafficking could be informative for the study of other pathologies of red blood cell hydration.
Introduction
Isolated familial pseudohyperkalemia (FP) is a dominant red cell trait characterized by an increase of plasma potassium ion concentration upon exposure of whole blood to temperatures below 37°C. Red blood cells from individuals with FP have an increased mean corpuscular volume and shape abnormalities. Cation leak in FP families described to date has shown several patterns of temperature-dependence.1–4
The causative gene in FP was previously mapped to 2q35–36.5 In three multigenerational FP families functional gene mapping and sequencing analysis of the candidate genes within the 2q35–q36 critical interval identified two novel heterozygous missense mutations in the ABCB6 gene which co-segregated with disease phenotype.6 The two genomic substitutions altered two adjacent nucleotides within codon 375 of ABCB6, a previously identified porphyrin transporter7 that in erythrocyte membranes8 bears the Langereis (Lan) blood group antigen system.9 We previously showed in three separate models of erythropoiesis that ABCB6 expression increased during erythroid differentiation and localized to the plasma membrane.6 The ABCB6 R375Q mutation did not alter levels of mRNA or protein, or subcellular localization in mature erythrocytes or erythroid precursor cells, but was predicted to have pathogenic consequences for protein function.
Recently, the ABCB6 substitution R723Q was found in healthy subjects from a family affected by FP, two of whom had been evaluated as regular blood donors.10 The blood of both exhibited increased potassium leak upon storage at temperatures below 37°C. This interesting finding encouraged further study on the implications for neonates and infants of transfusion of whole blood from unknown FP subjects.
In this report, we describe three novel missense mutations in ABCB6 identified in FP families with a non-dominant pattern of inheritance of the condition. The presence of these mutations in human variation databases confirms that the prevalence of asymptomatic FP is likely underestimated and, moreover, frequently undetected in blood donor populations. We also report the results of ABCB6 screening in a blood donor population, and present the first functional study of the effects of ABCB6 FP mutations on that component of red cell potassium cation (K+) efflux characterized by resistance to ouabain plus bumetanide.
Methods
Patients
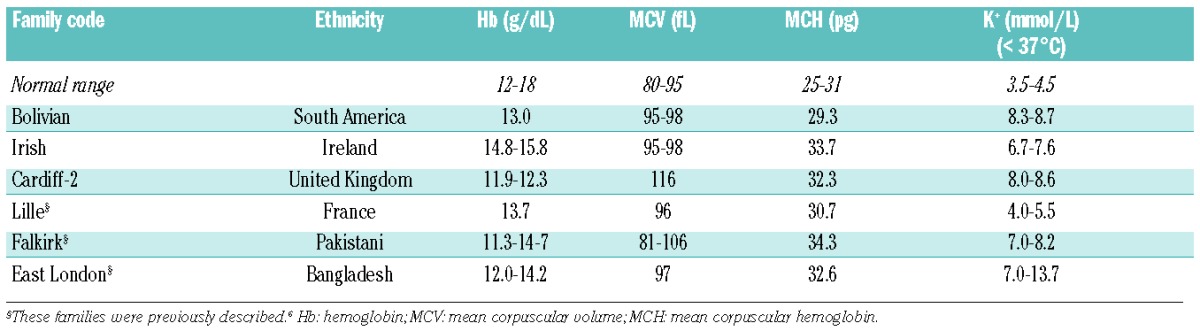
Three new patients from three independent pedigrees were enrolled in this study (Table 1), and blood samples were obtained from them. Whenever possible, relatives were also investigated. The diagnosis of FP was based on the patients’ history, clinical findings, routine laboratory data, peripheral blood smear, and genetic testing. Information about every clinical characteristic was not available for all cases. A cohort of 327 blood donors from the blood transfusion center of the Cardarelli Hospital in Naples was enrolled to undergo genetic screening for the ABCB6 mutations found in this study.
Table 1.
Clinical data of patients with familial pseudohyperkalemia.
Patients’ data and samples were collected by the clinicians responsible for the patients’ care, with informed consent according to the Declaration of Helsinki, and with approval by local university ethical committees.
Exome capture and sequencing
Blood was obtained for genetic analysis from affected and unaffected family members of the Irish family and from healthy controls, with signed informed consent according to the Declaration of Helsinki. Reads were aligned to the most recent version of the human genome (GRCh37/hg19) using the BWA software package v0.5.9 as previously described11 (see the Online Supplementary Material). Direct sequencing analysis of the additional families was performed (see the Online Supplementary Material).
ABCB6 screening in blood donors
Amplification-refractory mutation system (ARMS) analysis, using allele-specific tetra-primer ARMS-polymerase chain reaction primers designed by PRIMER1 (http://primer1.soton.ac.uk/primer1.html), was applied to screen for the ABCB6 variants R276W, V454A, and R723Q in a population of 327 blood donors.12
Bioinformatic modeling of ABCB6 protein structure
To assess the potential effects of the identified mutations on protein structure, we generated 3D structural models of dimeric human wild-type (WT) ABCB6 residues 231–827 and the corresponding regions of FP mutant ABCB6 polypeptides V454A, R276W, and R723Q, as described in the legends to Figure 1, and Online Supplementary Figures S1 and S2, and as previously described.6 Sequences were aligned in ClustalW2. MODELLER v9.913 was used for homology modeling in both inward- and outward-facing conformations. The best five structural models with lowest objective function values (as implemented in MODELLER) were subjected to energy minimization in GROMACSv4.5.4.14 Structural models were converged using steepest descent energy minimization with 1,000 steps of step size 0.01 nm. The stereochemical quality of each energy-minimized structure was assessed by PROCHECK.15 The average of three models of highest stereochemical quality was chosen for the ABCB6 structural models. Three dimensional structural models were visualized and aligned using MolMol16 and PyMOL 1.5.0.4 (Schrödinger, LLC).
Figure 1.
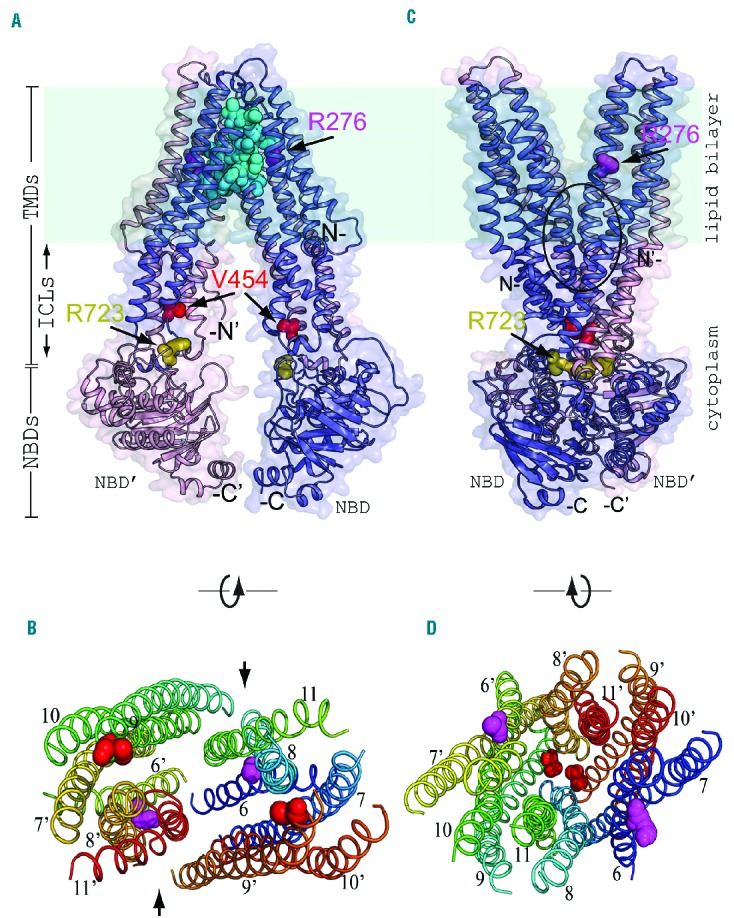
Three-dimensional structural model of human ABCB6 mutations. (A) Three-dimensional structural model of a portion of homodimeric WT human ABCB6 in an inward-facing conformation, as modeled on the aligned structure of M. musculus ABCB1A (PDB ID 3G5U). Monomer “a” (blue) of the homodimer represents ABCB6 amino acid (aa) residues 246 (N-) to 826 (-C), modeled on transmembrane helices 1–6 and NBD-1 of ABCB1A. Monomer “b” (pink) of the homodimer represents ABCB6 aa residues 237 (N’-) to 826 (-C’) modeled on ABCB1A transmembrane helices 7–12 and its NBD-2. A surface model is superposed on the modeled polypeptide backbone ribbon structure. The dehydrated hereditary stomatocytosis (DHSt) homozygous mutation site V454 (red spheres) is located between the membrane-spanning helices and the NBD, extending into the cytoplasmic vestibule of the dimer. Locations of the compound heterozygous DHSt mutation sites R276 (magenta spheres)/R723 (olive spheres) are also shown. Arrows mark R276 of monomer A (located within the lipid bilayer) and R723 of monomer B (located within the NBD region in the cytoplasmic vestibule) of the dimer. The cavity (cyan spheres) at the intermonomeric interface outlines a postulated intra-membrane binding site for inhibitors of ABCB6-mediated porphyrin transport,32 corresponding to the ABCB1 binding site of inhibitor QZ59.33 In this and subsequent figures, each modeled ABCB6 monomer lacks its ectofacial N-terminal tail and putative transmembrane spans 1–5, but includes putative transmembrane spans 6–11 followed by the single NBD. (B) Transverse intra-membrane profile of the modeled inward-facing conformation of dimeric WT ABCB6 (as in panel A), with transmembrane helices rotated 90° around the axis shown. The view (lacking NBD) looks outward from the ICL region, near the site of separated mutation site V454 (red) and further from mutation site R276. The colored M1 domain helices are numbered 6–11 for ABCB6 monomer “a”, and 6′–11′ for monomer “b” of the ABCB6 dimer. The arrows between helices 9 and 11 on one side, and helices 9′ and 11′ on the other side of the dimer mark the locations of side apertures proposed in mouse ABCB1 to mediate hydrophobic drug uptake from the inner leaflet of the lipid bilayer for subsequent efflux from the cell, or for flippase-like transfer to the outer leaflet.33 (C) Three-dimensional structural model of homodimeric WT human ABCB6 in an outward-facing conformation, as modeled on the aligned structure of S. aureus Sav1866 (PDB ID 2HYD). The black oval encloses a central cavity at the inter-monomeric interface, hypothesized to be an intra-membrane substrate binding site (as predicted for homodimeric Sav1866 of S. aureus34). Sites of homozygous and compound heterozygous mutations are shown using similar colored spheres as in panel A. (D) Transverse intramembranous profile of the modeled outward-facing conformation of dimeric ABCB6 (as in panel C), with the transmembrane helices rotated 90° around the axis shown. The view (lacking NBD) looks inward from the extracellular edge of the outer leaflet of the membrane bilayer towards the approximated mutation sites V454 (at the level of the ICL region) and R276 (further from the ICL region); color scheme as in panel C. Helices are labeled at ends closest to reader. The figure was prepared in PyMOL. NBD: nucleotide-binding domain; TMD: transmembrane domain; ICL: intracytoplasmic loop.
Cell culture and transfection assay
Human HEK-293 cells were maintained in DMEM (Sigma) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin (all from Life Technologies), in a humidified 5% CO2 atmosphere at 37°C.
pcDNA3.1-ABCB6-WT and pcDNA3.1-ABCB6 mutant constructs (5 μg) were transfected into HEK-293 cells using XtremeGENE HP DNA Transfection Reagent (Roche, Indianapolis, IN, USA). To phenocopy the heterozygous genotypes, cells were transfected with 2.5 μg of pcDNA3.1-ABCB6-WT plus 2.5 μg of pcDNA3.1-ABCB6 mutants R375Q, p.R276W or R375W. For the compound heterozygous genotypes, cells were transfected with 2.5 μg pcDNA3.1-ABCB6-R276W plus 2.5 μg pcDNA3.1-ABCB6-R723Q. For the homozygous genotype, cells were transfected with 5 μg pcDNA3.1-ABCB6-V454A. After 72 h, cells were harvested for analysis.
Immunofluorescence analysis
HEK-293 cells (2×106) on coverslips were transfected with ABCB6 cDNA as previously described6 (see the Online Supplementary Methods).
Measurements of potassium ion fluxes in red blood cells from the blood donor carrying the ABCB6 R276W variant
Blood samples from the donor carrying the ABCB6 R276W mutation and from two controls obtained from the transfusion center of the Cardarelli Hospital (Naples) were stored for 4 weeks at 4°C in citrate-phosphate-dextrose solution as anticoagulant, under blood bank conditions. During the 4 weeks of storage, plasma potassium levels were measured in triplicate by atomic absorption spectroscopy (ANALYST 2000, Perkin-Elmer) as previously described.17 The red blood cells were washed gently with a buffer containing 150 mM choline chloride, 1 mM MgCl2, and 10 mM Tris MOPS, then lysed to measure intracellular potassium by atomic absorption spectroscopy.17 The free hemoglobin levels were measured to evaluate the degree of hemolysis as for potassium.
Measurements of ouabain-plus-bumetanide-resistant rubidium and potassium ion fluxes in transfected HEK-293 cells
At 72 h after transfection, HEK-293 cells were maintained for 8 h under shear stress (rotary shaking at 0.12g at 30°C) in a K+-free medium containing 140 mM NaCl, 5 mM RbCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, to which 10 μM ouabain and 10 μM bumetanide were added. At the end of the incubation, cell viability was determined by trypan blue staining and medium was removed for determination of the extracellular K+ concentration. Cells were washed gently in buffer containing 150 mM choline chloride, 1 mM MgCl2, and 10 mM Tris MOPS, then lysed in order to determined intracellular Rb concentration. Intracellular Rb content and extracellular K+ concentration were measured in triplicate by atomic absorption spectroscopy (ANALYST 2000, Perkin-Elmer) as previously described.17
Results
Case reports
The Bolivian patient is a 41-year old female from a consanguineous family. Multiple outpatient blood samples indicated elevated plasma K+ (8.3 mmol/L). The patient’s renal and adrenal function and electrocardiogram were normal, as were hematologic indices except for macrocytosis (mean corpuscular volume 95-98 fL; Table 1). The patient’s past medical history included premature menopause and migraines. Physical examination was unremarkable.
Patient Cardiff-2 is a 35-year old female with a prior tentative diagnosis of hereditary spherocytosis with a positive family history, despite a normal red cell eosin maleimide binding test. Outpatient values of plasma K+ concentration ranged between 8.0 and 8.6 mmol/L, in contrast to hospital clinical values which were between 5.4 and 5.9 mmol/L (Table 1). The patient had been under outpatient care for diabetes and other conditions since 1999. Her peripheral blood smear revealed polychromasia, target cells, and a few spherocytes. Her past medical history included irritable bowel syndrome, oophorectomy in 2007, splenectomy secondary to trauma in 2008, and a diagnosis of depression in 2009. Medications included omeprazole, mebevirine and citalopram. The patient’s blood pressure was normal, as was the remainder of the physical examination. Her absolute reticulocyte count was 176.0 ×109 (4.10 %) with a mean reticulocyte volume of 118 fL (reference range, 90–110 fL).
The large Irish family was originally described by Stewart and colleagues18 as having autosomal dominant dehydrated hereditary stomatocytosis with FP. The propositus had several thrombotic episodes following splenectomy at the age of 40 years. An increased passive K+ leak was noted (Table 1).
The families Lille, Falkirk and East London have already been described.6 Affected individuals from the FP Lille family (of Flemish descent) had normal hematologic indices except for a slightly elevated mean corpuscular volume. Affected individuals from the FP Falkirk family (of Pakistani origin) also had macrocytosis. Affected individuals from the FP East London family (of Bangladeshi origin) were anemic and had hyperkalemia, but in the absence of reticulocytosis and jaundice were considered non-hemolytic.
None of the carriers had colobomatous abnormalities of iris or retina, also associated with missense ABCB6 mutations. Carriers were not tested for Lan−/− status, as this phenotype is caused by nonsense mutations that cause complete absence of ABCB6 polypeptide in circulating red cells.
ABCB6 mutational analysis in families with familial pseudohyperkalemia and blood donor screening
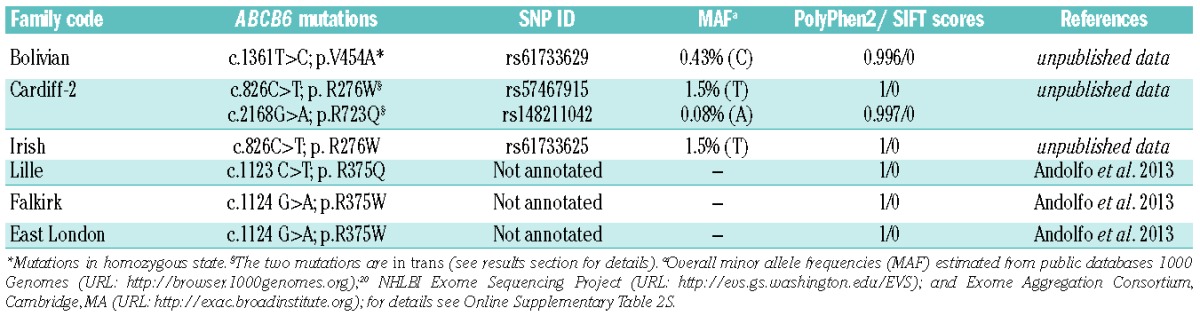
We sequenced the ABCB6 gene in two patients and one family with FP. In the Bolivian patient we found the homozygous mutation c.1361T>C; p.V454A, while in patient Cardiff-2 we found compound heterozygosity for the two mutations c.826G>T; p.R276W and c.2168G>A; p.R723Q (Table 2). The parents of both patients were unavailable for genetic analysis. To analyze the allelic pattern of the two mutations in patient Cardiff-2, we cloned the genomic region encompassing both ABCB6 variants (about 6 Kb) into a plasmid vector. DNA sequencing of this cloned region demonstrated that the two mutations are in trans in patient Cardiff-2. In the Bolivian patient, the presence of heterozygous single nucleotide polymorphisms excluded the possibility of a deletion within the region of the gene harboring the mutation.
Table 2.
Mutations found in patients with familial pseudohyperkalemia.
The abnormality in the Irish family originally diagnosed as having dominant dehydrated hereditary stomatocytosis plus FP was at first mapped to chromosome 1619 but subsequent analysis of the PIEZO1 gene was negative. We, therefore, subjected the Irish family to whole-exome analysis and identified ABCB6 variant c.826G>T; p.R276W, with subsequent confirmation by direct sequencing. Although there was probably a mis-assignment in the Irish pedigree linkage analysis, it cannot be ruled out that there are intronic mutations that could explain epistasis between PIEZO1 and ABCB6.
ABCB6 amino acid residues R276, R723 and V454 are conserved in all species analyzed and each have Polyphen2 scores of 1 (damaging) and SIFT scores of 0 (damaging).
Each of the three FP mutations is annotated in public databases: 1000 Genomes (URL:http://browser.1000genomes.org);20 NHLBI Exome Sequencing Project (URL:http://evs.gs.washington.edu/EVS); and Exome Aggregation Consortium, Cambridge, MA (URL: http://exac.broadinstitute.org). The minor allele frequency was 0.43% for the V454A variant, 0.08% for R723Q, and 1.5% for R276W (Table 2 and Online Supplementary Table S2).
The high frequency of the variants found in our patients prompted our genetic screening of a cohort of 327 blood donors. Of note, our analysis demonstrated the presence of variant R276W in 0.3% of this cohort (1/327) and the absence of the other two mutations V454A and R723Q, consistent with the minor allele frequencies reported to date.
ABCB6 mutations produce conformational changes in model structures
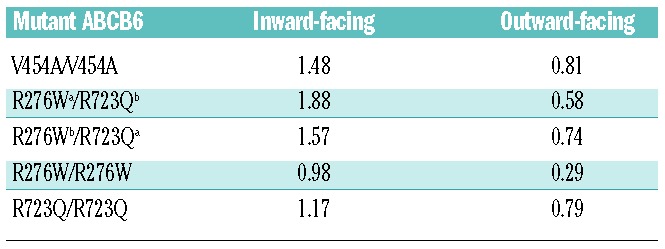
To analyze potential consequences of the identified mutations on protein structure, we generated three-dimensional structural models of the (putatively dimeric) human WT ABCB6 residues 231–827 and FP mutant polypeptides V454A, R276W, and R723Q (see Methods). Figure 1 shows three-dimensional structural models of homodimeric WT ABCB6 in inward- and outward-facing conformations, highlighting sites of the homozygous and compound heterozygous FP missense mutations investigated in this study. The structural models of ABCB6 homodimeric FP mutant V454A and heterodimeric mutant R276W (chain a)/R723Q (chain b) in both inward- and outward-facing conformations are presented in Online Supplementary Figures S1 and S2, respectively. Transverse views of intra-membrane bilayer regions are also presented. Comparison of WT with mutant models revealed that these mutations cause detectable conformational changes in regions on or near the missense substitution sites and at several more remote locations. In inward-facing models of homodimeric FP mutant V454A, the presence of Ala decreased by 2.3 Å the WT Cα-Cα interatomic distance between chain a residue 454 and chain b residue 454. In contrast, this change was minimal in the outward-facing conformation. The WT loop structure at amino acids 362–367 (packed adjacent to chain b residue 454 in the inward-facing conformation) underwent a partial loop-to-helix transition in the homodimeric FP mutant V454A. Furthermore, the WT Cα-Cα interatomic distance between chain a residue 276 and remote chain b residue 723 decreased by 3.3 Å in the heterodimeric FP mutant R276W (chain a)/R723Q (chain b). The inward-facing conformation of this heterodimeric mutant also induced a loop-to-helix transition of chain a residues 408–409 at the ecto-end of a transmembrane helix and spatially adjacent to chain a missense substitution R276W. These amino acid substitutions also modestly altered interhelical distances near the mutation sites. Structural superposition of modeled WT polypeptide with each modeled mutant polypeptide revealed larger global structural deviations of mutant polypeptides in inward-facing conformations than in outward-facing ones (Table 3 and Online Supplementary Table S1). Modeled heterodimeric R276W/R723Q and homodimeric V454A mutant polypeptides exhibited greater structural deviation from WT than did homodimeric mutants R276W or R723Q (Table 3, Online Supplementary Table S1).
Table 3.
Root-mean-square deviations (Å) of superposed homology-modeled structures of the indicated patient-derived mutant ABCB6 dimers with the modeled wild-type ABCB6 homodimer.
ABCB6 mutations cause no alteration of expression or cellular localization
We modeled our patients’ genotypes in vitro by transient transfection of WT and mutant ABCB6 expression plasmids into HEK-293 cells. No significant differences between mutant and WT mRNA accumulation were evident 72 h after transfection (Figure 2A). Similarly, immunoblot analysis of heterologous FLAG tag confirmed equivalent accumulation of WT and mutant heterologous ABCB6 polypeptides (Figure 2B).
Figure 2.
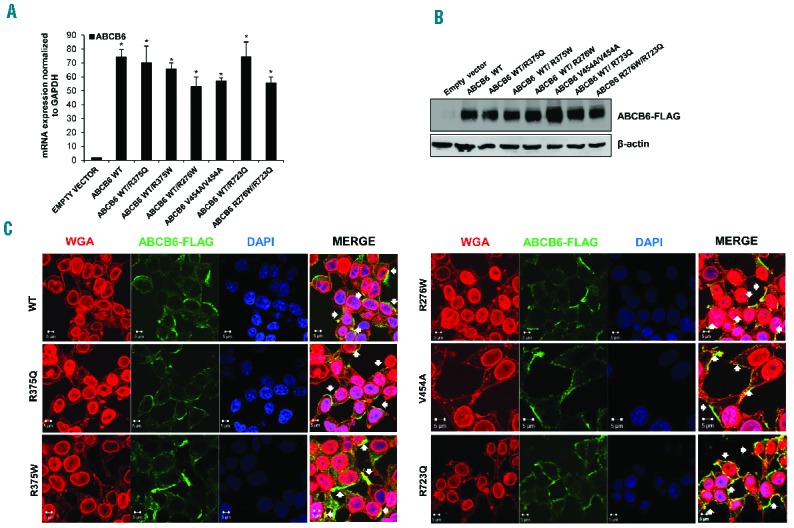
Expression and localization of ABCB6 mutants. (A) ABCB6 mRNA levels in HEK-293 cells transfected with ABCB6 WT and mutants and empty vector as control. Values are means ±SEM of three independent experiments. *P<0.001 WT, WT/R375Q, WT/R375W, WT/R276W, V454A/V454A, WT/R723Q, R276W/R723Q vs empty vector. (B) Immunoblot showing ABCB6 Flag protein expression in HEK-293 cells transfected with FLAG-tagged WT or mutant ABCB6 variants, or with empty vector as control and GAPDH as loading control. One of two similar experiments. (C) Laser-scanning confocal microscopy images of HEK-293 cells transfected with WT or mutant ABCB6 variants, or with empty vector as control, analyzed by immunofluorescence with rabbit polyclonal anti-ABCB6 antibody (green) and WGA (membrane marker, staining both the nuclear envelope and the plasma membrane, red), with the merged signal showing regions of co-localization in yellow (white arrows indicate the yellow regions in the merge). Cells were imaged with a Zeiss LSM 510 meta confocal microscope equipped with a 1.4 NA oil immersion plan Apochromat 100× objective. Intensity and contrast were adjusted with Axiovision software. Representative of three independent experiments.
We also tested effects of the mutations on ABCB6 membrane localization. Confocal microscopy analysis showed that all mutant polypeptides were expressed predominantly at the peripheral membrane of HEK-293 cells, as demonstrated by co-localization of ABCB6-FLAG with the lectin membrane marker, wheat germ agglutinin (Figure 2C).
ABCB6 mutation R276W increases potassium efflux from red blood cells of a blood donor
A blood sample from the blood donor heterozygous for ABCB6 variant and samples from two control donors were obtained and stored for 4 weeks at 4°C under blood banking conditions. Extracellular and intracellular potassium levels were measured throughout the storage period. As shown in Figure 3A, the potassium efflux of donor’s blood after 28 days of storage was about 3.5-fold higher than that of the controls. Correspondingly, the intracellular red blood cell potassium content was about 2.5-fold lower than that of the controls (Figure 3B). The degree of hemolysis over time for the blood samples was evaluated during storage by measurement of free hemoglobin levels and was the same for three samples (data not shown). The data demonstrated that the physiological consequences of the blood donor’s mutation is greater potassium efflux than in controls, and similar potassium efflux to that observed in FP patients.
Figure 3.

ABCB6 protein expression and potassium efflux in red blood cells of a blood donor carrying the ABCB6 R276W variant. (A) Plasma K+ content (expressed as mmol/L of whole blood) of blood from a donor heterozygous for the ABCB6 mutation R276W and from two healthy controls after 0, 7, 14, 21 and 28 days of cold storage under blood banking conditions. *P<0.01 donor vs. two healthy controls. (B) Intracellular K+ content (expressed as mmol*1013 cells) of blood from a donor heterozygous for R276W and from two healthy controls after 0, 7, 14, 21 and 28 days of cold storage as in (A). Ion contents in (A) and (B) were measured by atomic absorption spectrometry, and represent means ± SEM of three independent experiments. *P<0.01 donor vs. two healthy controls. (C) Immunoblot showing ABCB6 protein expression in RBC membranes from a blood donor heterozygous for mutation R276W and pooled membranes from two healthy controls. β-actin was used as the loading control. One of three similar experiments.
Moreover, immunoblot analysis of red blood cells from the blood donor carrying the R276W mutation demonstrated that the expression of ABCB6 did not differ between this donor with a mutation and healthy controls (Figure 3C).
ABCB6 mutations cause cation flux alterations
We next evaluated cell potassium content in HEK-293 cells over-expressing WT ABCB6 and different ABCB6 mutant variants. Preliminary experiments comparing cells maintained for 8 h at 37°C or 30°C revealed no differences (data not shown). To mimic shipping conditions (critical for the alteration in serum K+ concentrations observed in the FP patients, see the Case report section) we exposed HEK-293 cells over-expressing WT or mutant ABCB6 variants to 0.12 g rotary shaking at 30°C for 8 h. As shown in Figure 4A, levels of extracellular potassium in media from HEK-293 cells over-expressing mutant ABCB6 variants were significantly higher than the levels for cells expressing WT ABCB6. Correspondingly, residual intracellular Rb content was significantly reduced in cells expressing three of the ABCB6 mutant genotypes, WT/R375Q, WT/R375W, V454A/V454A, as well as in the double-mutant R276W/R723Q, compared to either WT ABCB6 or the other ABCB6 variants (WT/R273Q, WT/R276W) (Figure 4B). These data show different impacts of individual ABCB6 mutations on cellular K+ efflux insensitive to ouabain plus bumetanide, and an incrementally increased effect on cell K+ efflux of co-expression of the compound heterozygous ABCB6 mutations R276W/R723Q.
Figure 4.

Analysis of potassium efflux of ABCB6 mutants. (A) K+ content of extracellular medium sampled from cultures of cells overexpressing ABCB6 WT or ABCB6 FP mutants. Ion contents measured by atomic absorption spectrometry are expressed as mmol/mg protein. **P<0.001 R276W/R723Q vs. empty vector and WT; *P<0.05 for WT/R375Q, WT/R375W, WT/R276W, V454A/V454A, WT/R723Q vs. WT. (B) Rb content of cells overexpressing ABCB6 WT and ABCB6 FP mutants, expressed as mmol/mg protein. Values in (A) and (B) are means ±SEM of four independent experiments.
Discussion
Here we report three new mutations in the FP-disease gene ABCB6. FP had been described previously as a dominant condition, but, for the first time, we report two FP patients with homozygous or compound heterozygous mutations, both novel patterns of inheritance for FP. Of note, those patients homozygous and compound heterozygous for ABCB6 mutations showed higher plasma K+ concentrations than heterozygous patients. Moreover, the compound heterozygote also had a greater mean corpuscular volume than that of other patients. FP inheritance patterns thus constitute a crucial part of the diagnostic evaluation of patients.
ABCB6 variations are more common than previously predicted, as also reported for Lan- blood group carriers with ABCB6 nonsense mutations causing the ABCB6-null red cell phenotype. Through screening erythroid ABCB6 expression, Koszarska et al. found an unexpectedly high frequency of Lan mutations in healthy individuals.21 Indeed, in public databases (1000 Genomes, NHLBI Exome Sequencing Project, Exome Aggregation Consortium) the allele frequencies of V454A, R723Q and R276W are 0.43%, 0.08% and 1.5%, respectively. Moreover, the high frequency of ABCB6 variations in FP, including two FP patients found in a Cardiff blood donor cohort recently described by Bawazir et al.,22 has clinical implications for blood transfusion screening and practice. Our own screening of 327 blood donors of different geographical and ethnic origin corroborates this observation, since we found the R276W mutation in 0.3% of our cohort. Our analysis of potassium efflux from the blood donor red blood cells under blood banking storage conditions confirmed the cation leak shown by FP patients. Refrigerated storage of blood from FP patients causes rapid loss of potassium, while the extracellular potassium content of bags of stored cells increases during storage. This is of little consequence for the majority of transfusions, since the total amount of potassium transfused is relatively small compared to the total blood volume of the recipient. In contrast, this extracellular potassium can have serious or fatal consequences in neonates and infants given whole blood transfusions of large volume proportionate to body size. Several cases of whole blood transfusion leading to cardiac arrest and death in infants have been described.23–27
The ABCB6 FP mutants overexpressed in HEK-293 cells showed no difference in accumulation of mRNA or protein, or in peripheral membrane immunolocalization as compared to WT ABCB6, and as previously demonstrated for the ABCB6 FP variant R375Q.
Consistent with these findings, in silico modeled three-dimensional structures of these mutant ABCB6 polypeptide dimers predicted modest structural alterations of transmembrane and cytosolic ATP binding domains in both inward- and outward-facing conformations. Prediction of the consequences of these structural alterations to the cation leak process remains uncertain, since the relationship between the mutant cation leak and the (proposed but still debated) WT transport of porphyrins is still poorly understood. Future simulation studies of molecular dynamics in a model lipid bilayer across microsecond time-scales will expand our understanding of the impact of these FP mutants on the structure, and possibly the function, of ABCB6.
To further characterize the role of ABCB6 mutants, we tested the hypothesis that their expression could modify K+ transport in HEK-293 cells in a manner similar to the altered K+ efflux in FP red blood cells. We found that cells expressing each mutant variant tested exhibited increased K+ efflux compared to that of the WT cells. Co-expression in HEK-293 cells of the two mutant variants expressed by patient Cardiff in a compound heterozygous form produced the highest value of K+ efflux among all tested mutants. These data demonstrate that the new mutations, whether homozygous or compound heterozygous, act at the cellular level as gain-of-function mutations.
Among ABC proteins, only the cystic fibrosis transmembrane regulator CFTR/ABCC7 is known to mediate ion channel function. However, several ABC proteins, in addition to CFTR, function as ion channel regulators,28–30 including the Kir6 KATP channel regulatory subunits, sulphonylurea receptors SUR1, SUR2A, and SUR2B.31 It is unclear whether ABCB6 FP mutant polypeptides generate intrinsic cation leak pathways in membranes of red cells (or experimentally in HEK-293 cells), or might secondarily dysregulate one or more endogenous membrane cation permeability pathways in red cells (or HEK-293 cells). The negative results obtained to date in our electrophysiological studies conducted in HEK-293 cells and Xenopus laevis oocytes expressing WT or mutant ABCB6 variants (data not shown) encourage further consideration of dysregulated endogenous electroneutral (or low-level electrogenic) transporters as cation leak mediators in FP red cells or cell models of heterologous expression.
Our findings demonstrate that both heterozygous and homozygous missense mutations in ABCB6 lead to increased efflux of cellular K+ from HEK-293 cells, a property shared with red blood cells from FP patients. Screening for the most frequently found ABCB6 variant, R276W, confirmed that patients with FP are relatively common in the blood donor population. Storage of FP blood can cause a significant increase in whole blood K+ levels, with serious clinical implications for neonates and infants receiving large-volume transfusions of whole blood. For these reasons, we endorse the proposal to conduct genetic screening for ABCB6 FP mutations among potential blood donors, especially when whole blood is needed. Finally, investigation of ABCB6 may contribute to our understanding of other pathologies of red blood cell hydration, such as sickle cell anemia.
Acknowledgments
This work was supported by grants from the Italian Ministero dell’Università e della Ricerca, by grants MUR-PS 35-126/Ind, by grants from Regione Campania (DGRC2362/07), by EU Contract LSHM-CT-2006-037296, by PRIN to AI and LDF (20128PNX83), Italy.
The authors thank the CEINGE Service Facility platforms including the Sequencing Core, the Rheology Facility, the FACS Core Laboratory and the Dynamic Imaging Facility (particularly Dr Daniela Sarnataro for providing helpful technical support).
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/8/909
References
- 1.Stewart GW, Corrall RJ, Fyffe JA, et al. Familial pseudohyperkalemia. A new syndrome. Lancet. 1979;2(8135):175–177. [DOI] [PubMed] [Google Scholar]
- 2.Dagher G, Vantyghem MC, Doise B, et al. Altered erythrocyte cation permeability in familial pseudohyperkalemia. Clin Sci (Lond). 1989;77(2):213–216. [DOI] [PubMed] [Google Scholar]
- 3.Vantyghem MC, Dagher G, Doise B, et al. Pseudo-hyperkalemia. Apropos of a familial case. Ann Endocrinol (Paris). 1991;52(2):104–108. [PubMed] [Google Scholar]
- 4.Haines PG, Crawley C, Chetty MC, et al. Familial pseudohyperkalemia Chiswick: a novel congenital thermotropic variant of K and Na transport across the human red cell membrane. Br J Haematol. 2001;112(2): 469–474. [DOI] [PubMed] [Google Scholar]
- 5.Carella M, d’Adamo AP, Grootenboer-Mignot S, et al. A second locus mapping to 2q35-36 for familial pseudohyperkalemia. Eur J Hum Genet. 2004;12(12):1073–1076. [DOI] [PubMed] [Google Scholar]
- 6.Andolfo I, Alper SL, Delaunay J, et al. Missense mutations in the ABCB6 transporter cause dominant familial pseudohyperkalemia. Am J Hematol. 2013;88(1): 66–72. [DOI] [PubMed] [Google Scholar]
- 7.Krishnamurthy PC, Du G, Fukuda Y, et al. Identification of a mammalian mitochondrial porphyrin transporter. Nature. 2006;443 (7111):586–589. [DOI] [PubMed] [Google Scholar]
- 8.Kiss K, Brozik A, Kucsma N, et al. Shifting the paradigm: the putative mitochondrial protein ABCB6 resides in the lysosomes of cells and in the plasma membrane of erythrocytes. PLoS One. 2012;7(5):e37378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helias V, Saison C, Ballif BA, et al. ABCB6 is dispensable for erythropoiesis and specifies the new blood group system Langereis. Nat Genet. 2012;44(2):170–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bawazir WM, Flatt JF, Wallis JP, et al. Familial pseudohyperkalemia in blood donors: a novel mutation with implications for transfusion practice. Transfusion. 2014; 54(12):3043–3050. [DOI] [PubMed] [Google Scholar]
- 11.Andolfo I, Alper SL, De Franceschi L, et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood. 2013;121(19):3925–3935. [DOI] [PubMed] [Google Scholar]
- 12.Ye S, Dhillon S, Ke X, Collins AR, Day IN. An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res. 2001;29(17):E88–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23(3):318–326. [DOI] [PubMed] [Google Scholar]
- 14.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. [DOI] [PubMed] [Google Scholar]
- 15.Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 1):191–205. [DOI] [PubMed] [Google Scholar]
- 16.Koradi R1, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14(1):51–55. [DOI] [PubMed] [Google Scholar]
- 17.De Franceschi L, Ronzoni L, Cappellini MD, et al. K-CL co-transport plays an important role in normal and beta thalassemic erythropoiesis. Haematologica. 2007;92(10):1319–1326. [DOI] [PubMed] [Google Scholar]
- 18.Stewart GW, Amess JA, Eber SW, et al. Thrombo-embolic disease after splenectomy for hereditary stomatocytosis. Br J Haematol. 1996;93(2):303–310. [DOI] [PubMed] [Google Scholar]
- 19.Carella M, Stewart G, Ajetunmobi JF, et al. Genomewide search for dehydrated hereditary stomatocytosis (hereditary xerocytosis): mapping of locus to chromosome 16 (16q23-qter). Am J Hum Genet. 1998;63 (3):810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.1000 Genomes Project Consortium, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015; 526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koszarska M, Kucsma N, Kiss K, et al. Screening the expression of ABCB6 in erythrocytes reveals an unexpectedly high frequency of Lan mutations in healthy individuals. PLoS One. 2014;9(10):e111590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bawazir WM, Flatt JF, Wallis JP, et al. Familial pseudohyperkalemia in blood donors: a novel mutation with implications for transfusion practice. Transfusion. 2014;54(12):3043–3050. [DOI] [PubMed] [Google Scholar]
- 23.Hall TL, Barnes A, Miller JR, et al. Neonatal mortality following transfusion of red cells with high plasma potassium levels. Transfusion. 1993;33(7):606–609. [DOI] [PubMed] [Google Scholar]
- 24.Chen CH, Hong CL, Kau YC, et al. Fatal hyperkalemia during rapid and massive blood transfusion in a child undergoing hip surgery: a case report. Acta Anaesthesiol Sin. 1999;37(3):163–166. [PubMed] [Google Scholar]
- 25.Baz EM, Kanazi GE, Mahfouz RA, et al. An unusual case of hyperkalaemia-induced cardiac arrest in a paediatric patient during transfusion of a “fresh” 6-day-old blood unit. Transfus Med. 2002;12(6):383–386. [DOI] [PubMed] [Google Scholar]
- 26.Smith HM, Farrow SJ, Ackerman JD, et al. Cardiac arrests associated with hyperkalemia during red blood cell transfusion: a case series. Anesth Analg. 2008; 106(4):1062–1069. [DOI] [PubMed] [Google Scholar]
- 27.Lee AC, Reduque LL, Luban NL, et al. Transfusion associated hyperkalemic cardiac arrest in pediatric patients receiving massive transfusion. Transfusion. 2014;54(1):244–254. [DOI] [PubMed] [Google Scholar]
- 28.Higgins CF. The ABC of channel regulation. Cell. 1995;82(5):693–669. [DOI] [PubMed] [Google Scholar]
- 29.Welsh MJ, Anderson MP, Rich DP, et al. Cystic fibrosis transmembrane conductance regulator: a chloride channel with novel regulation. Neuron. 1992;8(5):821–829. [DOI] [PubMed] [Google Scholar]
- 30.Inagaki N, Gonoi T, Clement JP, et al. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270(5239):1166–1170. [DOI] [PubMed] [Google Scholar]
- 31.Cui Y, Giblin JP, Clapp LH, Tinker A. A mechanism for ATP-sensitive potassium channel diversity: functional coassembly of two pore-forming subunits. Proc Natl Acad Sci USA. 2001;98(2):729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polireddy K, Khan MM, Chavan H, et al. A novel flow cytometric HTS assay reveals functional modulators of ATP binding cassette transporter ABCB6. PLoS One. 2012; 7(7):e40005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aller SG, Yu J, Ward A, et al. Structure of Pglycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323(5922):1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443(7108):180–185. [DOI] [PubMed] [Google Scholar]