

Abstract
To circumvent donor-to-donor heterogeneity which may lead to inconsistent results after treatment of acute graft-versus-host disease with mesenchymal stromal cells generated from single donors we developed a novel approach by generating these cells from pooled bone marrow mononuclear cells of 8 healthy “3rd-party” donors. Generated cells were frozen in 209 vials and designated as mesenchymal stromal cell bank. These vials served as a source for generation of clinical grade mesenchymal stromal cell end-products, which exhibited typical mesenchymal stromal cell phenotype, trilineage differentiation potential and at later passages expressed replicative senescence-related markers (p21 and p16). Genetic analysis demonstrated their genomic stability (normal karyotype and a diploid pattern). Importantly, clinical end-products exerted a significantly higher allosuppressive potential than the mean allosuppressive potential of mesenchymal stromal cells generated from the same donors individually. Administration of 81 mesenchymal stromal cell end-products to 26 patients with severe steroid-resistant acute graft-versus-host disease in 7 stem cell transplant centers who were refractory to many lines of treatment, induced a 77% overall response at the primary end point (day 28). Remarkably, although the cohort of patients was highly challenging (96% grade III/IV and only 4% grade II graft-versus-host disease), after treatment with mesenchymal stromal cell end-products the overall survival rate at two years follow up was 71±11% for the entire patient cohort, compared to 51.4±9.0% in graft-versus-host disease clinical studies, in which mesenchymal stromal cells were derived from single donors. Mesenchymal stromal cell end-products may, therefore, provide a novel therapeutic tool for the effective treatment of severe acute graft-versus-host disease.
Introduction
Since the first clinical trial of mesenchymal stromal cells (MSC) in 1995,1 their use has expanded rapidly. To date, 561 registered clinical trials (retrieved from www.clinicaltrials.gov, 2nd December 2015) have been performed to examine an extremely wide spectrum of therapeutic MSC applications. Despite the general consensus that MSCs appear to be well-tolerated, safe and effective for the treatment of various diseases, there has been limited progress in this field due to inconsistencies in the outcome of clinical trials. These inconsistencies may be attributed to the lack of a standardized methodology for MSC generation2 and MSC dosing, the heterogeneity in MSC potency between donors3 and tissue sources,4 and the variable number of MSC progenitor cells between tissue samples.5 MSCs exhibit donor-specific variations in their immunosuppressive properties not only at the donor level,3,6 but also at the clonal level.7 A recent study demonstrated passage effect on the immunosuppressive effect of MSCs by obtaining an optimal immunosuppressive effect in patients with steroid-resistant acute graft-versus-host disease (aGvHD) after administration of MSCs at passage 1 or 2.8 In contrast, several other reports demonstrated that the immunosuppressive effect of MSCs remains unchanged for up to 7 or 8 passages in culture.9,10 Another important issue regarding the clinical application of MSCs is their culture under serum-free conditions. The majority of clinical studies have used MSCs that were expanded in media supplemented with fetal bovine serum (FBS).1,11–15 To avoid the risks associated with the use of FBS,16 platelet lysate (PL) was proposed as a supplement to tissue culture media for MSCs.17 Recently, several studies showed that MSCs that were expanded in PL exhibited the same efficacy as MSCs cultured in serum-containing media for the treatment of GvHD.18–22
To date, clinical studies have used MSCs that have been generated from several individual donors. Considering the aforementioned inter-donor heterogeneity and the need for a large number of “off-the-shelf” MSCs, the establishment of MSC banks appears to be an indispensable strategy for providing a continuous supply of MSCs with predictable potency. To our knowledge, there are few established MSC banks worldwide, and these MSC banks were generated by separately isolating, expanding, and freezing MSCs from up to 10 donors in FBS-containing media.23–26
In the current study, we report for the first time the establishment of a serum-free and GMP-compliant MSC bank generated from pooled bone marrow mononuclear cells (BM-MNCs) of multiple donors as a novel strategy to circumvent donor-to-donor variability. Clinical-grade MSC end-products (MEPs) derived from the MSC bank were thoroughly assessed for their proliferation, differentiation, and, in particular, for the allosuppressive potential in vitro. Importantly, 81 MEPs were administered as a rescue therapy to 26 pediatric patients with severe steroid-refractory aGvHD in seven transplantation centers. Safety and efficacy of MEPs was compared to MSCs generated from a single or several individual donors that have been used in the GvHD-clinical studies reported thus far.
Methods
Generation of MSC bank and clinical-grade MEPs
Bone marrow was collected from 8 healthy volunteers (age 21–45 years old) after written informed consent and after the approval of the local Ethics Committee (n. 275/09). BM-MNCs were enriched from the bone marrow aspirate by using the Sepax II NeatCell process (Biosafe, Eysins, Switzerland) and frozen individually. After thawing and washing these BM-MNCs were pooled. This pool of BM-MNCs from 8 donors was used to generate MSCs over 14 days in culture. After their detachment, passage 1 mesenchymal stromal cells (MSC-P1) were washed and aliquoted into 209 cryovials (each containing 1.5×106 MSC-P1). Cryopreserved vials with MSC-P1 were referred to as the MSC bank.
To generate clinical-grade MEPs, MSC-P1 aliquots from the MSC bank were thawed and after washing they were expanded in medium containing 10% PL till the end of passage 2. These MSCs were re-suspended in cryomedium (0.9% NaCl containing 5% HSA and 10% DMSO), distributed in cryobags (each containing 1–3×106 MSCs/mL in 45 mL of cryomedium) and frozen in liquid nitrogen until use.
Further details on methods as to generation of MSC bank from BM-MNC pool, including collection and testing of platelet lysates, validation of MEPs regarding their phenotype (flow cytometry), differentiation and allosuppressive potential, genetic analysis (cytogenetics, FISH, RT-PCR, STR-PCR), determination of senescence and its typical markers, characteristics of patients with severe aGvHD, assessment of the disease and its treatment with MEPs and statistics, are presented in the Online Supplementary Appendix.
Results
Collection of bone marrow from 8 healthy 3rd-party donors and isolation of BM-MNCs
After obtaining written informed consent, healthy donors donated 152–184 mL of bone marrow (Online Supplementary Table S1). Following isolation of the BM-MNCs using the Sepax method, a total of 10.82×109 BM-MNCs were collected from 8 donors. The absolute number of BM-MNCs per 1 mL of bone marrow after isolation was 4.1×106±7.8×105. All donors were equally represented in the BM-MNC pool, i.e. the relative contribution of BM-MNCs by each donor was 12.5±2.7% (Online Supplementary Table S1). The cells from each donor were re-suspended in cryomedium and frozen individually in bags. The total number of frozen BM-MNCs was 9.86×109.
Testing of the concentration of PL for the generation and expansion of MSC
Before the establishment of the MSC bank, we determined the optimal concentration of PL to support the adherence of MSC progenitors and to assess whether PL filtration was needed. We observed that unfiltered PL at a concentration of 10% (Online Supplementary Figure S1A) or 5% (Online Supplementary Figure S1B) was optimal for MSC generation compared with media supplemented with filtered PL at the same concentrations (P<0.002 and P<0.01, respectively). In addition, 10% PL was significantly more efficient for MSC generation by plastic adherence (P<0.02) (Online Supplementary Figure S1C). Evaluation of the capacity of PL to expand MSC revealed that unfiltered PL at a concentration of 10% (Online Supplementary Figure S1D) or 5% (Online Supplementary Figure S1E) induced significantly greater expansion of MSC than filtered PL at the corresponding concentration (P<0.0008 and P<0.003, respectively). Furthermore, unfiltered PL at 10% was significantly more effective for expansion of MSC than 5% unfiltered PL (P<0.0001) (Online Supplementary Figure S1F).
Establishment of the MSC bank and generation of clinical-scale MEP
After thawing, the number of pooled BM-MNCs was 2.8×109. Culture of these cells for 14 days resulted in 320×106 primary MSC which exhibited a viability of 98.9% and expressed consensus markers for MSC. These cells were designated as MSC-P1 (passage 1) and were aliquoted and frozen in 209 cryovials, representing the MSC bank (Online Supplementary Figure S2Bi–iii).
To generate clinical-scale MEP and validate their proliferative, differentiation and allosuppressive potential, three randomly selected MSC aliquots from the MSC bank were thawed after cryopreservation for 6–8 weeks. The mean cell recovery was 1.39×106 (range 1.23–1.48×106) viable cells/vial, and the viability of these cells was 95.25±1.73% (range 93.45%–96.9%). On average, the expansion of these MSC over two weeks until the end of passage 2 resulted in the generation of 4.7×108 viable MSC (range 4.2–5.48×108), in which the number of cumulative population doublings (CPD) was 8.5±0.04. These samples (units), referred to as clinical-scale MEP, were frozen in 4–7 bags containing 50–129×106 MSC until use. These units served as clinical doses for recipients of various body weights (Online Supplementary Figure S2Ci–iii). Before freezing, the MEP phenotypes were analyzed to assess whether the samples fulfilled the International Society for Cellular Therapy (ISCT) MSC criteria. They were sterile, were mycoplasma-negative and contained endotoxin levels below the limit of detection (<0.2 EU/mL). For quality control and release purposes, all bags generated from one aliquot were considered as one batch.
Validation of the MEP: phenotype, allosuppressive and differentiation potential
For quality assessment, the MEP from three different batches were thawed after 4–6 weeks of cryopreservation. After thawing, 87.9±3.6% of the cells were viable, representing a mean recovery of 78.5±9.8%. MEP were negative for HLA-DR and the hematopoietic markers CD45, CD14, and CD34. However, they expressed high levels of the consensus MSC markers CD73, CD90 and CD105 and HLA class I molecules (Online Supplementary Figure 3B). The mean inhibitory effect of thawed MEP on the proliferation of HLA-mismatched peripheral blood MNC (PB-MNC) was 36.7±3.2% (Online Supplementary Figure S3B). All thawed MEP differentiated into adipocytes, osteoblasts or chondrocytes (Online Supplementary Figure S3C–E).
Proliferation potential and senescence of MEP
Three MSC bank cryovials were expanded to end-products, and these samples demonstrated a similar number of population doublings (PD) in passages 1 and 2 (4.3 PD/passage). The number of cumulative PD (CPD) did not exceed 8.5±0.024 (Figure 1A). In the interim, we have expanded 19 MSC-aliquots up to the end-product. The mean viability of these thawed aliquots was 95.1±2.1% (range 88.8%–98.2%). As shown in Figure 1B, the mean cell number of all expanded MEP at the end of passage 2 was 5.64×108±0.78×108 MSC, indicating a predictable proliferation potential.
Figure 1.
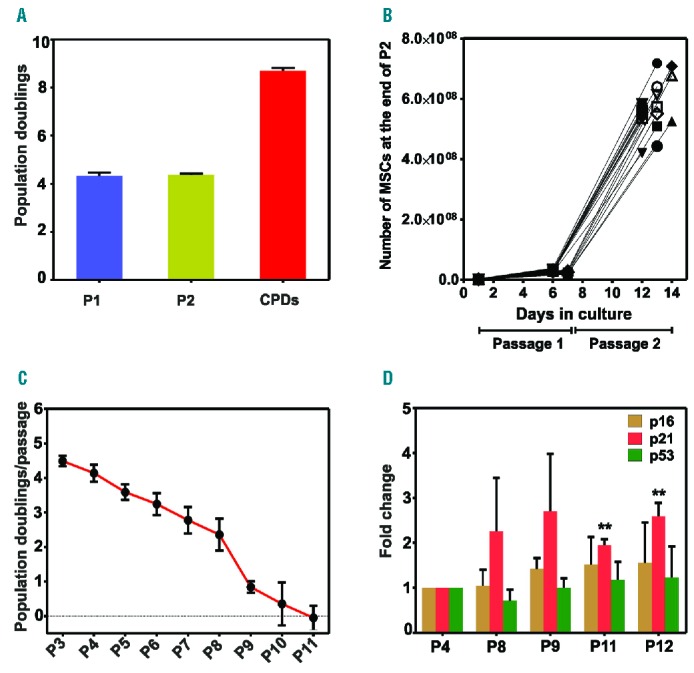
Proliferation potential and senescence of mesenchymal stromal cells (MSC) and MSC end-products (MEP). (A) MSC proliferated at a rate of approximately four population doublings (PD) per passage. The number of cumulative PD (CPD) was 8.5±0.4. Data presented as mean±SEM (n=3). (B) Ex vivo expansion of nineteen MEP through 2 passages. (C) Expansion of MEP for 11 passages and estimation of the number of PD (n=3). (D) RT-PCR analysis of genes related to cell senescence in three clinical-scale MEP. Data presented as mean±SEM (n=3). **P<0.003. Statistical analysis was performed using Student’s t-test.
To test whether the MSC were immortalized during expansion, we expanded three MEP for 13 passages. As shown in Figure 1C, at some point between passages 5 and 11, the MSC underwent replicative senescence, and the number of PD diminished rapidly. The three MEP that were expanded for 11 passages underwent 30.2 CPDs in 68 days. MEP expansion until passage 13 exhibited no post-senescence proliferation (data not shown).
Moreover, analysis of senescence marker expression in three clinical-scale MEP demonstrated no significant elevation in the levels of p16 and p53 gene expression. In contrast, p21 gene expression was significantly increased at passages 11 and 12 compared with passage 4 (Figure 1D). Consistent with the senescent behavior of the MSC from our bank, none of the three examined MEP expressed the oncogene c-myc or hTERT (data not shown).
Allosuppressive potential of MSC isolated from individual donors and MEP
Based on our preliminary data, we hypothesized that MSC generated from pooled BM-MNC of 8 donors may exhibit a higher allosuppressive potential than MSC generated from individual donors or pooled MSC from different donors. To test our hypothesis, we expanded MSC from the 8 individual donors from the start of passage 2, and pooled MSCs from the 8 donors (pooled-MSCs) or one MEP (MSC-140) until the end of passage 2. As expected, the allosuppressive potential of the MSC from individual donors in mixed leukocyte reaction (MLR) was highly heterogeneous, ranging from 20% (donors 1 and 8) to approximately 80% (donors 2 and 3) (Figure 2A). The allosuppressive potential of the pooled-MSC was equal to the mean allosuppressive potential of the MSC from the 8 individual donors. In contrast, the allosuppressive potential of the expanded MSC-140 end-product from the MSC bank was significantly greater than that of the pooled-MSC or the mean allosuppressive potential of the MSC from the 8 individual donors (P<0.001 and P<0.01, respectively). These results show the advantage of pooling BM-MNC for MSC generation. In addition, the allosuppressive potential of six freeze-thawed MEP (as usually administered to patients) demonstrated a consistent allosuppressive effect in vitro (Figure 2B), indicating the equipotency of MSC batches (mean 52±8.7%).
Figure 2.
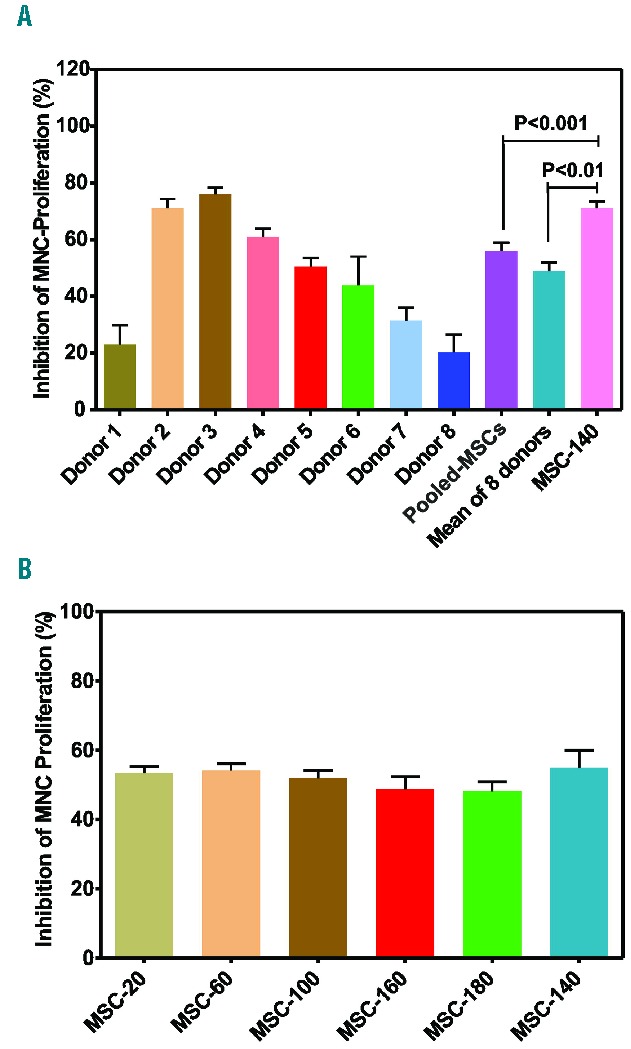
Allosuppressive potential of mesenchymal stromal cells (MSC) generated from individual donors and of MSC end-products (MEP). (A) MSC from the 8 individual donors and pooled MSC from these 8 donors (Pooled MSC); and one MEP (MSC-140) were expanded from the start of passage 2 to the end of passage 2. The MSC were evaluated for their allosuppressive effect in a mixed leukocyte reaction (MLR). (B) Six MEP (clinical doses) were thawed, washed, and directly tested in an MLR. Results presented as mean±SEM. Statistical analysis was performed using Student’s t-test.
Genetic characterization of MEP
Because in vitro culture may cause chromosomal aberrations in cells, we performed chromosomal analysis of 25 MSC undergoing mitosis with a resolution of approximately 350–400 bands; 21 of the 25 analyzed metaphases demonstrated a normal karyotype (Figure 3A). Four out of the 25 analyzed metaphases displayed balanced translocation between the short arms of chromosomes 5 and 19. Breakpoints were identified in bands 5p13 and 19p13.3. The karyotype was mos 46,XY[21]/46,XY,t(5;19)(p13;p13.3)[4]. FISH analysis using a 2-color probe for chromosome 5p15 (hTERT) and 5q35 (NSD1) and a 3-color break-apart probe for the MYC gene at chromosomal locus 8q24 demonstrated that the majority of MEP possessed a normal diploid pattern (Figure 3B and C). Interphase nuclei after 2-color hybridization of probe sets 5p15 (green) and 5q35 (red) revealed that 97.2% of the cells showed a normal diploid pattern for chromosome 5, and that only 2.8% of the cells showed a tetraploid hybridization pattern (Figure 3D). Similarly, visualization of interphase nuclei after 3-color hybridization of the MYC break-apart probe (Figure 3C) showed that 97% of the MSCs carried two normal fusion signals for chromosome 8q24 and that 3% of the MSCs displayed a tetraploid signal pattern (Figure 3E).
Figure 3.
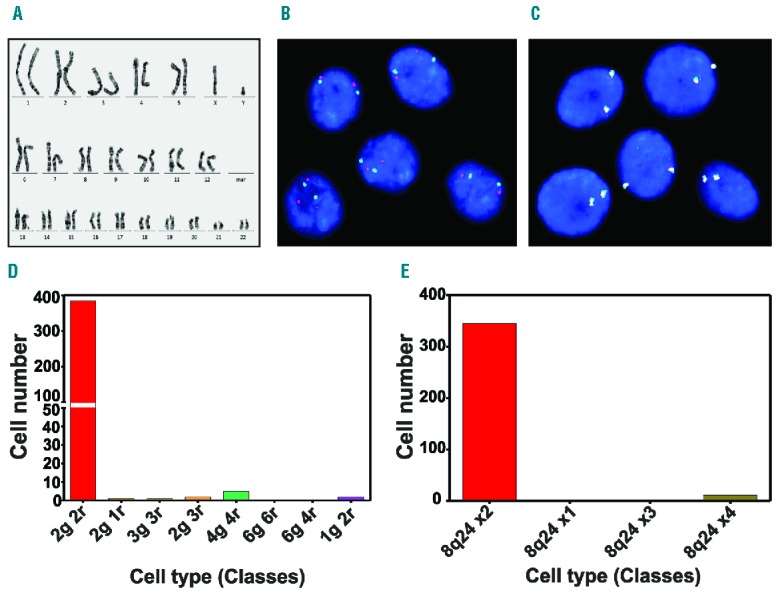
Genetic characterization of the clinical-grade mesenchymal stromal cells end-products (MEP). (A) Normal karyogram of MEP. (B) Interphase nuclei after 2-color hybridization of probe sets 5p15 (green) and 5q35 (red). (C) Interphase nuclei after 3-color hybridization of an MYC break-apart probe showed that almost all cells exhibited two normal fusion signals. (D) The number of MSC displaying a normal diploid or aneuploid pattern after 2-color hybridization of probe sets 5p15 and 5q35. (E) The number of MSC displaying a normal diploid or aneuploid pattern after 3-color hybridization of an MYC break-apart probe for chromosome 8q24.
Comparison of the proliferation potential of MSC from individual donors, pooled MSCs from the 8 donors and MEP
Before MSC bank generation, we tested the capacity of BM-MNC from each donor to generate MSC. The number of generated MSCs per 1×106 BM-MNCs after 13 days in culture varied by more than one order of magnitude, ranging from 0.5×105 to 5.4×105 MSC (Figure 4A).
Figure 4.
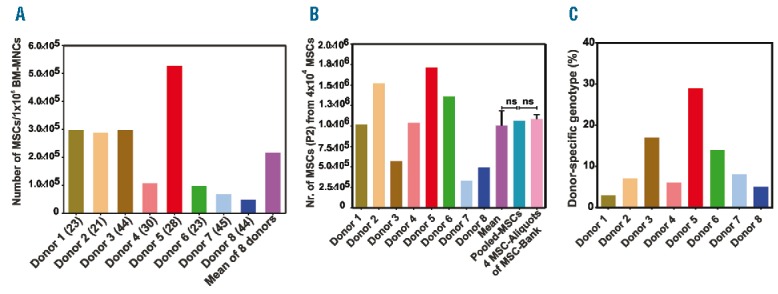
Capacity of bone marrow mononuclear cells (BM-MNC) from the 8 donors to generate mesenchymal stromal cells (MSC), their proliferation potential and chimeric analysis of the MSC end-products (MEP). (A) Data are expressed as the number of generated MSC per 1×106 BM-MNC from each donor cultured in 5% human platelet lysis (PL) for 13 days. Age of each donor (in years) is shown in brackets. (B) Comparison of the proliferation potential of MSC from the 8 individual donors with that of pooled MSC from the 8 individual donors and four MEP. (C) Determination of the relative proportion of each donor in MEP via STR-PCR; ns: not significant. Statistical analysis was performed using Student’s t-test.
Moreover, to validate the rationale of pooling BM-MNC from 8 donors to establish the MSC bank, we compared the in vitro proliferation capacity of the MSC from the 8 individual donors, the pooled MSC of the 8 individual donors, and the four MEP (Figure 4B). The MSC from each bone marrow donor showed different proliferation rates; these varied from 3×105 MSC (donor 7) to 1.7×106 MSC (donor 5). The mean of proliferation of the MSC from the 8 donors was 1×106±5×105 MSC, which correlated well with the number of expanded MSC generated from the pooled-MSC from the 8 donors (1.06×106 MSC). Interestingly, both values correlated very well with the mean number of MSC obtained from the expansion of four MSC bank aliquots within a passage (1.09×106±1×105 MSC). These results confirmed our hypothesis that pooling BM-MNC enables the generation of an “arithmetic mean” of high- and low-proliferating MSC.
Because the MSC in our bank were generated from a pool of BM-MNC from 8 “3rd-party” donors, we were interested in the contribution of the BM-MNC from each donor to the MEP. Chimeric analysis via STR-PCR using a series of genetic markers demonstrated the distinct proportions of the MEP derived from the 8 donor samples (Figure 4C). In principle, the relative contribution of each donor sample to the MEP did not strictly correlate with the proliferation potential of the MSC generated from the individual donors (Figure 4A). In addition, donor proportion in the MEP did not correlate with the relative donor proportion in the initially pooled BM-MNCs, which were used as a source for generation of our MSC bank (Online Supplementary Table S1).
Safety of MEP in severe aGvHD
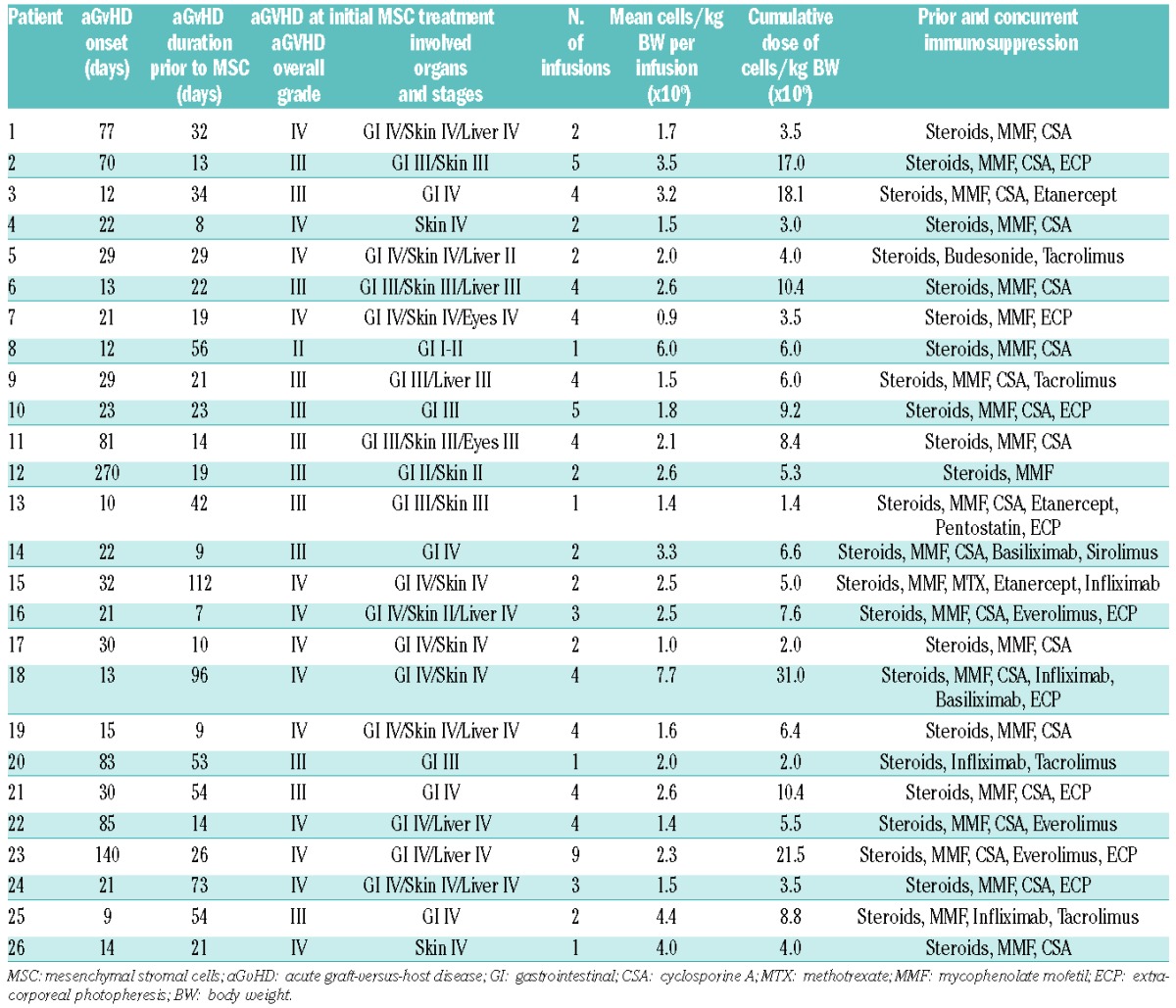
Twenty-six patients with severe aGvHD were enrolled in this compassionate use study (Table 1). They received a median of 2.2×106 MSCs per kg BW (range 0.9–4.4×106 MSC per kg BW in 24 patients). One patient received 6×106 MSCs per kg BW, and another patient received 7.7×106 MSCs per kg BW (Table 2). Overall, a median of 3 (range 1–9) MSC infusions were administered to each patient. Only 2 patients exhibited adverse effects to MSC infusion (one incident each of headache and nausea), which presumably may be attributed to the cryoprotectant.
Table 1.
Patients’ characteristics.
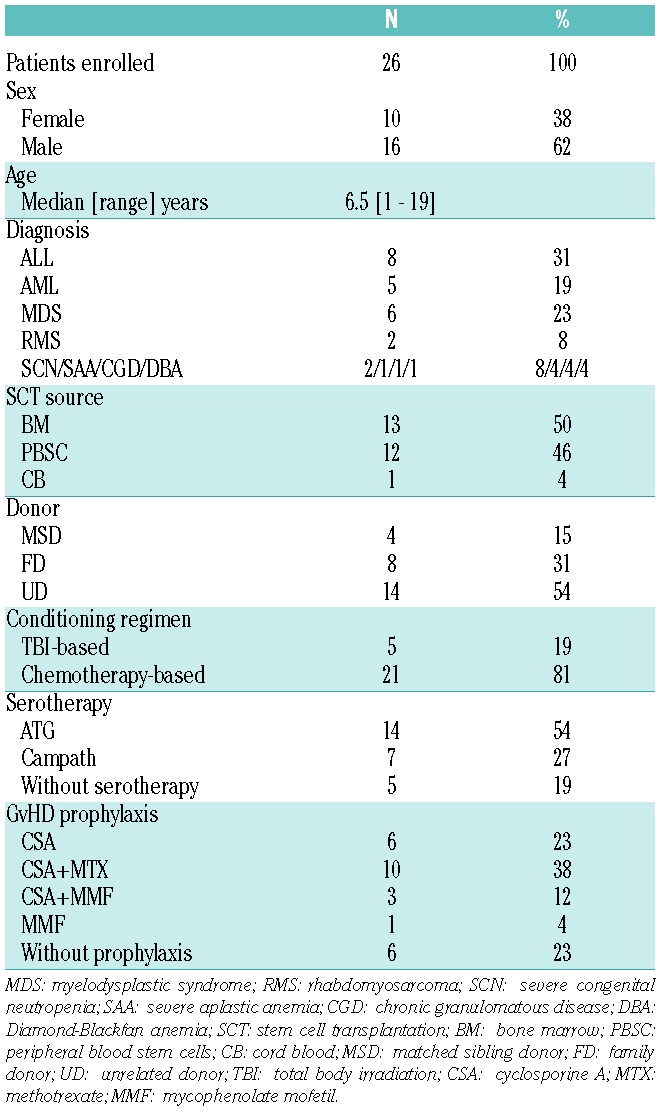
Table 2.
Acute graft-versus-host disease and mesenchymal stromal cell treatment.
Response to treatment with MSC and overall survival
Based on the assessment criteria on day 28 after the initial MSC infusion, 5 of 26 patients (19%) showed a complete response (CR), 15 of 26 patients (58%) a partial response (PR), 4 of 26 patients (15%) did not respond, and 2 of 26 patients (8%) died before day 28 and thus their response could not be evaluated. Overall response rate, defined as patients with CR or PR, was 20 of 26 patients (77%). Follow up for 15 months demonstrated an increase in the CR rate to 73.1% (19 of 26) and a decrease in the percentage of patients experiencing a PR to 11.5% (3 of 26). This treatment resulted in a 2-year overall survival (OS) estimate of 71±11% for the entire patient cohort (n=26) (Figure 5A), indicating the safety of this treatment and suggesting its efficacy in vivo. In addition, cumulative incidence (CI) of non-relapse mortality (NRM) estimate at two years in our patients was 15±7% (Figure 5B). All details concerning the responses to MSC treatment are presented in Table 3.
Figure 5.
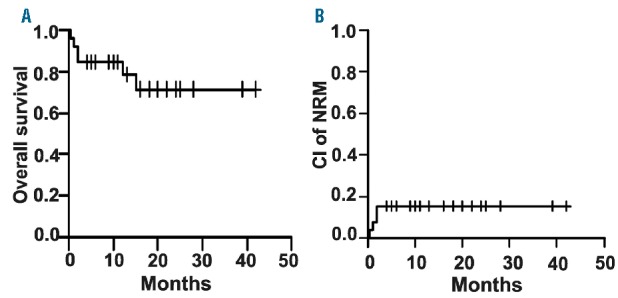
Overall survival (OS) and non-relapse mortality (NRM). (A) For all patients (n=26), OS estimate at two years was 71%±11%. (B) Cumulative incidence (CI) of NRM estimate at two years was 15%±7%. Estimates presented as mean±SE.
Table 3.
Response to the mesenchymal stromal cell treatment.
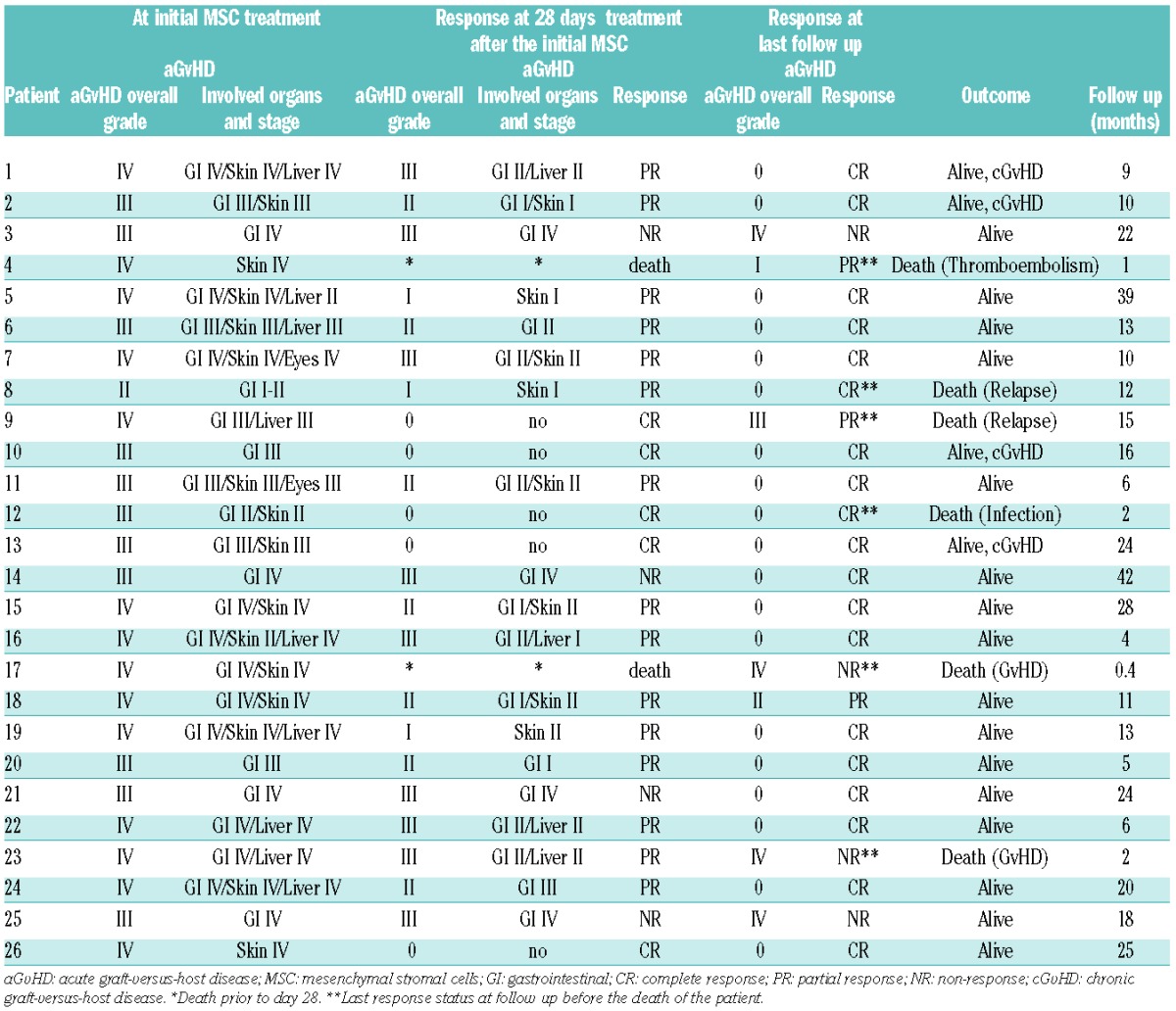
A total of 4 of 26 patients died due to non-relapse mortality (NRM). Two of these 4 patients died due to multi-organ failure based on progressive aGvHD. One of these 4 patients died because of cerebral thromboembolism of unknown origin, whereas the other patient died due to uncontrollable infection. Another 2 patients died due to relapse of their underlying leukemia.
Prognostic factors
Using Fisher’s exact test or the Kruskal-Wallis test, we found that none of the clinical factors (Online Supplementary Table S4), including sex, age, diagnosis, donor, conditioning regimen, graft source, GvHD prophylaxis, type and number of drugs used in the initial treatment, severity of aGvHD, and time of aGvHD onset, correlated with the response to MSC treatment.
Discussion
In the GvHD-related clinical studies reported so far, patients were treated with MSC generated from individual donors after their culture in either serum-containing13 or serum-free media.18–21 However, donor-to-donor heterogeneity27–29 and the lack of standardized manufacturing protocols may lead to inconsistent clinical results that cannot be compared. In addition, we have recently demonstrated for the first time at the clonal level the intra-donor heterogeneity of the allosuppressive potential of MSC. Interestingly, the net allosuppressive effect of MSC represented an “arithmetic mean” of the high and low allosuppressive clones composing the MSC population.7 Given these findings, the selection of an appropriate donor with potent MSC is challenging and, so far, those in the scientific community who are studying MSC have not offered any solution. In an attempt to resolve this issue, in this study, we developed a novel strategy to circumvent or at least to minimize donor-to-donor heterogeneity by establishing an MSC bank from pooled BM-MNCs of multiple “3rd-party” donors (in our case, 8 donors) for the generation of clinical-scale MSC. To validate the rationale of this approach, we tested the allosuppressive effect of MEP from the MSC bank and of MSC derived from 8 donors individually. As expected, the allosuppressive potential of individual MSC was highly heterogeneous. MSC derived from the individuals displayed effects on the alloantigen-induced proliferation of PB-MNC ranging from 20% (donors 1 and 8) to approximately 80% inhibition (donors 2 and 3), with a mean allosuppressive effect of 48%. These results correlated very well with the strikingly high inter-donor differences in the immunosuppressive effects of MSC (range 0–90%).30 In MLR, the allosuppressive potential of pooled MSC from the 8 donors was similar to the mean allosuppressive potential of the MSC from each individual donor. Remarkably, the allosuppressive potential of the expanded MEP, generated from pooled BM-MNC, was significantly higher than that of the pooled MSC and the mean allosuppressive potential of the MSC from each individual donor. Therefore, generation of MSC from pooled BM-MNC of multiple donors appears to be more efficient than pooling MSC from several donors, which was reported to generate greater and more stable suppression in vitro and in vivo.10,31,32 Importantly, all tested MEP demonstrated an equivalent allosuppressive effect in vitro after thawing (equipotent MSC doses) (mean 52±8.7%).
Although MSC banks provide a large number of “off-the-shelf” products, a few reports have cautioned that freeze-thawed MSC display lower therapeutic efficacy than fresh MSC.3,33,34 In contrast, other studies10,35–37 have demonstrated that cryopreserved MSC exhibit equivalent viability and immunosuppressive potential to freshly isolated MSC from cell culture. Consistent with these findings, we found that freeze-thawed MEP displayed a viability of 95% and retained the ability to effectively suppress lymphocyte proliferation in vitro.
One of the major criteria required for the clinical application of MSC is that the MSC should enter senescence without undergoing oncogenic transformation. Tarte et al.38 demonstrated that all bone marrow-derived MSC exhibit complete growth arrest at a PD between 35 and 52 and lack post-senescence proliferation even after long-term culture. Three of the thawed MEP, which were expanded for 13 passages, entered replicative senescence between passage 10 or 11 (30.2 CPD) after 68 days in culture. Their senescence was followed by increased levels of the cell cycle regulators p21 and p16 but no change in the TP53 gene expression. This finding is consistent with the data reported for lethally irradiated MSC.39 Importantly, none of the three MEP expressed the proto-oncogene c-myc or hTERT, and no post-senescence proliferation was observed, as previously demonstrated in other studies.28,40
After thorough phenotypic, genetic and functional characterization of our MSC bank, we administered a total of 81 MEP to 26 patients with severe refractory aGvHD on a compassionate use basis after individual approval by the regulatory authorities. All patients who received MSC infusions had exhibited failed responses to several other lines of treatment. It is known that the more drugs that fail in the treatment of aGvHD patients, the higher the risk that the patients succumb to GvHD. We did not observe any MSC-related side-effects during transfusion. However, 2 patients died due to progressive GvHD, 2 patients developed a relapse of their underlying leukemia, and 2 others died due to treatment complications. One of these died from infectious complications; as this patient was heavily immunosuppressed at the time of MSC infusion, it is impossible to attribute this event to any single treatment. As the relapse rate in our patients with malignant disease was only 9%, we found no evidence that MSC might hamper a graft-versus-leukemia effect.
In our cohort, 20 of 26 patients (77%) responded to the MSC treatment by day 28 (overall response), which is comparable to the results obtained in a randomized placebo-controlled study by using MSC product from the Osiris company (Prochymal) for the treatment of aGvHD (63%). Although the primary end point in that study was not achieved for the whole group of patients, there was a significant benefit over placebo group in the liver and GI tract.41 Response rate in our cohort of patients is also similar to that reported by Introna et al.20 in a cohort of 12 pediatric patients; that study demonstrated an overall response of 66.7%. However, in their patient cohort, only 25% of the patients exhibited aGvHD over grade III, whereas in our series, 96% of the patients exhibited aGvHD grade III or IV. Lucchini et al.19 observed a 62.5% overall response among 8 patients with aGvHD (50% grade I/II and 50% grade III/IV). Similar findings were reported by Prasad et al.14 in a compassionate use study (overall response of 66.7% at day 32 in 12 pediatric patients) and by Kurtzberg et al.15 (61.3% overall response in a large cohort of 75 pediatric patients after treatment of aGvHD with Prochymal). Our results are comparable to the results of the latter group considering the composition of our patient cohort (96% grade III/IV and only 4% grade II), which represents a very challenging patient population.
The CR rate increased from 19% at day 28 to 73.1% at the last follow up, whereas the primary PR rate decreased from 58% to 11.5%, indicating that tissue recovery requires time. The inversion of PR by the time in CR is similar to the data obtained by Le Blanc46 in their pediatric cohort. The increased CR rate in our cohort translated into a very favorable 2-year OS estimate of 71%. Kurtzberg et al.15 observed an OS of 57.3% (43 out of 75 patients) at day 100, and this value was similar to the OS rate at two years (40%) in a study by Prasad et al.14 and Le Blanc et al.42 who observed an OS rate at two years of 45% in their cohort of pediatric patients. Although our study was not randomized, considering that our patients were treated in very advanced stages of aGvHD the survival rate is very encouraging (Online Supplementary Table S5).
In summary, to our knowledge this is the first serum-free MSC bank generated from pooled BM-MNC of multiple donors as a source for bulk production of clinical-grade MSC with a predictable potency. Importantly, clinical data presented in this study demonstrated the in vivo safety and efficacy of MEP. Although the results of this single patient treatment are encouraging, a prospective randomized study is required to evaluate the beneficial effect of MEP as a novel cell-based therapy in the treatment of severe aGvHD.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/8/985
Funding
The authors would like to thank the Robert Pfleger Stiftung, DKMS and Else Kröner-Fresenius-Stiftung (2011_A186) for funding this study. HB, TK and PB are supported by the LOEWE Center for Cell and Gene Therapy Frankfurt/Main funded by Hessisches Ministerium für Wissenschaft und Kunst (HMWK) (funding reference number: III L 4-518/17.004, 2010). The authors also express their gratitude to Frankfurter Stiftung für krebskranke Kinder (Frankfurt, Germany) for the kind financial support of SK and Dr. Andrea Jochheim-Richter for the expert help in preparation of regulatory issues.
References
- 1.Lazarus HM, Haynesworth SE, Gerson SL, Rosenthal NS, Caplan AI. Ex vivo expansion and subsequent infusion of human bone marrow-derived stromal progenitor cells (mesenchymal progenitor cells): implications for therapeutic use. Bone Marrow Transplant. 1995;16(4):557–564. [PubMed] [Google Scholar]
- 2.Menard C, Pacelli L, Bassi G, et al. Clinical-grade mesenchymal stromal cells produced under various good manufacturing practice processes differ in their immunomodulatory properties: standardization of immune quality controls. Stem Cells Dev. 2013;22(12): 1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galipeau J. The mesenchymal stromal cells dilemma–does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy. 2013;15(1):2–8. [DOI] [PubMed] [Google Scholar]
- 4.Wegmeyer H, Broske AM, Leddin M, et al. Mesenchymal stromal cell characteristics vary depending on their origin. Stem Cells Dev. 2013;22(19):2606–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castro-Malaspina H, Gay RE, Resnick G, et al. Characterization of human bone marrow fibroblast colony-forming cells (CFU-F) and their progeny. Blood 1980;56(2):289–301. [PubMed] [Google Scholar]
- 6.Francois M, Romieu-Mourez R, Li M, Galipeau J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther. 2012;20(1):187–195. [DOI] [PubMed] [Google Scholar]
- 7.Kuçi Z, Seiberth J, Latifi-Pupovci H, et al. Clonal analysis of multipotent stromal cells derived from CD271+ bone marrow mononuclear cells: functional heterogeneity and different mechanisms of allosuppression. Haematologica. 2013;98(10):1609–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Bahr L, Sundberg B, Lonnies L, et al. Long-term complications, immunologic effects, and role of passage for outcome in mesenchymal stromal cell therapy. Biol Blood Marrow Transplant. 2012;18(4):557–564. [DOI] [PubMed] [Google Scholar]
- 9.Binato R, de Souza FT, Lazzarotto-Silva C, et al. Stability of human mesenchymal stem cells during in vitro culture: considerations for cell therapy. Cell Prolif. 2013; 46(1):10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samuelsson H, Ringden O, Lonnies H, Le BK. Optimizing in vitro conditions for immunomodulation and expansion of mesenchymal stromal cells. Cytotherapy. 2009; 11(2):129–136. [DOI] [PubMed] [Google Scholar]
- 11.Koc ON, Gerson SL, Cooper BW, et al. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J Clin Oncol. 2000;18(2):307–316. [DOI] [PubMed] [Google Scholar]
- 12.Kebriaei P, Isola L, Bahceci E, et al. Adult human mesenchymal stem cells added to corticosteroid therapy for the treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15(7):804–811. [DOI] [PubMed] [Google Scholar]
- 13.Le Blanc K, Rasmusson I, Sundberg B, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363(9419):1439–1441. [DOI] [PubMed] [Google Scholar]
- 14.Prasad VK, Lucas KG, Kleiner GI, et al. Efficacy and safety of ex vivo cultured adult human mesenchymal stem cells (Prochymal) in pediatric patients with severe refractory acute graft-versus-host disease in a compassionate use study. Biol Blood Marrow Transplant. 2011;17(4):534–541. [DOI] [PubMed] [Google Scholar]
- 15.Kurtzberg J, Prockop S, Teira P, et al. Allogeneic human mesenchymal stem cell therapy (remestemcel-L, Prochymal) as a rescue agent for severe refractory acute graft-versus-host disease in pediatric patients. Biol Blood Marrow Transplant. 2014;20(2):229–235. [DOI] [PubMed] [Google Scholar]
- 16.Spees JL, Gregory CA, Singh H, et al. Internalized antigens must be removed to prepare hypoimmunogenic mesenchymal stem cells for cell and gene therapy. Mol Ther. 2004;9(5):747–756. [DOI] [PubMed] [Google Scholar]
- 17.Doucet C, Ernou I, Zhang Y, et al. Platelet lysates promote mesenchymal stem cell expansion: a safety substitute for animal serum in cell-based therapy applications. J Cell Physiol 2005;205(2):228–236. [DOI] [PubMed] [Google Scholar]
- 18.Muller I, Kordowich S, Holzwarth C, et al. Animal serum-free culture conditions for isolation and expansion of multipotent mesenchymal stromal cells from human BM. Cytotherapy. 2006;8(5):437–444. [DOI] [PubMed] [Google Scholar]
- 19.Lucchini G, Introna M, Dander E, et al. Platelet-lysate-expanded mesenchymal stromal cells as a salvage therapy for severe resistant graft-versus-host disease in a pediatric population. Biol Blood Marrow Transplant. 2010;16(9):1293–1301. [DOI] [PubMed] [Google Scholar]
- 20.Introna M, Lucchini G, Dander E, et al. Treatment of graft versus host disease with mesenchymal stromal cells: a phase I study on 40 adult and pediatric patients. Biol Blood Marrow Transplant. 2014;20(3):375–381. [DOI] [PubMed] [Google Scholar]
- 21.von Bonin M, Stolzel F, Goedecke A, et al. Treatment of refractory acute GVHD with third-party MSC expanded in platelet lysate-containing medium. Bone Marrow Transplant. 2009;43(3):245–251. [DOI] [PubMed] [Google Scholar]
- 22.Te Boome LC, Mansilla C, van der Wagen LE, et al. Biomarker profiling of steroid-resistant acute GVHD in patients after infusion of mesenchymal stromal cells. Leukemia. 2015;29(9):1839–1846. [DOI] [PubMed] [Google Scholar]
- 23.Cooper K, Viswanathan C. Establishment of a mesenchymal stem cell bank. Stem Cells Int. 2011;2011:905621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabatino M, Ren J, David-Ocampo V, et al. The establishment of a bank of stored clinical bone marrow stromal cell products. J Transl Med. 2012;10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong W, Han Z, Zhao H, et al. Banking human umbilical cord-derived mesenchymal stromal cells for clinical use. Cell Transplant. 2012;21(1):207–216. [DOI] [PubMed] [Google Scholar]
- 26.Mamidi MK, Nathan KG, Singh G, et al. Comparative cellular and molecular analyses of pooled bone marrow multipotent mesenchymal stromal cells during continuous passaging and after successive cryopreservation. J Cell Biochem. 2012; 113(10):3153–3164. [DOI] [PubMed] [Google Scholar]
- 27.Phinney DG, Kopen G, Righter W, Webster S, Tremain N, Prockop DJ. Donor variation in the growth properties and osteogenic potential of human marrow stromal cells. J Cell Biochem. 1999;75(3):424–436. [PubMed] [Google Scholar]
- 28.Russell KC, Phinney DG, Lacey MR, Barrilleaux BL, Meyertholen KE, O’Connor KC. In vitro high-capacity assay to quantify the clonal heterogeneity in trilineage potential of mesenchymal stem cells reveals a complex hierarchy of lineage commitment. Stem Cells 2010;28(4):788–798. [DOI] [PubMed] [Google Scholar]
- 29.Muraglia A, Cancedda R, Quarto R. Clonal mesenchymal progenitors from human bone marrow differentiate in vitro according to a hierarchical model. J Cell Sci. 2000; 113:1161–1166. [DOI] [PubMed] [Google Scholar]
- 30.Moll G, Jitschin R, von Bahr L, et al. Mesenchymal stromal cells engage complement and complement receptor bearing innate effector cells to modulate immune responses. PLoS One. 2011;6(7):e21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ringden O, Le Blanc K. Mesenchymal stem cells for treatment of acute and chronic graft-versus-host disease, tissue toxicity and hemorrhages. Best Pract Res Clin Haematol. 2011;24(1):65–72. [DOI] [PubMed] [Google Scholar]
- 32.Ringden O, Keating A. Mesenchymal stromal cells as treatment for chronic GVHD. Bone Marrow Transplant. 2011;46(2):163–164. [DOI] [PubMed] [Google Scholar]
- 33.Francois M, Copland IB, Yuan S, Romieu-Mourez R, Waller EK, Galipeau J. Cryopreserved mesenchymal stromal cells display impaired immunosuppressive properties as a result of heat-shock response and impaired interferon-gamma licensing. Cytotherapy. 2012;14(2):147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moll G, Alm JJ, Davies LC, et al. Do cryopreserved mesenchymal stromal cells display impaired immunomodulatory and therapeutic properties? Stem Cells. 2014; 32(9):2430–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Lima PK, de Santis GC, Orellana MD, et al. Cryopreservation of umbilical cord mesenchymal cells in xenofree conditions. Cytotherapy. 2012;14(6):694–700. [DOI] [PubMed] [Google Scholar]
- 36.Al-Saqi SH, Saliem M, Quezada HC, et al. Defined serum- and xeno-free cryopreservation of mesenchymal stem cells. Cell Tissue Bank. 2015;16:181–93. [DOI] [PubMed] [Google Scholar]
- 37.Luetzkendorf J, Nerger K, Hering J, et al. Cryopreservation does not alter main characteristics of Good Manufacturing Process-grade human multipotent mesenchymal stromal cells including immunomodulating potential and lack of malignant transformation. Cytotherapy. 2015;17(2):186–198. [DOI] [PubMed] [Google Scholar]
- 38.Tarte K, Gaillard J, Lataillade JJ, et al. Clinical-grade production of human mesenchymal stromal cells: occurrence of aneuploidy without transformation. Blood. 2010;115(8):1549–1553. [DOI] [PubMed] [Google Scholar]
- 39.Fekete N, Erle A, Amann EM, et al. Effect of High-Dose Irradiation on Human Bone-Marrow-Derived Mesenchymal Stromal Cells. Tissue Eng Part C Methods. 2015; 21:112–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shibata KR, Aoyama T, Shima Y, et al. Expression of the p16INK4A gene is associated closely with senescence of human mesenchymal stem cells and is potentially silenced by DNA methylation during in vitro expansion. Stem Cells. 2007;25(9): 2371–2382. [DOI] [PubMed] [Google Scholar]
- 41.Martin PJ, Uberti JP, Soiffer RJ, et al. Prochymal improves response rates in patients with steroid-refractory acute graft versus host disease (SR-GVHD) involving the liver and gut: results of a randomized, placebo-controlled, multicenter phase III trial in GVHD. Biol Blood Marrow Transplant. 2010;16(2):169–170. [Google Scholar]
- 42.Le Blanc K, Frassoni F, Ball L, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008; 371(9624):1579–1586. [DOI] [PubMed] [Google Scholar]