

Abstract
Nicotinic acid adenine dinucleotide phosphate (NAADP) potently releases Ca2+ from acidic intracellular endolysosomal Ca2+ stores. It is widely accepted that two types of two‐pore channels, termed TPC1 and TPC2, are responsible for the NAADP‐mediated Ca2+ release but the underlying mechanisms regulating their gating appear to be different. For example, although both TPC1 and TPC2 are activated by NAADP, TPC1 appears to be additionally regulated by cytosolic Ca2+. Ion conduction and permeability also differ markedly. TPC1 and TPC2 are permeable to a range of cations although biophysical experiments suggest that TPC2 is slightly more selective for Ca2+ over K+ than TPC1 and hence capable of releasing greater quantities of Ca2+ from acidic stores. TPC1 is also permeable to H+ and therefore may play a role in regulating lysosomal and cytosolic pH, possibly creating localised acidic domains. The significantly different gating and ion conducting properties of TPC1 and TPC2 suggest that these two ion channels may play complementary physiological roles as Ca2+‐release channels of the endolysosomal system.
Abbreviations
- NAADP
nicotinic acid adenine dinucleotide phosphate
- TPC
two‐pore channel
- PI(3,5)P2
phosphatidylinositol 3,5‐bisphosphate
Introduction
In animal cells, the potent Ca2+‐releasing second messenger nicotinic acid adenine dinucleotide phosphate (NAADP) initiates Ca2+ release from the endolysosomal system (Cancela et al. 1999; Kinnear et al. 2004; Yamasaki et al. 2004; Brailoiu et al. 2005; Macgregor et al. 2007). The recently discovered family of proteins named two‐pore channels (TPCs) are associated with NAADP‐mediated Ca2+ signalling (Calcraft et al. 2009). Consistent with their role as the target channels of NAADP, overexpression of TPCs in cellular models potentiate NAADP‐evoked Ca2+ release (Brailoiu et al. 2009; Calcraft et al. 2009; Zong et al. 2009; Hooper et al. 2011), and electrophysiological studies using patch clamp or planar lipid bilayer methods provide direct evidence that TPCs are functional ion channels that are regulated by NAADP (Brailoiu et al. 2010; Pitt et al. 2010, 2014; Schieder et al. 2010; Rybalchenko et al. 2012; Jha et al. 2014; Sakurai et al. 2015). Furthermore gene knockdown, gene silencing or altered molecular function of TPCs abolishes NAADP‐induced Ca2+ signals (Brailoiu et al. 2010; Schieder et al. 2010; Yamaguchi et al. 2011; Grimm et al. 2014; Davidson et al. 2015; Ruas et al. 2015), providing further evidence that TPCs are intimately linked with NAADP‐mediated responses.
TPC‐mediated responses are becoming increasingly linked with disease, including myocardial ischaemia (Davidson et al. 2015), Parkinson's disease (Hockey et al. 2015), and Ebola infection (Sakurai et al. 2015). The function and dysfunction of TPCs in pathophysiology have been reviewed recently (Patel, 2015). The importance of TPCs in both physiology and pathophysiology makes them attractive therapeutic targets, but a better understanding of their biophysical properties and pharmacology is required before clinically relevant compounds can be designed.
Biophysical approaches have enabled us to study the conductance and gating properties of TPCs and these studies have revealed that the unique characteristics of NAADP‐induced Ca2+ release seen in mammalian cells are mirrored in experiments where TPCs are monitored. Measurements of channel activity following the addition of NAADP to purified TPC2 complexes incorporated into planar lipid bilayers provide an EC50 value for NAADP activation of approximately 5 nm (Pitt et al. 2010). This is consistent with K d values reported for NAADP binding to membranes isolated from cells overexpressing TPC2 and membranes isolated from mouse liver (Calcraft et al. 2009). In mammalian cells, NAADP‐mediated Ca2+ responses display a bell‐shaped concentration–response curve, whereby an optimum concentration of NAADP evokes maximal Ca2+ release. Increasing concentrations of NAADP above this optimum level cause a reduction in the Ca2+ response, and very high concentrations of NAADP prevent the release of Ca2+ (Cancela et al. 1999; Berg et al. 2000; Rosen et al. 2009; Zong et al. 2009). This pharmacological profile is echoed in biophysical studies of TPC2. Fusion of purified TPC2 complexes into artificial membranes (Pitt et al. 2010) and measurements of whole‐lysosomal currents from lysosomes overexpressing TPC2 (Schieder et al. 2010) show that NAADP‐mediated currents are abolished following the addition of NAADP in the micromolar to millimolar range. These data support the suggestion that TPC2 has at least two NAADP binding sites (Patel et al. 2001; Calcraft et al. 2009; Rosen et al. 2009), a high‐affinity activation site and a low‐affinity inhibition site (Rosen et al. 2009; Pitt et al. 2010). TPC1 is also activated by nanomolar concentrations of NAADP (Brailoiu et al. 2009; Rybalchenko et al. 2012; Pitt et al. 2014) but in contrast to TPC2, the gating of reconstituted human TPC1 channels fused into planar lipid bilayers is not significantly attenuated following the addition of NAADP up to a concentration of 1 mm (Pitt et al. 2014) suggesting that the regulation of TPC2 and TPC1 is markedly different.
Does NAADP activate TPCs directly?
There is growing evidence to support the idea that TPCs play a central role in mediating NAADP responses, but does NAADP bind directly to TPCs? Recombinant TPC2 proteins incorporated into artificial bilayers are consistently modulated by the addition of NAADP to the cytosolic face of the channel (Pitt et al. 2010), but conventional whole cell/excised patch recordings of TPCs from whole lysosomes show disparate results. Wang and co‐workers report a complete lack of sensitivity to NAADP (Wang et al. 2012) whereas other studies using an identical approach show robust TPC‐mediated currents in response to NAADP (Brailoiu et al. 2010; Schieder et al. 2010; Jha et al. 2014; Ruas et al. 2015). When recording, from plasma membrane patches, from TPC2 channels redirected to the plasma membrane there appears to be a loss of TPC function in approximately 45% of experiments (Jha et al. 2014). It is puzzling that these inconsistencies exist, but can we explain these irregularities by the presence or lack of an NAADP‐binding protein located within the TPC complex?
Photoaffinity labelling of NAADP revealed that NAADP targets proteins much smaller in size than TPCs (Lin‐Moshier et al. 2012; Walseth et al. 2012). This suggests that TPCs may exist within a larger protein complex and that NAADP may bind to individual accessory proteins within this complex rather than directly binding to the TPCs. These putative NAADP‐binding proteins are reported to co‐immunoprecipitate with TPCs (Ruas et al. 2010; Walseth et al. 2012) and to persist in transgenic mice lacking either TPC2 or TPC1 (Lin‐Moshier et al. 2012) or lacking both TPC1 and TPC2 (Ruas et al. 2015), inferring that TPCs are the pore‐forming subunits of a larger complex. The presence or absence of these proteins may therefore explain the variability of NAADP sensitivity across experimental systems (Marchant & Patel, 2013). It is feasible, therefore, that NAADP could bind to a secondary protein that is either tightly complexed with TPCs or that translocates to TPCs following NAADP binding to form part of the TPC complex. In both cases, the interaction of an NAADP‐bound binding protein with a TPC would be likely to modify TPC function as an ion channel. This does not rule out the possibility that NAADP could additionally interact directly with and regulate TPCs. The isolation, purification and identification of the proposed high‐affinity NAADP‐binding proteins are the next key experimental objectives in this field. Until the binding proteins are identified, we will not grasp whether NAADP exerts a long‐range induction of Ca2+ release from acidic stores involving multiple intracellular signalling steps and possibly multiple ion channels or whether a short range or direct effect of NAADP on TPCs is the primary mechanism that elicits Ca2+ release.
Is NAADP the only activator of TPC activity?
Although there is strong evidence that TPCs are required for NAADP‐induced Ca2+ release from lysosomal Ca2+ stores, recent reports argue that TPCs are not targeted by NAADP. Using conventional patch clamp of enlarged endolysosomes from TPC1 or TPC2 overexpressing cells, certain studies failed to observe any NAADP‐induced currents. Instead the endolysosome‐located lipid, phosphatidylinositol 3,5‐bisphosphate (PI(3,5)P2), robustly activated a current in these patches (Wang et al. 2012; Cang et al. 2013). Surprisingly in a double knockout mouse model created by disrupting the genes which code for TPC1 and TPC2, Wang et al. revealed that the NAADP‐mediated Ca2+ signal was unaffected in pancreatic β‐cells providing further evidence that TPCs do not respond to NAADP (Wang et al. 2012). Similarly, patch clamp studies of human TPC2 channels fused with an enhanced green fluorescent protein targeted to plant vacuoles showed insensitivity to NAADP but increased activity following application of nanomolar concentrations of PI(3,5)P2 (Boccaccio et al. 2014). Other studies using direct patch‐clamp of both enlarged endolysosomes overexpressing TPC2 or TPC1 and excised patches from HEK293 cells expressing TPC2 suggest that both NAADP and PI(3,5)P2 activate TPC‐mediated currents (Jha et al. 2014; Sakurai et al. 2015). When incorporated into artificial bilayers TPC1 is not activated by PI(3,5)P2 and PI(3,5)P2 does not potentiate NAADP‐mediated TPC1 responses (Pitt et al. 2014).
It is difficult to reconcile the contrasting data obtained by different groups. Recent work by Ruas and co‐workers may help shed some light on this conundrum (Ruas et al. 2015). When tpcn1/2 genes were disrupted by deletion of the first exon in order to reproduce the methods used by Cang et al. (2013) and Wang et al. (2012), the resultant transcript coded for a truncated but functional TPC protein, able to support NAADP‐induced Ca2+ release (Ruas et al. 2015). Production of a mouse line with demonstrable absence of both tpcn1 and tpcn2 expression, however, led to complete loss of any endogenous NAADP‐dependent Ca2+ responses as assessed by single‐cell Ca2+ imaging or patch‐clamp of single endolysosomes, supporting the theory that TPCs are required for NAADP‐mediated responses (Ruas et al. 2015). Nonetheless, this still does not fully explain why certain groups do not observe any effect of NAADP on TPC‐mediated currents, whether in wild‐type mouse cells or in cell lines overexpressing TPCs. To date, there are few studies describing the gating and conductance properties of TPC channels. When the mechanisms controlling these features are better understood, the reasons for the apparent discrepancies may be revealed.
Activation of TPCs by Ca2+
Although there is convincing evidence to support the idea that TPCs are NAADP‐regulated ion channels, there is evidence to suggest that multiple ligands regulate TPCs (Rybalchenko et al. 2012; Jha et al. 2014; Pitt et al. 2014). Recombinant human TPC1 channels incorporated into artificial membranes are activated by both NAADP and cytosolic Ca2+ (Pitt et al. 2014) but the simultaneous presence of both ligands is not required for channel opening. The same optimum level of channel activity appears to be induced by Ca2+ or NAADP. Thus, if TPC1 is activated first by an optimal concentration of Ca2+, presumably the subsequent presence of NAADP will have no further effect on the open probability of the channel. Perhaps this feature of TPC1 control may explain the reported inability of NAADP to activate TPC1 under certain experimental conditions (Wang et al. 2012; Cang et al. 2013, 2014 b). On the other hand the activity of TPC2 appears to be unaffected by cytosolic Ca2+ (Brailoiu et al. 2010; Pitt et al. 2011). Electrophysiological approaches have also uncovered a stimulatory effect of luminal Ca2+ on TPC2 channel activity (Pitt et al. 2010). Increasing the luminal [Ca2+] increases the sensitivity of TPC2 to NAADP (Pitt et al. 2010) and this may be an important mechanism for controlling release of Ca2+ from acidic stores. Since TPC2 is regarded as a Ca2+‐release channel it is easy to envisage that as Ca2+ is released from the lysosome, the resulting drop in the luminal Ca2+ concentration may serve as a feedback mechanism to regulate channel activity. The role of luminal Ca2+ on TPC1 activity is less clear. Fusion of microsomes prepared from HEK cells overexpressing TPC1 into artificial bilayers revealed that, in the presence of NAADP and using Ba2+ as the permeant ion, luminal Ca2+ increases channel activity in a dose‐dependent manner (Rybalchenko et al. 2012). In another study (Pitt et al. 2014) luminal Ca2+ had no effect on the activity of purified TPC1 channels incorporated into artificial bilayers regardless of whether the channel was activated by NAADP or cytosolic Ca2+.
Activation of TPCs by voltage
It has been proposed that endosomes and lysosomes are electrically excitable (Cang et al. 2014 a,b), but how these organelles sense and control changes in the membrane potential is not fully understood. Endolysosomes overexpressing TPC1 display a voltage‐dependent Na+ current that is absent from transgenic animals generated with a disrupted tpcn1 gene and restored following TPC1 transfection (Cang et al. 2014 b). These data suggest that TPC1 voltage‐gated endolysosomal Na+ currents enable endolyosomes to be electrically excitable. Importantly, voltage not only regulates TPC1 activity but also modifies the apparent affinity of NAADP for TPC1 (Rybalchenko et al. 2012). Here, TPC1 can be considered to be voltage‐regulated rather than voltage‐gated as the presence of NAADP or cytosolic Ca2+ is an absolute requirement for channel activation (Pitt et al. 2014). The lysosomal membrane potential has been reported to be about 20 mV lumen‐positive (Koivusalo et al. 2011). At rest, the apparent affinity for activation of TPC1 by NAADP will therefore be low, and the channel will reside in the closed state. As the membrane potential becomes more depolarised, this will increase the affinity of NAADP for TPC1 and also intrinsically increase channel activity through voltage regulation. This suggests that in cells, the membrane potential dynamically alters TPC1 channel activity (Rybalchenko et al. 2012; Pitt et al. 2014). TPC2 is voltage insensitive (Cang et al. 2014 b), which is surprising given that all TPCs have a putative voltage sensor. TPC1 may therefore contribute to excitability within the endolysosomal system.
Ned compounds as pharmacological tools
Ned‐19 is a molecular analogue of NAADP, discovered by a ligand‐based computational drug discovery approach (Naylor et al. 2009), that is now widely employed as a membrane‐permeant NAADP antagonist (Pereira et al. 2011; Davis et al. 2012; Aley et al. 2013; Lu et al. 2013; Ruas et al. 2015). In Ca2+‐release experiments, Ned‐19 selectively antagonises NAADP‐induced responses (Naylor et al. 2009; Rosen et al. 2009). Single channel experiments reveal that Ned‐19 has very different actions on TPC1 and TPC2 function. Ned‐19 at 1 μm antagonises NAADP‐mediated activation of TPC2 in a non‐competitive manner, but in the concentration range 1–100 nm, Ned‐19 potentiates NAADP‐mediated TPC2 responses (Pitt et al. 2010). Interestingly, Ned‐19 does not appear to modulate TPC1 activity (Pitt et al. 2014), but this work is still in the early stages. These data have important ramifications for interpreting cellular studies where Ned‐19 is used to reveal the presence or function of TPCs. Although further characterisation of the molecular mechanisms by which Ned‐19 influences TPC function is required, Ned‐19 may be a useful pharmacological tool to distinguish, at the cellular level, the specific physiological roles of TPC1 and TPC2. Recent data has revealed that a chemically modified form of Ned‐19 called Ned‐K, produced by replacing the fluoride with a cyano group, inhibits NAADP‐mediated Ca2+ oscillations thought to be dependent on TPC1 activity (Davidson et al. 2015). Although the direct pharmacological effects of Ned‐K on TPC1 and TPC2 are unknown, it appears that this compound may behave as an antagonist of both TPC isoforms.
Conductance properties of TPCs
It is widely accepted that NAADP is capable of initiating the release of Ca2+ from acidic stores and that it regulates many essential cellular processes. TPC1 and TPC2 are often thought of simply as ‘NAADP Ca2+ release channels’ rather than as two different ion channels with distinct mechanisms regulating gating and with distinct conductance and selectivity properties. The few reports where TPC1 and TPC2 ion channel function have been studied highlight that there are many important differences in their ability to conduct ions. Although TPC1 and TPC2 are both cation channels and are both permeable to Ca2+, their selectivity and relative permeability towards monovalent and divalent cations, including protons, are different.
In animals, several reports have demonstrated that TPCs are non‐selective cation channels displaying permeability to all of the major ions thought to play a role in the endolysosomal system including Ca2+, K+, Na+ and H+. Construction of single‐channel current–voltage relationships in biophysical studies have yielded a Ca2+ conductance between 15 and 40 pS (Brailoiu et al. 2010; Pitt et al. 2010) for TPC2 and 19 pS for TPC1 (Pitt et al. 2014). Although TPC1 and TPC2 show a similar Ca2+ conductance, TPC2 shows a much higher K+ conductance of 300 pS compared with TPC1, which has a K+ conductance of only 87 pS (Pitt et al. 2010, 2014). Both TPC1 and TPC2 are permeable to Na+ (Wang et al. 2012; Cang et al. 2013; Sakurai et al. 2015). To our knowledge there is no estimate of the single‐channel Na+ conductance for TPC2 but estimates for TPC1 suggest a Na+ conductance of 68 pS (Pitt et al. 2014). Interestingly, with Na+ as the only permeant ion, the open probability of TPC1 is reported to be much less than that with K+ as the permeant ion, suggesting that the permeant ion influences TPC1 activity (Pitt et al. 2014). When TPC1 is incorporated into artificial membranes, a permeability to protons is also revealed (Pitt et al. 2014).
Ion selectivity of TPCs
Biophysical studies indicate that when activated by NAADP, TPC2 is more selective for Ca2+ than K+ (Pitt et al. 2010; Schieder et al. 2010) whereas TPC1 is more permeable to monovalent cations (Rybalchenko et al. 2012; Pitt et al. 2014). Reconstitution of TPC1 into artificial membranes reveals a relative permeability sequence in the order H+ ≫ K+ > Na+ ≥ Ca2+ (Pitt et al. 2014). These data would implicate TPC1 with a role in maintaining or changing lysosomal pH alongside a leak of Ca2+, whereas TPC2 would be expected to release more Ca2+ from stores when activated. Experiments from other groups, however, suggest that the role of TPCs in the endolysosomal system is to support the overall process of Ca2+‐release by modulating monovalent cation flux rather than primarily acting as Ca2+‐release channels (Wang et al. 2012; Cang et al. 2013, 2014 b). The reports that TPCs are Na+‐selective channels that are activated by PI(3,5)P2 must be evaluated. In these studies, measurements of lysosomal currents from cells overexpressing TPC2 provided a Ca2+/Na+ relative permeability of approximately 0.1 (Wang et al. 2012; Cang et al. 2013). Several other groups have also reported that TPCs are permeable to Na+ (Jha et al. 2014; Pitt et al. 2014; Ruas et al. 2015; Sakurai et al. 2015), but the permeability of TPCs to Ca2+ or Na+ is of the same order of magnitude (Pitt et al. 2014; Ruas et al. 2015), suggesting that under specific ionic conditions, TPCs do act as Ca2+‐release channels. In single‐channel studies purified recombinant TPC1 is not activated or inhibited by PI(3,5)P2, but this lipid appears to alter the conducting properties of TPC1 by increasing the permeability of H+ and Na+ relative to Ca2+ (Pitt et al. 2014). Perhaps this action helps explain why a large Na+ current is observed in isolated lysosomal organelles overexpressing TPCs following the addition of PI(3,5)P2.
Can TPCs function as lysosomal Ca2+‐release channels?
On the basis of the rank order of ion selectivity displayed by both TPC2 and TPC1 in reconstitution studies, it would appear that TPC2 could support the release of Ca2+ from lysosomes or endolysosomes whereas TPC1 might only contribute a small fraction of the released Ca2+, especially in lysosomes where TPC2 is expressed in higher levels than TPC1 (Aley et al. 2010; Zhu et al. 2010). The capacity for TPC1 to participate in leaking Ca2+ may become more important in NAADP‐mediated Ca2+ responses when levels of TPC1 exceed those of TPC2. Studies suggest that TPC1 is capable of mediating endolysosomal Ca2+ currents when TPC2 expression is knocked out or where TPC1 is overexpressed (Brailoiu et al. 2009; Ruas et al. 2010; Davis et al. 2012), and in this respect both TPC1 and TPC2 may be considered as NAADP‐regulated Ca2+‐release channels (Fig. 1). Reconstitution studies also suggest that TPC1 would provide a proton flux that could be regulated both by NAADP and cytosolic Ca2+ but additionally by PI(3,5)P2 (Fig. 1). Such simplistic interpretation of the roles of TPC1 and TPC2 is all that is possible given the paucity of information available regarding ion conductance in TPCs. Since both TPC1 and TPC2 are relatively non‐selective towards cations, small changes in factors such as ionic composition of the acidic organelle, expression levels of the channels, intraluminal pH, membrane potential and concentrations of regulatory ligands (including NAADP, Ca2+ and PI(3,5)P2) may cause large changes to the current flux through the channels. A more comprehensive understanding of the ionic nature of the endolysosomal system is required before accurate predictions of the contributions made by TPC1 and TPC2 to NAADP‐ and non‐NAADP‐activated Ca2+ release from acidic stores can be calculated.
Figure 1. Schematic diagram showing regulation of TPC1 and TPC2 function in the endolysosomal system .
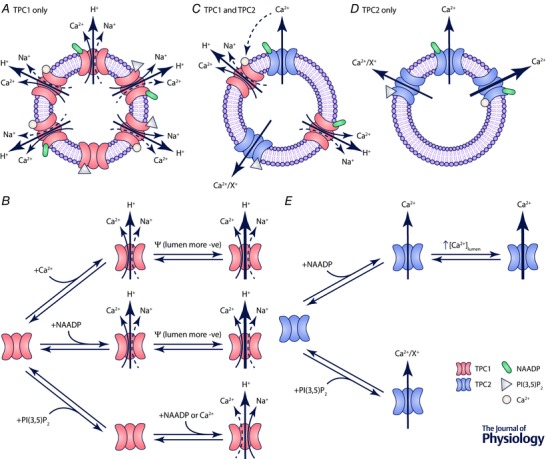
Representation of the predicted ion fluxes from endolysosomes via TPC1 (A), and channel state mechanism for TPC1 (B). Both NAADP and an increase in the cytosolic Ca2+ concentration activate TPC1. Given that the relative permeability sequence of TPC1 is in the order H+ ≫ K+ > Na+ ≥ Ca2+, this will result primarily in the release of H+ alongside smaller fluxes of Ca2+ and Na+. High intraluminal Ca2+ will favour Ca2+ flux over Na+. PI(3,5)P2 is unable to activate TPC1 directly but alters ion selectivity in favour of Na+ compared with Ca2+. Depolarisation of the endolysosome membrane (lumen becomes more negative compared to cytosol) increases TPC1 activity. C, potential interaction between TPC1 and TPC2. NAADP activates both TPC1 and TPC2. Ca2+ will flow outward primarily through TPC2 and H+ through TPC1. Ca2+ released from TPC2 can feed‐forward to recruit and activate TPC1 but does not affect TPC2 gating. As the luminal Ca2+ concentration falls, TPC2 becomes less active. As the membrane becomes depolarised TPC1 activity will increase (as shown in B). Representation of the expected ion fluxes from endolysosomes via TPC2 (D), and channel state mechanism of TPC2 (E). NAADP and PI(3,5)P2 activate TPC2. TPC2 releases Ca2+ from endolysosomes but also displays permeability to other monovalent cations (X+).
Outlook
Biophysical evidence indicates that TPC1 and TPC2, between them, allow movements of Ca2+, K+, Na+ and H+ across the membranes of acidic vesicles but that there are differences in their ion selectivity and regulation of gating (see Table 1). At face value, this suggests that the presence of these two ion channels on cellular acidic Ca2+ stores will provide a highly flexible system for regulating Ca2+ homeostasis in cells and for altering the biochemical environment within lysosomes and endolysosomes.
Table 1.
Basic biophysical properties comparing the activation mechanisms and ion selectivity recorded for TPC1 and TPC2
| Isoform | Activating ligand | Experimental technique | Ion selectivity | Reference |
|---|---|---|---|---|
| TPC1 | NAADP | Planar lipid bilayer | K+ > X2+ | Rybalchenko (2012) |
| H+ ≫ K+ > Na+ ≥ Ca2+ | Pitt (2014) | |||
| PI(3,5)P2 | Expanded endolysosome patch clamp | Na+ ≫ K+ > Ca2+ | Cang (2014) | |
| NAADP and PI(3,5)P2 | Planar lipid bilayer | H+ ≫ Na+ > Ca2+ | Pitt (2014) | |
| Ca2+ | Planar lipid bilayer | H+ ≫ K+ > Na+ ≥ Ca2+ | Pitt (2014) | |
| TPC2 | NAADP | Expanded endolysosome patch clamp (port‐a‐patch) | Ca2+ ⋙ K+ | Scheider (2010) |
| Planar lipid bilayer | Ca2+ > K+ | Pitt (2010, 2014) | ||
| Whole cell patch clamp | Na+ > K+ | Jha (2014) | ||
| PI(3,5)P2 | Expanded endolysosome patch clamp | Na+ > Li+ ≫ Ca2+ > K+ = Cs+ | Wang (2012) | |
| Na+ > Ca2+ | Sakurai (2015) |
The diverse properties of TPCs reported by different groups highlight the need for further experimentation. It is clear that we are missing crucial parts of the puzzle regarding the mechanisms linking NAADP to the release of Ca2+ from acidic stores. The identity, location and functional properties of the putative NAADP binding proteins are important missing pieces of information and we predict that the field will move forward quickly when armed with this information. Solving the structures of TPC1, TPC2 and the elusive binding proteins are long term aims that will reveal many key aspects to this puzzle, bringing ion conduction into focus and enabling investigation into the nature of the NAADP binding sites.
Additional information
Competing interests
None of the authors has any conflicts of interests.
Author contributions
S.J.P. and R.S. contributed equally to writing all sections of the manuscript. B.R.O.D. designed and prepared the figures and Table 1 and contributed towards writing the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
S.J.P. is supported by a Royal Society of Edinburgh Biomedical Fellowship (XRE013). R.S. is supported by the Department of Pharmacology, University of Oxford.
Biographies
Samantha Pitt gained her PhD degree from the University of Cambridge. Following two postdoctoral positions studying mechanisms of ion‐channel regulation, one at University College London (Department of Pharmacology) and one at the University of Bristol (Department of Pharmacology) she moved to her current position as a Royal Society of Edinburgh Biomedical Fellow in the School of Medicine, University of St Andrews. Her research group investigates intracellular calcium dynamics and the molecular function of intracellular ion channels. At present, she has a particular interest in understanding the molecular mechanisms of NAADP‐regulated signalling via two‐pore channels.

Benedict Reilly‐O'Donnell is a second year PhD student at the University of St Andrews. He is interested in the single channel properties of two‐pore channels and other intracellular calcium‐release channels. Using a combination of electrophysiological, molecular and biochemical methods, he is currently investigating the role of zinc in intracellular calcium release. Benedict completed his undergraduate studies in pharmacology at University College London in 2013.
Rebecca Sitsapesan obtained her PhD at the University of Strathclyde and following appointment as British Heart Foundation Basic Science Lecturer at Imperial College London moved to Bristol University and then to the University of Oxford where she is currently Professor of Pharmacology. Her group investigates the biophysical properties of RyR and other ion channels that are present on intracellular organelles and are involved in the process of intracellular Ca2+ release, particularly in regard to cardiac physiology and pathophysiology.
This review was presented at the symposium “Non‐selective cationic channels in chemical and physical stress”, which took place at Physiology 2015 in Cardiff, UK, 6–8 July 2015.
References
- Aley PK, Mikolajczyk AM, Munz B, Churchill GC, Galione A & Berger F (2010). Nicotinic acid adenine dinucleotide phosphate regulates skeletal muscle differentiation via action at two‐pore channels. Proc Natl Acad Sci USA 107, 19927–19932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley PK, Singh N, Brailoiu GC, Brailoiu E & Churchill GC (2013). Nicotinic acid adenine dinucleotide phosphate (NAADP) is a second messenger in muscarinic receptor‐induced contraction of guinea pig trachea. J Biol Chem 288, 10986–10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg I, Potter BVL, Mayr GW & Guse AH (2000). Nicotinic acid adenine dinucleotide phosphate (NAADP+) is an essential regulator of T‐Lymphocyte Ca2+‐signaling. J Cell Biol 150, 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaccio A, Scholz‐Starke J, Hamamoto S, Larisch N, Festa M, Gutla P, Costa A, Dietrich P, Uozumi N & Carpaneto A (2014). The phosphoinositide PI(3,5)P2 mediates activation of mammalian but not plant TPC proteins: functional expression of endolysosomal channels in yeast and plant cells. Cell Mol Life Sci 71, 4275–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu E, Churamani D, Cai XJ, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS & Patel S (2009). Essential requirement for two‐pore channel 1 in NAADP‐mediated calcium signaling. J Cell Biol 186, 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu E, Hoard JL, Filipeanu CM, Brailoiu GC, Dun SL, Patel S & Dun NJ (2005). Nicotinic acid adenine dinucleotide phosphate potentiates neurite outgrowth. J Biol Chem 280, 5646–5650. [DOI] [PubMed] [Google Scholar]
- Brailoiu E, Rahman T, Churamani D, Prole DL, Brailoiu GC, Hooper R, Taylor CW & Patel S (2010). An NAADP‐gated two‐pore channel targeted to the plasma membrane uncouples triggering from amplifying Ca2+ signals. J Biol Chem 285, 38511–38516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcraft PJ, Ruas M, Pan Z, Cheng XT, Arredouani A, Hao XM, Tang JS, Rietdorf K, Teboul L, Chuang KT, Lin PH, Xiao R, Wang CB, Zhu YM, Lin YK, Wyatt CN, Parrington J, Ma JJ, Evans AM, Galione A & Zhu MX (2009). NAADP mobilizes calcium from acidic organelles through two‐pore channels. Nature 459, 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancela JM, Churchill GC & Galione A (1999). Coordination of agonist‐induced Ca2+‐signalling patterns by NAADP in pancreatic acinar cells. Nature 398, 74–76. [DOI] [PubMed] [Google Scholar]
- Cang C, Aranda K & Ren D (2014. a). A non‐inactivating high‐voltage‐activated two‐pore Na+ channel that supports ultra‐long action potentials and membrane bistability. Nat Commun 5, 5015–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang C, Bekele B & Ren D (2014. b). The voltage‐gated sodium channel TPC1 confers endolysosomal excitability. Nat Chem Biol 10, 463–469. [DOI] [PubMed] [Google Scholar]
- Cang C, Zhou Y, Navarro B, Seo Y‐j, Aranda K, Shi L, Battaglia‐Hsu S, Nissim I, Clapham David E & Ren D (2013). mTOR regulates lysosomal ATP‐sensitive two‐pore Na+ channels to adapt to metabolic state. Cell 152, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Foote K, Kunuthur S, Gosain R, Tan N, Tyser R, Zhao YJ, Graeff R, Ganesan A, Duchen MR, Patel S & Yellon DM (2015). Inhibition of NAADP signalling on reperfusion protects the heart by preventing lethal calcium oscillations via two‐pore channel 1 and opening of the mitochondrial permeability transition pore. Cardiovasc Res 108, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LC, Morgan AJ, Chen JL, Snead CM, Bloor‐Young D, Shenderov E, Stanton‐Humphreys MN, Conway SJ, Churchill GC, Parrington J, Cerundolo V & Galione A (2012). NAADP activates two‐pore channels on T cell cytolytic granules to stimulate exocytosis and killing. Curr Biol 22, 2331–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm C, Holdt LM, Chen C‐C, Hassan S, Müller C, Jörs S, Cuny H, Kissing S, Schröder B, Butz E, Northoff B, Castonguay J, Luber CA, Moser M, Spahn S, Lüllmann‐Rauch R, Fendel C, Klugbauer N, Griesbeck O, Haas A, Mann M, Bracher F, Teupser D, Saftig P, Biel M & Wahl‐Schott C (2014). High susceptibility to fatty liver disease in two‐pore channel 2‐deficient mice. Nat Commun 5, 4699. [DOI] [PubMed] [Google Scholar]
- Hockey LN, Kilpatrick BS, Eden ER, Lin‐Moshier Y, Brailoiu GC, Brailoiu E, Futter CE, Schapira AH, Marchant JS & Patel S (2015). Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J Cell Sci 128, 232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper R, Churamani D, Brailoiu E, Taylor CW & Patel S (2011). Membrane topology of NAADP‐sensitive two‐pore channels and their regulation by N‐linked glycosylation. J Biol Chem 286, 9141–9149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Ahuja M, Patel S, Brailoiu E & Muallem S (2014). Convergent regulation of the lysosomal two‐pore channel‐2 by Mg2+, NAADP, PI(3,5)P2 and multiple protein kinases. EMBO J 33, 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnear NP, Boittin FX, Thomas JM, Galione A & Evans AM (2004). Lysosome‐sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin‐1. J Biol Chem 279, 54319–54326. [DOI] [PubMed] [Google Scholar]
- Koivusalo M, Steinberg BE, Mason D & Grinstein S (2011). In situ measurement of the electrical potential across the lysosomal membrane using FRET. Traffic 12, 972–982. [DOI] [PubMed] [Google Scholar]
- Lin‐Moshier Y, Walseth TF, Churamani D, Davidson SM, Slama JT, Hooper R, Brailoiu E, Patel S & Marchant JS (2012). Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J Biol Chem 287, 2296–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y‐Y, Hao B‐X, Graeff R, Wong CWM, Wu W‐T & Yue J (2013). TPC2 signaling inhibits autophagosomal‐lysosomal fusion by alkalizing lysosomal pH. J Biol Chem 288, 24247–24263. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Macgregor A, Yamasaki M, Rakovic S, Sanders L, Parkesh R, Churchill GC, Galione A & Terrar DA (2007). NAADP controls cross‐talk between distinct Ca2+ stores in the heart. J Biol Chem 282, 15302–15311. [DOI] [PubMed] [Google Scholar]
- Marchant JS & Patel S (2013). Questioning regulation of two‐pore channels by NAADP. Messenger 2, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, Rosen D, Thomas JM, Izumi M, Ganesan A, Galione A & Churchill GC (2009). Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol 5, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S (2015). Function and dysfunction of two‐pore channels. Sci Sig 8, re7. [DOI] [PubMed] [Google Scholar]
- Patel S, Churchill GC & Galione A (2001). Coordination of Ca2+ signalling by NAADP. Trends Biochem Sci 26, 482–489. [DOI] [PubMed] [Google Scholar]
- Pereira GJ, Hirata H, Fimia GM, do Carmo LG, Bincoletto C, Han SW, Stilhano RS, Ureshino RP, Bloor‐Young D, Churchill G, Piacentini M, Patel S & Smaili SS (2011). Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J Biol Chem 286, 27875–27881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt SJ, Funnell TM, Sitsapesan M, Venturi E, Rietdorf K, Ruas M, Ganesan A, Gosain R, Churchill GC, Zhu MX, Parrington J, Galione A & Sitsapesan R (2010). TPC2 is a novel NAADP‐sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+ . J Biol Chem 285, 35039–35046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt SJ, Funnell TM, Zhu MX, Parrington J, Ruas M, Galione A & Sitsapesan R (2011). TPC2 is a novel NAADP‐sensitive intracellular Ca2+‐release channel with unique gating characteristics. Biophys J 100, 433a. [Google Scholar]
- Pitt SJ, Lam AKM, Rietdorf K, Galione A & Sitsapesan R (2014). Reconstituted human TPC1 is a proton‐permeable ion channel and is activated by NAADP or Ca2+ . Sci Signal 7, ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen D, Lewis AM, Mizote A, Thomas JM, Aley PK, Vasudevan SR, Parkesh R, Galione A, Izumi M, Ganesan A & Churchill GC (2009). Analogues of the nicotinic acid adenine dinucleotide phosphate (NAADP) antagonist Ned‐19 indicate two binding sites on the NAADP receptor. J Biol Chem 284, 34930–34934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruas M, Davis LC, Chen C‐C, Morgan AJ, Chuang K‐T, Walseth TF, Grimm C, Garnham C, Powell T, Platt N, Platt FM, Biel M, Wahl‐Schott C, Parrington J & Galione A (2015). Expression of Ca2+‐permeable two‐pore channels rescues NAADP signalling in TPC‐deficient cells. EMBO J 34, 1743–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruas M, Rietdorf K, Arredouani A, Davis LC, Lloyd‐Evans E, Koegel H, Funnell TM, Morgan AJ, Ward JA, Watanabe K, Cheng X, Churchill GC, Zhu MX, Platt FM, Wessel GM, Parrington J & Galione A (2010). Purified TPC isoforms form NAADP receptors with distinct roles for Ca2+ signaling and endolysosomal trafficking. Curr Biol 20, 703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalchenko V, Ahuja M, Coblentz J, Churamani D, Patel S, Kiselyov K & Muallem S (2012). Membrane potential regulates nicotinic acid adenine dinucleotide phosphate (NAADP) dependence of the pH‐ and Ca2+‐sensitive organellar two‐pore channel TPC1. J Biol Chem 287, 20407–20416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai Y, Kolokoltsov AA, Chen C‐C, Tidwell MW, Bauta WE, Klugbauer N, Grimm C, Wahl‐Schott C, Biel M & Davey RA (2015). Two‐pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347, 995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieder M, Roetzer K, Brueggemann A, Biel M & Wahl‐Schott CA (2010). Characterization of two‐pore channel 2 (TPCN2)‐mediated Ca2+ currents in isolated lysosomes. J Biol Chem 285, 21219–21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walseth TF, Lin‐Moshier Y, Jain P, Ruas M, Parrington J, Galione A, Marchant JS & Slama JT (2012). Photoaffinity labeling of high affinity nicotinic acid adenine dinucleotide phosphate (NAADP)‐binding proteins in sea urchin egg. J Biol Chem 287, 2308–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang X, Dong XP, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, Zhu MX, Clapham DE, Ren D & Xu H (2012). TPC proteins are phosphoinositide‐activated sodium‐selective ion channels in endosomes and lysosomes. Cell 151, 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Jha A, Li Q, Soyombo AA, Dickinson GD, Churamani D, Brailoiu E, Patel S & Muallem S (2011). Transient receptor potential mucolipin 1 (TRPML1) and two‐pore channels are functionally independent organellar ion channels. J Biol Chem 286, 22934–22942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Masgrau R, Morgan AJ, Churchill GC, Patel S, Ashcroft SJH & Galione A (2004). Organelle selection determines agonist‐specific ca2+ signals in pancreatic acinar and β cells. J Biol Chem 279, 7234–7240. [DOI] [PubMed] [Google Scholar]
- Zhu MX, Ma J, Parrington J, Galione A & Evans AM (2010). TPCs: Endolysosomal channels for Ca2+ mobilization from acidic organelles triggered by NAADP. FEBS Lett 584, 1966–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong XG, Schieder M, Cuny H, Fenske S, Gruner C, Rotzer K, Griesbeck O, Harz H, Biel M & Wahl‐Schott C (2009). The two‐pore channel TPCN2 mediates NAADP‐dependent Ca2+‐release from lysosomal stores. Pflugers Arch 458, 891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]