

Abstract
Key points
Using an experimental rat intensive care unit (ICU) model, not limited by early mortality, we have previously shown that passive mechanical loading attenuates the loss of muscle mass and force‐generation capacity associated with the ICU intervention.
Mitochondrial dynamics have recently been shown to play a more important role in muscle atrophy than previously recognized.
In this study we demonstrate that mitochondrial dynamics, as well as mitophagy, is affected by mechanosensing at the transcriptional level, and muscle changes induced by unloading are counteracted by passive mechanical loading.
The recently discovered ubiquitin ligases Fbxo31 and SMART are induced by mechanical silencing, an induction that similarly is prevented by passive mechanical loading.
Abstract
The complete loss of mechanical stimuli of skeletal muscles, i.e. loss of external strain related to weight bearing and internal strain related to activation of contractile proteins, in mechanically ventilated, deeply sedated and/or pharmacologically paralysed intensive care unit (ICU) patients is an important factor triggering the critical illness myopathy (CIM). Using a unique experimental ICU rat model, mimicking basic ICU conditions, we have recently shown that mechanical silencing is a dominant factor triggering the preferential loss of myosin, muscle atrophy and decreased specific force in fast‐ and slow‐twitch muscles and muscle fibres. The aim of this study is to gain improved understanding of the gene signature and molecular pathways regulating the process of mechanical activation of skeletal muscle that are affected by the ICU condition. We have focused on pathways controlling myofibrillar protein synthesis and degradation, mitochondrial homeostasis and apoptosis. We demonstrate that genes regulating mitochondrial dynamics, as well as mitophagy are induced by mechanical silencing and that these effects are counteracted by passive mechanical loading. In addition, the recently identified ubiquitin ligases Fbxo31 and SMART are induced by mechanical silencing, an induction that is reversed by passive mechanical loading. Thus, mechano‐cell signalling events are identified which may play an important role for the improved clinical outcomes reported in response to the early mobilization and physical therapy in immobilized ICU patients.
Key points
Using an experimental rat intensive care unit (ICU) model, not limited by early mortality, we have previously shown that passive mechanical loading attenuates the loss of muscle mass and force‐generation capacity associated with the ICU intervention.
Mitochondrial dynamics have recently been shown to play a more important role in muscle atrophy than previously recognized.
In this study we demonstrate that mitochondrial dynamics, as well as mitophagy, is affected by mechanosensing at the transcriptional level, and muscle changes induced by unloading are counteracted by passive mechanical loading.
The recently discovered ubiquitin ligases Fbxo31 and SMART are induced by mechanical silencing, an induction that similarly is prevented by passive mechanical loading.
Abbreviations
- 4EBP‐1
4E binding protein‐1
- Akt
protein kinase B (PKB)
- CIM
critical illness myopathy
- CMV
controlled mechanical ventilation
- FbxO
F‐box protein
- FoxO
Forkhead Box O
- HDAC
histone deacetylaces
- ICU
intensive care unit
- Mgn
myogenin
- mTOR
(the mammalian (or mechanistic) target of rapamycin)
- MuRF1
muscle RING finger‐1
- NMB
neuromuscular blockade
- PGC‐1α
PPARγ coactivator‐1α
- RPKM
reads per kilobase per million
- S6
ribosomal protein S6
- S6K
p70 ribosomal protein S6 kinase
- SMART
specific of muscle atrophy and regulated by transcription
- UPS
ubiquitin‐proteasome system
Introduction
The acquired critical illness myopathy (CIM), frequently observed in in long‐term mechanically ventilated and immobilized intensive care unit (ICU) patients, is characterized by paralyses of limb and trunk muscles, a preferential myosin loss, muscle atrophy, reduced muscle membrane excitability and unbalanced muscle metabolism (MacFarlane & Rosenthal, 1977; Rich et al. 1997, 1998 a,b; Larsson et al. 2000; Sander et al. 2002). Several risk factors have been implicated in the development of CIM. Independent risk factor assessment studies show links between prolonged doses of glucocorticoids or non‐depolarizing neuromuscular blocking agents, sepsis and multiple organ failure. Exposure to either one of these factors is, however, not essential for the development of CIM (De Jonghe et al. 2002; Weber‐Carstens et al. 2010).
By using a unique experimental ICU model that mimics the basic ICU conditions of long term mechanical ventilation, sedation and muscle unloading, our group has previously shown that mechanical silencing is a dominant factor triggering the preferential loss of myosin, atrophy and decline in specific force in fast‐ and slow‐twitch muscles and muscle fibres in experiments ranging from 6 h to 14 days (Ochala et al. 2011 b). The ability of the muscle cell to sense, process and respond to mechanical stimuli, i.e. tensegrity, is an important regulator of gene expression and protein synthesis and accordingly also an important regulator of physiological and pathophysiological function (Raskin et al. 2009). Furthermore, we have previously shown that passive mechanical loading diminishes the severity of CIM in both experimental (Renaud et al. 2013) and clinical studies (Llano‐Diez et al. 2012). Passive mechanical loading alleviated the muscle wasting and the loss of force generation associated with the ICU intervention, resulting in a doubling of the functional capacity of the loaded versus the unloaded muscles after a 2 week ICU intervention (Renaud et al. 2013). The mechanism underlying the specific effects of this intervention on skeletal muscle structure and function, however, remains obscure.
Skeletal muscle is a highly adaptive tissue and responds to different stimuli, such as environmental factors, nutritional interventions, loading conditions, and contractile activity; it is capable of altering muscle fibre size, functional capacity and metabolism. Rapamycin has been shown to inhibit mechanically induced muscle growth, indicating that mTORC1 plays an important role for the increased protein synthesis and muscle growth in response to mechanical stimulation (Kubica et al. 2005; Hornberger et al. 2007). mTOR receives various types of informational input in the cell, such as energy and nutritional state, signals from membrane bound growth factor receptors and geno‐toxic stress levels. mTOR is therefore considered the master regulator of protein synthesis and cell growth (Weigl, 2012). The upstream activator of mTORC1, Akt, is also a modulator of FoxO which controls the activation of several degradation pathways involving alterations of multiple atrogenes, such as MuRF1, atrogin‐1, Fbxo31 (a novel ubiquitin E3 ligase involved in cyclin D degradation), and SMART (Fbxo21; a newly found ubiquitin ligase with so far unknown function) (Sandri, 2013; Milan et al. 2015).
The aim of this study is to gain insight into the gene expression signature and molecular pathways regulating the process of mechanical activation of skeletal muscle that are affected by the ICU condition. The mechanism underlying the effects of passive mechanical loading is part of a complex biological system involving activation and inhibition of different protein synthesis and degradation pathways. A full appreciation of this complexity cannot be fully explored in an in vitro system. We therefore aim to investigate the signalling pathways in an experimental ICU rat model allowing detailed studies of the effects of mechanical loading in the ICU condition, i.e. long‐term immobilization and mechanical ventilation. An improved understanding of the positive effects of mechanical loading during the ICU condition is of importance in deciphering the mechanisms underlying the positive effects of early mobilization and active physical therapy in immobilized ICU patients and to unravel potential targets for future pharmacological interventions.
Methods
Ethical approval
The ethical committees at Uppsala University and Karolinska Institutet approved all aspects of this study. All necessary steps were taken to minimize animal suffering, animals were deeply sedated by isoflorane inhalation from the beginning to the end of the experimental period and animals were monitored continuously to detect any pain reaction (EEG activity, heart rate and intra‐arterial blood pressure). At the end of the experiment, animals were killed by thoracotomy and removal of the heart, followed by tissue harvesting.
Experimental procedures
A total of five sham operated controls and 22 experimental female Sprague‐Dawley rats (Taconic Biosciences, Inc., Ejby, Lille Skensved, Denmark) were included in this study representing a subsample from a previous study in our laboratory (in Western blot experiments). Four controls were compared with 5, 4, and 6 rats in the 0.25–4 day, 5–8 day, and 9–14 day experimental groups, respectively. In the mRNA analyses, 5 controls were compared with 12, 4, and 6 rats in the 0.25–4 day, 5–8 day, and 9–14 day experimental groups, respectively). The experimental rats were anaesthetized, mechanically ventilated and treated with α‐cobratoxin for durations varying from 6 h to 14 days. The experimental model has previously been described in detail (Dworkin & Dworkin, 1990, 2004) and modified for the study of skeletal muscle (Ochala et al. 2011 b; Renaud et al. 2013; Corpeno et al. 2014). Briefly, all surgery was performed under sterile techniques. (1) Precordial silver wire electrocardiogram (ECG) electrodes were implanted subcutaneously. (2) An aortic catheter (28‐gauge Teflon) was inserted via the left carotid artery to record arterial blood pressure. (3) A 0.9 mm Renathane catheter was threaded into the left jugular vein to record venous blood pressure and administer parental solutions. (4) Three subcutaneous electroencephalogram needles were placed into the skull above the right and left temporal lobes and a third reference electrode was placed in the neck region. (5) Temperature was measured by a vaginal thermistor and servo regulated at 37°C. (6) A silicon cannula was inserted in the urethra to continuously record urine output. Animals were maintained in protein and fluid balance with an intra‐arterial solution (0.6 ml h−1) consisting of 50 ml H2O, 50 ml 0.5 n lactated Ringer solution, 1.25 g sodium oxacillin, 2.8 mg α‐cobratoxin, 0.3 mg vitamin K (Synkavite) and 20 mequiv K+ (as KCl), as well as with an intra‐venous solution (0.6 ml h−1) consisting of 50 ml H2O, 50 ml 0.5 n lactated Ringer solution, 20% glucose (Baxter, Deerfield, IL, USA) and 1.25 g sodium oxacillin. The sham operated control rats underwent the same intervention as the experimental group animals but were not pharmacologically paralysed with α‐cobratoxin. During surgery or at any possible irritating manipulation, the anaesthetic isoflorane level was > 1.5%. Isoflorane was delivered into the inspiratory gas stream by a precision mass‐flow controller. After the initial surgery, isoflorane was gradually lowered over 1–2 days and maintained at < 0.5% during the remaining experimental period. Rats were ventilated through a per os coaxial tracheal cannula at 72 breaths min−1 with an inspiratory and expiratory ratio of 1:2 and a minute volume of 180–200 ml. Neuromuscular blockade (NMB) was induced on the first day and maintained by continuous infusion. Mechanical ventilation was started immediately after the NMB induction. The left leg of the animal was activated for 6 h at the shortest duration and 12 h per day at duration 12 h and longer throughout the experiment, using a mechanical lever arm that produces a continuous passive maximal ankle joint flexion–extension at a speed of 13.3 cycle min−1.
Muscles
The plantaris, tibialis anterior (TA) and gastrocnemius muscles were dissected from the loaded (left leg) and from the unloaded (right leg) immediately after killing. The muscles were frozen, in liquid propane cooled by liquid nitrogen, and then stored in −160°C until further use.
Immunoblotting
For immunoblot analysis, the frozen tissue was pulverized by the use of a mikro dismembarator (Sartorius, Goettingen, Germany) at 2000 rpm for 1 min. The powdered tissue samples were reconstituted in ice‐cold lysis buffer containing 20 mm Tris HCl (pH 8.0), 138 mm NaCl, 2.7 mm KCl, 5 mm EDTA, 20 mm NaF, 5% glycerol, 1% Triton X‐100, complemented with Complete Mini Protease Inhibitor cocktail and PhosSTOP Phosphatase inhibitor cocktail (Roche, Basel Switzerland) according to the manufacturer's instructions. After 20 min incubation at 4°C lysates were cleared by centrifugation at 15,000 rpm at 4°C for 15 min. Supernatants were collected and protein concentration was determined using Pierce 660 nm protein assay (Thermo scientific, Waltham, MA, USA) according to the manufacturer's instructions. Protein was loaded at 7.5 μg per lane on 12% SDS‐PAGE gels before transfer to PVDF membrane (Millipore, Billerica, MA, USA) and incubation with the following antibodies: anti‐S6 ribosomal protein (No. 2217), anti‐phospho S6 (Ser240/244, No. 5364) ribosomal protein, anti‐4EBP1 (No. 9644), anti‐phospho 4EBP1 (Thr37/46, No. 2855), anti‐phospho Akt (Ser473, No. 9171), anti‐Akt (No. 9272, Cell Signalling Technology, Danvers, MA, USA) and anti‐α‐skeletal actin (Santa Cruz Biotechnology Inc., DE, USA). Membranes were subsequently incubated with IRDye 800 CW secondary antibody (LI‐COR Biosciences) according to the manufacturer's instructions. Proteins were detected by IR fluorescence; signal intensities were quantified with odyssey software (Odyssey Fc, infrared imaging systems, LI‐COR Biosciences, Lincoln, USA) and normalized to anti‐α‐actin.
Subcellular fractioning and immune blotting of cytochrome C and apoptosis inducing factor (AIF)
Proximal gastrocnemius muscle (50 mg) was minced in 1/10 (w/v) ice‐cold lysis buffer (210 mm mannitol, 70 mm sucrose, 5 mm Hepes, 1 mm EDTA, 0,2% BSA, and 0.01% protease inhibitor) using Teflon pestles operated at 16,000 rpm according to Frezza et al. (2007). At the end of homogenization, the samples were centrifuged at 1000 g for 10 min at 4°C to remove cellular debris and nuclei. The supernatant was centrifuged again at 14,000 g for 15 min at 4°C, the supernatant represented mitochondrial free cytoplasm and was stored at −80°C for further use, while the pellets represent the mitochondrial fraction. The pellets were washed with 1.5 ml ice‐cold washing buffer (210 mm mannitol, 70 mm sucrose, 5 mm Hepes, 1 mm EDTA) and centrifuged again at 14,000 g for 15 min at 4°C to pellet the mitochondrial fraction. At the end, the mitochondrial fraction was resuspended in 30 μl Ripa buffer, and stored at −80°C for further use (Marzetti et al. 2008).
Prior to loading, samples were boiled for 5 min at 95°C in Laemmli buffer (161‐0747, BIO‐RAD, USA) with 10% β‐mercaptoethanol. For both cytoplasmic and mitochondrial fractions, 60 μg of total protein was loaded per lane on a stacking gel with 4% acrylamide concentration, and a running gel of 12% acrylamide. Proteins were immediately transferred to PVDF membranes (Millipore). The membranes were incubated with the primary antibodies to detect AIF (SC‐9416, Santa Cruz Biotechnology Inc.), cytochrome C (SC‐13561, Santa Cruz Biotechnology Inc.), and caspase 3 (06‐735, Millipore). Furthermore, the membranes loaded with cytoplasmic proteins were also incubated with α‐actinin (A7732, Sigma‐Aldrich, MO, USA), while the membranes loaded with mitochondrial fraction were incubated with antibody for COX I subunit of complex IV (ab14705, abcam). The membranes were incubated with secondary antibodies NXA931 or NA9340 (GE Healthcare) or sc‐2020 (Santa Cruz Biotechnology Inc.) and processed using ECL Advance Western blotting detection kit (RPN 2135, Amersham Biosciences) according to manufacturer's instructions. The immunoblots were subsequently scanned in a soft laser densitometer (chemiDoc MP imaging system, Bio‐Rad Laboratories, Inc., Hercules, California, USA). The signal intensities were quantified using the volume integration function (arbitrary units) and normalized to actinin, or complex IV.
Gene expression analysis using real time PCR
Total RNA was isolated from the proximal part of gastrocnemius muscle using spin column purification RNAeasy (QIAGEN, Hilden, Germany). A 300 ng aliquot of total RNA from the proximal part of the gastrocnemius muscle was reversed transcribed to cDNA using Qscript cDNA super mix (Quanta Biosciences, Gaithersburg, MD, USA) and was diluted into a final volume of 100 μl. Primers were designed using the software Primer 3 (http://biotools.umassmed.edu/bioapps/primer3_www.cgi) and ordered from Sigma‐Aldrich (Sigma‐Aldrich). cDNA was amplified in triplicate using MyiQ single colour real time PCR detection system (Bio‐Rad Laboratories, Inc.). Gene expression analyses were performed using the ΔΔCt method based on correction to GAPDH.
IgG staining
To assess muscle damage, 10 μm sections of gastrocnemius were cut on a cryostat. Muscle sections were blocked in 5% goat serum, 2% BSA, and 0.2% Triton X‐100 in phosphate‐buffered saline and incubated with anti‐rat IgG Alexa Fluor 568. Muscle sections were visualized with an Axioskop 40 microscope (Zeiss).
RNA‐seq data generation and read mapping
Total RNA was extracted from muscle samples using MagMAX kit (Life tech, Carlsbad, CA, USA). mRNA was purified from 4 μg total RNA using Dynabeads mRNA Purification Kit (Invitrogen, Waltham, MA, USA). Strand‐specific RNA‐seq libraries were prepared using ScriptSeq mRNA‐Seq Library Preparation Kit (Epicentre). Twelve‐cycle PCR was performed to amplify libraries. Sequencing was performed on Illumina HiSeq2000 (Illumina) by multiplexed single‐read run with 33 cycles. Sequenced reads in Illumina Hiseq2000 image files (BCL files) were converted to FASTQ format via Illumina Casava 1.8.2. Reads were decoded based on their barcodes and merged for each individual samples. The overall read quality per sample was evaluated with FastQC to retain only samples with sufficient quality. Subsequently, the reads were mapped to rat genome (RGSC 5.0/rn5) using commercial software CLCBio with two allowed mismatches. For each gene, the reads mapped to the sense‐strand exons of the gene were identified and counted. Detectable genes are flagged based on their expression levels in reads per kilobase per million (RPKM) as well as the total exonic sense‐strand read counts summarized at the gene level. An empirical minimum RPKM of 0.5 was applied to determine the absence and presence of genes in each sample. For comparison between two groups of samples, genes were first eliminated that were not flagged as present in at least the number of samples that matched the smaller sample size of the two groups. Next, fold changes associated with the comparison were calculated as the ratio between the arithmetic mean RPKM values of the two groups. The statistical significance of the differential expressions was assessed by Student's t test (Student's paired t test in the case of loading vs. unloading comparisons). Genes with a fold change > 1.5 in either directions and with a P < 0.05 were selected as the significantly perturbed gene signatures. The differentially expressed genes in each comparison were subjected to pathway enrichment analysis using Running Fish exact test (NextBio). A P < 0.01 was used to extract significantly enriched canonical pathways from MSigDB. The raw fastq data are deposited to ArrayExpress, which is the RNAseq repertoire of EBI (accession number E‐MTAB‐4228).
Statistics
Means and standard error of means (SEM) were calculated according to standard procedures. One‐ and two‐way analyses of variance (ANOVA) and the Tukey post hoc test were used when comparing multiple groups and P < 0.05 was considered statistically significant.
Results
We performed transcriptome profiling on the plantaris muscle isolated from rats exposed to immobilization and mechanical ventilation (‘ICU intervention’) for 6 h to 9 days. We detected ∼15,000 expressed genes. By comparing non‐loaded right plantaris of rats under ventilation with those without ventilation, we found that short‐term (6 h to 4 days) intervention perturbed the expression of 1789 genes, whereas 8301 genes were affected following long‐term (5–9 days) ICU intervention. To study the impact of passive mechanical loading on gene expression, loaded left plantaris was compared with the unloaded right plantaris under either short‐ or long‐term intervention. A total of 528 or 540 genes were differentially expressed, respectively. Figure 1 A shows the heat map of the union of genes affected by short‐term or long‐term ventilation or loading (a total of 9420 genes) for each animal. The fact that many genes were affected by ventilation and significantly more genes affected by the long‐term ventilation than short‐term suggests the vast impact of ventilation on the plantaris muscle. The gene expression heat map also indicates that the impact of ventilation on gene expression is concordant with the ventilation duration at the individual animal level. To understand the biology of ventilation and mechanical loading, we identified the genes that were oppositely regulated by the two interventions and subjected them to MSigDB canonical pathway enrichment analysis. Figure 1 B shows that there is a significant overlap between the ICU intervention and the effect of passive mechanical loading. In the short‐term group, the overlap was 104 genes. Among them, passive mechanical loading had the opposite effect on 95 genes compared with the ICU intervention (unloading). In the long‐term intervention group the overlap was 269 genes, and loading and intervention had opposite effects on 177 genes. The nature of the genes oppositely regulated by ICU intervention and loading are shown in Table 1. These data show that a large number of genes and pathways are affected by immobilization and mechanical ventilation and increase with the duration of the intervention. Passive mechanical loading reverses the direction of expression change of many genes involved in mechanotransduction and protein synthesis, an effect that is more significant for short‐term ventilation. In the following sections, we focused our attention on key pathways controlling myofibrillar protein synthesis (Akt–mTORC1), myofibrillar protein degradation (ubiquitin E3 ligases), mitochondrial dynamics (Fis1, Drp1, Opa1, mitofusin (Mfn) 1 and 2), mitophagy (Bnip3, Parkin and Pink1), biogenesis (PGC‐1α) as well as apoptotic pathways.
Figure 1. Muscle gene expression profiling .

A, expression heatmap of genes affected by immobilization and mechanical ventilation alone left (L) and in combination with mechanical loading in the right (R) plantaris. Relative expression of 9420 genes (union of genes significantly affected by short‐ or long‐term ventilation or loading) is plotted. RPKM of each gene was normalized against the average of non‐ventilated and unloaded right leg controls followed by log2 transformation (colour scale is shown). B, Venn diagrams of genes affected by short‐ or long‐term ventilation and loading.
Table 1.
MSigDB canonical pathway analysis of genes oppositely regulated by ICU intervention and loading
| Number of | Direction of gene | ||||
|---|---|---|---|---|---|
| genes in | Common | expression level | |||
| Biogroup name | pathway | genes | change by loading | P value | |
| 95 genes in short term ICU intervention | TGF‐β signalling pathway | 19 | 2 | Both down | 0.0003 |
| Genes involved in xenobiotics | 11 | 1 | Down | 0.0003 | |
| Nuclear receptors in lipid metabolism and toxicity | 14 | 1 | Down | 0.0003 | |
| Pyruvate metabolism | 39 | 2 | Both down | 0.0008 | |
| 177 genes in long term ICU intervention | β5 β6 β7 and β8 integrin–cell surface interactions | 16 | 2 | Both up | 3.00E‐05 |
| Role of nicotinic acetylcholine receptors in the regulation of apoptosis | 16 | 2 | Both down | 6.60E‐05 | |
| Role of BRCA1, BRCA2 and ATR in cancer susceptibility | 21 | 2 | Both down | 0.0001 | |
| Mechanism of gene regulation by peroxisome proliferators via PPAR‐alpha | 33 | 3 | All down | 0.0002 |
Signalling impinging on protein synthesis
Key signalling markers of the Akt–mTORC1 pathway were analysed in predominantly fast‐twitch distal hindlimb muscles (mm. plantaris and gastrocnemius) to investigate the effects of passive mechanical loading on the protein synthesis pathway. Akt–mTORC1 activity was examined at three different time durations (0.25–4, 5–8 and 9–14 days) of the ICU intervention and compared with controls. Transcriptional levels of IGF‐1, an important upstream regulator of Akt, was investigated, together with the relative contents of total protein, phosphorylated protein and phosphorylated/total levels of Akt, S6 ribosomal protein (S6) and 4EBP‐1.
IGF‐1 was significantly down‐regulated in response to mechanical silencing after 6 h to 4 days (P < 0.05). Levels remained reduced after 5–8 days and returned to control levels at the longest duration (9–14 days). No significant effects on IGF‐1 were observed in response to passive mechanical loading (Fig. 2).
Figure 2. IGF‐1 transcription .

Transcription levels of IGF‐1 in the loaded (filled bars) and the unloaded (open bars) gastrocnemius muscle. Values are means + SEM and given as fold changes vs. controls. Significant differences according to two‐way ANOVA and Tukey post hoc test are indicated: *P < 0.05.
At the shortest duration (0.25–4 days), the active phosphorylated form (P‐Akt) and total Akt (T‐Akt) did not differ from controls. After 5–8 days, a slight decrease was observed in both P‐Akt and T‐Akt compared with controls (P < 0.05), but passive mechanical loading did not counteract the changes in P‐Akt during the initial 8 days. However, at the longest duration (9–14 days), a significant parallel up‐regulation was observed in both P‐Akt (P < 0.01) and T‐Akt (P < 0.05) in response to passive mechanical loading (Fig. 3 A). Thus, although mechanical loading had no significant effect on the relative content of phosphorylated Akt protein, the activation of Akt was significantly increased by the passive mechanical loading at the longest duration of 9–14 days (Fig. 3 A).
Figure 3. Akt–mTORC1 pathway .

Western blot analyses of total and phosphorylated Akt (A), ribosomal S6 (B) and 4EBP1 (C) protein normalized to actin contents in the loaded (filled bars) and the unloaded (open bars) plantaris muscle. Controls and rats exposed to sedation, postsynaptic blockade of neuromuscular transmission and mechanical ventilation for 0.25–4, 5–8, 9–14 day durations. Values are means + SEM and significant differences according to two‐way ANOVA and Tukey post hoc test are indicated: *P < 0.05, **P < 0.01, ***P < 0.001.
In order to understand whether the changes in Akt also affected the mTOR pathway, we analysed phosphorylated of S6 (P‐S6) representing an indicator of mTORC1 activation. A significant early (0.25–4 days) up‐regulation in response to the ICU intervention was observed in total S6 (T‐S6) (P < 0.05), but P‐S6 levels remained unchanged compared with controls. At durations longer than 5 days, T‐S6 was significantly down‐regulated (P < 0.01), but the relative amount of P‐S6 increased independently of loading condition. At the longest duration (9–14 days), T‐S6 remained down‐regulated, but P‐S6 was significantly increased in response to passive mechanical loading (P < 0.05; Fig. 3 B). This increase in S6 activity in response to mechanical loading mirrors the induction of Akt.
An additional downstream target of mTORC1, 4EBP‐1, was not affected by the passive mechanical loading in the fast twitch muscle. In fact, in plantaris muscle, total 4EBP‐1 (T‐4EBP‐1) was unaffected, while the phosphorylated 4EBP‐1 (P‐4EBP‐1) was up‐regulated at the longest duration (9–14 days; Fig. 3 C) in both the loaded and unloaded leg.
Signalling pathways controlling protein degradation
In previous studies we have been able to show that the ubiquitin proteasome pathway plays an integral role in the degradation of myofibrillar proteins in the experimental rat ICU model where animals develop the critical illness myopathy geno‐ and pheno‐type, i.e. the preferential loss of myosin, activation of proteolytic pathways and down‐regulation of contractile proteins at the transcriptional level (Norman et al. 2006; Ochala et al. 2011 b). Specific interest was therefore focused on the effects of passive mechanical loading on different E3 ligases.
Mechanical silencing had an early effect on MuRF1 expression; a significant up‐regulation was observed at the shortest duration (0.25–4 days; P < 0.05). Transcriptional levels increased further in the 5–8 day group (P < 0.001) and thereafter remained elevated in the 9–14 day group (P < 0.01). Slightly lower MuRF1 levels were observed in response to mechanical loading at short and long durations, but these differences were not statistically significant according to two‐way ANOVA (Fig. 4 A). Atrogin‐1 mRNA levels showed a similar pattern to MuRF1, i.e. atrogin‐1 transcription levels had the strongest significant up‐regulation, compared to controls, after 5–8 days (P < 0.01) and passive mechanical loading had no significant effect on its expression (Fig. 4 B).
Figure 4. MuRF1, atrogin‐1, Fbxo31 and SMART transcription .
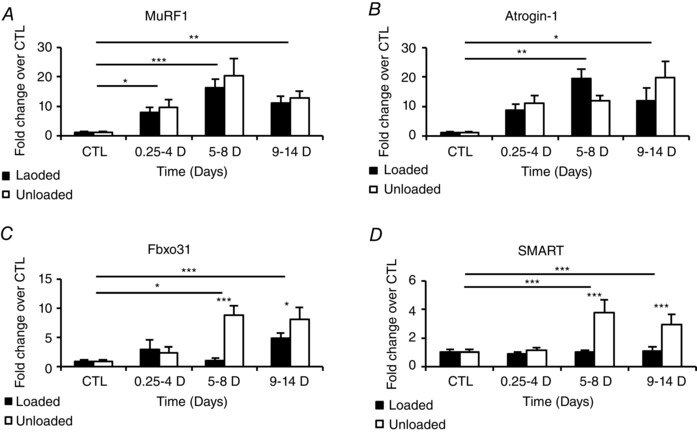
MuRF1 (A), atrogin‐1 (B), Fbxo31 (C) and SMART (D) transcription levels in the loaded (filled bars) and the unloaded (open bars) muscles from control animals and from animals exposed to sedation, NMB, mechanical ventilation and unilateral passive mechanical loading for 0.25–4, 5–8, 9–14 days. Values are means + SEM and given as fold changes vs. controls. Significant differences according to two‐way ANOVA and Tukey post hoc test are indicated: *P < 0.05, **P < 0.01, ***P < 0.001.
A novel set of ubiquitin ligases (E3s) regulated by the Forkhead Box (Fox) O family (FoxO 1, 3 and 4) have recently been described (Sartori et al. 2013; Milan et al. 2015). This novel set of ligases include Fbxo31, and Fbxo21. Fbxo21 is an ubiquitin ligase whose substrates are still unknown and has been given the name SMART (Specific of Muscle Atrophy and regulated by Transcription). These E3 ligases are induced in response to denervation and fasting (Milan et al. 2015). Therefore they were also of specific interest with regard to the pharmacological denervation in our experimental ICU model, i.e. irreversible neuromuscular blockade (NMB) by continuous infusion of α‐cobra toxin, and how they are affected by passive mechanical loading. Both NMB and mechanical loading had a strong impact on SMART and Fbxo31. Both were significantly up‐regulated after 5–8 days NMB (P < 0.001 and < 0.05, respectively) and stayed elevated after 9–14 days. Passive mechanical loading, on the other hand, counteracted these inductions completely (Fig. 4 C and D).
The expression of atrogenes is controlled by multiple signalling pathways, which play an important role in the regulation of muscle atrophy. Myogenin (Mgn) is an essential regulator of muscle development, but also a regulator of atrogene expression after denervation (Moresi et al. 2010). In accordance with this, an increase in Mgn expression was observed in response to pharmacological denervation, demonstrating that the denervation effect is primarily via motoneuron discharge pattern and not via neurotrophic factors. A 25‐ to 30‐fold increase in Mgn mRNA expression was observed at durations longer than 5 days, but Mgn expression was not affected by passive mechanical loading (Fig. 5 A).
Figure 5. Myogenin, Myo D and HDACs transcriptions .

Transcription levels of Mgn, Myo D, HDAC4 and HDAC5 in the loaded (filled bars) and the unloaded (open bars) gastrocnemius muscle, proximal part. Values are fold changes over controls and means + SEM. Significant differences according to two‐way ANOVA and Tukey post hoc test are indicated: *P < 0.05, **P < 0.01, ***P < 0.001.
Histone deacetylases (HDACs) act as negative regulators of Mgn expression via Dach2 (Moresi et al. 2010). The expression of class IIa HDACs, HDAC4 and HDAC5, was studied in response to the ICU intervention and passive mechanical loading. HDAC4 showed no significant changes during the initial 8 days of ICU intervention, but a significant (P < 0.001) HDAC4 up‐regulation was observed after 9–14 days. A small, but significant difference in the transcription levels of HDAC 4 was observed in response to loading in the 9–14 day group (Fig. 5 C). Transcription levels of HDAC5 showed an up‐regulation after 5–8 days of the ICU intervention and returned to control values after 9–14 days (Fig. 5 D).
In a subsample, rats (n = 6) were treated with an HDAC inhibitor (AR‐42) at three different time points (6 h, 4 days and 6 days). All rats treated with AR‐42 showed a strong negative effect on HDAC4, HDAC5 and Mgn expression (not detectable at any rtPCR amplification). However, the inhibition of HDAC and Mgn did not inhibit the MuRF1 up‐regulation (Fig. 6), suggesting that the Mgn pathway is not primarily involved in ICU‐mediated atrogene induction.
Figure 6. MuRF1 transcription in rats treated with AR42 .

Transcription levels of MuRF1 in the loaded (filled bars) and the unloaded (open bars) gastrocnemius muscle, proximal part from rats treated with AR42. Values are means + SEM and given as fold changes vs. controls. Significant differences according to two‐way ANOVA and Tukey post hoc test are indicated: *P < 0.05, **P < 0.01, ***P < 0.001.
Signalling pathways involved in mitochondria dynamics and function
There is a growing body of evidence showing that mitochondrial activity plays an important role in the regulation of muscle size. It was recently established that 4EBPs act as major mediators of the effects of mTORC1 on mitochondria biogenesis and function, i.e. the stimulation of biogenesis by antagonizing 4EBP‐dependent translation repression of mitochondria related mRNAs (Morita et al. 2013).
The transcriptional levels of PGC‐1α, the regulator of mitochondria biogenesis, were unchanged during the initial 4 days of the ICU intervention, followed by a down‐regulation (P < 0.01) at durations longer than 5 days of the ICU intervention (Fig. 7 A). Passive mechanical loading did not affect the total PGC‐1α expression levels or the PGC‐1α4 isoform, the isoform that is normally induced in response to exercise and is important for muscle growth, at durations longer than 5 days. Interestingly, a significant difference in the transcriptional levels of PGC‐1α4 isoform was observed in response to mechanical loading, but this difference was restricted to the early time point (0.25–4 days; P < 0.05; Fig. 7 B).
Figure 7. PGC‐1α transcription .

Transcription levels of total PGC‐1α and PGC‐1α isoform 4 (PGC‐1α4) in the loaded and unloaded gastrocnemius muscle from control animals and from animals exposed to sedation, NMB, mechanical ventilation and unilateral passive mechanical loading for 0.25–4, 5–8, 9–14 days. Values are means + SEM and given as fold changes vs. controls. Significant differences according to two‐way ANOVA and Tukey post hoc test are indicated: *P < 0.05, **P < 0.01, ***P < 0.001.
It has previously been demonstrated that an imbalance of mitochondrial dynamics contributes to muscle atrophy in response to denervation, fasting and overexpression of FoxO3 (Romanello et al. 2010). Mitochondrial fission and fusion were both affected by the ICU intervention. Fis1, an integral protein located in the outer mitochondrial membrane, was significantly increased after 5–8 days and remained elevated at the longest duration (9–14 days; P < 0.05), but this effect was completely counteracted by the passive mechanical loading (Fig. 8 A). However, another marker of mitochondrial fission (Drp1) showed a different pattern, i.e. slightly increased transcriptional levels were only observed at the longest duration (9–14 days) and lower Drp1 levels were restricted to the shortest duration on the unloaded side (Fig. 8 A). The discrepancy between the effects of the two markers of mitochondrial fission is not known, but it may be speculated that the Fis1 overexpression may represent a compensatory mechanism to the Drp1 down‐regulation during unloading since Fis1 is one of the receptors for Drp1. Opa1, Mfn1 and Mfn2, all mediators of mitochondrial fusion, followed the same pattern, i.e. remained unaffected during the initial 4 days and then increased after 5–8 days and remained elevated after 9–14 days (Fig. 8 B). On the loaded side, on the other hand, all mediators of mitochondrial fusion stayed at control levels during the whole experimental period (Fig. 8 B). These findings demonstrate that the mechanical silencing associated with the ICU intervention induces significant mitochondrial changes, which are blunted by mechanical loading.
Figure 8. Mitochondria dynamics and mitophagy .

Transcription levels of Fis1, Drp1, Opa1, Mfn1 Mfn2, Bnip3, Parkin and Pink1 in the loaded (filled bars) and the unloaded (open bars) leg of the plantaris muscle from control animals and from animals exposed to sedation, NMB, mechanical ventilation and unilateral passive mechanical loading for 0.25–4, 5–8, 9–14 days. Values are means + SEM and given as fold changes vs. controls. Significant differences according to two‐way ANOVA and Tukey post hoc test are indicated: *P < 0.05, **P < 0.01, ***P < 0.001.
Bnip3, Pink1, and Parkin were analysed at the transcriptional level to study the effects on the selective removal of damaged mitochondria via autophagy, mitophagy, in response to the ICU intervention and passive mechanical loading. Parkin was significantly induced after 5–8 days (P < 0.05) and 9–14 days (P < 0.001), but this up‐regulation was counteracted by mechanical loading (Fig. 8 C). Pink1 levels were also lower on the loaded side and identical to control levels after 5–8 days and 9–14 days (Fig. 8 C). A significant up‐regulation of Bnip3 was observed after 9–14 days, but no differences were seen in response to passive loading (Fig. 7 C). Thus, mitochondrial dynamics and mitophagy pathways were significantly affected by the ICU intervention at the gene level, but these changes were counteracted by passive mechanical loading.
Apoptosis and muscle fibre degradation
The apoptosis induced factor (AIF) and cytochrome C were investigated in cytosolic and mitochondrial fractions. No significant changes were detected in cytosolic AIF during the initial 8 days of the intervention, but an increase (P < 0.05) was observed on the unloaded side after 9–14 days. Cytosolic cytochrome C showed an early increase (P < 0.05; 0.25–4 days), but this increase was restricted to the unloaded side. AIF protein levels in the mitochondrial fraction were unchanged during the initial 8 days and then decreased at the longest duration (P < 0.05). Cytochrome C did not change significantly in the mitochondrial fraction in response to the ICU intervention irrespective loading condition. In addition, caspase 3 activation was evaluated, and caspase 3 was significantly activated in the 9–14 days (P < 0.05) group on the unloaded but not on the loaded side (Fig. 9).
Figure 9. Apoptosis .

Cytosolic cytochrome C and AIF normalized to actinin (upper left panel). Mitochondrial cytochrome C and AIF normalized to Complex IV (upper right panel). Inactive and active caspase normalized to actinin (lower panel). Western blot analyses done on loaded (filled bars) and unloaded (open bars) legs from gastrocnemius muscle taken from control animals and from animals exposed to sedation, NMB, mechanical ventilation and unilateral passive mechanical loading for 0.25–4, 5–8, 9–14 days. Values are mean + SEM. Significant differences are indicated: *P < 0.05, **P < 0.01, ***P < 0.001.
In parallel with the increase in active caspase, the percentage of IgG positive and necrotic muscle fibres increased in mechanically ventilated unloaded animals, indicative of muscle damage (Fig. 10). However, passive mechanical loading blunted this damage response to muscle fibres.
Figure 10. Muscle fibre necrosis .
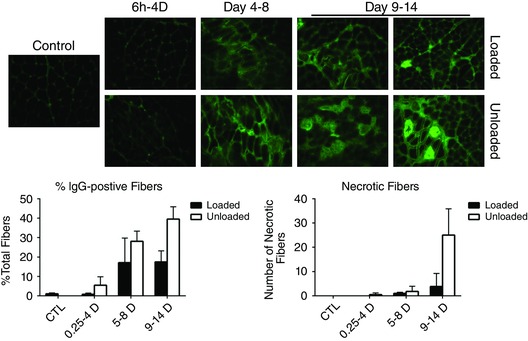
Upper panels, IgG staining from muscle sections from mechanically ventilated unloaded rats compared to sections from age‐matched control or loaded animals over a 14 day period. Lower panels, IgG positive muscle fibres from unloaded (open bars) vs. loaded (filled bars) animals were scored as a percentage of total fibres over a 14 day period.
Discussion
A complete or near‐complete mechanical silencing is uniquely observed in pharmacologically paralysed or deeply sedated mechanically ventilated ICU patients. Mechanical silencing has been proposed as an important factor triggering the critical illness myopathy associated with severe muscle wasting, muscle paralysis and preferential myosin loss in limb and trunk muscles (Ochala et al. 2011 b). Passive mechanical loading has been shown to reduce these effects in both experimental and clinical studies (Griffiths et al. 1995; Llano‐Diez et al. 2012; Renaud et al. 2013). The general aim of this study was to improve our understanding of the effects of the ICU intervention, mechanical silencing and passive mechanical loading on overall gene changes and intracellular signalling pathways involved in protein synthesis, and muscle protein breakdown. The major findings from this study are as follows: (1) a large number of genes and pathways are perturbed following immobilization and mechanical ventilation and the magnitude and extent of affected genes increase with duration of the intervention. Some of these changes at the gene level are reversed by mechanical loading. (2) Both mitochondrial fission and fusion events, as well as mitophagy, are induced by mechanical silencing at the gene level, and these effects are counteracted by passive mechanical loading. (3) The recently identified ubiquitin ligases Fbxo31 and SMART are induced by mechanical silencing, an induction that similarly is prevented by passive mechanical loading. (4) These effects are suggested to be secondary to the inhibition of FoxO by Akt since the total amount of Akt protein was significantly up‐regulated in response to long‐term loading and neuromuscular blockade. (5) The myogenin–HDAC4 axis is not critical for mechanical unloading.
To date most studies on the effects of muscle loading have been focused on the effects of removing the external load induced by weight bearing in response to hindlimb suspension or microgravity. However, in immobilized ICU patients the internal strain in response to activation of contractile proteins has been removed in addition to removal of weight bearing, resulting in the complete mechanical silencing unique to mechanically ventilated and deeply sedated or pharmacologically paralysed ICU patients. Microgravity or ground‐based microgravity models form the basis of research on the effect of muscle unloading–reloading, but the mechanisms and effects differ from the ICU condition. However, it is well established that muscle unloading induces a rapid decrease in protein synthesis which contributes to the loss of muscle mass (Thomason & Booth, 1990; Han et al. 2007). In order to understand how mechanical tension regulates the muscle mass, it is critical to know how muscles sense mechanical information and convert this stimulus into the biochemical events that regulate protein synthesis and degradation (Goldberg, 1968; Goldberg et al. 1975; Vandenburgh, 1987). The primary effect of mechanical stimulation appears to occur at the level of translational efficiency (Goldspink, 1977; Kimball et al. 2002). Components of the mTORC1 play a critical role in the mechanical regulation of protein synthesis and skeletal muscle mass (Jackman & Kandarian, 2004; Hornberger et al. 2007).
Signalling through mTOR has repeatedly been identified as the primary pathway involved in mechanically induced growth of skeletal muscle via activation of mTORC1 through a mechanism involving IGF‐1 and the Akt–mTORC1 pathway (Bodine et al. 2001; Schiaffino et al. 2013). That is, mechanical stimulation promotes an increase in IGF‐1 content and signalling through PI3K and Akt, which in turn induces mTORC1 signalling and promotes skeletal muscle growth (Bodine et al. 2001; Schiaffino et al. 2013). However, other studies have provided evidence of mechanical stimulation activating mTORC1 signalling through an IGF‐1–PI3K–Akt‐independent mechanism (Andrew et al. 2011; Hornberger, 2011). In support of a mechanical stimulation–IGF‐1–PI3K–Akt‐independent activation of mTORC1, our results show that IGF‐1 is slightly down‐regulated in response to passive mechanical loading compared to controls and compared to long‐term unloading in the experimental ICU model ranging from 6 h to 8 days. Results from this study show that the relative amount of phosphorylated Akt was not affected by passive mechanical loading, but both total and phosphorylated Akt protein was significantly up‐regulated in response to long‐term (9–14 days) loading, indicating that there was an increased amount of phosphorylated Akt compared to the unloaded leg to activate mTORC1. Further studies are needed to establish the upstream regulator underlying this effect on Akt. The activation of mTORC1 in response to passive mechanical loading was also confirmed in this study by the up‐regulation of the mTORC1‐downstream target, P‐S6 as well as the translational repressor 4EBP‐1. The phosphorylated form of S6 ribosomal protein, P‐S6, was up‐regulated compared to controls and in response to passive mechanical loading after 9–14 days of the ICU intervention.
Additionally, mTORC1 is a multidimensional kinase that has been shown to promote ATP production capacity by mitochondria as well as cell cycle progression. Thus, mTORC1 has been shown to drive cell proliferation and neoplastic growth by simultaneously activating translation of mRNAs that encode proteins involved in cellular energy and cell cycle progression (Dowling et al. 2010). Recently, Morita et al. (2013) established that 4EBPs act as major mediators of the mTORC1 effects on mitochondria biogenesis and function. The study demonstrates that 4EBPs stimulate the biogenesis by antagonizing 4EBP‐dependent translation repression of mitochondria related mRNAs. Further, mTORC1 also inhibits mitochondria degradation by suppressing autophagy. These novel findings suggest a coordinated control of translational and autophagy programmes that underpin important biological effects of mTORC1 signalling (Morita et al. 2013).
There is emerging evidence of a mitochondria‐related regulation of signalling pathways controlling muscle mass. Romanello et al. previously demonstrated that an imbalance of mitochondrial dynamics contributes to muscle atrophy in response to denervation, fasting and overexpression of FoxO3 (Romanello et al. 2010). In addition, a down‐regulation of metabolic enzymes and a decrease of mitochondrial oxidative capacity have been described in muscle disuse. Mitochondrial function plays a key role in the maintenance of cellular health while mitochondrial dysfunction is associated with various pathologies. To secure proper maintenance and repair of mitochondria, several systems of mitochondrial quality control have evolved at the organelle and cellular levels. Alteration of mitochondrial morphology allows exchange of mitochondrial content and discrimination of terminally damaged mitochondria to enable degradation by a controlled autophagy, i.e. mitophagy. Finally, substantial mitochondrial impairment can induce apoptosis via different pathways, for instance through the release of cytochrome C and apoptosis induced factor (AIF).
PGC‐1α is a central regulator of mitochondrial shape and of mitochondrial biogenesis. A decrease in PGC‐1α has been found in hindlimb unloading, denervation atrophy (Sandri et al. 2006) and in several catabolic states such as diabetes and ageing (Anderson & Prolla, 2009; Cannavino et al. 2014) and the down‐regulation of PGC‐1α could accordingly be responsible for the activation of degradation pathways through FoxO3 disinhibition and the consequent enhancement of ubiquitin ligase gene expression. It has also been shown that mitochondrial alterations induced by PGC‐1α overexpression can resist muscle protein breakdown during muscle disuse (Cannavino et al. 2014). mTOR controls mitochondrial gene expression through direct variation of PGC‐1α activity (Cunningham et al. 2007), indicating a way for the cell to connect nutrient pathways to activate mitochondrial function and ensure energy supply. The result from this study is in accordance with previous reports showing that PGC‐1α does not affect the Akt–mTORC1 pathway (Bonaldo & Sandri, 2013).
Mitochondria are constantly changing in number, shape and localization. This feature is a result of the continuous alteration between fusion and fission events. The result of mitochondrial fusion is a network of connected mitochondria that enables exchange of its content to maintain the overall integrity of the mitochondrial genome and proteome (Chen & Chan, 2005, 2010; Tondera et al. 2009). Fission events conversely result in smaller mitochondria that can function individually in a different position in the cell or be degraded by mitophagy (Elgass et al. 2013). Results from our study indicate that both fission (transcriptional levels of Fis1) and fusion (transcriptional levels of Opa1, Mfn1, and Mfn2) were induced by mechanical silencing after 5–8 days of the ICU intervention (Fig. 8 A and B), a response that was completely cancelled out by passive mechanical loading. In addition, Pink1 and Parkin, two important markers of mitophagy, were both exclusively increased on the unloaded side, demonstrating that mitophagy is controlled by mechanical signalling (Fig. 8 C). Thus, mitophagy, mitochondria fission and fusion are associated with mechanosensing.
The two degradation ligases, MuRF1 and atrogin‐1 were up‐regulated in response to mechanical silencing, but passive mechanical loading had no significant effect on the transcription levels of these two atrogenes, although a tendency was observed towards lower MuRF1 levels on the loaded side. The activation of these atrogenes and the ubiquitin degradation pathways play an important role in the degradation of myofibrils in different muscle wasting conditions, and the induction of these atrogenes precedes atrophy (Ikemoto et al. 2001). A novel set of ubiquitin E3 ligases, regulated by FoxO, were recently described by Milan et al. (2015). This novel set of ligases includes Fbxo31, and Fbxo21, which codes for a gene that so far has an unidentified function. Fbxo21 was given the name SMART (Specific of Muscle Atrophy and Regulated by Transcription), since inhibition of SMART was sufficient to partially prevent muscle atrophy after denervation (Milan et al. 2015). In this study we demonstrate that these two E3 ligases also are affected by pharmacological denervation by postsynaptic blockade of neuromuscular transmission. However, this effect was completely abolished by passive mechanical loading, indicating that mechanosensing plays a more important role than motoneuron discharge pattern in regulating the expression of these E3 ligases.
In this study, 9–14 days ICU intervention caused a significant release of AIF from mitochondria irrespective loading condition, but only the unloaded side had a significant elevation in cytosolic AIF levels. On the other hand, both mitochondrial and cytosolic cytochrome C fractions were unchanged except for an early increase in the cytosolic fraction on the unloaded side. Long duration of mechanical silencing was associated with caspase 3 activation on the unloaded side. Previous studies have shown that cytochrome C release from mitochondria into the cytosol interacts with caspase 9 and apoptotic protease activating factor 1 (Apaf‐1) to form the apoptosome. The apoptosome in turn activates caspase 3, leading to apoptotic cell death (Jiang & Wang, 2000; Acehan et al. 2002). Thus, the present results showing an early increase in the cytosolic cytochrome C levels on the unloaded leg followed by an activation of caspase 3 are in accordance with this. Furthermore, AIF released form mitochondria into the cytosol increases mitochondrial membrane permeability and additional release of cytochrome C and AIF. AIF has been shown to translocate into the nucleus causing chromatin condensation and to initiate apoptosis (Ferri et al. 2000; Susin et al. 2000).
Passive mechanical loading alleviates atrophy in the experimental ICU model via multiple signalling pathways. We demonstrate that passive mechanical loading did not affect the relative amount of phosphorylated versus total Akt content, but both were increased and higher levels of phosphorylated activated Akt were observed in response to mechanical loading at the longest duration (Fig. 3 A). Activated Akt not only prevents muscle atrophy by activating mTORC1, but also by inhibiting FoxO‐E3 ligases like Fbxo31 and SMART. Another interesting molecular network is the class II HDACs, Dach2 and Mgn signalling pathway, also regulating gene expression in denervated muscles (Tang & Goldman, 2006; Tang et al. 2009; Moresi et al. 2010) and the regulation of the expression of genes encoding E3 ubiquitin ligases (Moresi et al. 2010; Macpherson et al. 2011). In analogy with peripheral denervation, the pharmacological denervation induced a 20‐ to 25‐fold up‐regulation of Mgn mRNA expression. Moresi et al. (2010) demonstrated that Mgn controls the regulation of MuRF1 and atrogin‐1 and that class II HDACs control the up‐regulation of Mgn. We demonstrate that the HDAC inhibitor AR‐42 was able to inhibit both HDAC and Mgn expression, i.e. HDACs were repressed to undetectable levels, consequently inhibiting the transcriptional levels of Mgn. However, AR‐42 did not eliminate the increase in MuRF1 in response to mechanical silencing. Several reports have demonstrated that HDAC inhibitors induce dephosphorylation of Akt in different cancer cell lines (Nimmanapalli et al. 2003; Kodani et al. 2005). In a recent study, Yang et al. showed that the specific HDAC inhibitor, AR‐42, induced Akt dephosphorylation (Yang et al. 2013), suggesting that MuRF1 is induced via a FoxO activation with this inhibitor. In addition, both inactivation of mTORC1 and overexpression of Mgn (Moresi et al. 2010) in normal innervated muscle can induce E3 ubiquitin ligases, but neither of these manipulations alone result in muscle atrophy. Thus, a coordinated activation of both of these signalling cascades might be necessary for the development of muscle atrophy.
We have previously studied regulation of muscle contraction, muscle morphology, and myofibrillar protein synthesis/degradation in 46 rats exposed to neuromuscular blockade, isoflorane sedation and mechanical ventilation at durations varying between 6 h and 14 days (Ochala et al. 2011 b). Muscle fibre size, muscle mass, body weight and single muscle fibre force generation capacity (maximum force normalized to muscle fibre cross‐sectional area, specific force) were maintained during the initial 4 days in both fast‐ and slow‐twitch distal hindlimb muscles (Ochala et al. 2011 b). These results are in accordance with our observations using a porcine experimental ICU model, showing an unaltered limb muscle fibre size and specific force during 5 days mechanical ventilation, neuromuscular blockade and deep sedation (Ochala et al. 2011 a). At longer durations, a progressive decline was observed in both muscle fibre size and specific force in the rodent ICU model and was paralleled by a preferential myosin loss, transcriptional down‐regulation of contractile proteins, and activation of proteolytic pathways in both fast‐ and slow‐twitch distal hindlimb muscles (Ochala et al. 2011 b). An early transcriptional up‐regulation of MuRFs and atrogin‐1 was observed. However, at the protein level only MuRFs showed an increase as well as a translocation from the cytoplasm to the nucleus during the first 4 days, followed by perinuclear localization and co‐localizing with the serum response factor after 9 days (Ochala et al. 2011 b). A significant activation of the autophagy lysosomal and the calcium‐activated (calpains) protein degradation pathways were observed in response to the ICU condition. However, these degradation pathways followed a different temporal sequence and were activated later than the ubiquitin proteasome pathway and were preceded by muscle atrophy, a decline in specific force and preferential myosin loss (Ochala et al. 2011 b). It was originally hypothesized that the early up‐regulation of the MuRF‐mediated ubiquitination played an integral role for the preferential myosin loss and muscle atrophy associated with CIM (Ochala et al. 2011 b), but the preferential myosin loss and muscle atrophy in response to mechanical silencing may represent a more complex mechanism. Muscle atrophy is secondary to a series of biochemical and transcriptional changes leading to a highly coordinated adaptation of protein synthesis and activation of proteolytic pathways (Sandri, 2008) and there is reason to believe these coordinated processes are unique for different muscle wasting conditions and may vary according to muscle type. It is interesting to note that the transcriptional up‐regulation of the E3 ligases Fbox31 and SMART were preceded by the transcriptional up‐regulation of the atrogenes (MuRF1 and atrogin‐1) and paralleled the muscle atrophy, preferential myosin loss and decline in specific force in response to mechanical silencing. Thus, a coordinated activation of these different E3 ligases, and perhaps other E3 ligases, may be required in the development of the CIM phenotype. This is supported by our previous muscle loading experiments in the experimental rat ICU model, where a significant muscle sparing effect, a reduced loss in single muscle fibre size and specific force was observed in response to unilateral passive mechanical loading in muscle fibres from both fast‐ and slow‐twitch muscles (Renaud et al. 2013). A reduced preferential myosin loss was also observed in response to passive mechanical loading, albeit statistically significant only in the slow‐twitch soleus muscle (Renaud et al. 2013). Originally, loading was suggested to have a significant effect on MuRF11 expression (Renaud et al. 2013), but in this study only a trend towards lower MuRF1 transcript levels was observed on the loaded side, while Fbox31 and SMART levels were significantly down‐regulated in response to unilateral loading. Thus, the coordinated and temporal expression of different E3 ligases and their respective roles in the development of the CIM phenotype deserve further scientific attention in both clinical and experimental studies.
Conclusion
Mitochondrial dynamics have recently been shown to play a more important role in muscle atrophy than previously recognized. In this study we demonstrate that mitochondria gene expression is significantly affected by mechanosensing and the changes induced by unloading were counteracted by passive mechanical loading. In addition, we show that the recently identified ubiquitin ligases Fbxo31 and SMART are induced by mechanical silencing, an induction which was prevented by passive mechanical loading, and it suggested that this effect is due to inhibition of FoxO by Akt.
Additional information
Competing interests
There is no conflict of interest.
Author contributions
Design of the research study was done by L.L. All experimental work was done by L.L, R.C.K. and H.S. Immunoblotting was done by R.C.K. and H.S. Subcellular fractioning was done by H.S. Transcriptional expression analysis was done by R.C.K., V.R., R.S., S.G., W.F., Y.B. and J.G. IgG immunostainings were done by D.G. Data analysis was done by M.S., J.R., R.C., H.S., W.F., Y.B. and J.G. R.C.K. wrote the manuscript with assistance of M.S., D.G., H.S. and L.L. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was financially supported by the Swedish Research Council (8651), the Swedish Foundation for International Cooperation in Research and Higher Education (STINT), the European Commission (MyoAge, EC Fp7 CT‐223756 and COST CM1001), King Gustaf V and Queen Victoria's Foundation, and Karolinska Institutet to L.L., H.S. was supported by a European Commission fellowship. The Swedish Research Council (3074) to SG.
Acknowledgements
We are grateful to Yvette Hedström and Ann‐Marie Gustafson for excellent technical assistance and to Professor Mario Pende for valuable input during the initiation of this study. We are also grateful to Rr. Stefan Proniuk (Arno Therapeutics) for providing us with AR‐42.
References
- Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X & Akey CW (2002). Three‐dimensional structure of the apoptosome: implications for assembly, procaspase‐9 binding, and activation. Mol Cell 9, 423–432. [DOI] [PubMed] [Google Scholar]
- Anderson R & Prolla T (2009). PGC‐1alpha in aging and anti‐aging interventions. Biochim Biophys Acta 1790, 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew P, Hamilton DL & Keith B (2011). Signals mediating skeletal muscle remodeling by resistance exercise: PI3‐kinase independent activation of mTORC1. J Appl Physiol 110, 561–568. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ & Yancopoulos GD (2001). Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo . Nat Cell Biol 3, 1014–1019. [DOI] [PubMed] [Google Scholar]
- Bonaldo P & Sandri M (2013). Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavino J, Brocca L, Sandri M, Bottinelli R & Pellegrino MA (2014). PGC1‐alpha over‐expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J Physiol 592, 4575–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H & Chan DC (2005). Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet 14 (Suppl. 2), R283–R289. [DOI] [PubMed] [Google Scholar]
- Chen H & Chan DC (2010). Physiological functions of mitochondrial fusion. Ann N Y Acad Sci 1201, 21–25. [DOI] [PubMed] [Google Scholar]
- Corpeno R, Dworkin B, Cacciani N, Salah H, Bergman HM, Ravara B, Vitadello M, Gorza L, Gustafson AM, Hedstrom Y, Petersson J, Feng HZ, Jin JP, Iwamoto H, Yagi N, Artemenko K, Bergquist J & Larsson L (2014). Time course analysis of mechanical ventilation‐induced diaphragm contractile muscle dysfunction in the rat. J Physiol 592, 3859–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK & Puigserver P (2007). mTOR controls mitochondrial oxidative function through a YY1‐PGC‐1alpha transcriptional complex. Nature 450, 736–740. [DOI] [PubMed] [Google Scholar]
- De Jonghe B, Sharshar T, Lefaucheur JP, Authier FJ, Durand‐Zaleski I, Boussarsar M, Cerf C, Renaud E, Mesrati F, Carlet J, Raphael JC, Outin H & Bastuji‐Garin S (2002). Paresis acquired in the intensive care unit: a prospective multicenter study. JAMA 288, 2859–2867. [DOI] [PubMed] [Google Scholar]
- Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, Kozma SC, Thomas G & Sonenberg N (2010). mTORC1‐mediated cell proliferation, but not cell growth, controlled by the 4E‐BPs. Science 328, 1172–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin BR & Dworkin S (1990). Learning of physiological responses: I. Habituation, sensitization, and classical conditioning. Behav Neurosci 104, 298–319. [DOI] [PubMed] [Google Scholar]
- Dworkin BR & Dworkin S (2004). Baroreflexes of the rat. III. Open‐loop gain and electroencephalographic arousal. Am J Physiol Regul Integr Comp Physiol 286, R597–R605. [DOI] [PubMed] [Google Scholar]
- Elgass K, Pakay J, Ryan MT & Palmer CS (2013). Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta 1, 150–161. [DOI] [PubMed] [Google Scholar]
- Ferri KF, Jacotot E, Blanco J, Este JA, Zamzami N, Susin SA, Xie Z, Brothers G, Reed JC, Penninger JM & Kroemer G (2000). Apoptosis control in syncytia induced by the HIV type 1‐envelope glycoprotein complex: role of mitochondria and caspases. J Exp Med 192, 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S & Scorrano L (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc 2, 287–295. [DOI] [PubMed] [Google Scholar]
- Goldberg AL (1968). Protein synthesis during work‐induced growth of skeletal muscle. J Cell Biol 36, 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL, Etlinger JD, Goldspink DF & Jablecki C (1975). Mechanism of work‐induced hypertrophy of skeletal‐muscle. Med Sci Sports 7, 248–261. [PubMed] [Google Scholar]
- Goldspink DF (1977). The influence of denervation and stretch on the size and protein turnover of rat skeletal muscle. J Physiol 269, 87P–88P. [PubMed] [Google Scholar]
- Griffiths RD, Palmer TE, Helliwell T, MacLennan P & MacMillan RR (1995). Effect of passive stretching on the wasting of muscle in the critically ill. Nutrition 11, 428–432. [PubMed] [Google Scholar]
- Han B, Zhu MJ, Ma C & Du M (2007). Rat hindlimb unloading down‐regulates insulin like growth factor‐1 signaling and AMP‐activated protein kinase, and leads to severe atrophy of the soleus muscle. Appl Physiol Nutr Metab 32, 1115–1123. [DOI] [PubMed] [Google Scholar]
- Hornberger TA (2011). Mechanotransduction and the regulation of mTORC1 signaling in skeletal muscle. Int J Biochem Cell Biol 43, 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberger TA, Sukhija KB, Wang XR & Chien S (2007). mTOR is the rapamycin‐sensitive kinase that confers mechanically‐induced phosphorylation of the hydrophobic motif site Thr(389) in p70(S6k). FEBS Lett 581, 4562–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto M, Nikawa T, Takeda S, Watanabe C, Kitano T, Baldwin KM, Izumi R, Nonaka I, Towatari T, Teshima S, Rokutan K & Kishi K (2001). Space shuttle flight (STS‐90). enhances degradation of rat myosin heavy chain in association with activation of ubiquitin‐proteasome pathway. FASEB J 15, 1279–1281. [DOI] [PubMed] [Google Scholar]
- Jackman RW & Kandarian SC (2004). The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol 287, C834–C843. [DOI] [PubMed] [Google Scholar]
- Jiang X & Wang X (2000). Cytochrome c promotes caspase‐9 activation by inducing nucleotide binding to Apaf‐1. J Biol Chem 275, 31199–31203. [DOI] [PubMed] [Google Scholar]
- Kimball SR, Farrell PA & Jefferson LS (2002). Role of insulin in translational control of protein synthesis in skeletal muscle by amino acids or exercise. J Appl Physiol (1985) 93, 1168–1180. [DOI] [PubMed] [Google Scholar]
- Kodani M, Igishi T, Matsumoto S, Chikumi H, Shigeoka Y, Nakanishi H, Morita M, Yasuda K, Hitsuda Y & Shimizu E (2005). Suppression of phosphatidylinositol 3‐kinase/Akt signaling pathway is a determinant of the sensitivity to a novel histone deacetylase inhibitor, FK228, in lung adenocarcinoma cells. Oncol Rep 13, 477–483. [PubMed] [Google Scholar]
- Kubica N, Bolster DR, Farrell PA, Kimball SR & Jefferson LS (2005). Resistance exercise increases muscle protein synthesis and translation of eukaryotic initiation factor 2Bepsilon mRNA in a mammalian target of rapamycin‐dependent manner. J Biol Chem 280, 7570–7580. [DOI] [PubMed] [Google Scholar]
- Larsson L, Li XP, Edstrom L, Eriksson LI, Zackrisson H, Argentini C & Schiaffino S (2000). Acute quadriplegia and loss of muscle myosin in patients treated with nondepolarizing neuromuscular blocking agents and corticosteroids: Mechanisms at the cellular and molecular levels. Crit Care Med 28, 34–45. [DOI] [PubMed] [Google Scholar]
- Llano‐Diez M, Renaud G, Andersson M, Marrero HG, Cacciani N, Engquist H, Corpeno R, Artemenko K, Bergquist J & Larsson L (2012). Mechanisms underlying ICU muscle wasting and effects of passive mechanical loading. Crit Care 16, R209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane IA & Rosenthal FD (1977). Severe myopathy after status asthmaticus. Lancet 2, 615. [DOI] [PubMed] [Google Scholar]
- Macpherson PC, Wang X & Goldman D (2011). Myogenin regulates denervation‐dependent muscle atrophy in mouse soleus muscle. J Cell Biochem, 112, 2149–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzetti E, Wohlgemuth SE, Lees HA, Chung HY, Giovannini S & Leeuwenburgh C (2008). Age‐related activation of mitochondrial caspase‐independent apoptotic signaling in rat gastrocnemius muscle. Mech Ageing Dev, 129, 542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan G, Romanello V, Pescatore F, Armani A, Paik J‐HH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, Blaauw B, DePinho RA & Sandri M (2015). Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun, 6, 6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresi V, Williams AH, Meadows E, Flynn JM, Potthoff MJ, McAnally J, Shelton JM, Backs J, Klein WH, Richardson JA, Bassel‐Duby R & Olson EN (2010). Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 143, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, St‐Pierre J, Topisirovic I & Sonenberg N (2013). mTORC1 controls mitochondrial activity and biogenesis through 4E‐BP‐dependent translational regulation. Cell Metab 18, 698–711. [DOI] [PubMed] [Google Scholar]
- Nimmanapalli R, Fuino L, Bali P, Gasparetto M, Glozak M, Tao J, Moscinski L, Smith C, Wu J, Jove R, Atadja P & Bhalla K (2003). Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr‐Abl and induces apoptosis of imatinib mesylate‐sensitive or ‐refractory chronic myelogenous leukemia‐blast crisis cells. Cancer Res 63, 5126–5135. [PubMed] [Google Scholar]
- Norman H, Kandala K, Kolluri R, Zackrisson H, Nordquist J, Walther S, Eriksson LI & Larsson L (2006). A porcine model of acute quadriplegic myopathy: a feasibility study. Acta Anaesthesiol Scand 50, 1058–1067. [DOI] [PubMed] [Google Scholar]
- Ochala J, Ahlbeck K, Radell PJ, Eriksson LI & Larsson L (2011. a). Factors underlying the early limb muscle weakness in acute quadriplegic myopathy using an experimental ICU porcine model. PLoS One 6, e20876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochala J, Gustafson AM, Diez ML, Renaud G, Li M, Aare S, Qaisar R, Banduseela VC, Hedstrom Y, Tang X, Dworkin B, Ford GC, Nair KS, Perera S, Gautel M & Larsson L (2011. b). Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: underlying mechanisms. J Physiol 589, 2007–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin AM, Hoshijima M, Swanson E, McCulloch AD & Omens JH (2009). Hypertrophic gene expression induced by chronic stretch of excised mouse heart muscle. Mol Cell Biomech 6, 145–159. [PMC free article] [PubMed] [Google Scholar]
- Renaud G, Llano‐Diez M, Ravara B, Gorza L, Feng HZ, Jin JP, Cacciani N, Gustafson AM, Ochala J, Corpeno R, Li M, Hedstrom Y, Ford GC, Nair KS & Larsson L (2013). Sparing of muscle mass and function by passive loading in an experimental intensive care unit model. J Physiol 591, 1385–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich MM, Bird SJ, Raps EC, McCluskey LF & Teener JW (1997). Direct muscle stimulation in acute quadriplegic myopathy. Muscle Nerve 20, 665–763. [DOI] [PubMed] [Google Scholar]
- Rich MM, Pinter MJ, Kraner SD & Barchi RL (1998. a). Loss of electrical excitability in an animal model of acute quadriplegic myopathy. Ann Neurol 43, 171–179. [DOI] [PubMed] [Google Scholar]
- Rich MM, Teener JW, Raps EC & Bird SJ (1998. b). Muscle inexcitability in patients with reversible paralysis following steroids and neuromuscular blockade. Muscle Nerve 21, 1231–1232. [DOI] [PubMed] [Google Scholar]
- Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R & Sandri M (2010). Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J 29, 1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander HW, Golden M & Danon MJ (2002). Quadriplegic areflexic ICU illness: selective thick filament loss and normal nerve histology. Muscle Nerve 26, 499–505. [DOI] [PubMed] [Google Scholar]
- Sandri M (2008). Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 23, 160–170. [DOI] [PubMed] [Google Scholar]
- Sandri M (2013). Protein breakdown in muscle wasting: role of autophagy‐lysosome and ubiquitin‐proteasome. Int J Biochem Cell Biol 45, 2121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL & Spiegelman BM (2006). PGC‐1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy‐specific gene transcription. Proc Natl Acad Sci USA 103, 16260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A, Stricker S, Goldberg AL, Dupont S, Piccolo S, Amthor H & Sandri M (2013). BMP signaling controls muscle mass. Nat Genet 45, 1309–1318. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Dyar KA, Ciciliot S, Blaauw B & Sandri M (2013). Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 280, 4294–4314. [DOI] [PubMed] [Google Scholar]
- Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prevost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC & Kroemer G (2000). Two distinct pathways leading to nuclear apoptosis. J Exp Med 192, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H & Goldman D (2006). Activity‐dependent gene regulation in skeletal muscle is mediated by a histone deacetylase (HDAC)‐Dach2‐myogenin signal transduction cascade. Proc Natl Acad Sci USA 103, 16977–16982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Macpherson P, Marvin M, Meadows E, Klein WH, Yang XJ & Goldman D (2009). A histone deacetylase 4/myogenin positive feedback loop coordinates denervation‐dependent gene induction and suppression. Mol Biol Cell 20, 1120–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason DB & Booth FW (1990). Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol (1985) 68, 1–12. [DOI] [PubMed] [Google Scholar]
- Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, Alexander C, Bauer C, Youle R, Langer T & Martinou JC (2009). SLP‐2 is required for stress‐induced mitochondrial hyperfusion. Embo J 28, 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenburgh HH (1987). Motion into mass: how does tension stimulate muscle growth? Med Sci Sports Exerc 19, S142–S149. [PubMed] [Google Scholar]
- Weber‐Carstens S, Deja M, Koch S, Spranger J, Bubser F, Wernecke KD, Spies CD, Spuler S & Keh D (2010). Risk factors in critical illness myopathy during the early course of critical illness: a prospective observational study. Crit Care 14, R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigl LG (2012). Lost in translation: regulation of skeletal muscle protein synthesis. Curr Opin Pharmacol 12, 377–382. [DOI] [PubMed] [Google Scholar]
- Yang YL, Huang PH, Chiu HC, Kulp SK, Chen CS, Kuo CJ & Chen HD (2013). Histone deacetylase inhibitor AR42 regulates telomerase activity in human glioma cells via an Akt‐dependent mechanism. Biochem Biophys Res Commun 435, 107–112. [DOI] [PubMed] [Google Scholar]