

Abstract
Ion channels represent a large and growing family of target proteins regulated by gasotransmitters such as nitric oxide, carbon monoxide and, as described more recently, hydrogen sulfide. Indeed, many of the biological actions of these gases can be accounted for by their ability to modulate ion channel activity. Here, we report recent evidence that H2S is a modulator of low voltage‐activated T‐type Ca2+ channels, and discriminates between the different subtypes of T‐type Ca2+ channel in that it selectively modulates Cav3.2, whilst Cav3.1 and Cav3.3 are unaffected. At high concentrations, H2S augments Cav3.2 currents, an observation which has led to the suggestion that H2S exerts its pro‐nociceptive effects via this channel, since Cav3.2 plays a central role in sensory nerve excitability. However, at more physiological concentrations, H2S is seen to inhibit Cav3.2. This inhibitory action requires the presence of the redox‐sensitive, extracellular region of the channel which is responsible for tonic metal ion binding and which particularly distinguishes this channel isoform from Cav3.1 and 3.3. Further studies indicate that H2S may act in a novel manner to alter channel activity by potentiating the zinc sensitivity/affinity of this binding site. This review discusses the different reports of H2S modulation of T‐type Ca2+ channels, and how such varying effects may impact on nociception given the role of this channel in sensory activity. This subject remains controversial, and future studies are required before the impact of T‐type Ca2+ channel modulation by H2S might be exploited as a novel approach to pain management.
Abbreviations
- CBS
cystathionine β synthase
- CSE
cystathionine γ lyase
- DRG neuron
dorsal root ganglion neuron
- HEK293 cells
human embryonic kidney cells
- KATP
ATP‐sensitive K+ channel
- 3‐MST
3‐mercaptopyruvate sulfurtransferase
- TPEN
N,N,N′,N′‐tetrakis(2‐pyridylmethyl)ethane‐1,2‐diamine
Introduction
Ion channel activity is central to a vast and diverse array of cellular functions in both excitable and non‐excitable cell types. For example, ion channel activity controls gene expression, apoptosis, proliferation, contractility, fluid transport, exocytosis, excitability and action potential propagation. It is therefore unsurprising that the number of genes encoding ion channel proteins (either the pore‐forming proteins or auxiliary subunits) runs into the hundreds. This diversity is further increased by splice variation and also the fact that subunits of different channel types can form functional channels by combining with other (albeit related) subunits to form heteromeric complexes. Of the many functions regulated by ion channels one of the most important is the control of [Ca2+]i as this ion is a ubiquitous intracellular signalling molecule that mediates many of the ways in which channels influence the above‐mentioned fundamental cellular functions (Berridge et al. 2003; Clapham, 2007). Here, we discuss recently discovered new modes of regulation of one specific class of Ca2+ channel, the voltage‐gated T‐type Ca2+ channel.
Tailoring ion channel activity to serve specific and often co‐ordinated roles requires not only the availability of multiple channel types, but also that they can be dynamically regulated. This can occur by a plethora of means, for example by coupling to specific G proteins and via numerous post‐translational modifications, including phosphorylation, ubiquitination, sumoylation, nitrosylation, sulfhydration and S‐acylation (Rajan et al. 2005; Gonzalez et al. 2009; Mustafa et al. 2011; Lipscombe et al. 2013; Shipston, 2014). Such modifications can regulate ion channel trafficking and membrane insertion, as well as ongoing activity. In recent years, it has become apparent that ion channels are also modulated by endogenously generated, biologically active gases, often termed gasotransmitters, such as nitric oxide, carbon monoxide and hydrogen sulfide (NO, CO and H2S, respectively). The roles of these gases, especially NO, in diverse physiological and pathological settings have in many instances been long‐established, but the concept that they represent a new class of ion channel regulators is currently emerging (Wilkinson & Kemp, 2011; Peers et al. 2012, 2014). In this article, we focus on H2S specifically as a modulator of voltage‐gated T‐type Ca2+ channels.
T‐type Ca2+ channels
When first described at the single channel level, this class of voltage‐gated Ca2+ channel was named ‘T‐type’ since members had a relatively tiny conductance and gave rise to transient currents (Nowycky et al. 1985). These channels are also distinguished from other voltage‐gated Ca2+ channels by their rapid activation, slow deactivation and very negative activation thresholds, as low as −60 mV or even below (Carbone & Lux, 1984; Perez‐Reyes, 2003; Iftinca & Zamponi, 2009). Example currents are shown in Fig. 1 A, and a typical current–voltage relationship is presented in Fig. 1 B. Owing to this low threshold for activation, they also may display a significant window current (i.e. display tonic activity; Fig. 1 C, arrowed) at potentials close to the resting membrane potential (RMP), and so can provide a sustained route for Ca2+ entry into resting cells (exemplified in Fig. 1 D) and also contribute to setting the RMP (Perez‐Reyes, 2003).Three genes, CACNA1G, CACNA1H and CACNA1I give rise to pore‐forming α‐subunits of T‐type Ca2+ channels, which are nowadays referred to as Cav3.1–3.3, respectively (Catterall et al. 2005), with Cav3.3 showing relatively slower activation and inactivation kinetics than Cav3.1 or Cav3.2 (Fig. 1 A). Heterologous expression of individual Cav3 α‐subunits gives rise to currents which closely resemble native T‐type Ca2+ currents, suggesting that auxiliary subunits are not required for assembly of functional Ca2+ channels, although other channel properties and trafficking may indeed be affected by the auxiliary subunits (Dolphin et al. 1999).
Figure 1. Properties of T‐type Ca2+ channels .
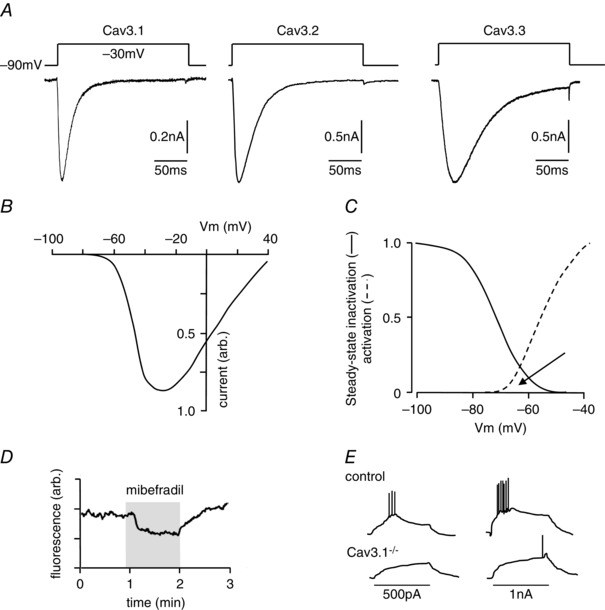
A, example currents evoked by step depolarizations applied (according to the protocol shown above each trace) to HEK293 cells expressing each of the 3 classes of T‐type Ca2+ channel, as indicated. Note the relatively slow activation and inactivation of Cav3.3 as compared with Cav3.1 and Cav3.2. B, schematic current–voltage relationship typical of T‐type Ca2+ channels; note the low threshold for activation. C, superimposition of steady‐state inactivation plot (continuous line) and activation profile (dashed line) typical of T‐type Ca2+ channels. Note the pronounced window current (region of overlap, arrowed). D, fluorimetric recording of [Ca2+]i in a Fura‐2‐loaded HEK293 cell expressing Cav3.2. For the period indicated by the shaded area, the T‐type channel blocker mibefradil (1 μm) was applied, causing a reduction in basal [Ca2+]i (N.T. Hettiarachchi and C Peers, unpublished observations). E, schematic diagram of membrane depolarizations evoked by current injections in thalamic neurones. Note the evoked burst activity is almost fully suppressed in cells lacking the T‐type channel Cav3.1 (see Kim et al. 2001).
T‐type Ca2+ channels are widely distributed, but their physiological functions have sometimes proved difficult to resolve. This is certainly true in vascular smooth muscle cells (VSMCs), where Cav3.1 and Cav3.2 are expressed. Numerous attempts have failed to record ‘classic’ T‐type Ca2+ channel activity in VSMCs (reviewed in Kuo et al. 2011, 2014). Recent studies suggest this is because VSMCs primarily express T‐type Ca2+ channel splice variants which do not activate at the low voltages normally associated with T‐type Ca2+ channels. Instead, they activate over more depolarized voltage ranges as are observed for L‐type Ca2+ channels. Splice variation accounting for these differences occurs in Cav3.1 (and similarly in Cav3.2) primarily around exons 25 and 26, corresponding to the intracellular linker region between domains III and IV (Chemin et al. 2001; Latour et al. 2004; Emerick et al. 2006). Four variants have been identified termed 25a, 25b, 25bc and 25ac, with variant 25bc predominating in both the juvenile and mature systemic vasculature. These variants all contribute to vasomotor tone (Kuo et al. 2011, 2014), and may also have a pacemaker role, controlling slow Ca2+ waves and contractile oscillations (Cribbs, 2006).
Despite continued uncertainty around the physiological roles of T‐type Ca2+ channels in the vasculature, it appears that in several vascular diseases the influence of T‐type Ca2+ channels, particularly Cav3.1, becomes more prominent. Their increased expression and activity appears to be instrumental in pathological vascular remodelling in both the systemic and pulmonary circulation, providing a route for Ca2+ entry which is required for proliferation (Tzeng et al. 2012; Chevalier et al. 2014). Interestingly, numerous cancers also rely on the expression of T‐type Ca2+ channels for proliferation, and hence tumour growth (Dziegielewska et al. 2014). These findings suggest that they represent a promising therapeutic target in the treatment of various cancers and cardiovascular diseases.
In the nervous system, T‐type Ca2+ channels serve better defined roles. Thus, for example, in thalamic and corticothalamic neurones T‐type Ca2+ channels are responsible for pacemaker activity and low threshold spikes (Huguenard & Prince, 1994), as illustrated in Fig. 1 E. They contribute to ‘rebound’ bursts of spikes following a hyperpolarizing postsynaptic potential. Indeed, evidence suggests that all three isoforms of T‐type Ca2+ channels can contribute to this excitability (Kim et al. 2001; Joksovic et al. 2006; Lee et al. 2014). In the peripheral nervous system, T‐type Ca2+ channels are prominent in somatosensory fibres including small, capsaicin‐sensitive (presumed nociceptive) dorsal root ganglion (DRG) neurons (Jevtovic‐Todorovic & Todorovic, 2006; Nelson et al. 2006; Rose et al. 2013) as well as in two distinct populations of low‐threshold mechanoreceptors (LTMRs), Aδ‐ and C‐LTMRs, innervating skin hair follicles (Francois et al. 2015). Cav3.2 is the dominant form in DRG and may even be the exclusive form in some mechanosensitive neurons (Shin et al. 2003). Cav3.2 channels can control burst firing in DRG neurons (White et al. 1989) and so strongly influence excitability (Nelson et al. 2005, 2007 a), implying they are of central importance to nociception since stimulus intensity correlates with burst frequency. Indeed, the role of T‐type Ca2+ channels in pain is well recognized and has been covered in depth in several recent reviews (Bourinet et al. 2014; Todorovic & Jevtovic‐Todorovic, 2014; Francois et al. 2014; Waxman & Zamponi, 2014; Zamponi et al. 2015). Thus, conditional genetic deletion (Francois et al. 2015), or downregulation of Cav3.2 in DRG using intrathecal injection of antisense oligonucleotides (Bourinet et al. 2005; Messinger et al. 2009) produced strong anti‐nociceptive effects in rodent pain models of neuropathic and inflammatory pain. Conversely, T‐type Ca2+ currents are often increased in pathological conditions associated with chronic pain, such as diabetic neuropathy (Jagodic et al. 2007; Messinger et al. 2009), peripheral nerve injury or inflammation (Jagodic et al. 2008; Marger et al. 2011; Garcia‐Caballero et al. 2014). Both the enhancement of channel trafficking (Orestes et al. 2013; Weiss et al. 2013) and enhanced deubiquitination (Garcia‐Caballero et al. 2014) were reported as underlying mechanisms for the latter phenomenon.
It is not entirely clear how exactly T‐type Ca2+ channels participate in the nociceptive transmission. Cav3.2 channel expression has been detected in different compartments of afferent fibres, including peripheral nociceptive terminals and axons of skin afferents (Rose et al. 2013; Francois et al. 2015), nodes of Ranvier of Aδ fibres (Francois et al. 2015), as well as in the presynaptic terminals of nociceptive fibres in the spinal cord (Jacus et al. 2012; however, cf. Francois et al. 2015). Therefore, multiple mechanisms (or their combinations) are conceivable, including setting the threshold for action potential generation (at the nerve terminals) and propagation (at the nodes of thinly myelinated fibres), supporting burst firing, or indeed promoting synaptic activity at the central terminals of afferent fibres. Finally, since T‐type Ca2+ channels are expressed in skin terminals of low threshold mechanoreceptors, including D‐hair cells (Coste et al. 2007; Francois et al. 2015), a more direct role of T‐type Ca2+ channels in mechanotransduction also cannot be excluded.
In accordance with strong evidence for the physiological role of T‐type Ca2+ channels in pain, recent data clearly demonstrated that pharmacological inhibition of T‐type Ca2+ channel activity produces strong anti‐nociceptive effects in various rodent pain models (Todorovic et al. 2001, 2002, 2004; Latham et al. 2009). Together with N‐type Ca2+ channels, T‐type Ca2+ channels are clinically validated drug targets for pain (Bourinet et al. 2014). Accordingly, intense search is currently underway for novel pharmacological tools targeting T‐type Ca2+ channel activity. These need to be more selective than widely used blockers such as mibefradil which, despite its ability to inhibit T‐type Ca2+ channels, is not highly selective (Bezprozvanny & Tsien, 1995). This new generation of more selective T‐type inhibitors (e.g. TTA‐A2, TTA‐P2, KYS‐05090S) shows promising analgesic properties in animal models of pain (Choe et al. 2011; Francois et al. 2013). Another novel, selective and orally bioavailable T‐type Ca2+ channel blocker, Z944, which showed analgesic activity in animal models of inflammatory and neuropathic pain, is currently being tested in clinical trials as a first‐in‐class novel oral analgesic (Lee, 2014). Moreover, Zamponi's lab has recently developed small molecule modulators that prevent deubiquitination of the Cav3.2 channels by the deubiquitinase USP5, which in turn can prevent channel upregulation in chronic pain conditions (Gadotti et al. 2015).
A redox‐ and Zn2+‐sensitive ‘module’ within the Cav3.2 subunit
Studies in both native DRG neurons and recombinant expression systems have revealed a regulatory site within Cav3.2 which confers high sensitivity to redox agents, distinguishing it functionally from both Cav3.1 and Cav3.3. Thus, for example, T‐type Ca2+ currents recorded in native nociceptive neurones and recombinant Cav3.2 currents are enhanced by reducing agents (dithiothreitol (DTT) or l‐cysteine) and inhibited by the oxidizing agent 5,5′‐dithio‐bis(2‐nitrobenzoic acid) (DTNB) (Nelson et al. 2007 a). Sensitivity to these agents was exploited in order to demonstrate the importance of Cav3.2 in nociception: hindpaw injections of DTT or l‐cysteine induces thermal and mechanical hyperalgesia (Todorovic et al. 2001), and such effects are prevented with the Ca2+ channel inhibitor mibefradil; furthermore, analgesic effects are observed with DTNB (Todorovic et al. 2001). Such findings were confirmed in a subpopulation of nociceptive neurons expressing high levels of Cav3 channels (Nelson et al. 2005). An important breakthrough in the field was achieved by the joint work of Perez‐Reyes’ and Lee's groups which allowed the characterization of specific Cav3.2 residues involved in the metal‐induced inhibition of the Ca2+ channel. The redox‐sensitive module is located extracellularly, involving interaction of the extracellular IS1–IS2 linker region with the IS3–IS4 linker region (Kang et al. 2010), the latter region containing a histidine residue (H191) which is key to conferring high redox sensitivity to this channel. Intriguingly, the same H191 residue that is involved in modulation of T‐type Ca2+ channel activity by redox mechanisms also mediates T‐type Ca2+ channel inhibition by low (submicromolar) concentrations of extracellular Zn2+ (Kang et al. 2006, 2010). The full Zn2+ binding site also includes residues that precede H191, namely D189 and G190, as well as the negatively charged residues at the outer portion of the IS2 segment (Kang et al. 2010). It is, as yet, unclear how exactly the redox‐ and Zn2+‐mediated modulatory pathways converge at the same site but one hypothesis suggests that H191 may represent a general binding site for transition metals (e.g. iron, copper, zinc) and can be subject to oxidation via a metal‐catalysed oxidation (MCO) reaction (Stadtman, 2001; Nelson et al. 2007 a). It is conceivable therefore that either binding of metal (e.g. Zn2+) or Zn2+‐independent MCO of H191 may result in a similar inhibition of channel activity. An alternative hypothesis is that oxidative modification (possibly but not necessarily at H191) can increase the effect of metal binding either by increasing the affinity at the binding site or by enhancing coupling efficiency between the metal binding and channel inhibition. In such a scenario, oxidative modification could result in sensitization of the channel to Zn2+ (and potentially other transition metals) making the channel sensitive to ambient concentrations in biological fluids. Total Zn2+ concentration in human plasma is reported to be within the range of 5–20 μm (Moran et al. 2012). Although it is likely that the free [Zn2+] is considerably lower, it is still quite plausible that the extracellular free [Zn2+] can reach the nanomolar range at which changes in Zn2+ affinity/sensitivity of Cav3.2 subunits may result in noticeable changes in channel activity. While the exact mechanism of regulation of Cav3.2 activity via the H191‐containing regulatory site remains to be elucidated, it is clear that redox modulation is involved in the action on the T‐type Ca2+ channels of some pharmacological agents and physiological signalling cascades including nitrous oxide (Orestes et al. 2011), CO and thioredoxin (Boycott et al. 2013), as well as GABAB receptors (Huang et al. 2015).
H2S as a gasotransmitter
H2S is regarded as the third ‘gasotransmitter’ since it is an endogenous, enzymatically generated, biologically active gas. Emerging evidence indicates that it has widespread physiological and pathophysiological importance throughout the body (Li et al. 2011). Unsurprisingly, therefore, it has received increasing interest as a potential therapeutic target over the past decade or so, as was the case previously for both NO and CO (Moore et al. 2003; Leffler et al. 2006; Szabo, 2007). H2S is generated primarily by the action of two widely distributed enzymes, cystathionine γ lyase (CSE) and cystathionine β synthetase (CBS; Fig. 2). Both synthesize the gas from l‐cysteine. More recently, the mitochondrially located 3‐mercaptopyruvate sulfurtransferase (3‐MST) has also been found to generate H2S in both the brain and vasculature (Fig. 2). 3‐MST generates H2S from 3‐mercaptopyruvate which is itself generated by cysteine aminotransferase (CAT; Kimura, 2010). Red blood cells can generate H2S non‐enzymatically from inorganic polysulfides, a finding which has prompted the idea that H2S may mediate the beneficial vascular effects of dietary garlic (Benavides et al. 2007). H2S is also known to be liberated in a redox‐ or pH‐sensitive manner from sulfur ‘stores’, i.e. sulfur bound to proteins in mitochondria or the cytosol (Kimura, 2010). Differential distribution of H2S‐generating enzymes results in a predomination of CBS in the central nervous system, along with 3‐MST (Leffler et al. 2006), whereas CSE is dominant peripherally, generating the majority of H2S elsewhere in the body, including the vasculature. More recently, H2S has been shown to be generated from d‐cysteine (Fig. 2), particularly in the cerebellum and kidney, via a novel pathway involving 3‐MST and d‐amino acid oxidase (Shibuya et al. 2013).
Figure 2. Hydrogen sulfide synthesis and effector ion channels .
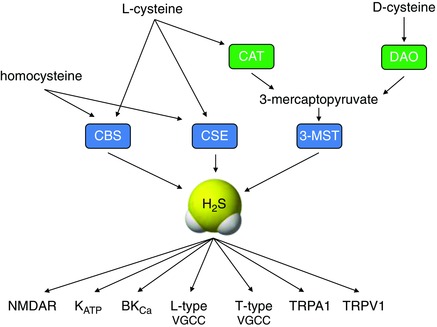
Schematic diagram illustrating the synthesis of H2S from homocysteine, l‐ and d‐cysteine via enzymes shown in boxes (CBS, cystathionine β synthase; CSE, cystathionine γ lyase; 3‐MST, 3‐mercaptopyruvate sulfurtransferase; CAT, cysteine aminotransferase; DAO, d‐amino acid oxidase). Some of the target ion channels modulated by H2S are also indicated (NMDAR, N‐methyl‐ d‐aspartate receptor; KATP, ATP‐sensitive K+ channel; BKCa, large conductance K+ channel; VGCC, voltage‐gated Ca2+ channel; TRP, transient receptor potential channel).
H2S as a regulator of ion channels
Ion channels (specifically NMDA receptors; Abe & Kimura, 1996) were amongst the first family of cellular proteins recognized as being molecular targets of H2S. Since then, reports of ion channel regulation by H2S have grown rapidly (Peers et al. 2012; Kuksis & Ferguson, 2015; Zhang et al. 2015), as indicated by Fig. 2. It is clear that modulation by H2S is not confined to any particular class of ion channels (such as voltage‐ or ligand‐gated channels) or to channels which are selective for specific ions (e.g. K+, Na+, Ca2+ or Cl−) (Zhang et al. 2015). H2S is also known to interfere with a number of intracellular signalling pathways (Wang, 2012), and it is via these pathways that H2S, in some cases, modifies ion channel activity. In other cases, direct post‐translational modification accounts for its effects. The best studied of these is the vascular smooth muscle ATP‐sensitive K+ channel (KATP channel), which becomes more active when sulfhydrated (i.e. when –SH groups within cysteine residues are altered to –SSH groups). This direct process (also known as persulfidation; see Paul & Snyder, 2015) contributes to vasorelaxation and thereby protects against hypertension (Mustafa et al. 2011; Paul & Snyder, 2012).
The list of ion channels regulated by H2S is long and continues to grow. However, some areas of contention have arisen along the way. Thus, for example, inhibition by H2S of the high‐conductance, Ca2+‐sensitive K+ channel has been described in detail (Telezhkin et al. 2010), yet others report activation of this channel by H2S (Sitdikova et al. 2010; Jackson‐Weaver et al. 2013). Similarly, inhibition of L‐type Ca2+ channels in cardiac myocytes (Sun et al. 2008; Avanzato et al. 2014) contrasts with the observed augmentation of L‐type Ca2+ channel activity reported in astrocytes (Nagai et al. 2004). Such discrepancies may have simple explanations, but these need to be identified if H2S modulation of ion channels is to be exploited therapeutically. The remainder of this review is focused on the known pro‐algesic effects of H2S and considers the recent, conflicting data concerning the role of T‐type Ca2+ channels in this process.
H2S as a regulator of Cav3.2
Our group has recently reported an inhibitory effect of micromolar concentrations of H2S on recombinant and native T‐type Ca2+ channels in sensory neurons (Elies et al. 2014). Among Cav3 subunits, this effect was selective to Cav3.2 while Cav3.1 and Cav3.3 were insensitive to H2S. In agreement with the predominant expression of Cav3.2 subunits in DRG neurons, H2S also strongly inhibited endogenous low‐voltage activated (LVA) currents in these neurons. Interestingly, the same extracellular histidine residue, H191, which is necessary for Cav3.2 modulation by Zn2+ and redox agents, was also found critical for the effect of H2S. Thus, the H191Q mutation (where histidine has been substituted by glutamine) in Cav3.2 abolished channel sensitivity to H2S and the analogous reciprocal mutation in Cav3.1 (Q172H) conferred sensitivity to H2S on this subunit (Elies et al. 2014). It is not entirely clear exactly how redox/Zn2+ binding of Cav3.2 is implicated in this H2S‐mediated effect but one theory is that H2S may increase channel sensitivity to extracellular Zn2+. Indeed, chelation of ambient Zn2+ with TPEN (N,N,N',N'‐tetrakis(2‐pyridylmethyl)ethane‐1,2‐diamine) fully reversed the H2S‐mediated T‐type Ca2+ channel inhibition. Moreover, pretreatment with TPEN abolished H2S‐mediated inhibition when H2S was applied in the continued presence of TPEN. However, subsequent washing out of TPEN in the presence of H2S allowed the inhibition to commence (Elies et al. 2014). Thus, the action of H2S on Cav3.2 seems to depend on ambient Zn2+, assumed to be present in extracellular solutions in trace amounts. Indeed, according to our atomic absorption spectroscopy measurements, total Zn2+ levels can reach micromolar levels in nominally zinc‐free laboratory solutions (D. Huang & N. Gamper, unpublished observations); likewise, micromolar concentrations of Zn2+ in plasma are also reported (Moran et al. 2012). While levels of free Zn2+ are likely to be much lower as compared to the total zinc, it is still likely that this metal is present in the extracellular milieu at levels sufficient to affect channel activity. We hypothesize that the action of H2S results in the potentiation of channel inhibition by such ambient Zn2+, via a mechanism which may share commonalities with the redox modulation of Cav3.2 (see Abstract figure).
Although our data strongly suggest that H2S inhibits Cav3.2 and the endogenous T‐type Ca2+ currents in DRG neurons, there is an alternative theory suggesting that H2S augments T‐type Ca2+ currents, possibly by chelating Zn2+, and that this augmentation underlies the pro‐algesic actions of this gasotransmitter (Kawabata et al. 2007; Maeda et al. 2009; Takahashi et al. 2010; Matsunami et al. 2011). Thus, injection of the H2S donor NaHS into the rat hindpaw produced hyperalgesia which was abolished by the oxidizing agent DTNB (5,5′‐dithiobis‐(2‐nitrobenzoic acid)) and by the pharmacological inhibition of T‐type Ca2+ channels (Kawabata et al. 2007). Inhibition of endogenous H2S production also produced an anti‐algesic effect. This and other studies by the same group led them to conclude that H2S could activate or ‘sensitize’ Cav3.2 channels in order to account for their pro‐algesic effects (Kawabata et al. 2007; Takahashi et al. 2010). Furthermore, H2S‐induced colonic pain could be mimicked by chelation of Zn2+ (Matsunami et al. 2011). Yet, most of the evidence suggesting that the pro‐algesic effects of H2S are mediated by T‐type Ca2+ channel augmentation is somewhat circumstantial and direct electrophysiological evidence is sparse and insubstantial. For example, pre‐incubation of NG108‐15 (mouse neuroblastoma and rat glioma hybridoma) cells with 1.5 mm NaHS resulted in ∼20% augmentation of endogenous T‐type Ca2+ current while at 0.5 mm the effect was not significant (Kawabata et al. 2007). Another study demonstrated that pre‐incubation of human embryonic kidney (HEK) 293 cells exogenously expressing Cav3.2 channels with the CSE inhibitor propargylglycine resulted in a reduction of the current, which was interpreted as tonic augmentation of recombinant Cav3.2 by endogenous H2S (Sekiguchi et al. 2014). Interestingly, only in the presence of this inhibitor were currents augmented by NaHS. These experiments are consistent with potential augmentation of T‐type Ca2+ currents by H2S but the following has to be taken into account: (i) only very high, millimolar concentrations of NaHS were efficacious; (ii) due to the experimental protocol used (pre‐incubation) these experiments did not directly assess the acute effect of H2S on T‐type Ca2+ channel activity.
In our hands, increasing the NaHS concentration to 3 mm did augment recombinant Cav3.2 currents, while even higher concentrations were needed to significantly augment native T‐type Ca2+ channel currents in DRG neurons (Elies et al. 2014). These experiments can, to some degree, reconcile conflicting experimental evidence from different laboratories and suggest a dual effect of H2S on T‐type Ca2+ channels: H191‐mediated inhibition at low (micromolar) concentrations and potentiation (due to an unknown mechanism) at high (several millimolar) concentrations. It is important to point out, however, that the presence of H2S at millimolar concentrations in mammalian tissues is highly unlikely. Initial reports estimated plasma H2S levels within the range of 20–100 μm (Li & Moore, 2008), but even these values are now regarded as overestimations (Li et al. 2011). Therefore, physiologically relevant concentrations of H2S are unlikely to be sufficiently high to produce T‐type Ca2+ channel augmentation. The expected effect of endogenous H2S is, therefore, inhibition of T‐type Ca2+ channel currents, at least in cells and tissues that express significant amounts of Cav3.2 (such as sensory neurons). Therefore, the hypothesis that physiological levels of H2S can trigger pro‐algesic effects via T‐type Ca2+ channel activation appears questionable at present. Furthermore, in our view, it is also unlikely that H2S can act as a Zn2+ chelator (as was suggested by Matsunami et al. 2011) as (i) the chemical properties of H2S do not suggest metal‐chelating properties (e.g. the ability to form polydentate complexes); (ii) acute application of H2S leads to T‐type Ca2+ channel current inhibition, whereas Zn2+ chelation with TPEN causes current augmentation (Nelson et al. 2007 b; Elies et al. 2014); (iii) application of TPEN completely reverses H2S‐induced T‐type Ca2+ channel inhibition; (iv) pre‐application of TPEN renders H2S unable to inhibit T‐type Ca2+ channels.
At present, whilst it is hard to envisage how the painful/hyperalgesic effects of H2S (especially those of endogenously produced H2S) can be mediated by its effect on T‐type Ca2+ channels, other possible targets may account for this action. Thus, H2S was suggested to produce some of its pro‐algesic effects via transient receptor potential channel TRPA1 activation (Andersson et al. 2012; Hsu et al. 2013). Additionally, H2S was suggested to inhibit voltage‐gated K+ channels in trigeminal sensory neurons (most likely Kv1.1 and Kv1.4), thus producing depolarizing and excitatory effects (Feng et al. 2013).
Conclusions and perspectives
Tonic H2S production clearly impacts a variety of both physiological and pathological processes, and probably does so in part via direct and indirect modulation of ion channel activity. The numerous and diverse pathways that can be modified by H2S suggest that interventional control of this gasotransmitter could be therapeutically beneficial (e.g. in the cardiovascular system and peripheral sensory neurones). However, conflicts have arisen and must be resolved. We believe that at least some issues could be rectified if the experimental conditions through which we study the effects of H2S are standardized (e.g. preparation of stock solutions of H2S donors, flow rates of solutions containing donors, exposure time and obligatory use of agar bridges for reference electrodes).
Twenty years after the discovery of H2S as an endogenously bioactive molecule (Abe & Kimura, 1996), one of the main challenges within the field remains the measurement of absolute concentrations of H2S, in real time, both intracellularly and in extracellular compartments. For instance, development of appropriate intracellular fluorescence probes will expand not only the knowledge regarding intracellular concentrations of H2S in space and time, but it will also help to correlate experimental data obtained from exogenously administered H2S with experimental data obtained from endogenously generated H2S. Additionally, future research regarding the physiological effects of H2S must bear in mind the crosstalk signalling with other gasotransmitters (NO and CO). Only then can we obtain more comprehensive and uniformly acceptable data on which to build in order to design modulators of H2S production and signalling for translational benefit.
Additional information
Competing interests
None declared.
Funding
This work was supported by grants from the British Heart Foundation (to C.P., J.P.B. and J.L.S.), the Medical Research Council (to C.P. and N.G.), and the Hebei Medical University (to N.G.).
Biographies
Jacobo Elies studied BSc Pharmacy and completed his PhD at the Department of Pharmacology of the University of Santiago de Compostela, Spain. In 2009 he joined Professor Chris Peers as a postdoctoral researcher. Since then he has been investigating the regulation of ion channels by gasotransmitters (CO and H2S) in cardiovascular disease.

Nikita Gamper obtained his PhD in Physiology at the Sechenov Institute, St Petersburg, Russia. After postdoctoral work in Tübingen (Germany) and the University of Texas at San Antonio (USA) he joined the University of Leeds where he studies molecular and cellular mechanisms of nociception. His group investigates regulation of ion channels and G protein coupled receptors that control or influence excitability of peripheral ‘pain’ neurons.
Chris Peers obtained his BSc in Physiology and his PhD in Pharmacology at the University of London. He entered the world of oxygen sensing as a postdoctoral researcher in Piers Nye’s laboratory in Oxford. He then moved to the University of Leeds. His interests in the effects of hypoxia have extended to incorporate effects of gasotransmitters on ion channels and how this impacts on the cardiovascular and neurodegenerative diseases.
This review was presented at the symposium “Gaseous regulation of Ca2+ homeostasis; for better or worse?”, which took place at Physiology 2015 in Cardiff, UK, 6–8 July 2015.
References
- Abe K & Kimura H (1996). The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DA, Gentry C & Bevan S (2012). TRPA1 has a key role in the somatic pro‐nociceptive actions of hydrogen sulfide. PLoS One 7, e46917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzato D, Merlino A, Porrera S, Wang R, Munaron L & Mancardi D (2014). Role of calcium channels in the protective effect of hydrogen sulfide in rat cardiomyoblasts. Cell Physiol Biochem 33, 1205–1214. [DOI] [PubMed] [Google Scholar]
- Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, Darley‐Usmar VM, Doeller JE & Kraus DW (2007). Hydrogen sulfide mediates the vasoactivity of garlic. Proc Natl Acad Sci USA 104, 17977–17982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD & Roderick HL (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4, 517–529. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I & Tsien RW (1995). Voltage‐dependent blockade of diverse types of voltage‐gated Ca2+ channels expressed in Xenopus oocytes by the Ca2+ channel antagonist mibefradil (Ro 40–5967). Mol Pharmacol 48, 540–549. [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A & Nargeot J (2005). Silencing of the Cav3.2 T‐type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J 24, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW & Zamponi GW (2014). Calcium‐permeable ion channels in pain signalling. Physiol Rev 94, 81–140. [DOI] [PubMed] [Google Scholar]
- Boycott HE, Dallas ML, Elies J, Pettinger L, Boyle JP, Scragg JL, Gamper N & Peers C (2013). Carbon monoxide inhibition of Cav3.2 T‐type Ca2+ channels reveals tonic modulation by thioredoxin. FASEB J 27, 3395–3407. [DOI] [PubMed] [Google Scholar]
- Carbone E & Lux HD (1984). A low voltage‐activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature 310, 501–502. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Perez‐Reyes E, Snutch TP & Striessnig J (2005). International Union of Pharmacology. XLVIII. Nomenclature and structure‐function relationships of voltage‐gated calcium channels. Pharmacol Rev 57, 411–425. [DOI] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Bourinet E, Nargeot J & Lory P (2001). Alternatively spliced α1G (CaV3.1) intracellular loops promote specific T‐type Ca2+ channel gating properties. Biophys J 80, 1238–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier M, Gilbert G, Roux E, Lory P, Marthan R, Savineau JP & Quignard JF (2014). T‐type calcium channels are involved in hypoxic pulmonary hypertension. Cardiovasc Res 103, 597–606. [DOI] [PubMed] [Google Scholar]
- Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, Jevtovic‐Todorovic V & Todorovic SM (2011). TTA‐P2 is a potent and selective blocker of T‐type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol 80, 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE (2007). Calcium signalling. Cell 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- Coste B, Crest M & Delmas P (2007). Pharmacological dissection and distribution of NaN/Nav1.9, T‐type Ca2+ currents, and mechanically activated cation currents in different populations of DRG neurons. J Gen Physiol 129, 57–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs LL (2006). T‐type Ca2+ channels in vascular smooth muscle: multiple functions. Cell Calcium 40, 221–230. [DOI] [PubMed] [Google Scholar]
- Dolphin AC, Wyatt CN, Richards J, Beattie RE, Craig P, Lee J‐H, Cribbs LL, Volsen SG & Perez‐Reyes E (1999). The effect of α2‐δ and other accessory subunits on expression and properties of the calcium channel α1G. J Physiol 519, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziegielewska B, Gray LS & Dziegielewski J (2014). T‐type calcium channels blockers as new tools in cancer therapies. Pflugers Arch 466, 801–810. [DOI] [PubMed] [Google Scholar]
- Elies J, Scragg JL, Huang S, Dallas ML, Huang D, MacDougall D, Boyle JP, Gamper N & Peers C (2014). Hydrogen sulfide inhibits Cav3.2 T‐type Ca2+ channels. FASEB J 28, 5376–5387. [DOI] [PubMed] [Google Scholar]
- Emerick MC, Stein R, Kunze R, McNulty MM, Regan MR, Hanck DA & Agnew WS (2006). Profiling the array of Cav3.1 variants from the human T‐type calcium channel gene CACNA1G: alternative structures, developmental expression, and biophysical variations. Proteins 64, 320–342. [DOI] [PubMed] [Google Scholar]
- Feng X, Zhou YL, Meng X, Qi FH, Chen W, Jiang X & Xu GY (2013). Hydrogen sulfide increases excitability through suppression of sustained potassium channel currents of rat trigeminal ganglion neurons. Mol Pain 9, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois A, Kerckhove N, Meleine M, Alloui A, Barrere C, Gelot A, Uebele VN, Renger JJ, Eschalier A, Ardid D & Bourinet E (2013). State‐dependent properties of a new T‐type calcium channel blocker enhance CaV3.2 selectivity and support analgesic effects. Pain 154, 283–293. [DOI] [PubMed] [Google Scholar]
- Francois A, Laffray S, Pizzoccaro A, Eschalier A & Bourinet E (2014). T‐type calcium channels in chronic pain: mouse models and specific blockers. Pflugers Arch 466, 707–717. [DOI] [PubMed] [Google Scholar]
- Francois A, Schuetter N, Laffray S, Sanguesa J, Pizzoccaro A, Dubel S, Mantilleri A, Nargeot J, Noel J, Wood JN, Moqrich A, Pongs O & Bourinet E (2015). The low‐threshold calcium channel Cav3.2 determines low‐threshold mechanoreceptor function. Cell Rep 10, 370–382. [DOI] [PubMed] [Google Scholar]
- Gadotti VM, Caballero AG, Berger ND, Gladding CM, Chen L, Pfeifer TA & Zamponi GW (2015). Small organic molecule disruptors of Cav3.2‐USP5 interactions reverse inflammatory and neuropathic pain. Mol Pain 11, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, Bladen C, Chen L, Hamid J, Pizzoccaro A, Deage M, Francois A, Bourinet E & Zamponi GW (2014). The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 83, 1144–1158. [DOI] [PubMed] [Google Scholar]
- Gonzalez DR, Treuer A, Sun QA, Stamler JS & Hare JM (2009). S‐Nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol 54, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CC, Lin RL, Lee LY & Lin YS (2013). Hydrogen sulfide induces hypersensitivity of rat capsaicin‐sensitive lung vagal neurons: role of TRPA1 receptors. Am J Physiol Regul Integr Comp Physiol 305, R769–R779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Huang S, Peers C, Du X, Zhang H & Gamper N (2015). GABAB receptors inhibit low‐voltage activated and high‐voltage activated Ca2+ channels in sensory neurons via distinct mechanisms. Biochem Biophys Res Commun 465, 188–193. [DOI] [PubMed] [Google Scholar]
- Huguenard JR & Prince DA (1994). Intrathalamic rhythmicity studied in vitro: nominal T‐current modulation causes robust antioscillatory effects. J Neurosci 14, 5485–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iftinca MC & Zamponi GW (2009). Regulation of neuronal T‐type calcium channels. Trends Pharmacol Sci 30, 32–40. [DOI] [PubMed] [Google Scholar]
- Jackson‐Weaver O, Osmond JM, Riddle MA, Naik JS, Gonzalez Bosc LV, Walker BR & Kanagy NL (2013). Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large‐conductance Ca2+‐activated K+ channels and smooth muscle Ca2+ sparks. Am J Physiol Heart Circ Physiol 304, H1446–H1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacus MO, Uebele VN, Renger JJ & Todorovic SM (2012). Presynaptic CaV3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J Neurosci 32, 9374–9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagodic MM, Pathirathna S, Joksovic PM, Lee W, Nelson MT, Naik AK, Su P, Jevtovic‐Todorovic V & Todorovic SM (2008). Upregulation of the T‐type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J Neurophysiol 99, 3151–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, Bayliss DA, Jevtovic‐Todorovic V & Todorovic SM (2007). Cell‐specific alterations of T‐type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci 27, 3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevtovic‐Todorovic V & Todorovic SM (2006). The role of peripheral T‐type calcium channels in pain transmission. Cell Calcium 40, 197–203. [DOI] [PubMed] [Google Scholar]
- Joksovic PM, Nelson MT, Jevtovic‐Todorovic V, Patel MK, Perez‐Reyes E, Campbell KP, Chen CC & Todorovic SM (2006). CaV3.2 is the major molecular substrate for redox regulation of T‐type Ca2+ channels in the rat and mouse thalamus. J Physiol 574, 415–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HW, Park JY, Jeong SW, Kim JA, Moon HJ, Perez‐Reyes E & Lee JH (2006). A molecular determinant of nickel inhibition in Cav3.2 T‐type calcium channels. J Biol Chem 281, 4823–4830. [DOI] [PubMed] [Google Scholar]
- Kang HW, Vitko I, Lee SS, Perez‐Reyes E & Lee JH (2010). Structural determinants of the high affinity extracellular zinc binding site on Cav3.2 T‐type calcium channels. J Biol Chem 285, 3271–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A, Ishiki T, Nagasawa K, Yoshida S, Maeda Y, Takahashi T, Sekiguchi F, Wada T, Ichida S & Nishikawa H (2007). Hydrogen sulfide as a novel nociceptive messenger. Pain 132, 74–81. [DOI] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW & Shin HS (2001). Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking α1G T‐type Ca2+ channels. Neuron 31, 35–45. [DOI] [PubMed] [Google Scholar]
- Kimura H (2010). Hydrogen sulfide: from brain to gut. Antioxid Redox Signal 12, 1111–1123. [DOI] [PubMed] [Google Scholar]
- Kuksis M & Ferguson AV (2015). Actions of a hydrogen sulfide donor (NaHS) on transient sodium, persistent sodium, and voltage gated calcium currents in neurons of the subfornical organ. J Neurophysiol 114, 1641–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo IY, Howitt L, Sandow SL, McFarlane A, Hansen PB & Hill CE (2014). Role of T‐type channels in vasomotor function: team player or chameleon? Pflugers Arch 466, 767–779. [DOI] [PubMed] [Google Scholar]
- Kuo IY, Wolfle SE & Hill CE (2011). T‐type calcium channels and vascular function: The new kid on the block? J Physiol 589, 783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham JR, Pathirathna S, Jagodic MM, Choe WJ, Levin ME, Nelson MT, Lee WY, Krishnan K, Covey DF, Todorovic SM & Jevtovic‐Todorovic V (2009). Selective T‐type calcium channel blockade alleviates hyperalgesia in ob/ob mice. Diabetes 58, 2656–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latour I, Louw DF, Beedle AM, Hamid J, Sutherland GR & Zamponi GW (2004). Expression of T‐type calcium channel splice variants in human glioma. Glia 48, 112–119. [DOI] [PubMed] [Google Scholar]
- Lee M (2014). Z944: a first in class T‐type calcium channel modulator for the treatment of pain. J Peripher Nerv Syst 19, Suppl. 2, S11–S12. [DOI] [PubMed] [Google Scholar]
- Lee SE, Lee J, Latchoumane C, Lee B, Oh SJ, Saud ZA, Park C, Sun N, Cheong E, Chen CC, Choi EJ, Lee CJ & Shin HS (2014). Rebound burst firing in the reticular thalamus is not essential for pharmacological absence seizures in mice. Proc Natl Acad Sci USA 111, 11828–11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler CW, Parfenova H, Jaggar JH & Wang R (2006). Carbon monoxide and hydrogen sulfide: gaseous messengers in cerebrovascular circulation. J Appl Physiol (1985) 100, 1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L & Moore PK (2008). Putative biological roles of hydrogen sulfide in health and disease: a breath of not so fresh air? Trends Pharmacol Sci 29, 84–90. [DOI] [PubMed] [Google Scholar]
- Li L, Rose P & Moore PK (2011). Hydrogen sulfide and cell signalling. Annu Rev Pharmacol Toxicol 51, 169–187. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Allen SE & Toro CP (2013). Control of neuronal voltage‐gated calcium ion channels from RNA to protein. Trends Neurosci 36, 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Aoki Y, Sekiguchi F, Matsunami M, Takahashi T, Nishikawa H & Kawabata A (2009). Hyperalgesia induced by spinal and peripheral hydrogen sulfide: evidence for involvement of Cav3.2 T‐type calcium channels. Pain 142, 127–132. [DOI] [PubMed] [Google Scholar]
- Marger F, Gelot A, Alloui A, Matricon J, Ferrer JF, Barrere C, Pizzoccaro A, Muller E, Nargeot J, Snutch TP, Eschalier A, Bourinet E & Ardid D (2011). T‐type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proc Natl Acad Sci USA 108, 11268–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunami M, Kirishi S, Okui T & Kawabata A (2011). Chelating luminal zinc mimics hydrogen sulfide‐evoked colonic pain in mice: possible involvement of T‐type calcium channels. Neuroscience 181, 257–264. [DOI] [PubMed] [Google Scholar]
- Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ, Orestes P, Latham JR, Todorovic SM & Jevtovic‐Todorovic V (2009). In vivo silencing of the CaV3.2 T‐type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin‐induced diabetic neuropathy. Pain 145, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PK, Bhatia M & Moochhala S (2003). Hydrogen sulfide: from the smell of the past to the mediator of the future? Trends Pharmacol Sci 24, 609–611. [DOI] [PubMed] [Google Scholar]
- Moran VH, Stammers AL, Medina MW, Patel S, Dykes F, Souverein OW, Dullemeijer C, Perez‐Rodrigo C, Serra‐Majem L, Nissensohn M & Lowe NM (2012). The relationship between zinc intake and serum/plasma zinc concentration in children: a systematic review and dose‐response meta‐analysis. Nutrients 4, 841–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE & Snyder SH (2011). Hydrogen sulfide as endothelium‐derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Tsugane M, Oka J & Kimura H (2004). Hydrogen sulfide induces calcium waves in astrocytes. FASEB J 18, 557–559. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Perez‐Reyes E & Todorovic SM (2005). The endogenous redox agent L‐cysteine induces T‐type Ca2+ channel‐dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J Neurosci 25, 8766–8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Su P, Kang HW, Van DA, Baumgart JP, David LS, Snutch TP, Barrett PQ, Lee JH, Zorumski CF, Perez‐Reyes E & Todorovic SM (2007. a). Molecular mechanisms of subtype‐specific inhibition of neuronal T‐type calcium channels by ascorbate. J Neurosci 27, 12577–12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Todorovic SM & Perez‐Reyes E (2006). The role of T‐type calcium channels in epilepsy and pain. Curr Pharm Des 12, 2189–2197. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Woo J, Kang HW, Vitko I, Barrett PQ, Perez‐Reyes E, Lee JH, Shin HS & Todorovic SM (2007. b). Reducing agents sensitize C‐type nociceptors by relieving high‐affinity zinc inhibition of T‐type calcium channels. J Neurosci 27, 8250–8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowycky MC, Fox AP & Tsien RW (1985). Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 316, 440–443. [DOI] [PubMed] [Google Scholar]
- Orestes P, Bojadzic D, Lee J, Leach E, Salajegheh R, Digruccio MR, Nelson MT & Todorovic SM (2011). Free radical signalling underlies inhibition of CaV3.2 T‐type calcium channels by nitrous oxide in the pain pathway. J Physiol 589, 135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orestes P, Osuru HP, McIntire WE, Jacus MO, Salajegheh R, Jagodic MM, Choe W, Lee J, Lee SS, Rose KE, Poiro N, Digruccio MR, Krishnan K, Covey DF, Lee JH, Barrett PQ, Jevtovic‐Todorovic V & Todorovic SM (2013). Reversal of neuropathic pain in diabetes by targeting glycosylation of CaV3.2 T‐type calcium channels. Diabetes 62, 3828–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD & Snyder SH (2012). H2S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol 13, 499–507. [DOI] [PubMed] [Google Scholar]
- Paul BD & Snyder SH (2015). H2S: A novel gasotransmitter that signals by sulfhydration. Trends Biochem Sci 40, 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C, Bauer CC, Boyle JP, Scragg JL & Dallas ML (2012). Modulation of ion channels by hydrogen sulfide. Antioxid Redox Signal 17, 95–105. [DOI] [PubMed] [Google Scholar]
- Peers C, Boyle JP, Scragg JL, Dallas ML, Al‐Owais MM, Hettiarachichi NT, Elies J, Johnson E, Gamper N & Steele DS (2014). Diverse mechanisms underlying the regulation of ion channels by carbon monoxide. Br J Pharmacol 172, 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E (2003). Molecular physiology of low‐voltage‐activated T‐type calcium channels. Physiol Rev 83, 117–161. [DOI] [PubMed] [Google Scholar]
- Rajan S, Plant LD, Rabin ML, Butler MH & Goldstein SA (2005). Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 121, 37–47. [DOI] [PubMed] [Google Scholar]
- Rose KE, Lunardi N, Boscolo A, Dong X, Erisir A, Jevtovic‐Todorovic V & Todorovic SM (2013). Immunohistological demonstration of CaV3.2 T‐type voltage‐gated calcium channel expression in soma of dorsal root ganglion neurons and peripheral axons of rat and mouse. Neuroscience 250, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi F, Miyamoto Y, Kanaoka D, Ide H, Yoshida S, Ohkubo T & Kawabata A (2014). Endogenous and exogenous hydrogen sulfide facilitates T‐type calcium channel currents in Cav3.2‐expressing HEK293 cells. Biochem Biophys Res Commun 445, 225–229. [DOI] [PubMed] [Google Scholar]
- Shibuya N, Koike S, Tanaka M, Ishigami‐Yuasa M, Kimura Y, Ogasawara Y, Fukui K, Nagahara N & Kimura H (2013). A novel pathway for the production of hydrogen sulfide from d‐cysteine in mammalian cells. Nat Commun 4, 1366. [DOI] [PubMed] [Google Scholar]
- Shin JB, Martinez‐Salgado C, Heppenstall PA & Lewin GR (2003). A T‐type calcium channel required for normal function of a mammalian mechanoreceptor. Nat Neurosci 6, 724–730. [DOI] [PubMed] [Google Scholar]
- Shipston MJ (2014). Ion channel regulation by protein S‐acylation. J Gen Physiol 143, 659–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitdikova GF, Weiger TM & Hermann A (2010). Hydrogen sulfide increases calcium‐activated potassium (BK) channel activity of rat pituitary tumor cells. Pflugers Arch 459, 389–397. [DOI] [PubMed] [Google Scholar]
- Stadtman ER (2001). Protein oxidation in aging and age‐related diseases. Ann N Y Acad Sci 928, 22–38. [DOI] [PubMed] [Google Scholar]
- Sun YG, Cao YX, Wang WW, Ma SF, Yao T & Zhu YC (2008). Hydrogen sulphide is an inhibitor of L‐type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc Res 79, 632–641. [DOI] [PubMed] [Google Scholar]
- Szabo C (2007). Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov 6, 917–935. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Aoki Y, Okubo K, Maeda Y, Sekiguchi F, Mitani K, Nishikawa H & Kawabata A (2010). Upregulation of Cav3.2 T‐type calcium channels targeted by endogenous hydrogen sulfide contributes to maintenance of neuropathic pain. Pain 150, 183–191. [DOI] [PubMed] [Google Scholar]
- Telezhkin V, Brazier SP, Cayzac SH, Wilkinson WJ, Riccardi D & Kemp PJ (2010). Mechanism of inhibition by hydrogen sulfide of native and recombinant BK(Ca) channels. Respir Physiol Neurobiol 172, 169–178. [DOI] [PubMed] [Google Scholar]
- Todorovic SM & Jevtovic‐Todorovic V (2014). Targeting of CaV3.2 T‐type calcium channels in peripheral sensory neurons for the treatment of painful diabetic neuropathy. Pflugers Arch 466, 701–706. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic‐Todorovic V, Meyenburg A, Mennerick S, Perez‐Reyes E, Romano C, Olney JW & Zorumski CF (2001). Redox modulation of T‐type calcium channels in rat peripheral nociceptors. Neuron 31, 75–85. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Meyenburg A & Jevtovic‐Todorovic V (2002). Mechanical and thermal antinociception in rats following systemic administration of mibefradil, a T‐type calcium channel blocker. Brain Res 951, 336–340. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Pathirathna S, Brimelow BC, Jagodic MM, Ko SH, Jiang X, Nilsson KR, Zorumski CF, Covey DF & Jevtovic‐Todorovic V (2004). 5β‐Reduced neuroactive steroids are novel voltage‐dependent blockers of T‐type Ca2+ channels in rat sensory neurons in vitro and potent peripheral analgesics in vivo. Mol Pharmacol 66, 1223–1235. [DOI] [PubMed] [Google Scholar]
- Tzeng BH, Chen YH, Huang CH, Lin SS, Lee KR & Chen CC (2012). The Cav3.1 T‐type calcium channel is required for neointimal formation in response to vascular injury in mice. Cardiovasc Res 96, 533–542. [DOI] [PubMed] [Google Scholar]
- Wang R (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92, 791–896. [DOI] [PubMed] [Google Scholar]
- Waxman SG & Zamponi GW (2014). Regulating excitability of peripheral afferents: emerging ion channel targets. Nat Neurosci 17, 153–163. [DOI] [PubMed] [Google Scholar]
- Weiss N, Black SA, Bladen C, Chen L & Zamponi GW (2013). Surface expression and function of Cav3.2 T‐type calcium channels are controlled by asparagine‐linked glycosylation. Pflugers Arch 465, 1159–1170. [DOI] [PubMed] [Google Scholar]
- White G, Lovinger DM & Weight FF (1989). Transient low‐threshold Ca2+ current triggers burst firing through an afterdepolarizing potential in an adult mammalian neuron. Proc Natl Acad Sci USA 86, 6802–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson WJ & Kemp PJ (2011). Carbon monoxide: an emerging regulator of ion channels. J Physiol 589, 3055–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A & Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xu C, Yang G, Wu L & Wang R (2015). Interaction of H2S with calcium permeable channels and transporters. Oxid Med Cell Longev 2015, 323269. [DOI] [PMC free article] [PubMed] [Google Scholar]