

Abstract
TRPM channels are a subgroup of the transient receptor potential (TRP) channel superfamily whose members have important roles in cell proliferation and survival. TRPM2, the second subfamily member to be cloned, is expressed in many tissues including brain, heart, vasculature and haematopoietic cells. TRPM2 is activated by oxidative stress and several other extracellular signals including tumour necrosis factor α (TNF‐α) and amyloid β‐peptide, which increase production of ADP‐ribose (ADPR). ADPR binds to the TRPM2 C‐terminal NUDT9‐H domain, activating the channel. Early studies support the paradigm that TRPM2 activation induces cell death by sustained Ca2+ influx or by enhancing cytokine production, aggravating inflammation and tissue injury. However, more recent data show that for a number of physiological processes, TRPM2 is protective. TRPM2 protects lungs from endotoxin‐induced injury by reducing reactive oxygen species (ROS) production by phagocytes. It protects hearts from oxidative damage after ischaemia–reperfusion or hypoxia–reoxygenation by maintaining better mitochondrial bioenergetics and by decreasing ROS. Sustained Ca2+ entry through TRPM2 is required to maintain cellular bioenergetics and protect against hypoxia–reoxygenation injury. TRPM2 also protects neuroblastoma from moderate oxidative stress by decreasing ROS through increased levels of forkhead box transcription factor 3a (FOXO3a) and a downstream effector, superoxide dismutase 2. TRPM2 is important for tumour growth and cell survival through modulation of hypoxia‐inducible transcription factor expression, mitochondrial function and mitophagy. These findings in cardiac ischaemia and in neuroblastoma suggest that TRPM2 has a basic role in sustaining mitochondrial function and in cell survival that applies to a number of physiological systems and pathophysiological processes including ischaemia–reperfusion injury.
Abreviations
- ADPR
ADP‐ribose
- cKO
cardiac‐specific knock‐out
- +dP/dt
first time derivative of the left ventricular pressure rise
- FOXO3a
forkhead box transcription factor 3a
- gKO
global knock‐out
- GFP
green fluorescent protein
- HIF‐1α
hypoxia inducible factor‐1α
- H/R
hypoxia–reoxygenation
- I/R
ischaemia/reperfusion
- mPTP
mitochondrial permeability transition pore
- NRVM
neonatal rat ventricular myocyte
- OCR
oxygen consumption rate
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TNF‐α
tumour necrosis factor α
- TRP
transient receptor potential
- WT
wild‐type
Introduction
Transient receptor potential (TRP) channels are members of a superfamily of monovalent and divalent cation‐permeable ion channels with six transmembrane domains (Ramsey et al. 2006; Nilius et al. 2007; Chen et al. 2014). The TRPM subfamily is named after the first member to be described, TRPM1 (M, melastatin), which is a putative tumour suppressor protein (Duncan et al. 1998). Other TRPM channel family members also have important roles in cell proliferation and cell survival including TRPM2 (Hara et al. 2002; Miller & Zhang, 2011), TRPM4 (Abriel et al. 2012; Simard et al. 2012), TRPM5 (Prawitt et al. 2000), TRPM7 (Aarts et al. 2003; Aarts & Tymianski, 2005; Guilbert et al. 2009) and TRPM8 (Lehen'kyi & Prevarskaya, 2011). The second member of the TRPM family to be cloned, the gene TRPM2 is located on human chromosome 21q22.3, has 32 exons and encodes a protein of 1503 amino acids (predicted molecular mass ∼170 kDa) (Nagamine et al. 1998). The mouse TRPM2 gene is highly homologous, with 34 exons encoding a protein of 1507 amino acids (Uemura et al. 2005).
TRPM2 channels are widely expressed in many cell types including brain, heart and haematopoietic, vascular, smooth muscle and endothelial cells and are permeable to Ca2+, Na+ and K+ (Perraud et al. 2003; Miller & Zhang, 2011). TRPM2 has been known since its identification to play an essential role in oxidative stress, but understanding its physiological role has become more complex with recent in vivo studies as well as in vitro mechanistic data. This review will focus on new data elucidating the role of TRPM2 in ischaemia–reperfusion (I/R) injury and the new concept that calcium entry via TRPM2 is protective rather than injurious to some tissues, including heart, when subjected to oxidative stress.
TRPM2 regulation
Extracellular signals which activate TRPM2 include oxidative stress, tumour necrosis factor α (TNF‐α), amyloid β‐peptide and concanavalin A (Hara et al. 2002; Wehage et al. 2002; Fonfria et al. 2005; Gasser et al. 2006; Chen et al. 2013). Stimulation with these extracellular signals results in production of ADPR, which binds to the TRPM2 C‐terminal NUDT9‐H domain (Perraud et al. 2001), activating the channel (Kolisek et al. 2005; Perraud et al. 2005; Gasser et al. 2006; Toth & Csanady, 2010). Cyclic adenosine diphosphoribose can potentiate the effects of ADPR at low concentrations and gates TRPM2 by itself. ADPR is produced primarily by mitochondria (Perraud et al. 2005) or by activation of poly(ADPR)polymerase (Fonfria et al. 2004; Buelow et al. 2008). TRPM2 is also positively regulated by intracellular Ca2+ and calmodulin (McHugh et al. 2003; Tong et al. 2006; Du et al. 2009 a). Interaction of ADPR with TRPM2 supports initial Ca2+ entry through TRPM2. The subsequent increase in Ca2+‐bound calmodulin enhances calmodulin binding to an IQ motif in the N terminus of TRPM2, providing positive feedback for TRPM2 activation and increased Ca2+ influx (Tong et al. 2006). TRPM2 with mutant ADPR binding sites can be directly activated by an increase in [Ca2+]i, and TRPM2 may be activated in a wide range of physiological situations through this mechanism (Du et al. 2009 b). TRPM2 has also been reported to be temperature sensitive (Togashi et al. 2006) and inhibited by acidification (Du et al. 2009 b; Starkus et al. 2010), thereby providing a mechanism for limiting Ca2+ entry during ischaemia.
In addition to full length TRPM2 (TRPM2‐L), physiological splice variants include TRPM2‐S (short) (Zhang et al. 2003), TRPM2‐ΔN, TRPM2‐ΔC (Wehage et al. 2002) and TRPM2‐TE (Orfanelli et al. 2008). TRPM2, like other TRP channels, is a tetramer, and association of splice variants with TRPM2‐L may modulate TRPM2 function. Indeed, TRPM2‐S, which lacks the entire C terminus and the putative Ca2+ pore, has been shown to inhibit Ca2+ influx through TRPM2‐L and functions as a dominant negative (Zhang et al. 2003). Little is known about the mechanisms by which alternative splicing to alter isoform expression is regulated in vivo, or the physiological functions and interactions of TRPM2 isoforms, other than that they influence and can disrupt full length channel function.
TRPM2 electrophysiology in adult cardiac cells
In the heart, intracellular ADPR elicited a large inward and outward current in wild‐type (WT) but not in TRPM2 global knock‐out (gKO) myocytes (Miller et al. 2014; Hoffman et al. 2015). ADPR‐activated currents displayed the characteristic TRPM2 linear current–voltage relationship with E rev close to 0 mV (Perraud et al. 2001; Miller et al. 2014). Flufenamic acid abolished the current elicited by ADPR in WT myocytes, consistent with the notion that the ADPR‐activated current was mediated by TRPM2. In WT myocytes, TRPM2 currents did not inactivate, consistent with observations in HEK293 cells stably expressing WT TRPM2 (Perraud et al. 2001). The ratio of calcium conductance to sodium conductance was 0.56 ± 0.02 (Miller et al. 2014) and 0.65 ± 0.08 (Hoffman et al. 2015) in adult cardiac myocytes and transiently transfected HEK293 cells, respectively.
Role of TRPM2 in inflamation and ischaemia–reperfusion injury
A number of TRP channels have been shown to mediate oxidative‐stress induced injury (Miller & Zhang, 2011), but in this review we will focus on TRPM2. Early studies on the function of the TRPM2 channel support the paradigm that activation of TRPM2 induced cell death by sustained increase in [Ca2+]i (Hara et al. 2002; Kaneko et al. 2006; Hecquet et al. 2008) or by enhanced cytokine production, which was associated with increased inflammation and tissue injury (Knowles et al. 2011; Knowles et al. 2013). In a model of human inflammatory bowel disease (dextran sulfate sodium‐induced colitis in mice), TRPM2‐mediated Ca2+ influx stimulated chemokine (CXCL2) production in monocytes, resulting in recruitment of neutrophils to the site of inflammation, and was also associated with tissue damage (Yamamoto et al. 2008). However, more recent reports suggest a different paradigm, that TRPM2 can be protective rather than deleterious in pathophysiological situations. In vitro, TRPM2‐L protected neuroblastoma cells from low to moderate oxidative stress through increased expression of forkhead box transcription factor 3a (FOXO3a) and superoxide dismutase (SOD)2, reducing reactive oxygen species (ROS) levels, whereas cells expressing the dominant negative TRPM2‐S isoform had reduced FOXO3a and SOD2 and enhanced ROS with increased susceptibility to oxidative stress (Miller et al. 2013). This is similar to studies on pyramidal neurons subjected to oxidant injury in which inhibition of TRPM2 enhanced cellular damage (Bai & Lipski, 2010), although this finding is controversial (Kaneko et al. 2006). The different results in the more recent in vitro studies may be accounted for by the lower, more physiological levels of oxidative stress (H2O2 dose), or potentially by the use of different cell types with different endogenous oxidant production and antioxidant defences. Recent in vivo studies confirmed these findings that TRPM2 activation in physiological or pathophysiological conditions can reduce tissue injury. In WT mice subjected to intraperitoneal injection of endotoxin, survial was 5 times higher than in gKO mice (Di et al. 2012). This was due to cation entry via TRPM2 channels, resulting in plasma membrane depolarization and decreased NOX‐mediated ROS production in WT phagocytes, thereby preserving viability. In humans, a TRPM2 mutant (P1018L) was identified in a subtype of Guamanian amyotropic lateral sclerosis and Parkinsonism dementia patients (Hermosura et al. 2008). Whereas WT TRPM2 channel did not inactivate, the P1018L mutant inactivated after channel opening by ADPR (Xia et al. 2008), effectively limiting Ca2+ entry and suggested that WT TRPM2 was necessary for normal neuronal function. In this review, the role of TRPM2 in oxidative stress‐induced injury and ROS production is largely derived from our experimental results of two models: cardiac I/R injury in WT and gKO and cardiac‐specific knock‐out (cKO) mice and in a neuroblastoma xenograft model treated with doxorubicin.
Role of TRPM2 in cardiac ischaemia–reperfusion injury
In the heart, ROS are produced physiologically during respiration by the mitochondrial electron transport chain, and increased ROS are observed after stimulation of myocytes with β‐adrenergic agonists and in pathological conditions including I/R and doxorubicin exposure. ROS play a major role in myocyte injury through protein oxidation, lipid peroxidation and mutagenesis. During hypoxia–reoxygenation (H/R, simulated I/R in vitro), a significant increase in intracellular ADPR occurs in cardiac myocytes, which may activate TRPM2 channels and leads to elevation in [Ca2+]i. We studied WT and TRPM2‐gKO mice to determine the impact of abrogation of TRPM2‐dependent Ca2+ influx on I/R injury. Using confocal microscopy, we first determined that TRPM2 channels were expressed in the sarcolemma and transverse tubules of adult cardiac myocytes (Miller et al. 2013). Exposure of myocytes to H2O2 resulted in a large increase in [Ca2+]i, which was significantly higher in WT compared with gKO myocytes; the [Ca2+]i increase was blocked by clotrimazole and was dependent on extracellular Ca2+, demonstrating that cardiac TRPM2 channels were functional. H2O2 did not increase [Na+]i in WT cardiac myocytes (Hoffman et al. 2015). At baseline, there were no differences in body weights, left venticular masses, heart rates, in vivo haemodynamics, fractional shortening, or response to isoproterenol stimulation between WT and gKO hearts (Miller et al. 2013). I/R injury was induced by occluding the left anterior descending coronary artery for 30 min followed by release, and cardiac function was evaluated 2–3 days post surgery. TRPM2 deficiency was associated with aggravation of in vivo cardiac contractile dysfunction, as demonstrated by significantly lower fractional shortening and first time derivative of the left ventricular pressure rise (+dP/dt). Similarly, worse cardiac contractility was observed in gKO hearts treated with doxorubicin (another oxidative cardiac injury model) compared to similarly treated WT hearts (Hoffman et al. 2015). To simulate I/R in vitro, freshly isolated myocytes from WT or gKO hearts were exposed in vitro to 30 min of hypoxia followed by 2 h of reoxygenation. Whereas there were no differences in ROS levels between WT and gKO myocytes incubated under normoxic conditions, after H/R ROS levels in gKO myocytes were significantly higher than in WT myocytes. SOD1 and SOD2 were significantly lower in I/R in KO hearts compared with WT‐I/R hearts. Upstream regulators of SOD expression including hypoxia inducible factor‐1α (HIF‐1α), FOXO1 and FOXO3a were also reduced in KO‐I/R hearts, suggesting that reduced Ca2+ influx in gKO myocytes may have a role in reduction of HIF‐1α, FOXOs and SODs (Liu et al. 2007; Chen et al. 2014), with the reduction in antioxidants leading to increased ROS. In addition, NOX4 was increased in KO‐I/R hearts. We could not distinguish whether increased ROS scavenging capacity or reduced ROS generation was the dominant method by which TRPM2 protected heart from I/R injury.
Mice with cardiac‐specific knock‐out (cKO) of TRPM2 were generated to differentiate whether the benefical effect of TRPM2 channels on cardiac contractility post‐I/R was due to the better capability of WT myocytes to withstand oxidative stress, or to the reduced inflammatory response secondary to reduced ROS production by infiltrating WT monocytes and macrophages (Hoffman et al. 2015). At 2 months of age, 64% of the floxed TRPM2 gene was deleted in cKO hearts as evaluated by qPCR and 79% of the floxed TRPM2 gene was deleted in cKO myocytes as determined by TRPM2 current measurements. Similar to gKO hearts, cKO hearts subjected to I/R exhibited significantly lower +dP/dt and maximal systolic pressure when compared to WT‐I/R hearts. This important observation strongly argues that it was the cardiac TRPM2 channels, rather than phagocytic TRPM2 channels (by inhibiting NOX‐mediated ROS production and thereby reducing inflammatory response) (Di et al. 2012), which mediated protection against I/R injury. In this context, it is relevant to note that the weight of evidence argues against a pivotal role of neutrophils as a causative factor in most forms of I/R injury in the heart and brain (Baxter, 2002). Finally, the role of TRPM2 in angiogenesis and ischaemic neovascularization in protection against cardiac I/R injury (Mittal et al. 2015) is likely to be small in view of our results obtained with cKO hearts.
A recent study using an independent gKO mouse (C57BL/6 background) reported very different results (Hiroi et al. 2012). Specifically, after 45 min of ischaemia followed by 24 h of reperfusion in vivo, neutrophil infiltration was less, infarct size was smaller and +dP/dt was higher in gKO compared to WT hearts. The authors speculated that increased neutrophil adhesion to endothelial cells mediated by TRPM2 channels caused increased damage post‐I/R. The proposed mechanism, however, is not compatible with the results from a recent study that found that Ca2+ entry via activated TRPM2 channels decreased NOX‐mediated ROS production in phagocytes (Di et al. 2012), thereby lessening the inflammatory response. The proposed mechanism would also have predicted similar post‐I/R myocardial dysfunction in WT and cKO mice since TRPM2 channels were present in bone marrow cells in cKO animals. This prediction is not supported by our observation that myocardial function was worse in cKO mice post‐I/R (Hoffman et al. 2015). The reasons for the different results between our studies (Miller et al. 2013; Miller et al. 2014; Hoffman et al. 2015) and those of Hiroi et al. (2012) may include the following: (1) TRPM2 was deleted by targeting different exons; (2) 45 vs. 30 min of ischaemia resulted in much larger infarcts (45% vs. 27% of area‐at‐risk); (3) cardiac function was examined at 24 h vs. 72 h of reperfusion; (4) different anaesthetics (phenobarbital vs. isoflurane); (5) different surgical techniques used in measuring in vivo haemodynamics (opening the chest followed by left ventricular puncture vs. catheterizing the right carotid artery in a closed‐chest preparation); and (6) increased heat dissipation in open‐chest mice compared with closed‐chest mice. Although the use of the cardioprotective volatile anaesthetic isoflurane during our surgical procedure may be a confounding factor (Roberge et al. 2014), our current results with cKO hearts, together with the positive rescue experiments by TRPM2 but not by the loss‐of‐function E960D mutant in gKO myocytes (Hoffman et al. 2015), strongly argue in favour of beneficial, rather than detrimental, effects of TRPM2 on cardiac bioenergetics and function, under both basal and stressed conditions. In addition, our observations on cKO hearts suggest the role played by phagocytic TRPM2 channels on cardiac I/R injury is likely to be small.
Another study utilizing neonatal rat ventricular myocytes (NRVMs) reported that TRPM2 exacerbated H2O2 injury (Yang et al. 2006). In this scenario, H2O2 (100 μm) activated TRPM2 through increased ADPR/NAD+ formation, leading to Ca2+ and Na+ overload in mitochondria, myocyte apoptosis through mitochondrial membrane disruption, cytochrome c release, and caspase‐3‐dependent chromatin condensation and fragmentation. It should be noted that NRVMs exhibit a very different phenotype from adult cardiac myocytes. For example, in culture NRVMs proliferate while mature adult cardiac cells do not divide. Another difference is that NRVMs spontaneously beat in culture whereas healthy adult left ventricular myocytes are quiescent. More relevant to our discussion is that while activation of TRPM2 by H2O2 resulted in increases in both [Ca2+]i and [Na+]i in NRVMs (Yang et al. 2006), in adult cardiac myocytes, H2O2 increased [Ca2+]i (Miller et al. 2013) but not [Na+]i (Hoffman et al. 2015). An explanation for the discrepancy may relate to the fact that NRVMs expressed α1‐ and α3‐isoforms (Lucchesi & Sweadner, 1991) whereas adult mouse cardiac myocytes expressed α1‐ and α2‐isoforms of Na+–K+‐ATPase (Berry et al. 2007). When expressed in Sf‐9 insect cells, the α3‐isoform exhibited > 2‐fold less affinity for Na+ when compared to the α2‐isoform (Blanco & Mercer, 1998) and may account for lower Na+ transport rate. In addition, adenovirus‐mediated overexpression of the α2‐isoform in adult rat cardiac myocytes led to greater affinity for Na+ and higher Na+–K+‐ATPase activity compared to control myocytes expressing β‐galactosidase (Correll et al. 2014). Finally, NRVMs do not possess the highly organized t‐tubules in which the α2‐isoform is preferentially localized in adult mouse cardiac myocytes (Berry et al. 2007). These fundamental differences in physiology may account for different results observed in NRVMs vs. adult cardiac myocytes.
Ca2+ influx via TRPM2 is essential for maintenance of cardiac bioenergetics and protection against H/R Injury
To examine if Ca2+ influx through TRPM2 was required for maintenance of bioenergetics, WT TRPM2, TRPM2 loss‐of‐function mutant (E960D) (Xia et al. 2008), inactivating mutant P1018L (Hermosura et al. 2008), or control green fluorescent protein (GFP) were expressed in gKO hearts by adenovirus‐mediated gene transfer (Hoffman et al. 2015) (Fig. 1). gKO‐GFP heart slices had significantly higher ROS levels and lower oxygen consumption rate (OCR) compared to WT‐GFP heart slices. WT TRPM2 but not loss‐of‐function E960D mutant decreased ROS and increased OCR when expressed in gKO hearts. These results indicate that TRPM2‐mediated Ca2+ influx was necessary for normal bioenergetics maintenance.
Figure 1. Ca2+ entry through TRPM2 is required to lower intracellular ROS in gKO myocytes .
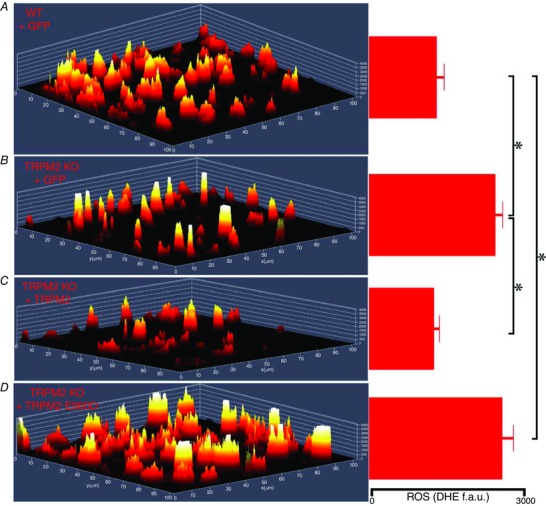
Hearts from WT and global TRPM2 KO mice were injected with adenovirus expressing either Adv‐GFP, Adv‐TRPM2, or Adv‐TRPM2‐E960D. Heart slices were generated from areas of left ventricle exhibiting GFP fluorescence after 5 days. Multiphoton confocal images of dihydroethidium (DHE)‐stained heart slices were obtained and a 2.5‐dimensional heatmap plot of mean DHE intensity is shown for WT+GFP (A), KO+GFP (B), KO+TRPM2 (C), and KO+TRPM2 E960D (D). Quantification of DHE fluorescence in fluorescence arbitrary units (f.a.u.) is shown on the right. DHE fluorescence was quantified from at least 3 slices from each heart. *P < 0.05.
To futher evaluate if TRPM2‐mediated Ca2+ entry was necessary for protection against H/R injury, we expressed GFP, WT TRPM2, E960D and P1018L mutants in gKO myocytes followed by H/R (Hoffman et al. 2015). After H/R, hearts with WT TRPM2 but not the loss‐of‐function E960D or inactivating P1018L mutants had significantly lowered mitochondrial superoxide levels in gKO myocytes, and this occurred only in the presence of extracellular Ca2+. TRPM2 but not E960D or P1018L also restored OCR. This indicates that sustained Ca2+ entry was necessary for TRPM2‐mediated reduction of ROS and for protection against H/R injury.
TRPM2, Ca2+ entry and I/R injury
Early studies showed diastolic [Ca2+]i increases progressively (to 3–4 μm) during ATP depletion resulting in contracture (Murphy et al. 1994). Ca2+ overload is proposed to lead to mitochondrial permeability transition pore (mPTP) opening and irreversible cell death. How then can TRPM2‐mediated Ca2+ entry during I/R be beneficial? Recent data suggest that in isolated cardiac myocytes subjected to I/R, Ca2+ overload is the consequence of bioenergetic failure after mPTP opening rather than its cause (Lemasters et al. 2009). In addition, in isolated perfused mouse hearts subjected to H/R, the mitochondrial‐targeted SOD mimetic MitoQ dramatically reduces Ca2+ wave‐associated mPTP opening (Davidson et al. 2012). Our data indicate that TRPM2‐mediated Ca2+ entry supports mitochondrial function and reduces mitochondrial ROS under both basal and H/R conditions (Hoffman et al. 2015). Maintenance of bioenergetics by TRPM2 may prevent mPTP opening and protect hearts from oxidative injury. Another possibility is that TRPM2‐mediated Ca2+ entry activates Ca2+‐dependent signalling pathways (e.g. Pyk2, RACK1) that result in reduction in ROS production and enhance pro‐survival signals. Finally, absence of TRPM2 results in a tenuous bioenergetics condition and sensitizes KO myocytes to further oxidative stress brought about by I/R or Doxo treatment (Miller et al. 2013; Hoffman et al. 2015). This second ‘hit’ leads to much worse bioenergetics (compared to WT) and higher ROS levels, resulting in impaired contractile performance.
TRPM2 and renal I/R injury
The ability of TRPM2 to protect against I/R injury did not extend to the kidney; rather, gKO exacerbated renal I/R injury (Gao et al. 2014). In the kidney, I/R was proposed to activate TRPM2, which physically interacted with Rac1, which in turn recruited NOX subunits to the membrane and increased ROS production. It is worth noting that while TRPM2 was expressed in the sarcolemma of the heart (Miller et al. 2013), in renal tubular epithelial cells TRPM2 was widely distributed in the cytoplasm and intracellular organelles but not on the plasma membrane (Gao et al. 2014). The role of Ca2+ permeation via activated TRPM2 in enhancing renal I/R injury is therefore not clear. Another factor contributing to different findings between heart and kidney may be the different levels of endogenous oxidants and antioxidants present in different cell types. Different levels of H2O2 affect different p53‐regulated gene expression patterns. We hypothesize that whereas exposure of TRPM2‐expressing cells to low levels of H2O2 may result in low levels of cation entry and activation of pathways including increased SOD2 which protect viability, exposure of TRPM2‐expressing cells to high levels of H2O2 results in significantly higher Ca2+ entry and leads to cell death. In the former case, inhibition of TRPM2 function is detrimental, whereas in the latter case it can preserve viability. Our experimental conditions in studies of heart and cardiac myocytes involved mild to moderate oxidative stress in which Ca2+ influx through TRPM2 would not be overwhelming but rather benefical, activating pathways which reduce ROS production and preserve viability.
TRPM2 enhances cell survival in cancer
TRPM2 is highly expressed in a number of malignancies including melanoma (Orfanelli et al. 2008), breast cancer (unpublished observations, Miller lab), and neuroblastoma (Chen et al. 2014). Neuroblastoma cells that expressed dominant negative TRPM2‐S had increased levels of ROS and increased sensitivity to doxorubicin (Chen et al. 2013). We demonstrated that TRPM2‐L protected neuroblastoma SH‐SY5Y cells from moderate oxidative stress through increased levels of FOXO3a and SOD2. In contrast, cells expressing the dominant negative TRPM2‐S had reduced FOXO3a and SOD levels, reduced Ca2+ influx in response to oxidative stress, and enhanced ROS, leading to reduced cell viability after exposure to a low or moderate level of H2O2. In vivo, growth of tumours expressing TRPM2‐S was also significantly reduced compared to tumours expressing TRPM2‐L, particularly following treatment with doxorubicin. These findings were confirmed in a second neuroblastoma cell line, SK‐N‐AS, and with the breast cancer cell line MCF‐7 (unpublished observations, Miller lab). Expression of HIF‐1/2α was significantly decreased in TRPM2‐S‐expressing tumour cells, as was expression of downstream target proteins regulated by HIF‐1/2α including proteins involved in glycolysis (lactate dehydrogenase), oxidant stress (FOXO3a), angiogenesis (VEGF), mitophagy, mitochondrial function (BNIP3 and NDUFA4L2), and mitochondrial electron transport chain activity (cytochrome oxidase 4.1/4.2 in complex IV) (Chen et al. 2014). Inhibition of TRPM2‐L by several approaches significantly increased sensitivity of cells to doxorubicin, and the reduced survival of TRPM2‐S expressing cells was rescued by gain of HIF‐1α or ‐2α function. These data show that TRPM2 activity and its modulation of HIF‐1/2α and its downstream effectors is important for cell viability and survival following doxorubicin treatment. Similar to the results observed with cardiac myocytes, TRPM2 played an important role in regulation of cellular ROS levels and cell survival in neuroblastoma and enhanced viability following doxorubicin.
TRPM2 in regulation of cellular bioenergetics
In order to identify the mechanisms through which TRPM2 protects heart from I/R injury, we analysed the proteomes of the left ventricles from WT‐I/R and gKO‐I/R mice to identify the major cellular mechanism which afford protection (Miller et al. 2014). Global label‐free proteomics analysis using GeLC‐MS/MS (in‐gel tryptic digestion followed by liquid chromatography‐tandem mass spectroscopy) technology revealed that the largest differences in canonical pathways between gKO‐I/R and WT‐I/R hearts were in mitochondrial function and the TCA cycle. In mitochondria, Complexes I, III and IV were down‐regulated, whereas Complexes II and V were upregulated in gKO‐I/R compared with WT‐I/R hearts. We then determined that both mitochondrial membrane potential (ΨM) and mitochondrial Ca2+ uptake were lower in gKO compared with WT myocytes post‐H/R. Reduced mitochondrial Ca2+ uptake in gKO myocytes was due to reduction in both the driving force ΨM and mitochondrial Ca2+ uniporter activity. OCR was lower in gKO myocytes, and ATP levels were markedly lower in gKO compared to WT myocytes. These studies demonstrated that loss of TRPM2‐mediated Ca2+ entry resulted in impaired cardiac mitochondrial bioenergetics. MitoSOX Red staining of heart slices demonstrated that the elevated ROS in KO heart was largely mitochondrial superoxide.
Similar results were observed in the neuroblastoma cell line SH‐SY5Y expressing the dominant negative TRPM2‐S. In cells expressing TRPM2‐S, ΨM and mitochondrial Ca2+ uptake, basal and maximal oxygen consumption, and ATP production were reduced compared to cells expressing only TRPM2‐L, mitochondrial ROS was increased, and mitochondria were dysmorphic (Chen et al. 2014). In cells expressing TRPM2‐S, the elevated ROS derived from mitochondria may be due to the lower levels of HIF‐1α, FOXO3a, SOD2, BNIP3 and NDUFA4L2, resulting in aberrant mitochondrial bioenergetics. Ca2+ has been shown to affect HIF‐1α translation and stability, and through this mechanism, Ca2+ influx through TRPM2 could affect a number of effectors downstream of HIF‐1/2α involved in cell survival. A critical role for low level mitochondrial Ca2+ uptake in regulation of bioenergetics has been recently demonstrated (Cardenas et al. 2010; Wallace, 2012), and reduced Ca2+ entry may directly affect cellular bioenergetics.
Conclusions
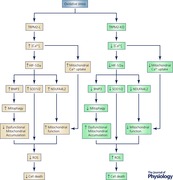
The findings that TRPM2 was critically important in protecting cells from cardiac I/R injury as well as in enhancing tumour cell survival after doxorubicin suggest that TRPM2 has a basic role in cell survival and mitochondrial function that may apply to a number of physiological systems. Our data demonstrate that Ca2+ influx through TRPM2 modulated cell viability, and we hypothesize that this may occur through mechanisms involving Ca2+ modulation of HIF‐1/2α expression. Ca2+ influx through TRPM2 enhances calcineurin activity and HIF stability (Liu et al. 2007). HIF‐1/2α enhances cell viability through regulation of a number of target genes including those involved in angiogenesis, glycolysis, energy metabolism, redox homestasis and mitophagy. In the TRPM2 KO or when TRPM2 function is inhibited, HIF‐1/2α expression is decreased, as is expression of downstream mitochondrial proteins including NDUFA4L2, and together with decreased mitochondrial Ca2+ uptake, this results in compromised mitochondria. Decreased HIF‐1/2α also results in reduced BNIP3, which contributes to reduced mitophagy, leading to an accumulation of dysfunctional mitochondria and increased ROS (Abstract Figure). With reduced SOD2, ROS levels are elevated and the cell has reduced tolerance to a further rise in ROS, for example from ischaemia–reperfusion or doxorubicin, leading to reduced cell survival and increased cell death in the absence of TRPM2. The work summarized here shows that TRPM2 channels protect cardiac myocytes from ischaemia–reperfusion injury and tumour cells from doxorubicin toxicity, and demonstrates that the mechanisms involve preservation of mitochondrial bioenergetics and modulation of ROS.
Additional information
Competing interests
None of the authors have a conflict of interest.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Biography
Barbara A. Miller is Professor of Pediatrics, Chief of the Division of Pediatric Hematology/Oncology and Vice Chair for Research in Pediatrics at the Pennsylvania State University College of Medicine. Her laboratory works on the role of calcium in growth factor signalling and cell survival, and recently has focused on the role of TRPM2 in oxidative stress. A major research interest is the role of TRPM2 in regulation of cell survival and mitochondrial function in cancer and following ischaemia–reperfusion injury. Joseph Y. Cheung is the Professor and Chairman of Medicine at Temple University School of Medicine. His research focuses on the cellular and molecular basis of cardiovascular disease and on the role of intracellular Ca2+ in ischaemic and hypertensive cardiomyopathy. More recently he has studied the role of TRPM2 channels in oxidative stress, bioenergetics and cardiac protection.

This review was presented at the symposium “Non‐selective cationic channels in chemical and physical stress?”, which took place at Physiology 2015 in Cardiff, UK, 6–8 July 2015.
References
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF & Tymianski M (2003). A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877. [DOI] [PubMed] [Google Scholar]
- Aarts MM & Tymianski M (2005). TRPMs and neuronal cell death. Pflugers Arch 451, 243–249. [DOI] [PubMed] [Google Scholar]
- Abriel H, Syam N, Sottas V, Amarouch MY & Rougier JS (2012). TRPM4 channels in the cardiovascular system: physiology, pathophysiology, and pharmacology. Biochem Pharmacol 84, 873–881. [DOI] [PubMed] [Google Scholar]
- Bai JZ & Lipski J (2010). Differential expression of TRPM2 and TRPV4 channels and their potential role in oxidative stress‐induced cell death in organotypic hippocampal culture. Neurotoxicology 31, 204–214. [DOI] [PubMed] [Google Scholar]
- Baxter GF (2002). The neutrophil as a mediator of myocardial ischemia‐reperfusion injury: time to move on. Basic Res Cardiol 97, 268–275. [DOI] [PubMed] [Google Scholar]
- Berry RG, Despa S, Fuller W, Bers DM & Shattock MJ (2007). Differential distribution and regulation of mouse cardiac Na+/K+‐ATPase α1 and α2 subunits in T‐tubule and surface sarcolemmal membranes. Cardiovasc Res 73, 92–100. [DOI] [PubMed] [Google Scholar]
- Blanco G & Mercer RW (1998). Isozymes of the Na‐K‐ATPase: heterogeneity in structure, diversity in function. Am J Physiol Renal Physiol 275, F633–650. [DOI] [PubMed] [Google Scholar]
- Buelow B, Song Y & Scharenberg AM (2008). The poly(ADP‐ribose) polymerase PARP‐1 is required for oxidative stress‐induced TRPM2 activation in lymphocytes. J Biol Chem 283, 24571–24583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR & Foskett JK (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SJ, Hoffman NE, Shanmughapriya S, Bao L, Keefer K, Conrad K, Merali S, Takahashi Y, Abraham T, Hirschler‐Laszkiewicz I, Wang J, Zhang XQ, Song J, Barrero C, Shi Y, Kawasawa YI, Bayerl M, Sun T, Barbour M, Wang HG, Madesh M, Cheung JY & Miller BA (2014). A splice variant of the human ion channel TRPM2 modulates neuroblastoma tumor growth through hypoxia‐inducible factor (HIF)‐1/2α. J Biol Chem 289, 36284–36302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SJ, Zhang W, Tong Q, Conrad K, Hirschler‐Laszkiewicz I, Bayerl M, Kim JK, Cheung JY & Miller BA (2013). Role of TRPM2 in cell proliferation and susceptibility to oxidative stress. Am J Physiol Cell Physiol 304, C548–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll RN, Eder P, Burr AR, Despa S, Davis J, Bers DM & Molkentin JD (2014). Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ Res 114, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Yellon DM, Murphy MP & Duchen MR (2012). Slow calcium waves and redox changes precede mitochondrial permeability transition pore opening in the intact heart during hypoxia and reoxygenation. Cardiovasc Res 93, 445–453. [DOI] [PubMed] [Google Scholar]
- Di A, Gao XP, Qian F, Kawamura T, Han J, Hecquet C, Ye RD, Vogel SM & Malik AB (2012). The redox‐sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol 13, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Xie J & Yue L (2009. a). Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc Natl Acad Sci USA 106, 7239–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Xie J & Yue L (2009. b). Modulation of TRPM2 by acidic pH and the underlying mechanisms for pH sensitivity. J Gen Physiol 134, 471–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan LM, Deeds J, Hunter J, Shao J, Holmgren LM, Woolf EA, Tepper RI & Shyjan AW (1998). Down‐regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res 58, 1515–1520. [PubMed] [Google Scholar]
- Fonfria E, Marshall IC, Boyfield I, Skaper SD, Hughes JP, Owen DE, Zhang W, Miller BA, Benham CD & McNulty S (2005). Amyloid β‐peptide(1‐42) and hydrogen peroxide‐induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J Neurochem 95, 715–723. [DOI] [PubMed] [Google Scholar]
- Fonfria E, Marshall ICB, Benham CD, Boyfield I, Brown JD, Hill K, Hughes JP, Skaper SD, Scharenberg AM & McNulty S (2004). TRPM2 channel opening in response to oxidative stress is dependent on activation of poly (ADP‐ribose) polymerase. Br J Pharmacol 143, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Wang W, Tadagavadi RK, Briley NE, Love MI, Miller BA & Reeves WB (2014). TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J Clin Invest 124, 4989–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser A, Glassmeier G, Fliegert R, Langhorst MF, Meinke S, Hein D, Kruger S, Weber K, Heiner I, Oppenheimer N, Schwarz JR & Guse AH (2006). Activation of T cell calcium influx by the second messenger ADP‐ribose. J Biol Chem 281, 2489–2496. [DOI] [PubMed] [Google Scholar]
- Guilbert A, Gautier M, Dhennin‐Duthille I, Haren N, Sevestre H & Ouadid‐Ahidouch H (2009). Evidence that TRPM7 is required for breast cancer cell proliferation. Am J Physiol Cell Physiol 297, C493–502. [DOI] [PubMed] [Google Scholar]
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K & Mori Y (2002). LTRPC2 Ca2+‐permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 9, 163–173. [DOI] [PubMed] [Google Scholar]
- Hecquet CM, Ahmmed GU, Vogel SM & Malik AB (2008). Role of TRPM2 channel in mediating H2O2‐induced Ca2+ entry and endothelial hyperpermeability. Circ Res 102, 347–355. [DOI] [PubMed] [Google Scholar]
- Hermosura MC, Cui AM, Go RC, Davenport B, Shetler CM, Heizer JW, Schmitz C, Mocz G, Garruto RM & Perraud AL (2008). Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc Natl Acad Sci USA 105, 18029–18034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroi T, Wajima T, Negoro T, Ishii M, Nakano Y, Kiuchi Y, Mori Y & Shimizu S (2012). Neutrophil TRPM2 channels are implicated in the exacerbation of myocardial ischemia/reperfusion injury. Cardiovasc Res 97, 271–281. [DOI] [PubMed] [Google Scholar]
- Hoffman NE, Miller BA, Wang J, Elrod JW, Rajan S, Gao E, Song J, Zhang XQ, Hirschler‐Laszkiewicz I, Shanmughapriya S, Koch WJ, Feldman AM, Madesh M & Cheung JY (2015). Ca2+ entry via Trpm2 is essential for cardiac myocyte bioenergetics maintenance. Am J Physiol Heart Circ Physiol 308, H637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, Takada Y, Kume T, Katsuki H, Mori Y & Akaike A (2006). A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci 101, 66–76. [DOI] [PubMed] [Google Scholar]
- Knowles H, Heizer JW, Li Y, Chapman K, Ogden CA, Andreasen K, Shapland E, Kucera G, Mogan J, Humann J, Lenz LL, Morrison AD & Perraud AL (2011). Transient receptor potential melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes . Proc Natl Acad Sci USA 108, 11578–11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles H, Li Y & Perraud AL (2013). The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol Res 55, 241–248. [DOI] [PubMed] [Google Scholar]
- Kolisek M, Beck A, Fleig A & Penner R (2005). Cyclic ADP‐ribose and hydrogen peroxide synergize with ADP‐ribose in the activation of TRPM2 channels. Mol Cell 18, 61–69. [DOI] [PubMed] [Google Scholar]
- Lehen'kyi V & Prevarskaya N (2011). Oncogenic TRP channels. Adv Exp Med Biol 704, 929–945. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z & Nieminen AL (2009). Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 1787, 1395–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YV, Hubbi ME, Pan F, McDonald KR, Mansharamani M, Cole RN, Liu JO & Semenza GL (2007). Calcineurin promotes hypoxia‐inducible factor 1α expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J Biol Chem 282, 37064–37073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi PA & Sweadner KJ (1991). Postnatal changes in Na,K‐ATPase isoform expression in rat cardiac ventricle. Conservation of biphasic ouabain affinity. J Biol Chem 266, 9327–9331. [PubMed] [Google Scholar]
- McHugh D, Flemming R, Xu SZ, Perraud AL & Beech DJ (2003). Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J Biol Chem 278, 11002–11006. [DOI] [PubMed] [Google Scholar]
- Miller BA, Hoffman NE, Merali S, Zhang XQ, Wang J, Rajan S, Shanmughapriya S, Gao E, Barrero CA, Mallilankaraman K, Song J, Gu T, Hirschler‐Laszkiewicz I, Koch WJ, Feldman AM, Madesh M & Cheung JY (2014). TRPM2 channels protect against cardiac ischemia‐reperfusion injury: role of mitochondria. J Biol Chem 289, 7615–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BA, Wang J, Hirschler‐Laszkiewicz I, Gao E, Song J, Zhang XQ, Koch WJ, Madesh M, Mallilankaraman K, Gu T, Chen SJ, Keefer K, Conrad K, Feldman AM & Cheung JY (2013). The second member of transient receptor potential‐melastatin channel family protects hearts from ischemia‐reperfusion injury. Am J Physiol Heart Circ Physiol 304, H1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BA & Zhang W (2011). TRP channels as mediators of oxidative stress. Adv Exp Med Biol 704, 531–544. [DOI] [PubMed] [Google Scholar]
- Mittal M, Urao N, Hecquet CM, Zhang M, Sudhahar V, Gao XP, Komarova Y, Ushio‐Fukai M & Malik AB (2015). Novel role of reactive oxygen species‐activated Trp melastatin channel‐2 in mediating angiogenesis and postischemic neovascularization. Arterioscler Thromb Vasc Biol 35, 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C, Levy LA, Gabel S & London RE (1994). Measurement of cytosolic free calcium in perfused rat heart using TF‐BAPTA. Am J Physiol Cell Physiol 266, C1323–1329. [DOI] [PubMed] [Google Scholar]
- Nagamine K, Kudoh J, Minoshima S, Kawasaki K, Asakawa S, Ito F & Shimizu N (1998). Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics 54, 124–131. [DOI] [PubMed] [Google Scholar]
- Nilius B, Owsianik G, Voets T & Peters JA (2007). Transient receptor potential cation channels in disease. Physiol Rev 87, 165–217. [DOI] [PubMed] [Google Scholar]
- Orfanelli U, Wenke AK, Doglioni C, Russo V, Bosserhoff AK & Lavorgna G (2008). Identification of novel sense and antisense transcription at the TRPM2 locus in cancer. Cell Res 18, 1128–1140. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP & Scharenberg AM (2001). ADP‐ribose gating of the calcium‐permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411, 595–599. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Schmitz C & Scharenberg AM (2003). TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium 33, 519–531. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, Stoddard BL & Scharenberg AM (2005). Accumulation of free ADP‐ribose from mitochondria mediates oxidative stress‐induced gating of TRPM2 cation channels. J Biol Chem 280, 6138–6148. [DOI] [PubMed] [Google Scholar]
- Prawitt D, Enklaar T, Klemm G, Gartner B, Spangenberg C, Winterpacht A, Higgins M, Pelletier J & Zabel B (2000). Identification and characterization of MTR1, a novel gene with homology to melastatin (MLSN1) and the trp gene family located in the BWS‐WT2 critical region on chromosome 11p15.5 and showing allele‐specific expression. Hum Mol Genet 9, 203–216. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Delling M & Clapham DE (2006). An introduction to TRP channels. Annu Rev Physiol 68, 619–647. [DOI] [PubMed] [Google Scholar]
- Roberge S, Roussel J, Andersson DC, Meli AC, Vidal B, Blandel F, Lanner JT, Le Guennec JY, Katz A, Westerblad H, Lacampagne A & Fauconnier J (2014). TNF‐α‐mediated caspase‐8 activation induces ROS production and TRPM2 activation in adult ventricular myocytes. Cardiovas Res 103, 90–99. [DOI] [PubMed] [Google Scholar]
- Simard C, Salle L, Rouet R & Guinamard R (2012). Transient receptor potential melastatin 4 inhibitor 9‐phenanthrol abolishes arrhythmias induced by hypoxia and re‐oxygenation in mouse ventricle. Br J Pharmacol 165, 2354–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus JG, Fleig A & Penner R (2010). The calcium‐permeable non‐selective cation channel TRPM2 is modulated by cellular acidification. J Physiol 588, 1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, Mori Y & Tominaga M (2006). TRPM2 activation by cyclic ADP‐ribose at body temperature is involved in insulin secretion. EMBO J 25, 1804–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Q, Zhang W, Conrad K, Mostoller K, Cheung JY, Peterson BZ & Miller BA (2006). Regulation of the transient receptor potential channel TRPM2 by the Ca2+ sensor calmodulin. J Biol Chem 281, 9076–9085. [DOI] [PubMed] [Google Scholar]
- Toth B & Csanady L (2010). Identification of direct and indirect effectors of the transient receptor potential melastatin 2 (TRPM2) cation channel. J Biol Chem 285, 30091–30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Kudoh J, Noda S, Kanba S & Shimizu N (2005). Characterization of human and mouse TRPM2 genes: identification of a novel N‐terminal truncated protein specifically expressed in human striatum. Biochem Biophys Res Commun 328, 1232–1243. [DOI] [PubMed] [Google Scholar]
- Wallace DC (2012). Mitochondria and cancer. Nat Rev Cancer 12, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehage E, Eisfeld J, Heiner I, Jungling E, Zitt C & Luckhoff A (2002). Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP‐ribose. J Biol Chem 277, 23150–23156. [DOI] [PubMed] [Google Scholar]
- Xia R, Mei ZZ, Mao HJ, Yang W, Dong L, Bradley H, Beech DJ & Jiang LH (2008). Identification of pore residues engaged in determining divalent cationic permeation in transient receptor potential melastatin subtype channel 2. J Biol Chem 283, 27426–27432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H & Mori Y (2008). TRPM2‐mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 14, 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KT, Chang WL, Yang PC, Chien CL, Lai MS, Su MJ & Wu ML (2006). Activation of the transient receptor potential M2 channel and poly(ADP‐ribose) polymerase is involved in oxidative stress‐induced cardiomyocyte death. Cell Death Differ 13, 1815–1826. [DOI] [PubMed] [Google Scholar]
- Zhang W, Chu X, Tong Q, Cheung JY, Conrad K, Masker K & Miller BA (2003). A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem 278, 16222–16229. [DOI] [PubMed] [Google Scholar]