

Abstract
Sphingosine-1-phosphate (S1P) is a lysophospholipid mediator carried by the HDL-associated apoM protein in blood, regulating many physiological processes by activating the G protein-coupled S1P receptor in mammals. Despite the solved crystal structure of the apoM-S1P complex, the mechanism of S1P release from apoM as a part of the S1P pathway is unknown. Here, the dynamics of the wild type apoM-S1P complex as well as of mutants were investigated by means of atomistic molecular dynamics simulations. The potential of mean force for S1P unbinding from apoM reflected a large binding strength of more than 60 kJ/mol. This high unbinding free energy for S1P underlines the observed specificity of the physiological effects of S1P as it suggests that the spontaneous release of S1P from apoM is unlikely. Instead, S1P release and thus the control of this bioactive lipid probably requires the tight interaction with other molecules, e.g. with the S1P receptor. Mutations of specific S1P anchoring residues of apoM decreased the energetic barrier by up to 20 kJ/mol. Moreover, the ligand-free apoM protein is shown to adopt a more open upper hydrophilic binding pocket and to result in complete closure of the lower hydrophobic cavity, suggesting a mechanism for adjusting the gate for ligand access.
Sphingosine-1-phosphate (S1P) is a pluripotent, zwitterionic lysophospholipid mediator. S1P binding to one of its G protein-coupled receptors (S1P1-S1P5) in the cell membrane regulates a variety of cell and tissue functions, including vascular maturation1,2, lymphocyte trafficking3, barrier functions4, calcium mobilization5, cell proliferation6,7,8, differentiation5, and survival7. S1P is synthesized locally in most organs but also circulates with a high level in blood to fully stimulate S1P receptors3,9. Plasma S1P is transported by plasma proteins, which are comprised of ~55% high density lipoprotein (HDL), ~40% albumin, and <10% low density lipoprotein (LDL) and very low density lipoprotein (VLDL)10,11,12.
A previous study demonstrated that apolipoprotein M (apoM) is the direct carrier of S1P in HDL and the crystal structure of recombinantly expressed apoM in complex with S1P was solved at 1.7 Å resolution13. ApoM belongs to the lipocalin family characterized by an eight-stranded antiparallel β-barrel enclosing a ligand binding pocket for small lipophilic molecules. Human apoM is a 188-residue-long protein, synthesized primarily in liver and kidney and mainly bound to HDL14,15,16,17. The N-terminal 21 amino acid residues form a hydrophobic signal peptide which is essential for anchoring the lipoprotein to the phospholipid layer14,18,19. ApoM has been shown to influence lipoprotein and cholesterol metabolism20,21,22, atherosclerosis20,21, diabetes23,24,25, and rheumatoid arthritis (RA)26,27 as recently shown in a number of experimental studies. Still, the molecular mechanisms of apoM action are largely unknown. ApoM can bind different ligands including diverse fatty acids and lipids, containing 14, 16, or 18 carbon atoms in their hydrophobic tail28. S1P is one of the apoM ligands, its binding to apoM was shown to efficiently quench the apoM intrinsic fluorescence with an IC50 of 0.90 μM at 345 nm28. This indicates that the apoM-S1P complex could play a key role in the physiological S1P pathway.
In 2011, Christoffersen et al.13 reported the crystal structure of the apoM-S1P complex. The S1P headgroup was observed to bind to the hydrophilic upper moiety of apoM while the hydrophobic chain was buried in the hydrophobic lower cavity of the protein, similar to the before reported position of myristic acid in apoM28. The N-terminal signal peptide of apoM, which anchors apoM into HDL, was not resolved in the structure.
In apoM, S1P binds at the center of the calyx-like binding pocket through direct hydrogen bonds with Arg98, Trp100, Arg116, and Glu136, as well as via water-mediated hydrogen bonds to Tyr102, Arg143, and Tyr14728. The recognition of the phosphate group of S1P by the arginines appears to be highly specific. Despite the crystal structure of the apoM-S1P complex, the biological process of S1P (un-)binding to the carrier is still widely debated.
Using atomistic molecular dynamics (MD) simulations, we characterized the human-apoM binding pocket for the ligand S1P and the changes in S1P anchoring to apoM upon mutating residues involved in the polar S1P binding. Enforced S1P dissociation molecular dynamics (MD) simulations29 and potential of mean force (PMF) calculations for S1P unbinding were applied to shed light on the mechanism of ligand unbinding. The observed high binding free energy for S1P binding to apoM of >60 kJ/mol and the dominant interactions of polar apoM residues to binding suggest that the S1P release depends on the interaction of the apoM calyx opening with other molecules. The interactions between S1P and apoM attached to a lipid bilayer displayed similar characteristics as those in solution.
Results
Protonation of S1P
Possible protonation changes of S1P upon binding to apoM were addressed by analyzing the pKa of S1P both in its free and apoM-bound states, as well as during unbinding from apoM.
During a 200 ns MD simulation, the pKa values of the apoM-bound S1P phosphate oxygens were stable with values of ≈4, supporting the chosen protonation of the phosphate group (PO42−). Additionally, the pKa value of the amino group fluctuated around 14, consistent with the protonated state of NH3+. The total charge of bound S1P is thus −1 e, which is in line with a previous study30.
The pKa values of an isolated solvated S1P molecule were calculated to be ≈6 for the phosphate oxygens and 11 for the amino group. Taken together, this change in S1P protonation upon solvation sums up to a net release of 0.16 protons during S1P unbinding from apoM at pH 7 (see Equation 3), i.e. the assumption of an unchanged total charge of −1 e for S1P during unbinding is well justified.
The change in pKa was furthermore followed during the enforced unbinding simulation (see Supplementary Fig. S2). The change in the S1P pKa values correlates with the rupture of intermolecular hydrogen bonds between apoM and S1P. The isoelectric point (pI) of apoM increased from 6.2 to 6.7 during S1P unbinding. This estimated change in pI by 0.5 pH units is in very good agreement with the experimental data reporting a decrease of the pI by 0.2 pH units upon S1P addition28.
Characteristics of S1P binding to apoM
The wt apoM structures both in solution and attached to POPC remained stable on the 200 ns timescale with a root mean square deviations (RMSD) of ≈2 Å (see below). ApoM overall kept its local structure with the exception of β-sheet E both in solution and attached to POPC. The short β-sheet E region (six residues) partially unfolded, thereby increasing the solvent exposure of the E/F loop. Noteworthy, the hydrogen bond between S1P and Arg116 that is located in the E/F loop was lost (Fig. 1a). However, S1P was still stable within the binding pocket. Arg116 showed a large flexibility, increasing its interaction with the rest of the protein (Fig. 1b). Due to this flexibility, Arg116 is suggested to easily adapt to different ligands and to be involved in the recognition of S1P and possibly also of interaction partners of apoM.
Figure 1.

(a) The conformational switch of Arg116 after 80 ns of simulation time. (b) The interaction energy computed as the sum of short range Lennard-Jones and electrostatic interactions between Arg116 and S1P (red) and between Arg116 and the protein (blue).
The S1P binding environment was characterized by computing the averaged residue-wise forces between the ligand and apoM residues. Residues strongly contributing to S1P binding are easily identified by their large interaction forces (>70 pN, see Fig. 2a, visualized in Fig. 3a). The largest forces were recorded for the residues Arg98, Trp100, Arg116, and Glu136. These residues are located at the opening of apoM13 and interact mainly electrostatically with S1P. In the crystal structure, Arg143 was reported to form a hydrogen bond with S1P via a water molecule13. During the simulation, Arg143 moved closer to S1P and formed a direct hydrogen bond with the hydroxyl group of S1P. This position of Arg143 is stabilized by backbone hydrogen bonding to Glu136 (Fig. 3a, left). Residues His43, Trp47, Phe71, Met73, Leu84, Tyr102, Leu134, and Tyr147 primarily form the hydrophobic lower binding pocket for the hydrocarbon chain of the substrate (Fig. 3a, right). These interactions with S1P are accordingly dominated by Lennard-Jones interactions.
Figure 2. Pairwise interaction forces between apoM and S1P.
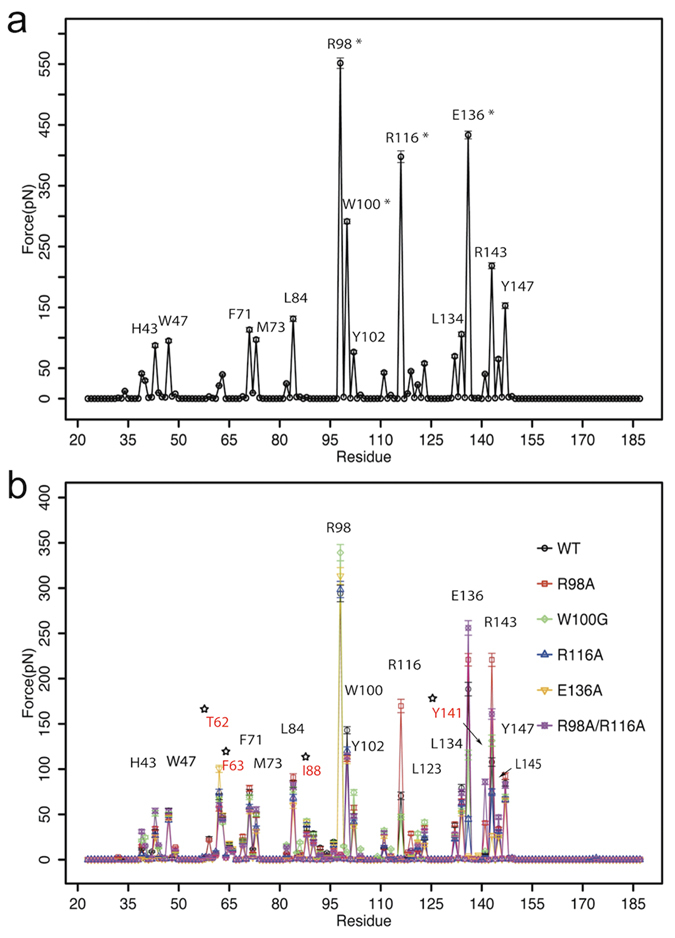
(a) Interaction forces for the apoM-S1P complex. The pairwise forces between S1P and each residue of apoM were calculated using the force distribution analysis (FDA, see Methods). The forces were averaged over simulations of the apoM WT both in solution and anchored to POPC. The residues with largest forces (>70 pN) are labeled. The four residues with the highest values (marked by asterisk) are in accordance with the S1P binding residues reported in the crystal structure. (b) Interaction forces during enforced S1P unbinding. Each line was averaged over three pulling simulations for each complex. Residues with forces >40 pN are marked. Residues with forces significantly exceeding those from wt equilibrium MD simulation are labeled in red and marked with an asterisk.
Figure 3. The S1P binding pocket of apoM.

(a) A snapshot of wt apoM-S1P after 200 ns of equilibration. Left panel: The residues highlighted in yellow mainly contribute to the upper hydrophilic part of the binding pocket and stabilize the charged moiety of S1P. Right panel: The residues colored orange form the hydrophobic binding groove of apoM. (b) Three main conformational changes were observed for the equilibrium MD simulations of the studied mutants (colored green):① Arg116 switched to stabilize S1P in the R98A mutant, and occasionally also in the W100G and E136A mutants; ② The flexibility of Arg98 increased in the W100G mutant due to the loss of the cation-π interaction; ③ The distance between Cα atoms of residues 98 and 136 indicating calyx structure loosening increased for all mutants as compared to the WT protein. (c) The distances between Cα atoms of residues 98 and 136 as a function of simulation time for the WT (black), the WT anchored to a POPC bilayer (brown), the R98A (red), W100G (green), R116A (blue), E136A (orange), and R98A/R116A mutants (magenta).
In all five mutant equilibrium simulations, the RMSD values were smaller than 3 Å (Fig. 4a) and the protein secondary structure was maintained (apart from partial unfolding of the β-sheet E, not shown) similar to the wt simulation. Large deviations as well as large fluctuations (root mean square fluctuation, RMSF) were observed for the loop regions (Fig. 4b). For the R98A mutant, Arg116 replaced Arg98 in stabilizing the phosphate group of S1P (no conformational switch as discussed above for the WT). I.e., S1P may be stabilized in the binding groove of apoM either by Arg98 or Arg116 (preferentially Arg98). In the W100G mutant, hydrogen bonding between Arg98 and S1P was slightly diminished as compared to the wt (hydrogen bond existence time decreased from 97% to 88%). This suggests that Trp100 can steady Arg98 in its binding position through a cation-π interaction, thus stabilizing the ligand binding. The width of the binding pocket slightly increased for all mutants as evidenced by the increased distance between the Cα of residues 98 and 136 for the mutant simulations (Fig. 3c). This loosening of the calyx structure resulted in significant reduction of the S1P binding strength (see below). This was in particular observed for R116A and R98A/R116A as well as for E136A (connection between the Glu136 and the amine group of S1P got disrupted). The flexibility of S1P was largest in E136A, suggesting a key role of Glu136 in stabilizing the ligand motion in the binding pocket (Fig. 4c). An overall reduced S1P flexibility is seen for the membrane-bound form of apoM.
Figure 4. Stability and fluctuations of apoM and S1P.
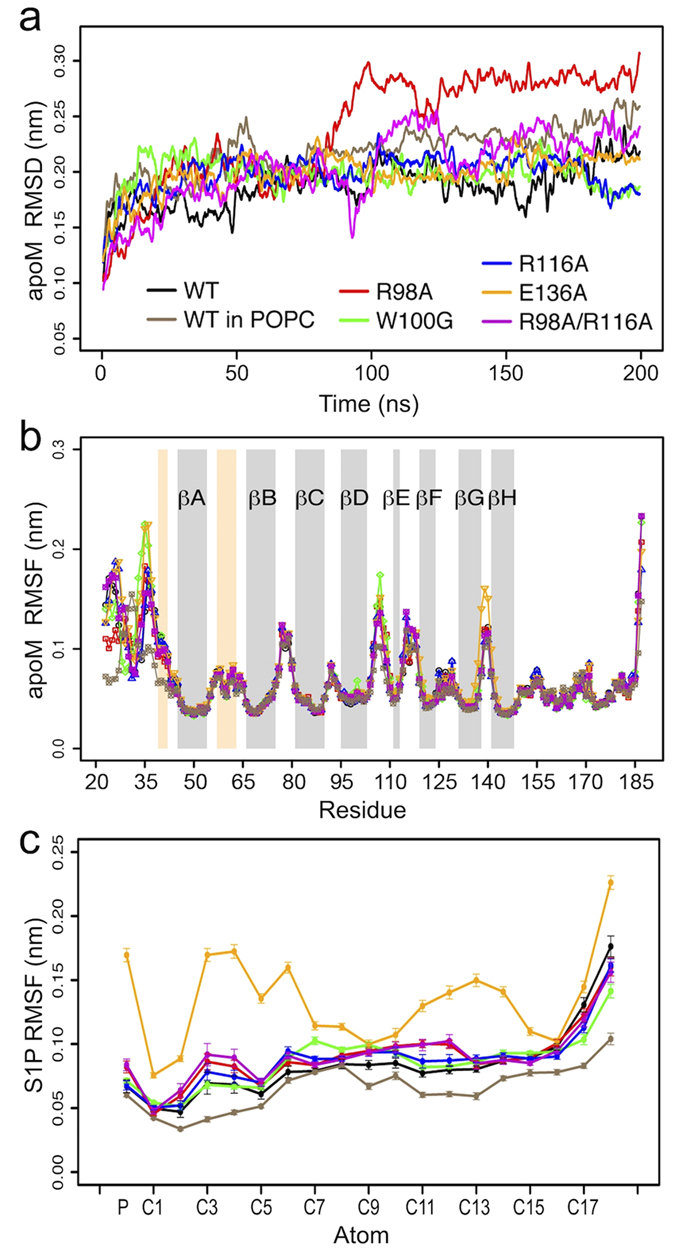
RMSD (a) and RMSF (b) values of apoM were analyzed for the Cα-atoms of the seven studied complexes (WT, black; WT attached to POPC, brown; R98A, red; W100G, green; R116A, blue; E136A, orange; R98A/R116A, magenta). The values were averaged over 40 windows of 5 ns length each. Error bars show the standard error. (c) RMSF values of the phosphorus atom and carbon atoms of S1P. The values were averaged over 20 windows of 10 ns length each. Error bars show the standard error.
ApoM-S1P complex associated with lipid layer
ApoM was suggested to be associated via the N-terminal signal peptide to HDL particles21. Sevvana et al. modeled the anchoring and suggested that hydrophobic residues facing the HDL lipid layer, including L2 (76-GSAPM), L6 (124-FSSSCPGG), L8 (150-SPHPP), and a patch (29-LTTLGV) at the N-terminus, further facilitate the interaction between apoM and HDL28. Here, in the apoM-membrane simulation, the loop of apoM before β-sheet A (41-EVHLGQW), L6 (128-CPG), L8 (150-SPHPP), and a C-terminal patch (178-RNQEAC) (Fig. 5d, green color) interacted with the POPC bilayer, similar to the suggested model. The hydrophobic residues interacted with the lipid chains, while the charged residues were engaged in interactions with the phosphatidyl choline headgroup of POPC, which is a major constituent of HDL31. Despite this association, the apoM-S1P complex behaved similarly to that in solution, which suggests an HDL-independent ligand binding and release mechanism. A previously suggested release of S1P due to the flexibility of the peptide segment preceding β-strand A appears unlikely, the flexibility of this segment was decreased for the membrane-bound apoM as compared to free apoM (Fig. 4b), and an uncovering of the bottom of the calyx was not observed in the simulations.
Figure 5.

(a) Sequence of human apoM along with the secondary structure elements gained from the crystal structure. (b) S1P structure. Oxygen atoms, phosphorus atom, and nitrogen atom are shown in red, yellow and blue, respectively. (c) ApoM structure from residue 27 to residue 187, containing a β-barrel with eight anti-parallel β-strands displayed in cartoon representation. (d) The S1P-bound apoM embedded in a POPC bilayer by the N-terminal anchor helix. The region of apoM interacting with the bilayer is highlighted in green.
Enforced S1P unbinding
The un-/binding process of S1P to apoM was addressed in enforced unbinding simulations by pulling the ligand out of apoM (compare Fig. 5). A constant velocity was applied to a spring attached to S1P (the pulling force, apoM-S1P interaction energy, and number of hydrogen bonds during unbinding are provided in Supplementary Fig. S3). In all pulling simulations, interactions to apoM were lost for distances between the phosphorus atom of S1P and the center of mass (COM) of apoM above 2.30 nm. Upon pulling, the RMSD of the Cα atoms of apoM remained below 3 Å for all systems (not shown), i.e. the protein did not undergo larger conformational changes related to S1P unbinding. Key residues involved in the unbinding process were identified using the force distribution analysis (FDA) analysis32: Comparison to equilibrium simulations (see Fig. 2a) shows that forces between S1P and the residues Thr62, Phe63, Ile88, and Tyr141 were markedly increased during S1P unbinding (Fig. 2b, labeled red). In turn, these residues are probably also involved in ligand recognition.
Five steps can be distinguished for the S1P unbinding pathway (compare Fig. 6): In the initial stage (stage a), the pulling force increased until reaching Fmax (Supplementary Fig. S3), the so-called unbinding force. The height of this force typically depends on the pulling or unbinding velocity33,34. Concomitant with the main force barrier the number of hydrogen bonds between S1P and apoM drastically decreased (Fig. S3), suggesting the polar interactions between S1P and the hydrophilic opening of the calyx as main opposing force to unbinding.
Figure 6. S1P unbinding process from WT apoM.

The residues located in the hydrophobic lower part are colored wheat. The five residues forming the hydrophilic entrance for S1P are colored yellow. Residues interacting strongly with S1P during enforced unbinding are colored violet. Residues 23–46, 74–83 and 101–126 were omitted in figures (b–e) for clarity.
Following the initial breakage of hydrogen bonds to the S1P headgroup, the motion of the phosphate headgroup was temporarily restricted by interactions with Thr62, Phe63, and Glu59 in the flanking 310 helix (step b). Simultaneously, the hydrocarbon chain of S1P left the lower hydrophobic cavity. Finally, S1P binding is restricted to interactions between the hydrocarbon tail and residues on the top of apoM, especially from the flanking 310 helix (d). In a different pathway, a prior flipping of the S1P headgroup to Arg98 was observed (c). In the final step, the S1P tail glided out of the apoM binding groove without exhibiting any further significant energetic barriers (e).
The potential of mean force (PMF) was analyzed for both pathways, i.e. a–b–c–d–e (path 1) and a–b–d–e (path 2), using umbrella sampling simulations. The PMF analysis followed the local samplings along each pathway, without restraints on the sampling orthogonal to the pulling direction. The degree of orthogonal sampling is displayed in Supplementary Fig. S4. Slightly different regions in phase space are sampled for the paths, differing in particular for the intermediate binding to Arg98 along path 1. The PMF curves for both pathways show a monotonous increase (Fig. 7), the energetic barrier for path 1 was determined to 75 kJ/mol and for path 2 to 64 kJ/mol.
Figure 7. Potential of mean force (PMF) for S1P unbinding.
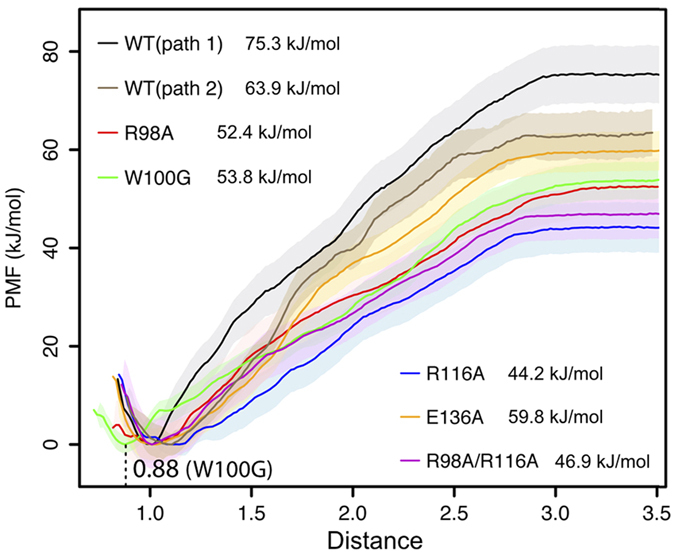
The PMF was calculated from between 42 and 88 umbrella simulations for each system.
The chosen apoM mutations in particular impacted the electrostatic contribution to S1P headgroup binding. For all mutants, unbinding of S1P followed the energetically favored path 2. In agreement with the selection of mutants to decrease the S1P binding affinity, the PMFs of all mutant complexes for S1P unbinding were significantly decreased by 4 kJ/mol (E136A) to 20 kJ/mol (R116A) as compared to wt apoM. In W100G, Arg98 lost the cation-π interaction with Trp100, resulting in weakened ligand binding (by 10 kJ/mol). Additionally, S1P was observed to interact with Tyr102 (side chain directly beneath Trp100), resulting in a shift of the lowest energy state of S1P by about 1 Å into the apoM interior. This finding suggests that Trp100 is important for keeping a certain binding position of S1P within apoM. The largest reduction of binding strength was caused by the R116A and R98A/R116A mutations. Though Arg116 is flexible it is indispensable to integrate the binding pocket in the calyx structure.
ApoM microenvironment changes during ligand unbinding
Sevvana et al. reported the distance of the broadest opening of the fatty-acid-bound-apoM to 29 Å (distance between the Cα atoms of Gly93 and Tyr141), tapering to 19 Å in the middle of apoM (Lys99 to Phe144) and 20 Å at the bottom (His103 to Tyr147)28. The corresponding values in the equilibrated S1P-bound apoM wild type complex are in close agreement with the reported data. However, following S1P removal both the diameter of the opening of the calyx increased by ≈2 Å as well as the flexibility in particular of the middle region of the barrel (see Supplementary Fig. S5). The structure of the ligand-free mutants resembled the one of the ligand-free apoM structure.
The two tyrosine residues in positions 102 and 147 separate the hydrophobic lower cavity from the hydrophilic upper portion of the binding pocket. The minimum distance between Tyr102 and Tyr147 exhibited a slight decrease during the S1P pulling simulation, hindering the ligand from release (data not shown). After the tail of S1P left the hydrophobic pocket, these two residues restored their positions and showed an increased flexibility as compared to the S1P-bound state (Supplementary Fig. S4). This indicates that the two tyrosines form a gate, similar to the arrangement of the corresponding residues Tyr83 and Tyr131 reported for the human complement protein C8γ35.
ApoM cavity volume
The opening of the calyx as well as the above reported gating of the tyrosines 102 and 147 influenced the form and size of the apoM binding pocket. The total volume of the binding pocket is ≈430 Å3 in the S1P-bound crystal structure, which may be divided into an upper part with ≈285 Å3 and a lower pocket with ≈145 Å3 (gate formed by residues Tyr102 and Tyr147, see Fig. 8 & Supplementary Table S1). Both simulations of ligand-free apoM, i.e. a simulation starting from the crystal apoM after S1P deletion and a simulation of apoM after enforced S1P unbinding, showed a widened opening and a significantly increased pocket volume of ≈455 Å3 for the upper cavity, while the lower part was completely closed due to rearrangement of the hydrophobic residues Phe71, Met73, Ile132, and His43 (Fig. 8c): The re-oriented Phe71 (Fig. 3a) contributed most to the shielding of the binding cavity from the solvent, a function that has been observed in other lipocalins as well (e.g. in bovine odorant-binding protein36 and in aphrodoisin37). Our results suggest that the empty apoM structure adopts a more open conformation that is prone to ligand recognition and binding. Preceding ligand binding, the hydrophobic lower cavity is shielded.
Figure 8. S1P binding pocket of apoM.

(a,b) The binding pockets of S1P-bound apoM (orange) and of ligand-free apoM (grey) are compared to each other. The blue surface borders the measured pocket volumes. The upper pocket, as well as the distance between the Cα atoms of residues G93 and Y141 are increased for the ligand-free apoM while the lower part of the cavity is closed. (c) The gating is achieved by Tyr102 and Tyr147 and the pocket occluded by Phe71, Met73, His43, and Ile132 (grey sticks). For bound S1P (cyan sticks), the sidechains (orange sticks) slightly reorient to accomodate the ligand.
The bottom part of the calyx is formed by His43 and Trp47. His43 is replaced by phenylalanine in some other lipocalins with the side chain pointing either towards the inside of the calyx or positioned parallel to the bottom of the calyx. Here, His43 adopts the latter conformation in the apoM-S1P complex, enabling to accomodate the long S1P hydrocarbon chain within the calyx (Fig. 8c). In both ligand-free and ligand-bound complexes, His43 was very stable and close to Trp47, which forms π-π interaction with His43 and cation-π interaction with Arg149 to stabilize the bottom of the calyx. It was suggested before that Trp47 stabilizes the lipocalin fold and supports ligand binding28,38.
Discussion
S1P is a lipidic mediator in mammalian bodies. In order to be able to bind to its receptor and trigger the signaling pathway it has to be first transported by its carrier apoM protein to the GPCR. For receptor binding and activation, S1P unbinding from the carrier apoM is required first. Here, we characterized by means of MD simulations the S1P binding strength to wt apoM as well as to a number of apoM mutants and investigated the plasticity of the apoM carrier upon S1P dissociation.
The apoM binding pocket was seen to be gated by the tyrosines in positions 102 and 147. These tyrosines were observed to be more flexible in the ligand-free apoM, shielding together with Phe71 the lower hydrophobic cavity of apoM in the apo state. A similar rearrangement for closure was seen for the hydrophobic amino acids lining this cavity.
The upper part of the binding cavity, which opens significantly in the absence of the ligand, is mostly formed by hydrophilic residues. These play an important role in stabilizing the charged S1P headgroup in the bound state. The unbinding energy for S1P was estimated to more than 60 kJ/mol. It was determined along a path through the calyx opening at the top of apoM. Mutation(s) of apoM residues anchoring the S1P headgroup (Arg98, Trp100, Arg116, and Glu136) decreased the potential of mean force by up to 20 kJ/mol. Thus, the main energetic barrier for S1P unbinding was due to hydrogen bonding of the polar S1P headgroup. Our results moreover suggest that the 310 helix is involved in ligand recognition upon binding.
ApoM was found to be stably anchored in the POPC bilayer via the N-terminal signal peptide and by interactions between the loops at the bottom of the calyx and the lipid bilayer. Interestingly, the anchoring of apoM to a lipid bilayer did hardly affect the S1P bound conformation or ligand unbinding from apoM, suggesting an HDL-independent ligand binding mechanism. Previously discussed alternative ligand release mechanisms either through the bottom of the calyx or a lateral release between strands D and G13 are unlikely given the overall structural stability of apoM observed in the simulations.
In summary, our results suggest (i) that S1P binds to and leaves apoM via the calyx opening, and, (ii) that a specific mechanism decreasing the energetic barrier is essential for the transfer of S1P to the S1P receptor. This could be achieved through other proteins, like the endothelial lipase that was suggested to mediate S1P release from HDL39 or by direct binding of apoM to the S1P1 receptor.
Methods
Starting Structures
All molecular dynamics simulations of apoM in complex with S1P were based on the crystal structure (PDB ID 2YG213). Not resolved residues were built using the PyMOL40 software (see Fig. 5a for the apoM sequence). The total complex comprised 165 residues of apoM (Fig. 5c) and one S1P molecule (Fig. 5b).
Based on the crystal structure, four residues (Arg98, Trp100, Arg116 and Glu136) of apoM can form direct hydrogen bonds to S1P via their side chains. Here, five mutants (R98A, W100G, R116A, E136A, and the double mutant R98A/R116A) were designed in order to study the influence of the specific S1P headgroup binding on the total binding free energy.
Additionally, the N-terminal membrane anchor region of apoM, ‘1-MFHQI WAALL YFYGI ILNSI YQ-22’, was modeled in α-helical conformation using PyMOL and inserted into a lipid bilayer by the tool ‘g_membed’41 (Fig. 5d). The membrane consisted of 332 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) molecules mimicking the HDL lipid layer.
Force field parameters
All simulations were performed with the GROMACS 4.5 package42 using a combination of the AMBER99sb-ildn protein force field43 and the Slipids force field for S1P and POPC44,45,46. Force field parameters for the S1P molecule were prepared based on the parameters for sphingomyelin available within the Slipids force field by deleting the tail connected to the nitrogen atom. The nitrogen atom was accordingly changed into a 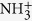 group. In order to keep the charges for this amino group equal to the amino group of phosphatidylethanolamine (PE) in the Slipids force field, the charge of the adjacent carbon (C2) had to be altered from 0.09 e to 0.07 e.
group. In order to keep the charges for this amino group equal to the amino group of phosphatidylethanolamine (PE) in the Slipids force field, the charge of the adjacent carbon (C2) had to be altered from 0.09 e to 0.07 e.
In the crystal structure, the phosphate group of S1P forms an intramolecular hydrogen bond with Arg98 of apoM. Shin et al. reported that the phosphomonoester of PA is deprotonated upon formation of a hydrogen bond with the primary amine of the headgroup of PE lipids, or to arginines and lysines of proteins30. These findings suggest that the phosphate group of S1P should be modeled with a charge of −2 e (PO42−) if bound to apoM. This assumption is further validated by pKa calculations for S1P (Results Section). Overall, S1P thus has a total charge of −1 e. Charges of the phosphate group were gained from restrained electrostatic potential (RESP) calculations47 using Gaussian0948. In detail, the charges of methyl phosphate (CH3-PO42−) were calculated using the Hartree-Fock method with the 6-31G* basis set as is typical for charge estimation in the Amber force field49. The charges of chemically equivalent oxygen atoms were symmetrized. The force field parameters for S1P are summarized in Table S2 of the Supplementary Information.
MD simulation parameters
The wild type (wt) apoM-S1P complex and selected mutants were solvated using the TIP3P water model50. Na+ and Cl− ions were randomly placed in the solvent phase at a physiological concentration of 0.15 M. Additional ions were added to neutralize the system.
A 200 steps energy minimization using the steepest descent algorithm was performed, followed by a 200 ps position restrained simulation with harmonic restraints (k = 1000 kJ/mol/nm2) on all protein heavy atoms to relax the water and ions. Subsequently, the system was simulated for 200 ns at constant pressure and temperature (NpT ensemble). The temperature was controlled to stay at 310 K using the v-rescale thermostat51 with a coupling time of 0.1 ps. The pressure was kept constant at 1.013 bar by applying an isotropic coupling using the Parrinello-Rahman barostat52,53 with a coupling time of 4 ps. All bonds were constrained using the LINCS algorithm54,55. Short-range interactions were cut-off at 1 nm, and the long-range electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method56,57. An integration timestep of 2 fs was used in all simulations. The equilibrated apoM-S1P structure was used to prepare the mutant structures. The same simulation procedure was used for the equilibrium MD simulations of the five investigated mutants.
For the simulation of apoM anchored to a POPC bilayer, the same simulation parameters were used except for the coupling set. Here, the temperature was coupled using the Nosé-Hoover thermostat58,59 with a coupling time of 0.5 ps and the pressure was kept constant at 1.013 bar by applying a semi-isotropic coupling using the Parrinello-Rahman barostat52,53 with a coupling time of 10 ps.
Enforced S1P Unbinding and Umbrella Sampling
Force probe simulations or steered molecular dynamics (SMD) simulations33,60 are frequently employed to study unbinding processes or steered conformational changes in proteins.
Here, an external force was applied to the phosphorus atom of the S1P headgroup to drive the S1P unbinding from apoM. In order to avoid co-pulling of apoM, the protein was kept in place by keeping the positions of the apoM residues Gln81 and Gly131 fixed. These are located at the bottom of apoM with side chains pointing outwards and are thus expected not to influence S1P binding. No restraint was applied for apoM anchored to a POPC bilayer.
The spring attached to S1P was pulled at a constant velocity of 0.01 nm/ns (spring constant k = 100 kJ/mol/nm2), applied along the preset pulling direction, i.e. the axis of the binding pocket (see Fig. 5c). Motion of the ligand orthogonal to the pulling direction was not restricted.
During enforced unbinding, the total distance between the phosphorus atom of S1P and the center of mass (COM) of apoM increased from 1 nm (bound state) to 3.3 nm (free state). In order to grasp possibly varying unbinding pathways, the enforced unbinding of each mutant was repeated three times.
The potential of mean force (PMF) for ligand unbinding was calculated using umbrella sampling along the S1P unbinding pathway. As start structures for the umbrella sampling simulations, starting structures were selected from the enforced unbinding simulations with a spacing of the phosphorus atom positions of S1P along the pulling axis of 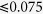 nm.
nm.
In the umbrella simulations a spring constant k = 1000 kJ/mol/nm2 was used. Each umbrella was simulated for at least 10 ns. Some samples were equilibrated for longer times in order to obtain sufficient sampling (umbrella histograms shown in Supplementary Information Fig. S1). In total, between 41 and 88 umbrella simulations were performed for each system (total of 482 umbrella simulations). The weighted histogram analysis method (WHAM, GROMACS tool g_wham) was used to extract the PMF from the obtained distributions61. The statistical error was estimated using bootstrap analysis with 200 bootstraps as implemented in g_wham, treating complete histograms as independent data points.
In order to study the conformational flexibility of the ligand-free apoM structure, both the wild type apoM after S1P pulling, as well as the crystal apoM structure with removed S1P, were equilibrated for 200 ns.
pKa calculation
Changes in the protonation state of S1P may be estimated from an in silico pKa analysis. Here, the PROPKA (version 3.1) workflow62,63 was used: PROPKA determines the pKa of ionizable groups through application of an environmental perturbation:
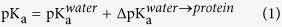 |
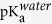 is the pKa value of the isolated titratable group in water (also often referred to as
is the pKa value of the isolated titratable group in water (also often referred to as 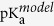 ), and
), and 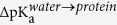 is the protein environmental contribution to the energy difference between protonated and deprotonated forms of the amino acid or a molecule, i.e. the influence of the protein surrounding. This protein contribution predicted by PROPKA 3.1 consists of (1) desolvation effects, (2) hydrogen bond interactions (HB), (3) unfavorable electrostatic reorganization energies (RE), and (4) Coulombic interactions (QQ):
is the protein environmental contribution to the energy difference between protonated and deprotonated forms of the amino acid or a molecule, i.e. the influence of the protein surrounding. This protein contribution predicted by PROPKA 3.1 consists of (1) desolvation effects, (2) hydrogen bond interactions (HB), (3) unfavorable electrostatic reorganization energies (RE), and (4) Coulombic interactions (QQ):
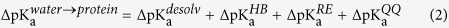 |
In order to estimate whether S1P dissociation from apoM results in a protonation change of S1P, the overall net change n of S1P was calculated based on the pKa change:
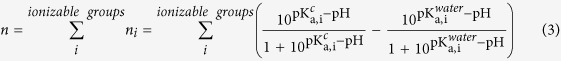 |
The pKa values of all titratable groups (two oxygen atoms of the phosphate group and one nitrogen atom of the amino group) in the ligand were summed. 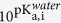 and
and 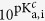 refer to the pKa values of S1P in water and within the apoM-S1P complex, respectively. The isoelectric point (pI) of the protein can as well be obtained using PROPKA 3.1. The change of pI was recorded as a function of simulation time.
refer to the pKa values of S1P in water and within the apoM-S1P complex, respectively. The isoelectric point (pI) of the protein can as well be obtained using PROPKA 3.1. The change of pI was recorded as a function of simulation time.
Force distribution analysis
The propagation of internal strain through apoM by S1P un-/binding was followed using force distribution analysis (FDA)32 implemented in GROMACS 4.5.3.
The time-averaged residue-pair wise force between S1P and individual apoM residues was analyzed in order to pinpoint key residues involved in the unbinding process. The residue-pair wise force 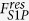 at every time point was obtained by summing up all atom-pair wise forces Fij from all pairs of atoms i of S1P and j of each residue of apoM.
at every time point was obtained by summing up all atom-pair wise forces Fij from all pairs of atoms i of S1P and j of each residue of apoM.
Additional Information
How to cite this article: Zhang, H. et al. Binding Characteristics of Sphingosine-1-Phosphate to ApoM hints to Assisted Release Mechanism via the ApoM Calyx-Opening. Sci. Rep. 6, 30655; doi: 10.1038/srep30655 (2016).
Supplementary Material
Acknowledgments
This work was supported by a scholarship from the China Scholarship Council and by the DFG Research Training Group 1962/1, Dynamic Interactions at Biological Membranes. We acknowledge computational support from the Computer Center of the University of Erlangen-Nürnberg (RRZE).
Footnotes
Author Contributions R.A.B. conceived research, H.Z. performed simulations and analysed the results, K.P. set up the force field for S1P, Z.J. performed force distribution analysis. All authors wrote the manuscript.
References
- Lee M. J. et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99, 301–312 (1999). [DOI] [PubMed] [Google Scholar]
- Liu Y. et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Invest. 106, 951–961 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappu R. et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 316, 295–298 (2007). [DOI] [PubMed] [Google Scholar]
- Prager B., Spampinato S. F. & Ransohoff R. M. Sphingosine 1-phosphate signaling at the blood-brain barrier. Trends Mol. Med. 21, 354–363 (2015). [DOI] [PubMed] [Google Scholar]
- Spiegel S. & Milstien S. Sphingosine-1-phosphate: signaling inside and out. FEBS Lett. 476, 55–57 (2000). [DOI] [PubMed] [Google Scholar]
- Paik J. H., Chae S. S., Lee M. J., Thangada S. & Hla T. Sphingosine 1-phosphate-induced endothelial cell migration requires the expression of EDG-1 and EDG-3 receptors and Rho-dependent activation of αvβ3- and β1-containing integrins. J. Biol. Chem. 276, 11830–11837 (2001). [DOI] [PubMed] [Google Scholar]
- Spiegel S. Sphingosine 1-phosphate: a prototype of a new class of second messengers. J. Leukoc. Biol. 65, 341–344 (1999). [DOI] [PubMed] [Google Scholar]
- Zhang H. et al. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell. Biol. 114, 155–167 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänel P., Andréani P. & Gräler M. H. Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB J. 21, 1202–1209 (2007). [DOI] [PubMed] [Google Scholar]
- Aoki S. et al. Sphingosine 1-phosphate-related metabolism in the blood vessel. J. Biochem. 138, 47–55 (2005). [DOI] [PubMed] [Google Scholar]
- Murata N. et al. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem. J. 352 Pt 3, 809–815 (2000). [PMC free article] [PubMed] [Google Scholar]
- Argraves K. M. & Argraves W. S. HDL serves as a S1P signaling platform mediating a multitude of cardiovascular effects. J. Lipid Res. 48, 2325–2333 (2007). [DOI] [PubMed] [Google Scholar]
- Christoffersen C. et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 108, 9613–9618 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N. & Dahlbäck B. A novel human apolipoprotein (apoM). J. Biol. Chem. 274, 31286–31290 (1999). [DOI] [PubMed] [Google Scholar]
- Faber K., Axler O., Dahlbäck B. & Nielsen L. B. Characterization of apoM in normal and genetically modified mice. J. Lipid Res. 45, 1272–1278 (2004). [DOI] [PubMed] [Google Scholar]
- Pan G. et al. Molecular characterization, expression profile and association analysis with fat deposition traits of the porcine APOM gene. Mol. Biol. Rep. 37, 1363–1371 (2010). [DOI] [PubMed] [Google Scholar]
- Zhang X. Y. et al. Specific tissue expression and cellular localization of human apolipoprotein M as determined by in situ hybridization. Acta Histochem. 105, 67–72 (2003). [DOI] [PubMed] [Google Scholar]
- Axler O., Ahnström J. & Dahlbäck B. Apolipoprotein M associates to lipoproteins through its retained signal peptide. FEBS Lett. 582, 826–828 (2008). [DOI] [PubMed] [Google Scholar]
- Liu M. et al. Uncleaved apoM signal peptide is required for formation of large apoM/S1P-enriched HDL particles. J. Biol. Chem. 290, 7861–7870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum C., Poy M. N. & Stoffel M. Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat. Med. 11, 418–422 (2005). [DOI] [PubMed] [Google Scholar]
- Christoffersen C. et al. Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J. Biol. Chem. 283, 1839–1847 (2008). [DOI] [PubMed] [Google Scholar]
- Mulya A. et al. Apolipoprotein M expression increases the size of nascent pre beta HDL formed by ATP binding cassette transporter A1. J. Lipid Res. 51, 514–524 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N., Nilsson-Ehle P. & Ahrén B. Suppression of apolipoprotein M expression and secretion in alloxan-diabetic mouse: Partial reversal by insulin. Biochem. Biophys. Res. Commun. 342, 1174–1177 (2006). [DOI] [PubMed] [Google Scholar]
- Zhang X., Jiang B., Luo G., Nilsson-Ehle P. & Xu N. Hyperglycemia down-regulates apolipoprotein M expression in vivo and in vitro. Biochim. Biophys. Acta 1771, 879–882 (2007). [DOI] [PubMed] [Google Scholar]
- Kurano M. et al. Induction of insulin secretion by apolipoprotein M, a carrier for sphingosine 1-phosphate. Biochim. Biophys. Acta 1841, 1217–1226 (2014). [DOI] [PubMed] [Google Scholar]
- Singal D. P., Li J. & Zhu Y. HLA class III region and susceptibility to rheumatoid arthritis. Clin. Exp. Rheumatol. 18, 485–491 (2000). [PubMed] [Google Scholar]
- Hu H. J. et al. Common variants at the promoter region of the APOM confer a risk of rheumatoid arthritis. Exp. Mol. Med. 43, 613–621 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevvana M. et al. Serendipitous fatty acid binding reveals the structural determinants for ligand recognition in apolipoprotein M. J. Mol. Biol. 393, 920–936 (2009). [DOI] [PubMed] [Google Scholar]
- Grubmüller H., Heymann B. & Tavan P. Ligand binding: Molecular mechanics calculation of the streptavidin-biotin rupture force. Science 271, 997–999 (1996). [DOI] [PubMed] [Google Scholar]
- Shin J. J. & Loewen C. J. Putting the pH into phosphatidic acid signaling. BMC Biol. 9, 85 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguchi H., Fernandez J. A., Hackeng T. M., Banka C. L. & Griffin J. H. Cardiolipin is a normal component of human plasma lipoproteins. Proc. Natl. Acad. Sci. USA 97, 1743–1748 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costescu B. I. & Gräter F. Time-resolved force distribution analysis. BMC biophys. 6, 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubmüller H., Heymann B. & Tavan P. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science 271, 997–999 (1996). [DOI] [PubMed] [Google Scholar]
- Izrailev S., Stepaniants S., Balsera M., Oono Y. & Schulten K. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72, 1568–1581 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiswell B. et al. Structural features of the ligand binding site on human complement protein C8γ: A member of the lipocalin family. Biochim. Biophys. Acta 1774, 637–644 (2007). [DOI] [PubMed] [Google Scholar]
- Vincent F. et al. Crystal structures of bovine odorant-binding protein in complex with odorant molecules. Eur. J. Biochem. 271, 3832–3842 (2004). [DOI] [PubMed] [Google Scholar]
- Vincent F. et al. Crystal structure of aphrodisin, a sex pheromone from female hamster. J. Mol. Biol. 305, 459–469 (2001). [DOI] [PubMed] [Google Scholar]
- Greene L. H. et al. Role of conserved residues in structure and stability: tryptophans of human serum retinol-binding protein, a model for the lipocalin superfamily. Protein Sci. 10, 2301–2316 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzer A., Giral H. & Landmesser U. High-density lipoproteins as modulators of endothelial cell functions: Alterations in patients with coronary artery disease. Cardiovasc. Res. 103, 350–361 (2014). [DOI] [PubMed] [Google Scholar]
- DeLano W. L. The PyMOL Molecular Graphics System. Schrödinger, LLC. http://www.pymol.org (2002). [Google Scholar]
- Wolf M. G., Hoefling M., Aponte-SantamarA C., Grubmüller H. & Groenhof G. G-membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 31, 2169–2174 (2010). [DOI] [PubMed] [Google Scholar]
- Pronk S. et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K. et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jämbeck J. P. M. & Lyubartsev A. P. Derivation and systematic validation of a refined all-atom force field for phosphatidylcholine lipids. J. Phys. Chem. B 116, 3164–3179 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jämbeck J. P. M. & Lyubartsev A. P. An extension and further validation of an all-atomistic force field for biological membranes. J. Chem. Theory Comput. 8, 2938–2948 (2012). [DOI] [PubMed] [Google Scholar]
- Jämbeck J. P. M. & Lyubartsev A. P. Another piece of the membrane puzzle: Extending slipids further. J. Chem. Theory Comput. 9, 774–784 (2013). [DOI] [PubMed] [Google Scholar]
- Bayly C. C. I., Cieplak P., Cornell W. D. & Kollman P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10269–10280 (1993). [Google Scholar]
- Frisch M. J. et al. Gaussian~09 Revision E.01. Gaussian Inc. Wallingford CT (2009). [Google Scholar]
- Cornell W. D. et al. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117, 5179–5197 (1995). [Google Scholar]
- Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W. & Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926 (1983). [Google Scholar]
- Bussi G., Donadio D. & Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 14101–14107 (2008). [DOI] [PubMed] [Google Scholar]
- Parrinello M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- Nosé S. & Klein M. L. Constant pressure molecular dynamics for molecular systems. Molecular Physics 50, 1055–1076 (1983). [Google Scholar]
- Ryckaert J. P., Ciccotti G. & Berendsen H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977). [Google Scholar]
- Hess B., Bekker H., Berendsen H. J. C. & Fraaije J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997). [Google Scholar]
- Darden T., York D. & Pedersen L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089 (1993). [Google Scholar]
- Essmann U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995). [Google Scholar]
- Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511 (1984). [Google Scholar]
- Hoover W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985). [DOI] [PubMed] [Google Scholar]
- Isralewitz B., Gao M. & Schulten K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 11, 224–230 (2001). [DOI] [PubMed] [Google Scholar]
- Hub J. S., de Groot B. L. & van der Spoel D. g_wham – Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 6, 3713–3720 (2010). [Google Scholar]
- Søndergaard C. R., Olsson M. H. M., Rostkowski M. & Jensen J. H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 7, 2284–2295 (2011). [DOI] [PubMed] [Google Scholar]
- Olsson M. H. M., Søndergaard C. R., Rostkowski M. & Jensen J. H. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.