

Abstract
RING‐in‐between‐RING (RBR) ubiquitin (Ub) ligases are a distinct class of E3s, defined by a RING1 domain that binds E2 Ub‐conjugating enzyme and a RING2 domain that contains an active site cysteine similar to HECT‐type E3s. Proposed to function as RING/HECT hybrids, details regarding the Ub transfer mechanism used by RBRs have yet to be defined. When paired with RING‐type E3s, E2s perform the final step of Ub ligation to a substrate. In contrast, when paired with RBR E3s, E2s must transfer Ub onto the E3 to generate a E3~Ub intermediate. We show that RBRs utilize two strategies to ensure transfer of Ub from the E2 onto the E3 active site. First, RING1 domains of HHARI and RNF144 promote open E2~Ubs. Second, we identify a Ub‐binding site on HHARI RING2 important for its recruitment to RING1‐bound E2~Ub. Mutations that ablate Ub binding to HHARI RING2 also decrease RBR ligase activity, consistent with RING2 recruitment being a critical step for the RBR Ub transfer mechanism. Finally, we demonstrate that the mechanism defined here is utilized by a variety of RBRs.
Keywords: HHARI, HOIP, Parkin, RBR, ubiquitin E3 ligase
Subject Categories: Post-translational Modifications, Proteolysis & Proteomics
Introduction
Ubiquitin (Ub) is a small protein involved in the regulation of a wide variety of cellular processes. Ub signaling occurs through the covalent attachment of the Ub C‐terminus to protein substrates via a trio of enzymes: a Ub‐activating enzyme (E1), a Ub‐conjugating enzyme (E2), and a Ub ligase (E3). E1s activate the C‐terminus of Ub in an ATP‐dependent manner and facilitate the transfer of Ub onto the E2 active site cysteine (Cys) via transthiolation to generate an E2~Ub conjugate (“~” denotes a thioester bond). Most E2~Ub conjugates can pair with one or more E3 Ub ligases to facilitate Ub transfer onto a substrate amino group, usually the side chain of a lysine (Lys). There are three major types of eukaryotic E3 Ub ligases: Really Interesting New Gene (RING)‐type E3s (including Ubox E3s), Homologous to E6AP C‐Terminus (HECT)‐type E3s, and RING‐in‐between‐RING (RBR) E3s.
A majority of E3s are RING‐type ligases that use a conserved RING/Ubox domain defined by a characteristic cross‐brace fold to bind the E2~Ub. These E3s bind both the substrate and E2~Ub to facilitate Ub transfer from the E2~Ub directly onto a substrate amino group, usually a lysine side chain. In addition to this scaffolding role, RING‐type E3s activate the E2~Ub to transfer Ub onto Lys residues (an aminolysis reaction) by promoting closed conformations of the E2~Ub 1, 2, 3, 4, 5. A crucial feature of the RING‐type activation mechanism is a conserved basic residue (Lys/Arg) that contacts both the E2 and the Ub, referred to as the “linchpin” residue 1. HECT‐type E3s bind the E2~Ub via a conserved HECT domain that is structurally distinct from a RING domain. Importantly, HECT‐type E3 ligases contain a conserved Cys residue on which an obligatory E3~Ub intermediate is formed prior to Ub transfer onto a substrate Lys. This mechanism requires two types of chemical reactions: (i) a transthiolation reaction transfers Ub from E2~Ub to the E3 active site Cys to generate a reactive E3~Ub species, and (ii) a subsequent aminolysis reaction transfers the C‐terminus of Ub from the E3 to a substrate Lys to form a stable isopeptide bond. Unlike the mechanism utilized by RING‐type E3 ligases that induce closed E2~Ub conformations, available data on HECT ligases suggest that they do not need a closed E2~Ub for activity 6, 7.
The third class of E3 ligases was discovered more recently when two RBR E3s, HHARI (human homologue of Ariadne) and Parkin, were shown to function via a RING/HECT hybrid mechanism 6. Despite containing an eponymous E2‐binding RING domain, RBR E3s proceed through an E3~Ub thioester intermediate similar to HECT‐type E3s. The functional importance of an active site Cys for catalytic activity has since been confirmed for other RBR E3 ligases including HOIP, HOIL‐1L, TRIAD1, and RNF144 8, 9, 10, 11. Although a small class with only 12–14 members in the human genome, RBR E3s are involved in many essential cellular pathways. The most studied RBR E3 is Parkin for which mutations have been linked to autosomal recessive juvenile Parkinson's disease 12, 13. HOIP and HOIL‐1L are distinctive for their unique ability to generate linear Ub chains that are crucial regulators of the NF‐κB signaling pathway 14, 15, 16, 17. HHARI and TRIAD1 belong to the Ariadne family of RBR E3 ligases defined by their C‐terminal auto‐inhibitory Ariadne domain. HHARI and its homologues in Drosophila and C. elegans have been implicated in processes including regulation of translation via the translation initiation factor 4EHP, cellular proliferation, and development 18, 19, 20, 21.
RBR E3s are defined by three characteristic domains each of which coordinates two Zn2+ ions: RING1, in‐between RING (IBR), and RING2. An RBR unit may be found at any position relative to other domains within an RBR E3, but its subdomains RING1‐IBR‐RING2 always appear in order (N‐terminal to C‐terminal). RING1 is the E2‐binding domain and RING2 contains the active site Cys that when mutated to an alanine (Ala) renders RBRs inactive 6, 8, 9, 10, 11, 22, 23. While RING1 domains adopt structures similar to canonical RING‐type E3 ligases, RING2 domains do not resemble canonical RING domains, either structurally or functionally. The IBR domain adopts a structure that is similar to that of RING2, though its function remains elusive.
As mentioned above, canonical RING‐type E3s activate an E2~Ub conjugate by promoting closed E2~Ub conformations that exhibit increased reactivity toward the amino groups of Lys residues. A signature of closed conformations are non‐covalent interactions between a surface on the E2 formed by the “crossover” helix (also known as helix‐2) and the hydrophobic patch or “I44 surface” of Ub. Mutations of residues within the E2:Ub interface abrogate activation by RING‐type E3 ligases 1, 2, 3, 4, 5. Paradoxically, HHARI RING1 fails to activate the E2~Ub for Ub transfer by aminolysis despite its close structural resemblance to canonical RING domains 6. Furthermore, mutations of the E2 crossover helix that decrease in vitro Ub transfer activity with canonical RING‐type E3s do not affect Ub transfer activity of the RBR E3 HHARI 1. These observations imply that HHARI RING1 is mechanistically distinct from its canonical RING relatives in ways yet to be defined.
RBR E3s such as Parkin, HOIP, and HHARI display activity with a variety of E2s; in particular, they are all active with two well‐characterized human E2s, UbcH5 and UbcH7 6, 8, 9. While most E2s, including UbcH5, are able to perform both transthiolation reactions and aminolysis reactions, UbcH7 solely performs transthiolation reactions 6. This suggests that UbcH7 can function with HECT‐type and RBR‐type E3s, but not with RING‐type E3s. Notably, C. elegans orthologs of HHARI and UbcH7 act together in pharyngeal development, suggesting that UbcH7 is a biologically relevant E2 for HHARI 20, 24. But HHARI, Parkin, and HOIP are also known to work with E2s that are able to transfer Ub directly to amino groups. Importantly, HOIP specifically generates linear Ub chains regardless of the chain linkage preference of the E2 with which it works 6, 8, 9, 15, 25, 26, 27, 28. This suggests a dichotomy in the determining factor for product formation: in the case of RING‐type E3s, the identity of the E2 determines the type of product, while in RBR E3s, the E3 determines the type of product. This leads to the question: How do RBR E3s ensure that transfer of Ub occurs via the E3 active cite Cys to maintain control of product formation?
Here, we report the mechanistic strategies used by RBR‐type E3s to transfer ubiquitin from the E2 onto the E3. First, we show that RING1 domains of HHARI and RNF144 specifically inhibit closed E2~Ub conformations. This strategy ensures that Ub transfer occurs via the RBR active site by preventing off‐target Ub transfer events. Second, we identify a weak but functionally important interaction between HHARI RING2 and Ub which serves to recruit RING2 to the RING1:E2~Ub complex. In structures of auto‐inhibited RBR E3s, RING1 and RING2 are far from each other and a large domain rearrangement is required to bring the RING2 active site close to the E2~Ub active site bound to RING1 29, 30, 31, 32, 33, 34. Consistent with this notion, mutations in either Ub or RING2 at the Ub:RING2 interface substantially reduce Ub transfer from E2~Ub to RING2. Finally, we demonstrate that the mechanism defined here is utilized by a variety of RBR E3 ligases, indicating its generality for this important class of enzymes.
Results
RBR E3 ligases do not require closed states of E2~Ub for ubiquitin transfer
In the absence of an E3, UbcH5~Ub is highly dynamic, with the Ub moiety populating mainly open states relative to the E2 35. Upon binding a canonical RING/Ubox, UbcH5~Ub is biased toward closed states that are activated for aminolysis reactions which constitute the final step in Ub transfer by RING‐type E3s. Closed E2~Ub states bound to RING/Ubox domains have been visualized in several co‐crystal structures and by NMR 1, 2, 3, 4. Mutation of a residue on the crossover helix of UbcH5 (L104Q) disrupts formation of closed states and dramatically decreases ubiquitination activity with canonical RINGs such as BRCA1/BARD1 (Fig 1A) 1. Though RBRs contain an E2‐binding RING domain (RING1), UbcH5L104Q shows robust activity with HHARIRBR, TRIAD1ΔAri, ParkinRBR, and HOIPRBR‐LDD (Figs 1A and EV1; information regarding constructs used in this study is included in Appendix Table S1). This observation indicates that RBR E3s do not require closed E2~Ub conformations for Ub transfer activity. Therefore, we wondered whether RING1s of RBRs are able to induce closed E2~Ub conformations. To address this question, we used an active site Cys‐to‐Ser E2 mutant to generate a stable oxyester mimic of E2~Ub (“E2‐O‐Ub”). In a previous study, the NMR spectrum of 15N‐UbcH5c‐O‐15N‐Ub exhibited chemical shift perturbations (CSPs) in the presence of a canonical RING/Ubox that are hallmarks of the closed state 1. We therefore performed similar NMR binding experiments of 15N‐UbcH5c‐O‐15N‐Ub with the HHARI RING1 domain (residues 177–270). Comparable to previous observations for canonical RINGs, binding of HHARI RING1 to UbcH5‐O‐Ub occurs in fast‐to‐intermediate exchange (peaks shift and broaden), indicating that the interaction is of fairly modest affinity (Fig 1B). CSP analysis of free UbcH5‐O‐Ub compared to HHARI RING1‐bound UbcH5‐O‐Ub revealed residues that are perturbed upon HHARI RING1 binding. These define a surface composed of residues in helix 1, loops 4 and 7—those known to be central to binding of UbcH5 by canonical RING‐type E3s 2, 3, 36, 37, 38. These results indicate that HHARI RING1 binds UbcH5 in a manner similar to that used by canonical RINGs (Fig 1C). However, CSPs are not observed for most residues of the UbcH5 crossover helix nor are they observed for the Ub moiety upon HHARI RING1 binding to UbcH5‐O‐Ub (Fig 1C). Together, these observations are strong evidence that HHARI RING1 does not promote UbcH5‐O‐Ub closed conformations. This finding provides a basis for understanding two previous observations: (i) HHARI RING1 does not enhance UbcH5~Ub reactivity toward free Lys 6 and (ii) HHARIRBR exhibits robust activity with the crossover helix mutant UbcH5L104Q (1 and Fig 1A).
Figure 1. RBR E3 ligases do not require closed states of E2~Ub for ubiquitin transfer.
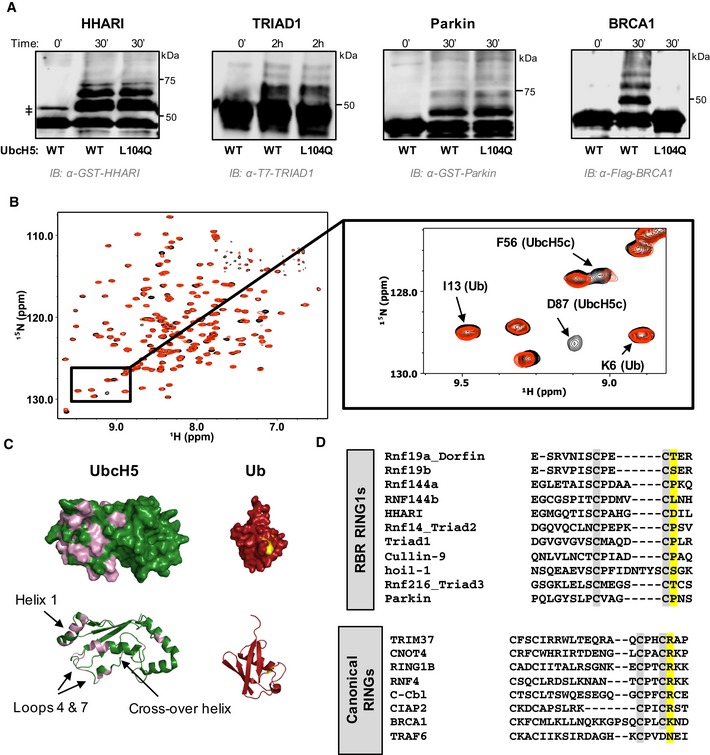
- Auto‐ubiquitination assays in which the E3 acts both as an E3 and as proxy substrate were performed with GST‐HHARIRBR, T7‐Triad1ΔAri, GST‐ParkinRBR , and Flag‐BRCA1/BARD1 and either UbcH5WT or UbcH5L104Q as the E2. Products were visualized by Western blotting against the indicated tags on E3s. Times given are post‐ATP addition.
- Overlay of (1H,15N)‐HSQC‐TROSY spectra of 15N‐UbcH5‐O‐15N‐Ub in the absence (black) and presence (red) of 0.5 mol equiv. HHARI RING1. A subset of UbcH5 peaks, but not Ub peaks, shift and broaden upon HHARI RING1 binding.
- Chemical shift perturbations (CSPs) from (B) are mapped onto the structures of UbcH5c (PDB 2fuh) and Ub (PDB 1ubq). Residues that exhibit loss of intensity > 1 stdv upon RING1 binding (intensity < 0.47) are colored on each structure: UbcH5 residues 5, 6, 7, 16, 20, 22, 56, 62, 74, 87, 90, 91, 96, 97, 99–103, 137, 138 (pink) and Ub residues 48, 50 (yellow). The lack of CSPs on the UbcH5 crossover helix and on the surface of Ub signifies that HHARI RING1 does not induce closed UbcH5˜Ub.
- CLUSTAL OMEGA alignments of the C‐terminal sequences of RBR RING1 domains (top) and canonical RING domains (bottom). Each alignment includes the 7th and 8th Zn2+ coordinating positions (gray). The allosteric linchpin position critical for E2˜Ub activation by RING‐type E3s is highlighted in yellow. RBR RING1 domains lack the allosteric linchpin.
Figure EV1. HOIP does not require closed states of E2~Ub for linear chain formation.
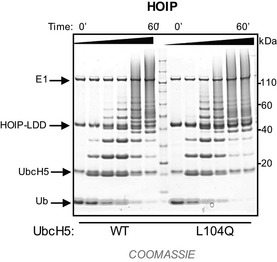
Linear Ub chain building reactions were performed using HOIPRBR ‐ LDD and either UbcH5WT or UbcH5L104Q. Product formation (free linear Ub chains) was visualized by Coomassie blue staining.
A highly conserved position in canonical RINGs, called the linchpin residue, is largely responsible for the ability to promote closed E2~Ubs by forming hydrogen bonds to both E2 and Ub 1, 2, 3, 4. In RINGs, the linchpin is most often a basic residue (Arg or Lys) and is occasionally a neutral H‐bonding residues such as Asn. HHARI contains an Asp residue in the structurally analogous position to the linchpin, and RBRs in general do not share a common residue that could fulfill the function of a hydrogen‐bonding linchpin (Fig 1D). This provides one possible explanation for HHARI RING1's failure to induce closed states of UbcH5~Ub.
HHARI RING1 promotes open E2~Ub conformations
UbcH7 is a specialized E2 that can only perform transthiolation reactions, making it an RBR/HECT‐specific E2 6. UbcH7 is active in vitro with many, if not all, RBR‐type E3 ligases. Notably, C. elegans orthologs of UbcH7 and HHARI are vital partners in vivo 20, prompting us to examine the effects of HHARI RING1 binding on UbcH7~Ub.
Unexpectedly, we discovered that in the absence of an E3, UbcH7‐O‐Ub populates closed conformations to a considerable extent. NMR CSP analysis of UbcH7‐O‐Ub compared to free UbcH7 reveals perturbed residues in the crossover helix of UbcH7 in addition to residues around the active site (Fig 2A). The perturbations identify a surface similar to that seen in closed states of other E2~Ubs 1, 2, 3, 4, 35. To verify this conclusion, we conjugated I44A‐Ub to 15N‐UbcH7 because UbI44A disrupts closed states of other E2~Ubs 1, 2, 3. Indeed, residues of the UbcH7 crossover helix as exemplified by Q106 and S107 resonate at positions more similar to free UbcH7 than to UbcH7‐O‐Ub when I44A‐Ub is the conjugated species (Fig 2B and Appendix Fig S1). Altogether, these data are consistent with UbcH7~Ub adopting closed conformations in the absence of an E3 that require surfaces that include the crossover helix of UbcH7 and the I44 surface of Ub (Fig 2A and B, and Appendix Fig S1).
Figure 2. UbcH7~Ub populates E3‐independent closed conformations.
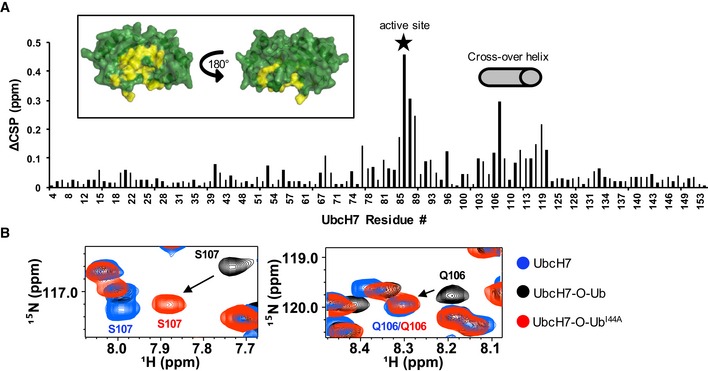
- Histogram of CSPs between free 15N‐UbcH7 and 15N‐UbcH7‐O‐Ub identifies UbcH7 residues affected by conjugation to Ub. Active site (Ser86) is indicated with a star and crossover helix residues (101–113) are marked with a gray cylinder. Inset: CSPs > 1 stdv (> 0.115 ppm) are highlighted in yellow on a surface representation of UbcH7 (PDB 1fbv). The view on the left shows the surface that contains the UbcH7 crossover helix.
- Region of 1H‐15N‐HSQC‐TROSY spectra containing resonances of the crossover helix residues S107 and Q106 is overlaid: unconjugated 15N‐UbcH7 (blue), 15N‐UbcH7‐O‐Ub (black), and 15N‐UbcH7‐O‐UbI44A (red). Peaks representing S107 and Q106 in the context of UbcH7˜Ub‐I44A (red) are closer to peaks observed in free UbcH7(blue) than UbcH7˜Ub‐WT (black), indicating that I44A of Ub disrupts interactions with the crossover helix of UbcH7.
We next asked what effect HHARI RING1 binding has on UbcH7~Ub. Due to the high affinity of UbcH7 for HHARI RING1 29, residues affected by complex formation exhibit slow exchange behavior in 1H‐15N‐HSQC‐type NMR experiments. This property leads to peak intensity loss rather than peak shifting in binding experiments (Fig EV2A). First, the effect of HHARI RING1 binding to unconjugated UbcH7 was analyzed to map the RING1‐binding surface. Comparison of peak intensities between the spectrum of free 15N‐UbcH7 and of RING1‐bound 15N‐UbcH7 reveals that the RING1‐binding surface on UbcH7 is very similar to that mapped for UbcH5 bound to RING1 (Fig EV2B and C). To simplify analysis of the UbcH7‐O‐Ub conjugate, either 15N‐labeled UbcH7 or 15N‐labeled Ub was incorporated into conjugates for HHARI RING1 binding experiments. Turning to the Ub moiety, an overlay of NMR spectra of free UbcH7‐O‐15N‐Ub and RING1‐bound UbcH7‐O‐15N‐Ub shows several perturbed Ub resonances, as exemplified in Fig 3A. Notably, a majority of Ub residues (47,49,71–74) that are affected by HHARI RING1 binding to UbcH7~Ub are among those identified in a comparison of UbcH7~Ub versus free Ub (Appendix Fig S2). For example, when UbcH7~Ub binds to RING1, the Ub Q49 resonance, which experiences the largest chemical shift upon Ub conjugation to UbcH7, moves back close to its position in the spectrum of free Ub (Fig 3A and Appendix Fig S2). The simplest explanation for this observation is that RING1 binding alters or disrupts contacts between Ub and UbcH7 in UbcH7~Ub. Importantly, chemical shifts for Ub residues within both RING1‐bound UbcH7~Ub and UbcH5~Ub (Figs 1B and 3A, and Appendix Fig S2) are similar to those seen for free Ub. We propose that RING1 promotes open conformations of E2~Ub. We note that while Ub resonances of RING1‐bound UbcH7~Ub are similar to those of free Ub, they are not identical, indicating that the environment of the Ub moiety may be affected by its proximity to RING1 in the complex (Fig 3A and Appendix Fig S2).
Figure EV2. NMR mapping of HHARI RING1 binding to UbcH7.
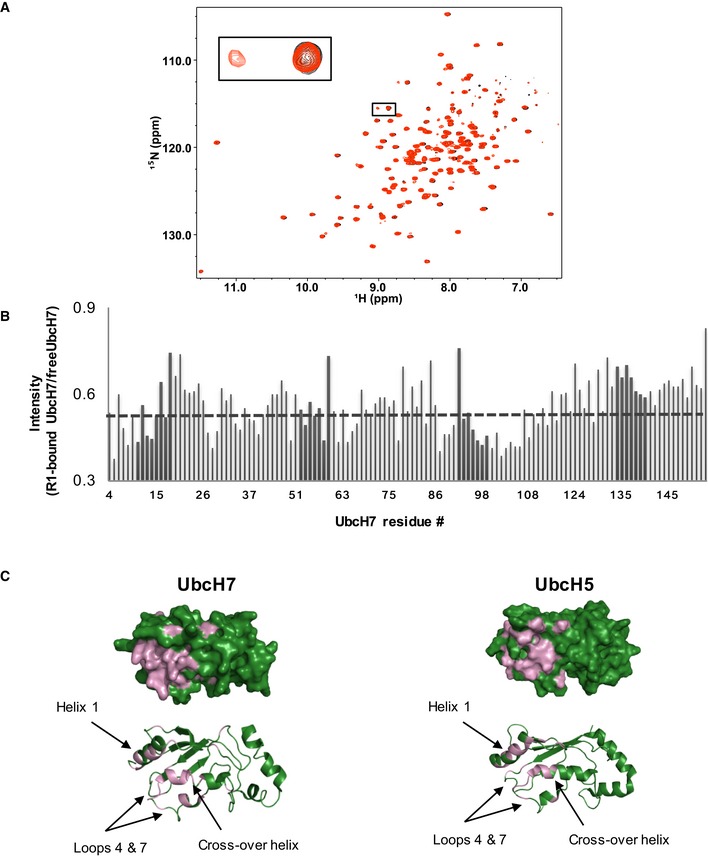
- Spectral overlay of 15N‐UbcH7 in the absence (black) and presence (red) of 0.5 mol equiv. HHARI RING1. Affected resonances show peak doubling, characteristic of a high‐affinity complex, where a new set of resonances that correspond to resonances from the complex appear simultaneously with resonances of the unbound species.
- Histogram displays the ratio of peak intensity of UbcH7 residues (bound/free) measured from the spectra shown above. The average intensity ratio is indicated with a gray dashed line.
- Left: Surface (top) and cartoon (bottom) representation of UbcH7 (PDB 1fbv). Residues that exhibit loss of intensity > 1 stdv from the average upon RING1 binding (intensity < 0.448) are colored in pink (residues 6, 11, 13, 14, 28, 40, 57, 61, 64, 78, 87, 89, 90, 96, 98–100, 102–106, 108). Right: For comparison, UbcH5 residues (PDB 2fuh) that are perturbed (either CSP or intensity loss) upon RING1 binding are indicated in pink (CSPs: > 0.028 ppm and intensity < 0.44).
Figure 3. HHARI RING1 disfavors closed E2~Ub conformations.
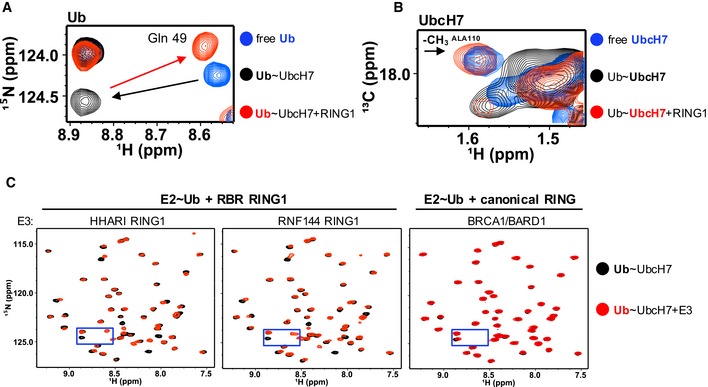
- Region of 1H‐15N‐HSQC‐TROSY spectra that contains Gln49 Ub resonance is overlaid: free 15N‐Ub (blue), UbcH7‐O‐15N‐Ub (black), and HHARI RING1‐bound UbcH7‐O‐15N‐Ub (red). Black arrow highlights perturbation upon conjugation to UbcH7 and red arrow highlights perturbation of conjugated Ub upon HHARI RING1 binding to UbcH7.
- Region of 1H‐13C‐HSQC spectra of 13C‐UbcH7 (blue), 13C‐UbcH7‐O‐Ub (black), and HHARI RING1‐bound 13C‐UbcH7‐O‐Ub (red) that includes the methyl (13 CH 3) resonance of the surface‐exposed UbcH7 crossover helix residue, Ala110 (black arrow), is shown. The 13 CH 3 peak of Ala110 either broadens dramatically or shifts to an unknown position in the spectrum of 13C‐UbcH7‐O‐Ub (black) in the presence of HHARI RING1 it reappears at its position in free UbcH7, consistent with disruption of closed UbcH7˜Ub conformations. Pairwise overlays and larger spectra are provided in Appendix Fig S3.
- Regions of 1H‐15N‐HSQC‐TROSY spectra of UbcH7‐O‐15N‐Ub in the absence (black) and presence (red) of a RING1 domain from the RBR E3s HHARI (left) or RNF144 (middle) or a canonical RING domain of BRCA1/BARD1 (right). The perturbations on Ub due to binding of HHARI RING1 and RNF144 RING1 are remarkably similar, while binding of the canonical RING domain of BRCA1/BARD1 has no observable effect on the Ub spectrum. Blue boxes mark area expanded in (A).
For additional evidence of disruption of UbcH7~Ub closed conformations by RING1 binding, we used 1H‐13C‐HSQC‐type experiments and published assignments to observe and interpret side chain resonances of 13C‐UbcH7 39. Side chain resonances of UbcH7 crossover helix residues shift upon conjugation to Ub, consistent with closed UbcH7~Ub states (as exemplified in Fig 3B and Appendix Fig S3). Figure 3B compares three spectra: free 13C‐UbcH7 (blue) and 13C‐UbcH7‐O‐Ub in the absence (black) and presence of HHARI RING1 (red). Notably, the methyl resonance of the surface‐accessible crossover helix residue Ala110 is significantly perturbed upon conjugation of Ub to UbcH7, as evidenced by the lack of a (black) peak on or near the blue peak labeled Ala110 (Fig 3B), indicating that the side chain of Ala110 interacts with Ub. Upon the addition of RING1 (red spectrum), the Ala110‐CH3 peak reappears at the chemical shift observed for Ala110‐CH3 in free 13C‐UbcH7 (blue), evidenced by the red peak that overlays the blue Ala110 peak (Fig 3B and Appendix Fig S3). These data corroborate the notion that UbcH7~Ub populates closed conformations in the absence of an E3 and that these are disrupted by HHARI RING1 binding.
To test whether disruption of UbcH7~Ub closed state is specific to HHARI RING1, we assessed the effects of binding of other RING domains to UbcH7‐O‐15N‐Ub. Though all domains tested bind to UbcH7, neither the canonical RING heterodimer BRCA1/BARD1 nor the Ubox E4BU disrupted the closed conformation of UbcH7~Ub while RNF144 RING1 produced effects similar to HHARI RING1 upon binding to UbcH7‐O‐15N‐Ub (Fig 3C and Appendix Fig S4). Thus, RBR‐type RING1 domains share the ability to discourage closed UbcH7~Ub conformations, making them functionally distinct from canonical RING‐type domains despite their structural similarities. Finally, we wondered whether HHARI RING1 can disrupt closed conformations of other E2~Ub species. Ubc13~Ub detectably populates closed conformations in the absence of E3 35. Indeed, addition of HHARI RING1 to Ubc13‐O‐15N‐Ub leads to chemical shifts of Ub resonances back toward their positions in free Ub, along the same trajectory as the “open” to “closed” perturbations (Fig EV3). Thus, the ability of HHARI RING1 to disrupt closed E2~Ub states is not limited to UbcH7.
Figure EV3. HHARI RING1 disruption of (E3‐independent) closed E2~Ub conformations.
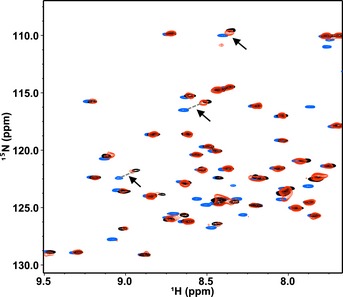
Similar to UbcH7, Ubc13~Ub populates closed states in the absence of an E3 35. Shown here are three 1H‐15N‐HSQC‐TROSY spectra of Ub overlaid: free 15N‐Ub (blue), Ubc13‐O‐15N‐Ub (black), and Ubc13‐O‐15N‐Ub in the presence of 3 mol equiv. HHARI RING1 (red). Due to the weak affinity binding of Ubc13 to HHARI RING1, the complex is far from saturation and the spectrum exhibits fast‐to‐intermediate exchange behavior. Therefore, the observed CSPs for the effects of RING1 binding to Ubc13‐O‐15N‐Ub are quite small (black versus red). Black arrows highlight several Ub resonances that shift along the same trajectory (but opposite direction) upon binding HHARI RING1 as experienced when Ub is conjugated to Ubc13, consistent with Ubc13~Ub populating more open‐like states when in complex with HHARI RING1.
Closed E2~Ubs are associated with increased reactivity toward Lys amino groups 1, 2, 3, 4, 5, 6.Therefore, a possible corollary to increased open E2~Ub conformations is an increase in transthiolation (reactivity toward Cys) as this is the relevant nucleophile in the context of RBR‐type E3 ligases. However, we did not observe a difference between UbcH7~Ub reactivity toward free Cys in the absence versus presence of HHARI RING1 (Appendix Fig S5).
Based on these data, we propose a mechanism in which not only does the RING1 of an RBR fail to promote closed E2~Ub states, but also it actively discriminates against them.
RING1 opening of E2~Ub enforces Ub transfer via the RING2 active site Cys
RBR E3s are active with E2s that are also active with canonical RING‐type E3s 6, 8, 9, 15, 25, 26, 27, 28. This implies that the E2~Ub must distinguish between its two reaction modes (transthiolation and aminolysis) based on the type of E3 ligase with which it interacts. Open E2~Ub states are minimally reactive toward Lys and require activation for canonical RING‐type Ub transfer 1, 2, 3, 4. We wondered whether there would be negative consequences if an RBR RING1 could also stabilize closed, aminolysis‐activated E2~Ubs as seen with canonical RINGs. However, our efforts to generate a HHARI RING1 that activates the E2~Ub for aminolysis by mutating positions known to be critical in canonical RING domains failed. We took an alternative approach to assess the consequence of having aminolysis activity in the context of an RBR E3 by replacing the RING1 domain within the HHARI RING1‐IBR‐RING2 (WT‐RBR domain) construct with the Ubox domain of E4BU to generate a Ubox‐IBR‐RING2 (UBR) hybrid (Fig 4A). Ubox domains are E2‐binding domains that contain RING‐like folds, but do not ligate Zn2+ and are therefore more likely to fold successfully within a hybrid construct. Importantly, the Ubox domain of E4BU has been shown to activate UbcH5~Ub for aminolysis via promotion of closed E2~Ub conformations 1.
Figure 4. RING1 opening of E2~Ub enforces transfer via the RING2 active site.
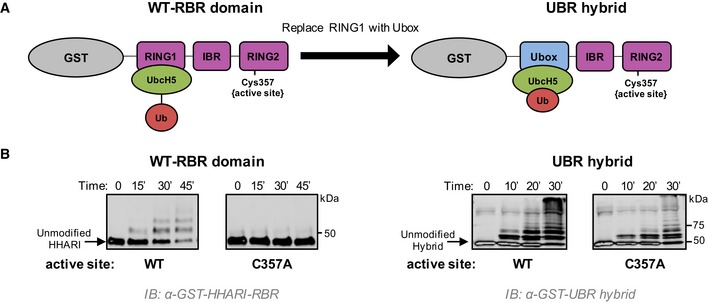
- Schematic representation of the two constructs used. Left: GST‐tagged HHARIRBR with its native RING1 domain that does not induce closed conformations of UbcH5˜Ub. Right: GST‐tagged HHARI construct in which the RING1 domain was replaced with the Ubox domain of E4BU to generate the UBR hybrid that promotes closed conformations of UbcH5˜Ub.
- Left: Auto‐ubiquitination assays were performed with wild‐type HHARIRBR or an active‐site‐dead mutant (C357A‐HHARIRBR). Products were visualized by Western blotting against GST. The zero time point was taken immediately prior to ATP addition; all other times are post‐ATP addition. Right: Identical assays as shown on left were performed with the UBR hybrid. The active‐site‐dead mutant (C357A) retains substantial auto‐ubiquitination activity signifying that UbcH5˜Ub is able to transfer Ub directly onto the GST‐UBR hybrid construct, bypassing the RING2 active site Cys.
We posited that an RBR with the ability to activate E2~Ub for aminolysis might circumvent the obligate transthiolation reaction through the active site Cys residue in RING2. In in vitro auto‐ubiquitination assays where the RBR E3 acts as a proxy substrate, Ub transfer occurs through the active site Cys as the active site mutation C357A essentially abrogates ubiquitination activity of HHARIRBR (Fig 4B, left panel). Remarkably, this is not the case for reactions carried out by the UBR hybrid as the active‐site‐dead (C357A) version retains substantial auto‐ubiquitination activity (Fig 4B, right panel). The result implies that UbcH5~Ub transfers its Ub directly to Lys residues when bound to the hybrid E3 construct as it does not require the active site Cys. Our UBR hybrid construct demonstrates that the presence of an E2‐binding domain that can activate E2~Ub for aminolysis creates an RBR that no longer undergoes obligate transfer to and through the active site Cys on RING2. Altogether, we conclude that by favoring open E2~Ub conformations, HHARI RING1 (and likely other RING1 domains) prevents off‐target ubiquitination events catalyzed by the E2~Ub and consequently enforces Ub transfer through the RING2 active site Cys.
The hydrophobic surface of Ub is required for transfer of Ub to the RBR active site
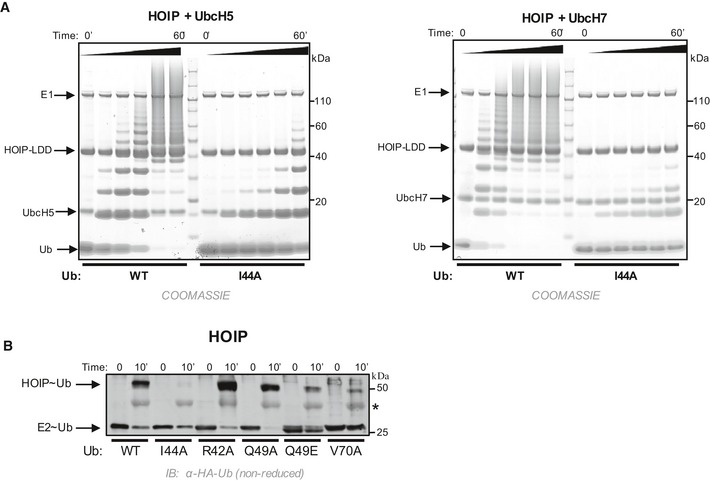
Mechanistically speaking, there is no need for RBR E3s to enhance E2~Ub ability to transfer Ub through aminolysis. Furthermore, as we demonstrated above, induction of closed E2~Ub can lead to undesirable off‐target Ub transfer (Fig 4). These two rationales may be sufficient to explain why RBR E3s prefer to keep E2~Ubs in open states. However, we noted that the hydrophobic (“I44”) surface of Ub that plays a critical role in protein–protein interactions is sequestered in closed E2~Ub but is exposed in open states. Furthermore, mutations of the Ub hydrophobic patch have previously been shown to decrease auto‐ubiquitination activity in Parkin 40. This raises the question whether the exposed Ub surface has a specific role in Ub transfer by RBR E3s. In in vitro ubiquitination assays with the RBR E3s HHARIRBR, ParkinRBR, and HOIPRBR‐LDD, use of Ub species that carry a mutation in a single hydrophobic patch residue led to reduced activity (Figs 5A and B, and EV4A). These results indicate that the Ub hydrophobic patch is required for Ub transfer by RBRs.
Figure 5. The Ub hydrophobic patch plays a role in Ub transfer from E2~Ub onto RING2 of RBR E3 ligases.
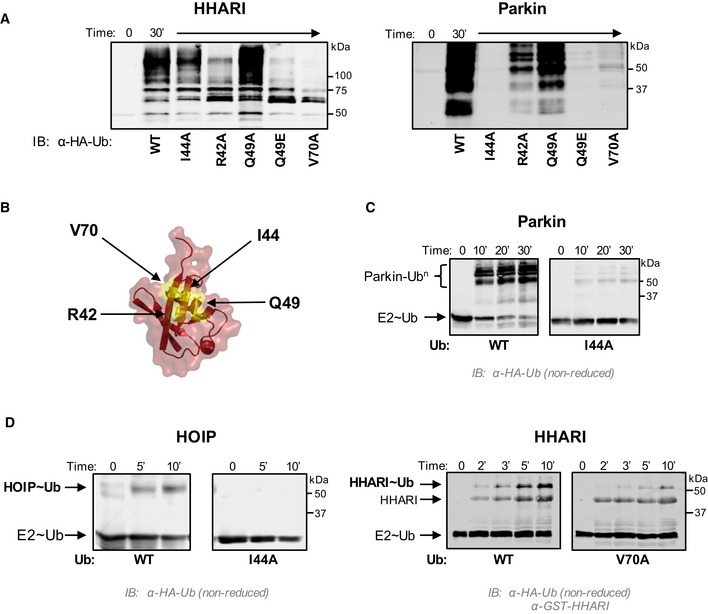
- E3 auto‐ubiquitination assays were performed using various Ub mutants (I44A, R42A, Q49A, Q49E, and V70A) and the RBR E3s HHARIRBR (left) and ParkinRBR (right). Products were visualized by Western blotting against HA‐Ub. Samples were analyzed 30 min after ATP addition.
- The hydrophobic patch of Ub (PDB 1ubq) is colored yellow on a surface representation and positions of each mutation are noted.
- UbcH7˜Ub conjugates were preformed with either WT‐Ub or I44A‐Ub. After the addition of apyrase to quench the charging reaction, UbcH7˜UbWT (left) or UbcH7˜UbI44A (right) was incubated with ParkinRBR. The disappearance of each UbcH7˜Ub species and appearance of auto‐ubiquitinated E3 were visualized under non‐reducing conditions by Western blotting for HA‐Ub. Time was recorded post‐addition of ParkinRBR. E2˜Ub conjugated with I44A‐Ub does not disappear over the time course of the reaction.
- UbcH7 conjugated with WT‐Ub, I44A‐Ub, or V70A Ub as indicated was incubated with either H887A‐HOIPRBR ‐ LDD (left) or H359A‐HHARIRBR (right) mutants that allow trapping of the E3˜Ub thioester with WT‐Ub 29, 41. While a HOIP˜Ub thioester species is observed when UbcH7 was charged with WT‐Ub, no detectable transfer occurs with the I44A‐Ub conjugate (left). Similarly, V70A‐Ub shows reduced formation of the HHARI˜Ub thioester (right). In addition to blotting for HA‐Ub, the blot on the right was also blotted for GST‐HHARI.
Figure EV4. Ub hydrophobic patch involvement in linear chain building by the RBR E3 HOIP .
- Time courses of linear chain building assays using HOIPRBR ‐ LDD and either UbcH5 (left) or UbcH7 (right) conjugated with WT‐Ub or I44A‐Ub as followed by Coomassie‐stained gels. The I44A mutation in the donor Ub does not impair the formation of di‐Ub 9, yet I44A‐Ub significantly reduces linear chain building ability by HOIPRBR ‐ LDD.
- UbcH7 was pre‐conjugated with either WT‐Ub or mutant Ub (as indicated) and the reaction was quenched with apyrase. Each UbcH7˜Ub species was incubated with H887A‐HOIPRBR ‐ LDD to enable detection of the E3˜Ub intermediate 41. Samples were analyzed on SDS–PAGE under non‐reducing conditions and subsequently visualized by Western blotting for the HA‐tag on Ub. Time was recorded post‐addition of H887A‐HOIPRBR ‐ LDD . Note: The HA antibody cross‐reacts with free HOIP (see *).
Current models of RBR‐type mechanisms assume a two‐step process for Ub transfer: First, Ub is transferred from the E2~Ub to the active site Cys on RING2 to form an E3~Ub, and second, Ub is transferred from the E3~Ub to a substrate amino group. In the case of HOIPRBR‐LDD, it has been shown that the I44 surface is not required for binding of the acceptor Ub 9. We therefore postulated that the I44 surface might be required for the first step of the RBR Ub transfer mechanism. To test this hypothesis, we compared discharge rates for E2~Ub charged with either WT‐Ub or mutant Ub prior to incubation with ParkinRBR. In this assay, the disappearance of E2~Ub can be observed directly by SDS–PAGE (in the absence of reducing agent). UbcH7~UbWT disappears rapidly when incubated with Parkin, whereas the UbcH7~UbI44A is stable in the presence of Parkin indicating that Ub transfer from the E2~Ub to Parkin RING2 is impaired when the Ub to be transferred is I44A‐Ub (Fig 5C). Because E3~Ub intermediates for RBR E3s are often short‐lived and therefore difficult to detect, it is not possible to say whether the observed disappearance of the E2~Ub is via transfer to the Parkin RING2 active site Cys or merely hydrolysis. To address this question, we took advantage of RING2 mutations in HOIP and HHARI (H887A and H359A, respectively) that stabilize the E3~Ub 29, 41. In assays with HOIPH887A or HHARIH359A as the E3, both the UbcH7~Ub and the E3~Ub conjugates are detected. Single mutations in the Ub hydrophobic surface show both reduced disappearance of the E2~Ub (similar to Parkin) and, importantly, decreased generation of E3~Ub (Figs 5D and E, and EV4B). These experiments provide strong evidence that the hydrophobic patch of Ub plays a key role in the first step of the RBR mechanism, namely for Ub transfer from the RING1‐bound E2 to the Cys on RING2. Whether the Ub hydrophobic patch is also required for the second step of Ub transfer by RBR E3s remains to be addressed in future studies.
HHARI RING2 binds to the hydrophobic patch of Ub
A possible explanation for the observation that the hydrophobic patch of Ub is required for transfer of Ub from the E2 onto the E3 active site is that the Ub surface binds to the RING2 domain to recruit its active site to the E2~Ub. Consistent with this notion, some RBR RING2 domains retain weak but detectable Ub transfer activity independent of RING1, suggesting that when protein concentrations are high enough in vitro, RING2 can recruit an E2~Ub on its own 9, 41, 42, 43, 44. We reasoned that an interaction between free Ub and RING2 is likely to be of very low affinity. Because Ub is extremely soluble, we performed NMR binding experiments using 100 μM 15N‐HHARI RING2 (residues 325–396) and high concentrations of Ub (1 mM; Fig 6A). A subset of resonances disappear or shift in the presence of WT‐Ub but not of V70A‐Ub, the Ub mutant that most affects HHARI activity (Figs 5A and 6A top versus middle panels). The result has two implications: (i) The effects observed with WT‐Ub are not merely due to the very high Ub concentrations used and (ii) there is a direct, albeit low affinity interaction between RING2 and the hydrophobic surface of Ub. An NMR solution structure of HHARI RING2 has been solved, and therefore, the NMR spectrum is assigned 45, allowing perturbed residues to be identified and mapped onto the RING2 domain structure. The resulting surface forms a contiguous patch in proximity to the active site Cys357 and extends into the linker between IBR and RING2 (Fig 6B). We were surprised to see involvement of the linker which is not observed in the crystal structure 29, so we repeated the binding experiment using a RING2 construct that lacks most of the RING2‐IBR linker (HHARI RING2‐ΔL, residues 336–395). Remarkably, binding of Ub to 15N‐HHARI RING2‐ΔL is substantially reduced, consistent with the linker serving as part of the binding interface (Fig 6A, bottom panel).
Figure 6. HHARI RING2 binds to Ub.
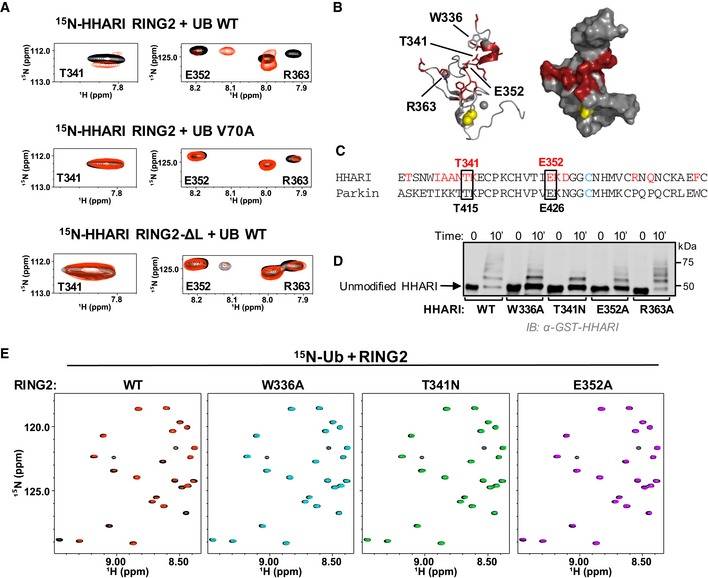
- Top panel: Regions of 1H‐15N‐HSQC‐TROSY spectra of 15N‐HHARI RING2 in the absence (black) and presence (red) of excess Ub. Middle panel: Identical spectral region, but in the presence of Ub‐V70A (red). Bottom panel: Identical spectral region, but spectra are from a truncated HHARI RING2 construct that lacks linker residues 325–335 (HHARI RING2‐ΔL).
- Residues with CSPs > 1 stdv (HHARI residues 333, 337–342, 352–354, 363, 365, 371) are colored red in a cartoon (left) and surface (right) representation of HHARI RING2 (PDB 2m9y). The active site C357 is shown in yellow.
- CLUSTAL OMEGA sequence alignment of HHARI and Parkin RING2 domains. HHARI residues perturbed by Ub binding (A) are in red. HHARI residues that are conserved in Parkin and that decrease auto‐ubiquitination activity when mutated in HHARIRBR are indicated with black boxes.
- Mutations in the Ub‐binding surface of RING2 decrease activity in E3 auto‐ubiquitination assays. Time points (10 min) are visualized by Western blotting for the GST‐tag on HHARI. Relative activity of HHARI WT and mutant forms is clearest when the intensity of the unmodified HHARI band is compared.
- 1H‐15N‐HSQC‐TROSY spectra of 15N‐Ub demonstrate binding by WT‐HHARI RING2, but not by mutant HHARI RING2 constructs that exhibit decreased auto‐ubiquitination activity. Overlay of 15N‐Ub spectra in the absence (black) and presence of WT‐HHARI RING2 (red), W336A‐HHARI RING2 (blue), T341N‐HHARI RING2 (green), or E352A‐HHARI RING2 (purple). HHARI mutations that exhibited reduced binding to Ub also show decreased ubiquitination activity in (D).
Mutations in HHARI residues most highly perturbed by Ub binding were tested for functional consequences in auto‐ubiquitination assays. In the context of HHARIRBR, Trp336, Glu352, and Arg363 were each mutated to Ala, and Thr341 was changed to an Asn to mimic the pathogenic mutation T415N in the analogous conserved residue in Parkin (Fig 6C). Strikingly, T341N decreases HHARI's ligase activity substantially and both W336A and E352A show a moderate reduction of ubiquitination activity while R363A has no observable effect (Fig 6D). NMR binding experiments confirm that the mutations that decrease ligase activity also decrease Ub binding to HHARI RING2 (Fig 6E). Together, the results corroborate that RING2 and linker residues are important for both Ub binding and HHARI activity, leading us to propose that the hydrophobic surface on the donor Ub binds and recruits RING2 and that this step is crucial for overall RBR activity.
Discussion
Until a few years ago, RBR E3 ligases were considered to be RING‐type E3 ligases because they were thought to contain two RING domains based on primary sequence analysis. Today, we know that only RING1 domains adopt a fold similar to canonical RING domains whereas RING2 domains fold into a different domain architecture that is shared by IBR domains 29, 30, 31, 32, 41, 43, 46. In common with canonical RING domains, RING1 binds an E2~Ub, but RING2 contains an active site Cys, similar to HECT‐type E3 ligases, leading to the proposal that RBR E3 ligases act via a RING/HECT hybrid mechanism 6 (Fig 7A). We sought here to define the mechanistic details of the RBR E3 HHARI and in so doing have discovered unique strategies that are shared among RBR E3s. Previously reported results had hinted that RBR E3s differ in essential functional ways from their eponymous RING‐type E3 cousins, despite the structural similarity of RING1 domains and canonical RING domains. For example, HHARI retains the ability to transfer Ub using the UbcH5L104Q mutant that abrogates Ub transfer activity with canonical RING‐type E3 ligases 1. Also, HHARI RING1 does not increase the Lys reactivity of UbcH5~Ub 6. Together, these results suggested that HHARI RING1 does not function like a canonical RING domain. Here, we demonstrate that RING1 domains not only fail to induce closed E2~Ub conformations—a mechanistic hallmark of canonical RING‐type E3 ligases—but instead RING1s actively favor open E2~Ub conformations. This strategy ensures that the transfer of Ub proceeds via the active site Cys on RING2 and therefore that the type of product generated (e.g. mono‐ or a specific poly‐Ub chain) is determined by the RBR E3 and not by the E2 (Fig 7B).
Figure 7. Model of RBR E3 Ub transfer mechanisms.
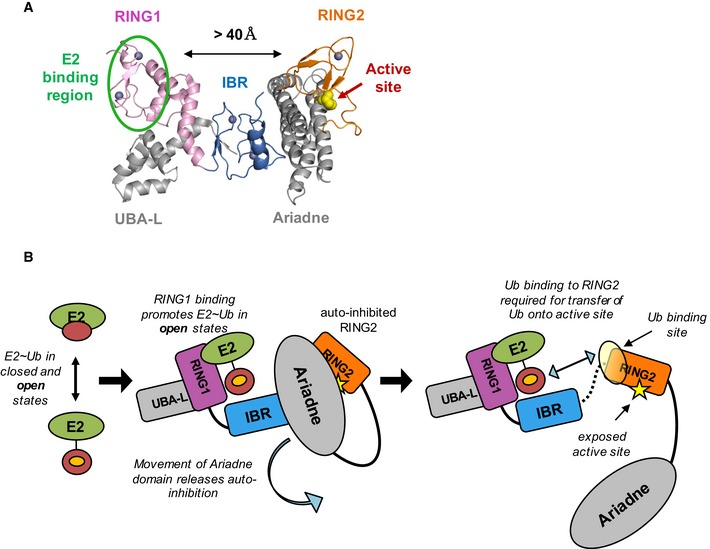
- Auto‐inhibited HHARI structure is shown in cartoon representation (PDB 4KC9). The E2‐binding site (green oval) on RING1 (pink) is separated from the active site Cys (yellow spheres) on RING2 (orange) by more than 40 Å. The IBR domain (dark blue) connects RING1 and RING2 and the Ariadne domain (gray) occludes the active site Cys.
- A model depicts the events required for Ub transfer by HHARI (with no intended order). E2˜Ub binding to RING1 favors open E2˜Ub conformations (middle panel) that expose the Ub hydrophobic patch (orange circle). Movement of the Ariadne domain (middle panel) exposes the active site Cys (right panel, yellow star) and may allow a disorder‐to‐helix transition of the IBR‐RING2 linker (dotted black line in right panel, not seen in structure in A) which completes the Ub‐binding site on RING2 (yellow sphere, right panel). The hydrophobic patch of Ub (orange circle) recruits RING2 to ensure transfer of Ub to the active cite Cys.
We propose that the disfavoring of closed E2~Ub states by RING1 is a common mechanistic feature of RBR E3s for several reasons. First, we demonstrate by NMR that the RING1 domain from another RBR, RNF144, also promotes open UbcH7~Ub states (Fig 3C). Second, the RBR E3s HHARI, Parkin, TRIAD1, and HOIP all exhibit robust Ub transfer activity with UbcH5L104Q in vitro (Figs 1A and EV1). Third, not promoting closed E2~Ub states is functionally important as it prevents off‐target ubiquitination events catalyzed by the E2~Ub (Fig 4). In sum, RING1 domains act in an opposite manner to canonical RINGs in that they actively inhibit E2~Ub closed states and consequently, suppress E2~Ub aminolysis reactivity and therefore E2 specificity. Such a strategy can be rationalized in the context of the overall RBR mechanism in which an E2~Ub must transfer Ub specifically onto a Cys (the active site of RING2) and not to a Lys residue. RBR E3 ligases are rendered catalytically inactive when their active site Cys is mutated to an Ala proving that the Ub transfer occurs through an obligate covalent E3~Ub conjugate 6, 8, 9, 10, 11, 22, 23. Therefore, Ub transfer from the E2~Ub to Lys residues on either the E3 itself or another protein in its vicinity would be off‐target and likely detrimental to the cellular processes that RBR E3s regulate. This unique feature of RBR RING1 domains allows RBR E3s to accept Ub from a variety of E2~Ubs including E2s that are able to transfer Ub onto either Lys or Cys, while ensuring that Ub transfer occurs through the active site Cys on RING2. Such discrimination is crucial, as in all cases known to date, it is the enzyme that carries out the final aminolysis reaction that determines the type of Ub modification on a given substrate. In the case of RING‐type E3s, it is the E2 that determines whether poly‐Ub chains are built as well as the chain topology (e.g. K48‐ versus K63‐linked Ub chains). But in the case of E3 ligases that utilize an E3~Ub intermediate, the E3 controls the final Ub product formation. For example, the E2 E2‐25K possesses intrinsic ability to build K48‐linked Ub chains in the absence of an E3, but this preference is suppressed when paired with the linear chain building complex that contains HOIL‐1L/HOIP known to build linear Ub chains 15. In such reactions, the E2~Ub acts as a supplier of Ub for the E3, but must not modify substrates directly. Altogether, these properties enable use of the same E2s by both RING‐type and RBR E3s to follow the adage “The last guy holding the activated Ub/Ubl gets to determine the product” 47.
Consistent with this general principle, the only available structure of a HECT/E2~Ub complex shows the E2~Ub in an extended conformation with non‐covalent interactions between Ub and the HECT domain C‐lobe, suggesting that HECT‐type E3s may also promote open E2~Ub states 7. However, the open E2~Ub state appears to be stabilized by interactions between Ub and the HECT active‐site‐containing C‐lobe, while our NMR results show that the E2‐binding domain (RING1) of RBR E3s actively disfavors E2~Ub closed states on its own. Likely, this feature is particularly important for RBR E3s like HHARI that can bind E2~Ub in their auto‐inhibited states where RING1 and RING2 are very far removed as it serves to inhibit premature Ub transfer from the bound E2~Ub before an activation event allows recruitment of RING2 to occur.
Given the structural similarities between canonical RINGs and RBR RING1s, their opposite effects upon binding E2~Ub is surprising. As pointed out above, RING1 domains lack the basic linchpin residue responsible for stabilizing closed E2~Ub states, but this does not adequately explain why RING1 binding actively promotes open E2~Ubs. One possibility is that RING1 binds Ub in the context of E2~Ub as has been suggested for Parkin 33, but our NMR data for HHARI and RNF144 RING1 binding to E2~Ub presented here do not indicate Ub binding by RING1. Instead, NMR binding experiments with either UbcH5 or UbcH7 identified an E2 surface that includes residues of the crossover helix that are not perturbed by canonical RING binding, suggesting that RING1 binds to a shifted or expanded surface on the E2. Notably, this “new” E2‐binding surface overlaps with that contacted by Ub in closed E2~Ub states, suggesting that RING1 binding may effectively compete with Ub binding, thereby disfavoring closed conformations. The details of HHARI RING1:E2~Ub must await further structural characterization.
Our finding that mutation of Ub hydrophobic patch residues affects the first Ub transfer step for HHARI, Parkin, and HOIP led to our subsequent discovery of a Ub‐binding surface on RING2. Although there are differences in severity among different Ub hydrophobic patch mutations for different RBR E3 ligases, all three E3s tested exhibit reduced activity with V70A‐Ub and Q49E‐Ub. Consistent with the modest degree of sequence conservation in RBR RING2s, we speculate that the Ub‐binding surface on RING2 is composed of some different residues in each RBR E3 (Figs 5A and EV4B). That the composition of the Ub‐binding regions on RING2s varies is not particularly surprising. Various Ub‐binding domains that use the same Ub hydrophobic patch differ in primary and tertiary structures, indicating that there are many possible binding modes between the Ub hydrophobic patch and a binding motif. We therefore propose that RING2 binding to the hydrophobic patch of Ub serves to recruit RING2 to the RING1‐bound E2~Ub and that this feature is shared among RBR E3 ligases.
While this study was under review, a crystal structure of HOIPRBR‐LDD bound to UbcH5~Ub was published that is consistent with the main tenets of the model presented here 48. The structure reveals a myriad of interactions among the E3, E2, and Ub that involve multiple copies of HOIP, UbcH5, and Ub in the asymmetric unit. Nevertheless, a number of features and interactions observed in the crystal are analogous to what we report here from solution measurements. First, the E2~Ub bound to RING1 of the HOIPRBR‐LDD is in an open conformation. In the crystal, this Ub moiety forms an extensive interface with the E3 including residues from the RING1‐IBR linker and the IBR. Our work demonstrates that HHARI and RNF144 RING1 constructs (which include the RING1‐IBR linker) are sufficient to induce open E2~Ub conformations, so the importance of additional contacts with the IBR during the first step of ubiquitin transfer remains to be addressed. Second, the hydrophobic patch of this Ub makes contacts to the IBR‐RING2 linker and RING2 of another HOIP molecule in the crystal and this interaction is centered around Ub residues I44 and V70, in agreement with our assays showing that I44A‐Ub and V70A‐Ub significantly impair Ub transfer with HOIPRBR‐LDD (Fig EV4B). As the HOIP/E2~Ub structure is of UbcH5~Ub which is already in open states when unbound to an E3, our study provides an important additional insight that cannot be inferred from the crystal structure: RING1 binding actively opposes closed states, rather than just failing to promote them.
Structural and biochemical investigations have provided rationales for the effects of many patient mutations in Parkin, but the T415N mutation has remained enigmatic 49. Substitution of Thr415 with Asn decreases ligase activity substantially though it alters neither the structure nor solubility of Parkin 43, 50, 51. Thr415 is proposed to be involved in a hydrogen‐bonding network around the active site that is required for catalysis 43. In our study, we found that the analogous mutation in HHARI, T341N, also decreases ligase activity and, importantly, decreases Ub binding to HHARI RING2 (Figs 6D and E, and EV5B). Thus, our discovery of a Ub‐binding site composed of residues from RING2 and its proximal linker provides another possible mechanistic explanation for the loss of function associated with this Parkin mutation. Our results support an earlier report that peptides spanning the Parkin IBR‐RING2 linker bind Ub in a peptide array assay 40. Thr415 is conserved in orthologs of Parkin as well as orthologs in HHARI. Altogether, the observations lead us to propose that the pathogenic Parkin T415N mutation disrupts Ub binding and consequently disables the recruitment of RING2 to the bound E2~Ub conjugate to enable the transfer of Ub onto the Parkin active site. It remains possible that the detrimental effect of T415N on Parkin activity is due to a combination of the loss of Ub binding to RING2 and the loss of a critical hydrogen‐bonding network around the active site 43.
Figure EV5. Comparison of RBR RING2 domains.
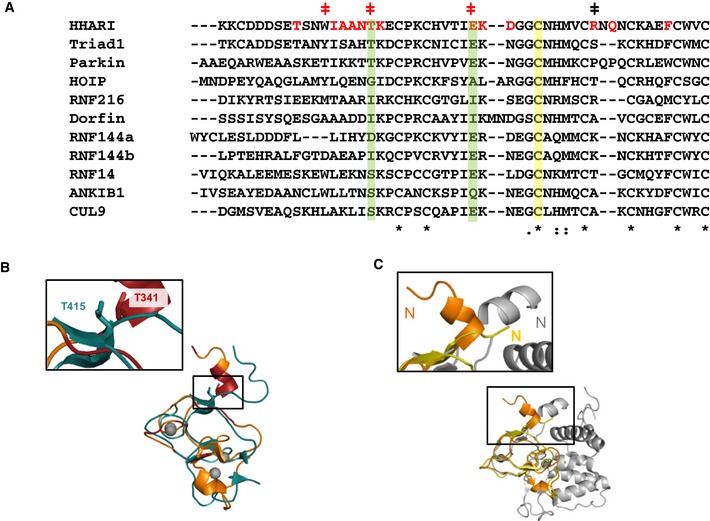
- RING2 sequence alignments were generated using CLUSTAL OMEGA. Residues observed to be perturbed upon Ub binding to HHARI RING2 in NMR titration experiments are highlighted in red. HHARI residues that were mutated and tested for change in activity are marked with a (ǂ) with red indicating decreased and black indicating no observable change in activity (see Fig 6C). Green bars highlight residues that are important for Ub binding of HHARI RING2 that are well conserved among RBR RING2s. The active site Cys is marked with a yellow bar. 7th and 8th Zn2+ coordinating residues are not shown.
- Structural overlay of NMR solution structures of Parkin (teal, PDB 2lwr) and HHARI (orange, PDB 2m9y) RING2 domains. RING2 residues perturbed upon Ub binding (Fig 6A) are colored dark red on HHARI RING2. The HHARI RING2 mutation T341N abrogates Ub binding and decreases ligase activity (Fig 6D and E). Inset: The analogous mutation in Parkin, T415N, has been linked to early‐onset Parkinson's disease 49.
- Structural overlay of three RING2 domains: a HHARI NMR solution structure (orange, PDB 2m9y), a HHARI crystal structure (yellow, PDB 4kc9), and a HOIP crystal structure (gray, PDB 4ljq). An enlargement of the N‐terminal region that is part of the IBR‐RING2 linker shows a helical disposition of the linker in the HOIP and HHARI NMR, but not in the HHARI crystal structure.
A common feature of RBR E3 ligases is that they exist in auto‐inhibited states characterized by low activity 8, 10, 29, 30, 31, 32, 33, 34, 40, 52. Structures of HHARI and Parkin reveal two common features that define the auto‐inhibited states 29, 30, 31, 32, 33, 34. First, the active site Cys on RING2 is at least partially buried by another domain. Second, the E2‐binding RING1 domain and the RING2 domain are separated by large distances (Fig 7A). Both features imply that RBR E3 ligases must undergo major rearrangements to become active enzymes. Details of release of auto‐inhibition are specific for each RBR E3 and are not yet fully defined 8, 10, 29, 30, 31, 32, 33, 34, 40, 52. However, they share one common feature: The E2‐binding RING1 domain and the active‐site‐containing RING2 domain must come together for transfer of Ub from the E2~Ub onto the E3 active site. In auto‐inhibited conformations of HHARI and Parkin, the domains of the RBR are connected by long flexible linkers that presumably allow the domains to adopt positions that are quite remote from one another. Other than a short helix referred to as the REP (repressor) element in Parkin, linkers between IBR and RING2 domains are either not observed or are unstructured in existing crystal structures of HHARI and Parkin 29, 30, 31, 32, 33, 34, 52. However, the linker proximal to HHARI RING2 is observed in solution by NMR and in crystal structures of HOIPRING2‐LDD and HOIPRBR‐LDD bound to E2~Ub where in all cases it forms a short helix (Fig EV5C and 41, 45, 48). Importantly, several of the HHARI residues that we observed to be perturbed upon Ub binding are in the part of the IBR‐RING2 linker that is seen to form a helix. We propose that the linker is extended and/or disordered in auto‐inhibited states, resulting in an incomplete RING2 Ub‐binding surface that will have little or no ability to bind Ub. Upon release of the inhibitory domain(s), the linker could undergo a coil‐to‐helix transition to complete the Ub‐binding site on RING2, enabling it to be recruited to the conjugated Ub moiety bound at RING1. We propose that open E2~Ub conformations induced by RING1 binding expose the hydrophobic patch on the conjugated Ub to allow Ub within the RING1‐bound E2~Ub to contact RING2 and ensure proper transfer of Ub onto the RING2 active. Although the structural details by which this mechanism is carried out may vary among RBRs, we believe that the main features defined here will be shared among them.
In closing, we note that our original proposal that RBR E3s are RING/HECT hybrids remains true in a structural sense. But findings reported here show clearly that the RBRs have evolved their own distinctive mechanistic strategies to achieve their function. Most remarkably, the centerpiece of the canonical RING allosteric mechanism for Ub transfer has been turned on its head by the RBRs.
Materials and Methods
Cloning and constructs
The following constructs were used in this study. If not stated otherwise, the following constructs are human and full‐length: HHARI‐RBR (aa 177–395), HHARI‐RING2 (aa 325–396), HHARI‐RING2‐∆L (aa 336–395), HHARI‐RING1 (aa 177–270), GST‐Parkin‐RBR (aa 217–465, rat), TRIAD1‐∆ARI (aa 1–348), RNF144 RING1 (aa 2–108), HOIP‐RBR‐LDD (aa 697–1,072), HOIP‐RING2 (aa 853–1,072), E4BU (aa 1,142–1,173, mouse), BRCA1/BARD1 (aa 1–100/26–140), UbcH7WT or UbcH7C86S, UbcH5cWT or UbcH5cC85S, His6‐Ubc13C87S, E4BU‐HHARI hybrid (mouseE4BU aa 1,142–1,173, HHARI aa 271–396) with either WT‐HHARI RING2 active site C357 or C357A. HHARI RING1 and E4BU‐HHARI hybrid were cloned into pGEX‐4T in‐frame with thrombin‐cleavable, N‐terminal GST‐tag. RNF144 RING1 was cloned into a His6‐SUMO vector (N‐terminal tag). TRIAD1‐∆ARI was cloned into pet28a in‐frame with His6‐T7 at the N‐terminus.
Expression and purification of recombinant proteins
Proteins were expressed either in LB or minimal M9 medium supplemented with [15N]‐ammonium chloride or 13C‐glucose in Escherichia coli (BL21 DE3 cells) and induced with 200 μM IPTG at 16°C for 18–22 h. Media for E3s (except E4BU) was supplemented with 0.2 mM Zn2+. UbcH5c, UbcH7, His6‐Ubc13, BRCA1/BARD1, E4BU, GST‐HHARI‐RBR, GST‐Parkin‐RBR, HOIP‐RBR‐LDD, HOIP‐RING2 were purified as previously described 6, 8, 35, 36, 41, 53. GST‐E4BU‐HHARI hybrid was purified as GST‐HHARI‐RBR.HHARI RING1 and RING2 constructs were purified using GST columns (GE Healthcare and Life Sciences) in 50 mM Tris, 200 mM NaCl, pH 8.0, and eluted with 10 mM glutathione. GST‐tags cleaved with thrombin (Sigma‐Aldrich) and removed by size‐exclusion chromatography (25 mM NaPO4, 150 mM NaCl, pH 7.0). His6‐T7‐TRIAD1‐∆ARI and His6‐SUMO‐RNF144‐RING1 were purified using Ni2+ affinity chromatography. His6‐SUMO‐tag was removed from RNF144‐RING1 using SUMO protease in 50 mM Tris, 200 mM NaCl, 1 mM DTT, pH 8.0. Finally, size‐exclusion chromatography in 25 mM NaPO4, 150 mM NaCl, pH 7.0, was performed on His6‐T7‐TRIAD1‐∆ARI and cleaved RNF144‐RING1.
E2~Ub discharge assays
2 μM wheat E1, 20 μM UbcH7 or UbcH5, 10 μM of Ub or HA‐Ub (WT or mutant), 5 mM ATP were mixed at 37°C in 25 mM NaPO4, 150 mM NaCl, pH 7.0 for 20 min. Charging reactions were quenched with the addition of 0.1 units of apyrase (Sigma‐Aldrich)/10 μl reaction for 5 min. A zero‐minute time point was taken using non‐reducing SDS–PAGE load dye prior to incubation with either free nucleophile or E3. Cys reactivity assays: Charging reactions were diluted twofold and incubated at 37°C with a final concentration of either 5 mM Cys in the absence or presence of 75 μM E3 (HHARI RING1). Reactions were quenched at given time points by addition of non‐reducing SDS–PAGE load dye. Products were visualized with Coomassie blue stain. Parkin discharge assays: Charging reactions were diluted twofold and incubated at 37°C with a final concentration of 4 μM GST‐Parkin‐RBR. Reactions were quenched at given time points by addition of non‐reducing SDS–PAGE load dye. UbcH7~Ub discharge and E3‐Ub product formation were visualized by Western blotting for HA‐Ub (HA antibody from Life Tein, LT0422). HOIP˜Ub/HHARI˜Ub capture assay: 50 μl of charging reactions was quenched with addition of 5 μl apyrase for 5 min at room temperature (RT). Time point zero was taken immediately prior to addition of H887A‐HOIPRBR‐LDD or H359A‐HHARIRBR to a final concentration of 5 μM E3. Reactions were performed at RT and quenched at given time points by addition of non‐reducing SDS–PAGE load dye. Product formation was visualized by Western blotting for HA‐Ub and GST (HHARI).
Ubiquitination assays
For E3 auto‐ubiquitination assays, 0.5 μM wheat E1, 2 μM E2, 2 μM E3 (GST‐HHARIRBR, T7‐TRIAD1ΔARI, GST‐Parkin‐RBR, Flag‐BRCA1/BARD1) and 20 μM Ub were incubated at 37°C in 25 mM NaPO4, 150 mM NaCl, pH 7.0. Reactions were initiated with 10 mM ATP and quenched with SDS–PAGE reducing buffer. Samples were run on SDS–PAGE gel and visualized on Western blots (blotting for tags on E3). Antibodies used were as follows: GST antibody—Life Tein, LT0423 (Figs 1, 4 and 6); Flag antibody—Sigma‐Aldrich, F3165 (Fig 1); and T7 antibody—EMD Millipore, 69522 (Fig 1). Free linear chain building assays using HOIPRBR‐LDD were done as described previously 41.
Generation of stable E2‐O‐Ub
10 μM human E1, 250 μM E2 (UbcH7C86S, UbcH5cC85S or His6‐Ubc13C87S), 750 μM Ub, 12.5 mM ATP were incubated at 37°C for 8 h in 25 mM NaPO4, 150 mM NaCl, pH 7.0. Charged species were separated from uncharged species using size‐exclusion chromatography.
NMR experiments
The same buffer (25 mM NaPO4, 150 mM NaCl, pH 7.0, 10% D2O) and temperature (298 K) were used for all experiments. All (1H,15N)‐HSQC‐TROSY experiments were acquired at field strengths of 500 MHz except for data collected for Fig 6A where 600 MHz was used instead. The following concentrations were used for each figure. Fig 1: 220 μM 15N‐UbcH5‐O‐15N‐Ub; Fig 2: 250 μM of 15N‐UbcH7C86S and 15N‐UbcH7‐O‐UbI44A, 200 μM of 15N‐UbcH7‐O‐Ub; Fig EV2: 250 μM free 15N‐UbcH7C86S and 250 μM 15N‐UbcH7C86S + 125 μM HHARI RING1; Fig 3A, left panel: 50 μM 15N‐Ub, 200 μM free UbcH7‐O‐15N‐Ub, 160 μM UbcH7‐O‐15N‐Ub + 200 μM HHARI RING1; Fig 3C: 200 μM free UbcH7‐O‐15N‐Ub, 160 μM UbcH7‐O‐15N‐Ub + 200 μM HHARI RING1, 70 μM UbcH7‐O‐15N‐Ub + 200 μM RNF144 RING1, 220 μM UbcH7‐O‐15N‐Ub + 220 μM BRCA1/BARD1; Appendix Fig S4: 160 μM UbcH7‐O‐15N‐Ub + 510 μM E4BU; Fig EV3: 50 μM 15N‐Ub, 100 μM UbcH13‐O‐15N‐Ub, 100 μM Ubc13‐O‐15N‐Ub + 300 μM HHARI RING1; Fig 6A: 100 μM free 15N‐HHARI RING2(‐ΔL), 100 μM 15N‐HHARIRING2(‐ΔL) + 1 mM Ub (either WT or V70A); Fig 6E: 50 μM free 15N‐Ub, 50 μM 15N‐Ub + 500 μM HHARI RING2 (WT or mutants).
(1H,13C)‐HSQC‐TROSY experiments for Fig 3B and Appendix Fig S3 were acquired at 500 MHz (200 μM free 13C‐UbcH7C86S, 200 μM 13C‐UbcH7‐O‐Ub, 125 μM 13C‐UbcH7‐O‐Ub + 150 μM HHARI RING1) or 600 MHz (300 μM 13C‐UbcH7C86S + 360 μM HHARI RING1). NMRPipe/NMRDraw 54 was used to process NMR data. NMRViewJ (One Moon Scientific) was used for data visualization 55. The equation Δδ = [(Δδ15N/5)2 + (Δδ 1H)2]1/2 was used to calculate chemical shift perturbations of 2D TROSY‐HSQC NMR experiments.
Author contributions
KKD, KR, and REK conceived the experiments and wrote the manuscript. KKD performed the NMR and biochemical experiments with crucial support by EDD BS performed linear chain building assays with HOIP.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank M. Stewart and P. DaRosa for insightful discussions and critical reading of the manuscript; P. Brzovic, S. Delbecq, and L. Martino for useful discussion; and M. Cook and K. Reiter for proof reading the manuscript. This work was supported by National Institute of General Medical Sciences grant R01 GM088055 and 5T32 GM007270 (KKD), the Francis Crick Institute (grant number FCI01) which receives its core funding from Cancer Research UK, the UK Medical Research Council, and the Wellcome Trust (KR), and University of Washington Hurd Fellowship Fund (KKD).
EMBO Reports (2016) 17: 1221–1235
References
- 1. Pruneda JN, Littlefield PJ, Soss SE, Nordquist KA, Chazin WJ, Brzovic PS, Klevit RE (2012) Structure of an E3:E2~Ub complex reveals an allosteric mechanism shared among RING/U‐box ligases. Mol Cell 47: 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT (2012) BIRC7‐E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat Struct Mol Biol 19: 876–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Plechanovova A, Jaffray EG, Tatham MH, Naismith JH, Hay RT (2012) Structure of a RING E3 ligase and ubiquitin‐loaded E2 primed for catalysis. Nature 489: 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Branigan E, Plechanovova A, Jaffray EG, Naismith JH, Hay RT (2015) Structural basis for the RING‐catalyzed synthesis of K63‐linked ubiquitin chains. Nat Struct Mol Biol 22: 597–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saha A, Lewis S, Kleiger G, Kuhlman B, Deshaies RJ (2011) Essential role for ubiquitin‐ubiquitin‐conjugating enzyme interaction in ubiquitin discharge from Cdc34 to substrate. Mol Cell 42: 75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wenzel DM, Lissounov A, Brzovic PS, Klevit RE (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474: 105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamadurai HB, Souphron J, Scott DC, Duda DM, Miller DJ, Stringer D, Piper RC, Schulman BA (2009) Insights into ubiquitin transfer cascades from a structure of a UbcH5B approximately ubiquitin‐HECT(NEDD4L) complex. Mol Cell 36: 1095–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stieglitz B, Morris‐Davies AC, Koliopoulos MG, Christodoulou E, Rittinger K (2012) LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep 13: 840–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smit JJ, Monteferrario D, Noordermeer SM, van Dijk WJ, van der Reijden BA, Sixma TK (2012) The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING‐IBR‐RING domain and the unique LDD extension. EMBO J 31: 3833–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kelsall IR, Duda DM, Olszewski JL, Hofmann K, Knebel A, Langevin F, Wood N, Wightman M, Schulman BA, Alpi AF (2013) TRIAD1 and HHARI bind to and are activated by distinct neddylated Cullin‐RING ligase complexes. EMBO J 32: 2848–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ho SR, Mahanic CS, Lee YJ, Lin WC (2014) RNF144A, an E3 ubiquitin ligase for DNA‐PKcs, promotes apoptosis during DNA damage. Proc Natl Acad Sci USA 111: E2646–E2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608 [DOI] [PubMed] [Google Scholar]
- 13. Matsumine H, Saito M, Shimoda‐Matsubayashi S, Tanaka H, Ishikawa A, Nakagawa‐Hattori Y, Yokochi M, Kobayashi T, Igarashi S, Takano H et al (1997) Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2‐27. Am J Hum Genet 60: 588–596 [PMC free article] [PubMed] [Google Scholar]
- 14. Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz‐Wachtel M, van Wijk SJ, Goswami P, Nagy V, Terzic J et al (2011) SHARPIN forms a linear ubiquitin ligase complex regulating NF‐kappaB activity and apoptosis. Nature 471: 637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, Sano S, Tokunaga F, Tanaka K, Iwai K (2006) A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J 25: 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WW et al (2011) Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471: 591–596 [DOI] [PubMed] [Google Scholar]
- 17. Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, Tanaka K, Nakano H, Iwai K (2011) SHARPIN is a component of the NF‐kappaB‐activating linear ubiquitin chain assembly complex. Nature 471: 633–636 [DOI] [PubMed] [Google Scholar]
- 18. Aguilera M, Oliveros M, Martinez‐Padron M, Barbas JA, Ferrus A (2000) Ariadne‐1: a vital Drosophila gene is required in development and defines a new conserved family of ring‐finger proteins. Genetics 155: 1231–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tan NG, Ardley HC, Scott GB, Rose SA, Markham AF, Robinson PA (2003) Human homologue of ariadne promotes the ubiquitylation of translation initiation factor 4E homologous protein, 4EHP. FEBS Lett 554: 501–504 [DOI] [PubMed] [Google Scholar]
- 20. Qiu X, Fay DS (2006) ARI‐1, an RBR family ubiquitin‐ligase, functions with UBC‐18 to regulate pharyngeal development in C. elegans . Dev Biol 291: 239–252 [DOI] [PubMed] [Google Scholar]
- 21. Elmehdawi F, Wheway G, Szymanska K, Adams M, High AS, Johnson CA, Robinson PA (2013) Human Homolog of Drosophila Ariadne (HHARI) is a marker of cellular proliferation associated with nuclear bodies. Exp Cell Res 319: 161–172 [DOI] [PubMed] [Google Scholar]
- 22. Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ (2013) PINK1 drives Parkin self‐association and HECT‐like E3 activity upstream of mitochondrial binding. J Cell Biol 200: 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng X, Hunter T (2013) Parkin mitochondrial translocation is achieved through a novel catalytic activity coupled mechanism. Cell Res 23: 886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mani K, Fay DS (2009) A mechanistic basis for the coordinated regulation of pharyngeal morphogenesis in Caenorhabditis elegans by LIN‐35/Rb and UBC‐18‐ARI‐1. PLoS Genet 5: e1000510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fiesel FC, Moussaud‐Lamodiere EL, Ando M, Springer W (2014) A specific subset of E2 ubiquitin‐conjugating enzymes regulate Parkin activation and mitophagy differently. J Cell Sci 127: 3488–3504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lim GG, Chew KC, Ng XH, Henry‐Basil A, Sim RW, Tan JM, Chai C, Lim KL (2013) Proteasome inhibition promotes Parkin‐Ubc13 interaction and lysine 63‐linked ubiquitination. PLoS ONE 8: e73235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM (2000) Parkin functions as an E2‐dependent ubiquitin‐protein ligase and promotes the degradation of the synaptic vesicle‐associated protein, CDCrel‐1. Proc Natl Acad Sci USA 97: 13354–13359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haddad DM, Vilain S, Vos M, Esposito G, Matta S, Kalscheuer VM, Craessaerts K, Leyssen M, Nascimento RM, Vianna‐Morgante AM et al (2013) Mutations in the intellectual disability gene Ube2a cause neuronal dysfunction and impair parkin‐dependent mitophagy. Mol Cell 50: 831–843 [DOI] [PubMed] [Google Scholar]
- 29. Duda DM, Olszewski JL, Schuermann JP, Kurinov I, Miller DJ, Nourse A, Alpi AF, Schulman BA (2013) Structure of HHARI, a RING‐IBR‐RING ubiquitin ligase: autoinhibition of an Ariadne‐family E3 and insights into ligation mechanism. Structure 21: 1030–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L, Shaler T, Walker D, Yang Y, Regnstrom K et al (2013) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun 4: 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al‐Abdul‐Wahid S, Krett J, Wong K, Kozlov G et al (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340: 1451–1455 [DOI] [PubMed] [Google Scholar]
- 32. Wauer T, Komander D (2013) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J 32: 2099–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kumar A, Aguirre JD, Condos TE, Martinez‐Torres RJ, Chaugule VK, Toth R, Sundaramoorthy R, Mercier P, Knebel A, Spratt DE et al (2015) Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J 34: 2506–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sauve V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, Sprules T, Wang J, Trempe JF, Gehring K (2015) A Ubl/ubiquitin switch in the activation of Parkin. EMBO J 34: 2492–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pruneda JN, Stoll KE, Bolton LJ, Brzovic PS, Klevit RE (2011) Ubiquitin in motion: structural studies of the ubiquitin‐conjugating enzyme approximately ubiquitin conjugate. Biochemistry 50: 1624–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D III, Fukuda M, Ohta T, Klevit R (2003) Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin‐ligase complex. Proc Natl Acad Sci USA 100: 5646–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DaRosa PA, Wang Z, Jiang X, Pruneda JN, Cong F, Klevit RE, Xu W (2015) Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP‐ribosyl)ation signal. Nature 517: 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu Z, Kohli E, Devlin KI, Bold M, Nix JC, Misra S (2008) Interactions between the quality control ubiquitin ligase CHIP and ubiquitin conjugating enzymes. BMC Struct Biol 8: 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Serniwka SA, Shaw GS (2008) 1H, 13C and 15N resonance assignments for the human E2 conjugating enzyme, UbcH7. Biomol NMR Assign 2: 21–23 [DOI] [PubMed] [Google Scholar]
- 40. Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, Walden H (2011) Autoregulation of Parkin activity through its ubiquitin‐like domain. EMBO J 30: 2853–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stieglitz B, Rana RR, Koliopoulos MG, Morris‐Davies AC, Schaeffer V, Christodoulou E, Howell S, Brown NR, Dikic I, Rittinger K (2013) Structural basis for ligase‐specific conjugation of linear ubiquitin chains by HOIP. Nature 503: 422–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Capili AD, Edghill EL, Wu K, Borden KL (2004) Structure of the C‐terminal RING finger from a RING‐IBR‐RING/TRIAD motif reveals a novel zinc‐binding domain distinct from a RING. J Mol Biol 340: 1117–1129 [DOI] [PubMed] [Google Scholar]
- 43. Spratt DE, Martinez‐Torres RJ, Noh YJ, Mercier P, Manczyk N, Barber KR, Aguirre JD, Burchell L, Purkiss A, Walden H et al (2013) A molecular explanation for the recessive nature of parkin‐linked Parkinson's disease. Nat Commun 4: 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rankin CA, Galeva NA, Bae K, Ahmad MN, Witte TM, Richter ML (2014) Isolated RING2 domain of parkin is sufficient for E2‐dependent E3 ligase activity. Biochemistry 53: 225–234 [DOI] [PubMed] [Google Scholar]
- 45. Spratt DE, Mercier P, Shaw GS (2013) Structure of the HHARI catalytic domain shows glimpses of a HECT E3 ligase. PLoS ONE 8: e74047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beasley SA, Hristova VA, Shaw GS (2007) Structure of the Parkin in‐between‐ring domain provides insights for E3‐ligase dysfunction in autosomal recessive Parkinson's disease. Proc Natl Acad Sci USA 104: 3095–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS (2016) E2 enzymes: more than just middle men. Cell Res 26: 423–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lechtenberg BC, Rajput A, Sanishvili R, Dobaczewska MK, Ware CF, Mace PD, Riedl SJ (2016) Structure of a HOIP/E2~ubiquitin complex reveals RBR E3 ligase mechanism and regulation. Nature 529: 546–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R et al (1999) A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet 8: 567–574 [DOI] [PubMed] [Google Scholar]
- 50. Matsuda N, Kitami T, Suzuki T, Mizuno Y, Hattori N, Tanaka K (2006) Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro . J Biol Chem 281: 3204–3209 [DOI] [PubMed] [Google Scholar]
- 51. Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, Dawson VL, Dawson TM (2005) Familial‐associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet 14: 2571–2586 [DOI] [PubMed] [Google Scholar]
- 52. Wauer T, Simicek M, Schubert A, Komander D (2015) Mechanism of phospho‐ubiquitin‐induced PARKIN activation. Nature 524: 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nordquist KA, Dimitrova YN, Brzovic PS, Ridenour WB, Munro KA, Soss SE, Caprioli RM, Klevit RE, Chazin WJ (2010) Structural and functional characterization of the monomeric U‐box domain from E4B. Biochemistry 49: 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293 [DOI] [PubMed] [Google Scholar]
- 55. Johnson BA, Blevins RA (1994) NMR view: a computer program for the visualization and analysis of NMR data. J Biomol NMR 4: 603–614 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File