

Abstract
DNA synthesis and chromatin assembly are coordinated and well regulated and comprise the two most critical processes of eukaryotic cell division, Although the interplay between DNA and its higher-order chromatin state is integral for many processes, including cell survival and genome stability, little is known about the reestablishment of chromatin structure during the cell cycle. Moreover, the extent to which the fidelity of the newly synthesized chromatin plays a role in the maintenance of cellular identity is still under debate. Here, we present a novel approach to purify nascent chromatin from the replication fork. In this protocol, we take advantage of click chemistry, a method that allows efficient conjugation of azide-containing biotin molecules to ethynyl-labeled nucleic acids. Using this approach, we selectively enrich biotin-nucleic acid conjugates via streptavidin affinity purification to pull down and assess chromatin states as well as chromatin-bound complexes from newly replicated DNA fragments with western blot and/or mass spectrometry.
Keywords: Epigenetic inheritance, cell cycle, nascent chromatin, EdU, Click Chemistry
INTRODUCTION
In eukaryotes, nuclear DNA is packed into a nucleoprotein complex called chromatin and all DNA-templated processes occur in the context of this structure. Histone variant content, post-translational modifications of histones and positioning of nucleosomes along with other chromatin-associated factors dictate chromatin organization which in turn control DNA accessibility and therefore influence all DNA-templated processes. Therefore, the maintenance of the established chromatin organization during transcription, replication and repair, is essential to preserve transcriptional programs and epigenetic memory.
Of these DNA-templated tasks, replication is among the most challenging for the faithful reorganization of chromatin since, during replication the entire genome disassembles, duplicates and reassembles into two daughter sets. Therefore, much attention has focused on this process. For example, it has been established that DNA synthesis and canonical histone synthesis are strictly coordinated; chromatin assembly progresses simultaneously with the replication fork (Marzluff et al., 2008; Mejlvang et al., 2014). Moreover, this coordination has been shown to be essential for genome integrity, since defects in chromatin assembly causes replication fork stalling and DNA damage (Ye et al., 2003). We also have some information on how parental and newly assembled nucleosomes are distributed between daughter strands and which specific protein complexes have roles in these processes (Groth, 2009; Budhavarapu et al., 2013). But beyond that, not much is known about chromatin maturation and how cell type-specific chromatin landscapes are re-established.
To shed light on these well-regulated processes and to have a better understanding of replication dynamics, chromatin maturation and epigenetic inheritance, new technologies are in demand to study nascent DNA and associated factors at or behind the replication fork. Here we describe a protocol utilizing EdU or EdC (Ethynyl deoxy Cytidine) metabolic labeling and click chemistry to isolate nascent chromatin.
BASIC PROTOCOL 1
ISOLATION of NASCENT CHROMATIN WITH CLICK CHEMISTRY
This protocol describes labeling and isolation of newly synthesized DNA-associated factors after release in the S phase. This protocol utilizes click chemistry on formaldehyde cross-linked whole cells with commercially available ingredients.
Cells are synchronized at the G1/S border. At different stages after release into the S-phase, medium is changed to EdU-containing medium for 1 or 2 hours to label newly synthesized DNA. EdU-labeled DNA is then biotinylated with click chemistry to allow the isolation of nascent chromatin-associated factors through biotin-streptavidin affinity purification.
Materials
Cultured hTERT immortalized RPE1 cells: 8 × 106 –10 × 106 cells for each experiment
5-Ethynyl deoxy Uridine (Cat.No. PY7562, Berry & Associates)
5-Ethynyl deoxy Cytidine (Cat.No. CLK-N003, Jena Bioscience)
L-Mimosine (Cat.No. M0253, Sigma)
Nuclease-free water (Cat.No. AM9932, Ambion)
100% Ethanol
Formaldehyde (Cat.No.28908, ThermoScientific)
Glycine (Cat.No. 50046, Sigma)
Triton X-100 (Cat.No. T93443, Sigma)
N-Lauroyl Sarcosine (Cat.No. 61743, Sigma)
Glycerol (Cat.No. G5516, Sigma)
Sodium-L-Ascorbate (Cat.No. A4034, Sigma)
Copper Sulfate (Cat.No. 451657 Sigma)
Biotin dPEG®7-Azide (Cat.No. 10825, Quanta Biodesign Ltd.)
THPTA (Cat.No. CLK-1010, Jena Bioscience)
Glycogen (Cat.No. AM9510, Ambion)
Dynabeads MyOne Streptavidin C1 magnetic beads (Cat.No. 65001, Life Technologies)
3M Sodium Acetate, pH 5.5 (Cat.No. AM9740, Ambion)
Mammalian Protease Inhibitor Cocktail (Cat.No. P8340, Sigma)
RNasin Plus Rnase inhibitor (Cat.No. PRN2615, Fisher Scientific)
50× Danhard’s blocking buffer (Cat.No. 750018, Life Technologies)
2× Tween Wash buffer (see recipe)
Click Reaction Mixture (see recipe)
Cytoplasm Wash Buffer (see recipe)
DTT Wash Buffer (see recipe)
Pull Down Sonication Buffer (see recipe)
RIPA Buffer (see recipe)
5× Reducing sample buffer (Cat.No. 39000, Thermo Scientific)
RNAse A (Cat.No., Life Technologies)
Nuclease-free BSA (Cat.No. B2518, Sigma-Aldrich)
Dyna Mag, magnetic stand (Cat.No. 12321D, Life Technologies)
QSonica, sonicator (Cat.No. Q800R1, QSonica)
Cell lifter (Cat.No. 3008, Corning)
Low binding nuclease free 1.7mL tubes (Cat.No. 022431021, Eppendorf)
Thermo mixer (Cat.No. 022670107, Eppendorf)
Rotator (Cat.No. 1217H25, Thomas Scientific)
Note: All solutions advised to be prepared with Nuclease free water.
Synchronization and Metabolic Labeling of Cultured Cells with EdU (or EdC)
-
1Grow hTERT immortalized RPE1 cells to 80–85% confluence. Trypsinize and plate 3 × 106 cells per 15cm dish in RPE1 medium containing a final concentration of 450 µM mimosine to arrest cells at G1/S border.This protocol can work with other cell types once the cell type-specific synchronization conditions are determined. We chose to use hTERT-RPE1 cells for two reasons. First, they are diploid immortalized cells; second, they can be very efficiently synchronized at the G1/S border of the cell cycle.)
-
2After 21hrs of mimosine synchronization, wash cells twice with 15mL PBS and release them into S phase in regular culture medium.It takes approximately 3 hours to have 20% of the population enter S phase. This timing is specific for the hTERT-RPE1 cells. We recommend testing different timing and conditions followed by FACS to determine the best conditions for desired synchronization (Figure A). Also, note that RPE1 cells have contact inhibition and their release from G1 to S phase is delayed when they are grown to confluence.
-
3At different time points after release into S phase, change culture medium to medium containing 10 µM EdU.Time points should be chosen to capture the desired phases of cell cycle for the purpose of the investigator. This will be determined in part by the rate at which S phase is traversed in the individual cell type. In hTERT-RPE1 cells we examine 3–4 hours as early S phase, 5–6 hours as mid S phase, 7–8 hours as late S phase after release.
-
4
After one or two hours (labeling time can vary according to which stage of replication is desired to be captured) of labeling newly synthesized DNA with EdU, discard EdU medium, wash cells with PBS and add 15 mL 0.05%PBS-T containing 1% formaldehyde. Incubate at room temperature for 10 minutes while shaking gently.
-
5
Quench formaldehyde crosslinking by adding 750ul of 2.5M Glycine and incubate another 2 minutes.
-
6
Wash crosslinked cells with 0.05% PBS-Tween20 with protease inhibitor complex (PIC), then collect cells by centrifugation in 15ml conical tubes with 0.05% PBS-T with PIC using cell lifter.
-
7
Remove supernatant from cells and flash-freeze samples in liquid nitrogen and store them at −80C.
FIGURE 1.
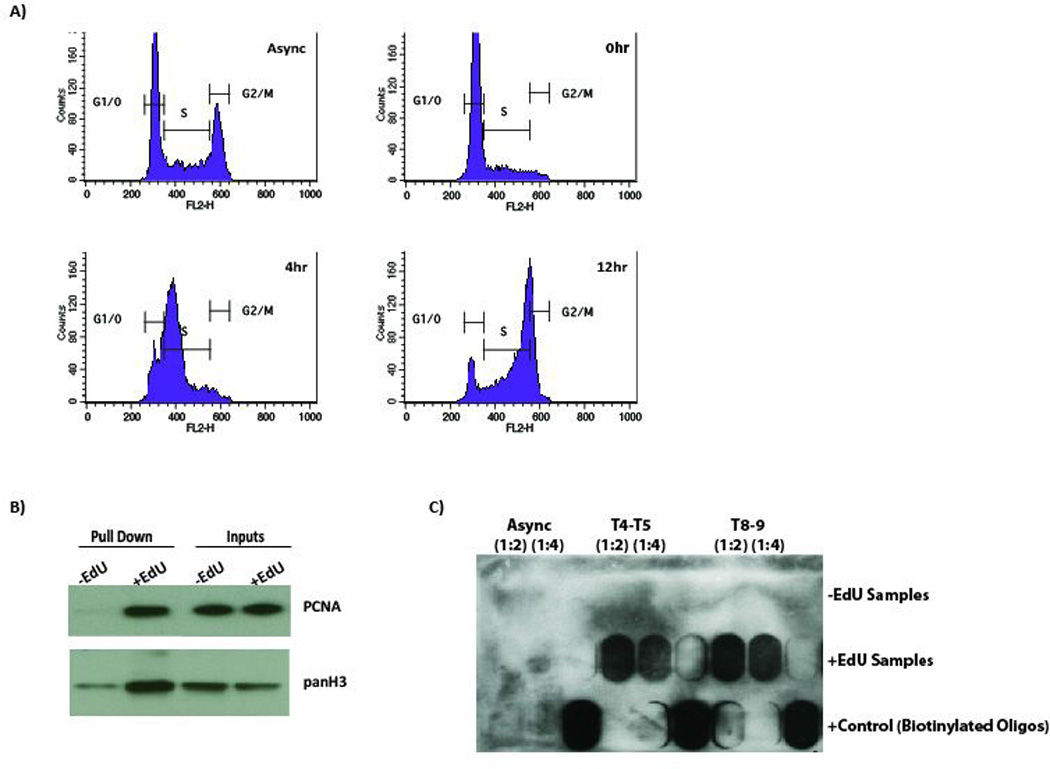
A) At a given time unsynchronized cells (Async) contain a population of all three states, about 50% at G1, 20% at S and 30% at G2/M phases. A successful synchronization requires over 80% of the cells at G1/S border at T0. Somewhat synchronous progress of cells in S phase after the release is also an important factor. Mimosinesynchronization of RPE1 cells fulfills these criteria.
B) PCNA western blot is a good positive control for nascent chromatin isolation at the replication fork. Pan H3 western blot is a good control for the specificity of the pull down step.
C) DNA dot/slot blotting followed by streptavidin-hrp probing is a fast and simple way to check biotin-click efficiency.
Whole Cell Click Reaction
-
8
Permeabilize cells on ice in 1 mL 0.3% PBS-Triton X-100 containing PIC for 15 minutes. Spin cells down at 1000g for 5 minutes, 4°C
-
9Resuspend permeabilized cell pellets in 500ul freshly made Click reaction mixture (See recipe) and incubate in dark (foil wrap thermo mixer to protect samples from light) for 45 min at room temperature while mixing at 800rpm in thermo mixerThis reaction can be applied directly to nuclei instead of the permeabilized whole cells.
-
10
Add 1mL of 2mg/ml BSA in 0.05%TritonX-100-PBS, incubate in dark at room temperature for 15 minutes.
-
11
Spin cells down at 1000g for 5 minutes, 4°C
-
12
Resuspend cells in 500ul Cytoplasm Wash Buffer with PIC to wash off cytoplasmic proteins. Incubate at 4°C rotating for 20 minutes.
-
13
Spin cells down at 500g for 5 minutes, 4°C
-
14
Resuspend pellets in 500ul DTT Wash Buffer with PIC. Incubate samples at 4°C rotating for 20 minutes.
-
15
Spin cells down at 500g for 5 minutes, 4°C
-
16
Resuspend pellets in 300ul Pull Down Sonication Buffer with PIC and RNAsin, incubate on ice for 10 minutes.
-
17Transfer samples into QSonica 1.5 mL sonication tubes. Sonicate samples to an average fragment size between 300 bps to 800 bps.Sonication efficiency is affected by cell type, cell number/density and detergent amount in the sonication buffer. Therefore, optimal sonication conditions should be determined before the actual experiment is executed.
-
18
Calculate approximate concentration of each sample with Abs260 reading after 1:50 dilution in water.
-
19
Pipet 10ug of each sample into a new tube and flash freeze the rest in liquid nitrogen and store at −80°C until both shearing and biotinylation efficiencies are confirmed.
-
20
Bring the 10ug DNA-containing samples’ volume to 300ul with nuclease-free TE. RNase treat samples by adding 1ul RNAse A, incubate at 37°C for at least 30 minutes.
-
21
Add 6ul 10% SDS and 10ul Proteinase K (20mg/ml) incubate overnight at 65°C to reverse the cross-link.
-
22
Phenol/Chloroform extract and ethanol precipitate the DNA.
-
23
Check shearing efficiency via loading 2 µg on a 2% agarose gel. [Fragments size should run between 500bp and 800bp ideally]
-
24
Check click efficiency with biotin streptavidin dot/slot blotting (See support protocol)
Block Streptavidin Beads
-
25
Prepare 50ul of Dynabeads® MyOne™ Streptavidin C1 beads per 250–300ug clicked-chromatin sample.
-
26
Wash beads once with 700ul PBS for 5 min at room temperature. Place tubes in magnetic stand for 2 minutes at room temperature and remove PBS.
-
27
Wash beads once with 500ul 2× Danhardt’s Solution. Place tubes in magnetic stand for 2 minutes at room temperature and remove wash buffer.
-
28
Resuspend beads in 500ul 2× Danhardt’s Solution and block beads for 2hr at room temperature in thermomixer.
Pull-down of Nascent DNA With Streptavidin Beads
-
29
Thaw samples on ice and transfer 10ug of each sample as input to a new 1.5mL tube, bring volume to 40ul with RIPA buffer and place the tubes on ice until the experimental pull-down samples are ready.
-
30
Wash blocked beads with 300ul pull-down sonication buffer
-
31
Add 250–300ug of each sample to the beads in pull-down sonication buffer with a final volume of 500ul.
-
32
Incubate the tubes with the blocked beads and the biotin clicked samples in a thermo mixer at 18°C while mixing vigorously (1100rpm) in the dark (foil wrap thermo mixer to protect samples from light) for 20 min.
-
33
Wash beads once with 500ul pull-down sonication buffer
-
34
Wash beads once with 500ul 2× Tween20 wash buffer. Vortex samples at medium to high setting then apply them to magnetic stand.
-
35
Wash beads three times with 500ul 1× Tween wash buffer. Vortex samples at medium to high setting then apply them to magnetic stand.
-
36
Wash beads once with 250ul 1× RIPA buffer without removing them from the magnetic stand.
-
37
Resuspend beads in 40ul RIPA buffer
-
38
Add 10ul 5× Reducing sample buffer to both beads and input material
-
39
Incubate samples at 95°C for 20 minutes mixing vigorously (1200rpm) in thermo mixer
-
40
Put samples on ice for 1 minute then apply them to magnetic stand.
-
41
Transfer pull-down eluates to new tubes.
-
42
Resuspend beads in 30ul 1× loading buffer, incubate at 95°C for 10 minutes
-
43
Put them on ice then apply them to magnetic stand.
-
44
Combine eluates with previous ones, and continue analysis by performing Western blots or doing a mass spectrometry based protocol to identify proteins (Figure B.
SUPPORT PROTOCOL 1 (optional)
ASSESSING CLICK EFFICIENCY WITH DNA DOT/SLOT-BLOT
Additional Materials (also see Basic Protocol)
20× SSC (AM9763, Ambion)
High Sensitivity Streptavidin-HRP (Cat.No. 21134, Pierce)
Nucleic Acid Detection Blocking Buffer (Cat.No. 89880A, Thermo Scientific)
Ammonium acetate (Cat.No. 09689, Sigma)
Streptavidin-HRP Wash Buffer (See recipe)
Biodyne® Modified Nylon Membrane (Cat.No. 77016, Thermo Scientific)
Whatman 3MM paper
Bio-Dot apparatus (Bio-Rad)
UV crosslinker (UV Stratalinker 1800)
Prepare Membrane and Assemble Manifold For Transfer
-
1
Clean dot blot manifold with 10 % bleach and rinse well with Milli-Q water
-
2
Cut Biodyne membrane to the same size as the manifold and, pre-wet the membrane in a clean tray with 6× SSC for 5–10 min.
-
3
Assemble the membrane and the manifold according to the manufacturer’s instructions. Mark the membrane for orientation with a pencil, without damaging the membrane
-
4
Fill each well with 700ul nuclease free water or TE. Check wells for even suction, make sure there are no air bubbles in the wells or air leaks in the manifold assembly. Be careful with the amount of vacuum applied since too strong suction can damage the membrane. Use manifold suction setting 1.
Denature DNA samples and immobilize them on the membrane
-
5
Prepare 2-fold serial dilutions of Biotinylated DNA samples in 116ul final volume with water or TE buffer
-
6
Denature them in by 80ul 1M NaOH and 4ul 0.5M EDTA to reach a final concentration of 0.4 M NaOH/10 mM EDTA and incubating at 95°C for 10 min
-
7
Add an equal volume of ice-cold 2M Ammonium acetate (pH 7.0) to denatured samples, mix well, and transfer mixture to the wells. Fill empty wells with same amount of nuclease free water or TE and then apply gentle suction.
-
8
Wash wells with 800ul 2×SSC buffer and apply suction
-
9
Disassemble manifold rinse membrane with 2×SSC in a glass tray and air dry the membrane; place it on a clean 3M-Whattmann paper.
-
10
UV crosslink membrane in UV Stratalinker 2400- auto crosslink setting.
Streptavidin-HRP detection
-
11
Place membrane facing up on a clean plastic tray and wash membrane once with TBST
-
12
Block membrane with 12ml Nucleic Acid Detection Blocking Buffer for 30 min at room temperature on an orbital shaker
-
13
Dilute Pierce-High Sensitivity Streptavidin-HRP 1:100 with 12ml 3% nuclease free BSA in TBST and incubate with the membrane for 15 min at room temperature on an orbital shaker
-
14
Wash membrane once with 1:10 dilution of Nucleic Acid Detection Blocking Buffer for 10 min at room temperature
-
15
Then wash twice with streptavidin-hrp wash buffer for 5 min at room temperature
-
16
Then wash once with TBST for 5 min at room temperature
-
17
Detect biotin-streptavidin-hrp complex with enhanced chemiluminescence Figure C
REAGENTS AND SOLUTIONS
Mimosine stock solution
Dissolve in 0.1N sodium hydroxide (cell culture tested). Aliquot and store at −80°C indefinitely
2× Tween20 Wash buffer
10mM Tris-HCl pH 8.0, 1mM EDTA, 2M NaCl and 0.1% Tween20 in nuclease free water (Add Tween20 fresh each time. Store –Tween20 Buffer up to a year at room temperature)
Click Reaction Mixture
15mM NaAscorbate, 200uM Biotin TegAzide, 2mM CuSO4 and 1mM THPTA in 0.05%Triton-PBS (Prepare fresh, Add 60ul of 250mM NaAscorbate, 2ul of 100mM Biotin TegAzide, 20ul of 100mM CuSO4 and 10ul of 100mM THPTA in 0.05% TritonX-100 containing PBS with a final volume of 1mL)
Cytoplasm Wash Buffer
50mM HEPES pH 7.8, 150mM NaCl, 0.5%NP40, 0.25%TritonX-100 and 10% Glycerol (Add 2.5mL of 1M HEPES pH 7.8, 1.5mL of 5M NaCl, 2.5mL of 10% NP-40, 1.25mL of 10% TritonX-100 and 5mL of 100% glycerol. Bring volume to 50mL with nuclease free water.) (Store solution at 4°C up to 6 months)
DTT Wash Buffer
20mM Tris-HCl pH 7.8, 200mM NaCl, 0.5mM DTT and 5% glycerol (Add 100ul of 1M Tris-HCl pH 7.8, 200ul of 5M NaCl, 25ul of 100mM DTT and 250ul glycerol and bring volume to 5mL with nuclease free water) (Always add DTT fresh, Store solution at 4°C up to 6 months)
Pull Down Sonication Buffer
10mM Tris-HCl pH 7.5, 200mM KoAc, 1mM EDTA, 0.5mM EGTA (Add 500ul of 1M Tris-HCl pH 7.5, 5mL of 2M KoAc, 100ul of 0.5M EDTA, 50ul of 0.5M EGTA. Bring volume to 50mL with nuclease free water. Keep buffer up to a year at room temperature) Add fresh following with final concentration as indicated: 0.5% Lauroyl Sarcosine, 0.125mM Spermine, 0.05mM Spermidine, 1mM DTT, protease inhibitor complex).
RIPA Buffer
50mM Tris-HCl pH 7.4, 150mM NaCl, 1mM EDTA, 1%NP-40, 1%Na Deoxycholate, 0.1% SDS (Add 2.5mL of 1M Tris-HCl pH 7.4, 1.5mL of 5M NaCl, 100ul of 0.5M EDTA 5mL of 10% NP-40, 5mL of 10%Na-Deoxycholate, 500ul of 10%SDS, bring volume up to 50mL with nuclease free water) (Store solution at room temparature up to 6 months).
Streptavidin-HRP Wash Buffer
10× stock solution contains 100mM Tris-HCl, pH 8.0, 100mM NaCl, 20mM MgCl2 (Store solution at room temperature up to 6 months)
COMMENTARY
Background Information
The single-cell zygote gives rise to more than 200 different cell types in the mammalian body. In an organism, all these cell types essentially carry the same genome, but still manage to establish and maintain unique cellular identities. This is the result of unique genome organization of each cell type and their selective transcriptional programs. Importantly, these differentially expressed regions of the genome with distinct chromatin organizations are maintained from one generation to the next. Such maintenance of specific phenotypes by non-genetic means is called epigenetic inheritance. It has been proposed that much of the heritable epigenetic information is carried by chromatin structure (Henikoff et al., 2004; Rando, 2007). However, little is known about the specific factors and mechanisms governing the inheritance of chromatin states through several rounds of cell division.
Recently, advances have been made to develop novel technologies that would allow metabolic labeling and isolation of nascent nucleic acid. Copper catalyzed azide-alkyne cyclo-addition CuAAC) also known as “click” reaction (Rostovtsev et al., 2002; Kolb et al., 2001) enables efficient conjugation of azide containing molecules to ethynyl labeled nucleic acids both in vitro (Paredes and Das, 2010; Gramlich et al., 2008) and in vivo (Salic and Mitchison, 2008). Alternatively, these labeled nucleotides can further be selectively enriched via affinity purification through biotin-streptavidin interactions. Using this approach, a few groups have recently developed techniques similar to the one described here to analyze nascent chromatin. Sirbu et. al., labeled replicated DNA with EdU and tagged nascent DNA with a biotinylated azide probe using click chemistry. This technology, called iPOND (isolation of protein on nascent DNA), was shown to successfully isolate newly replicated chromatin and associated proteins, both at the replication fork and upon DNA damage response (Sirbu et al., 2011). In parallel, Dm-ChP (DNA mediated chromatin pull-down) was established as a protocol to isolate nascent chromatin (Kliszczak et al., 2011). Finally, pre-biotinylated dUTP was introduced into permeabilized cells during replication to label and isolate newly replicated chromatin (Alabert et al., 2014). These protocols hold the potential to study not only replication fork and DNA damage response but also chromatin maturation behind the replication fork more thoroughly. Expansion of these protocols in combination with other chromatin-based protocols such as chromatin immuno precipitation will provide a better understanding of complex questions such as cellular memory and epigenetic inheritance in development and disease.
Critical Parameters
Click Efficiency
The basic CuAAC reaction requires copper ions at +1 oxidation state. Instead of using Cu(I) salts such as cuprous bromide or iodide, Cu(II)-salts such as copperII sulfate (CuSO4) reduced to Cu(I) by ascorbic acid during the reaction is preferred to avoid undesired byproducts (Rostovtsev et al., 2002). Sodium ascorbate not only allows safer reactive copper but also extends the half-life of the reactive Cu(I) state in the reaction. It is crucial to maintain higher Cu(I) levels longer during the reaction for high click efficiency. Using at least 5 fold excess ascorbate over copper II salt is important for maintaining required +1 oxidation state of copper and hence the reaction efficiency. Importantly, sodium ascorbate needs to be prepared fresh.
If the reaction is done on whole cells rather then cell nuclei, the permeabilization step is important to ensure an efficient click reaction.
Sonication Efficiency
Sonication efficiency is important to avoid non-specific pull down of non-nascent DNA fragments and associated factors. On the other hand, over shearing of the chromatin should also be avoided, to prevent disruption of chromatin composition. We recommend an average fragment size of 500bp–800bps.
Control Samples
Processing −EdU samples prepared in parallel with +EdU samples for the same stages of S phase analyzed is an important control for biotinylation specificity. In addition, another useful negative control is to have the exact same reaction for each sample, with the exception of the biotin azide component, to assess pull-down specificity. The PanH3 antibody is not only a good readout for pull-down specificity but also serve as an appropriate loading control to compare +EdU samples.
Troubleshooting
After the streptavidin pull-down step −Biotin azide or −EdU samples should not produce any detectible bands. Detection of any protein from these samples suggests a non-specific pull down or false positive. The bead blocking step and/or wash steps can be extended to minimize any nonspecific pull down. Extended bead sample incubation increases background drastically and should be avoided.
In our hands, boiling samples longer than 20 minutes increases the degradation of particularly high molecular weight proteins and should be avoided.
Anticipated Results
This protocol allows the isolation of nascent chromatin at the desired stages of the cell cycle either at the replication fork itself or behind it, depending on the experimental design. Following the nascent chromatin pull down, Western blot can be used to assess the association of the protein of interest with the daughter strand. When studying the replication fork itself, short pulses of EdU and blotting for replication fork specific proteins such as PCNA and/or CAF1 can be used as positive control. If nucleosome assembly behind the replication fork is of interest, post-translational modifications specific to newly synthesized histones such as H4K5 or H4K12 deacetylation may be used (Sirbu et al., 2011).
Time Considerations
Cell cycle synchronization, EdU labeling and sample collection can be done in total of 2 days. Click reaction on permeabilized cells and solubilizing cytoplasmic components fallowed by the cell lysis and sonication requires 5–6 hours. Pull-down of biotinylated samples with streptavidin beads and elution step takes about 2 hours. Then protein of interest can be examined in nascent chromatin pull down eluate with Western blotting, 1 day.
Acknowledgments
Ozlem Yildirim is an HHMI Fellow of The Damon Runyon Cancer Research Foundation (DRG-2156-13). Authors would like to acknowledge Kingston Lab members for stimulating discussions; particularly, Dr. Aaron Plys and Dr. Patrick Schorderet for critical reading during the preparation of the manuscript. REK acknowledges NIH funding (GM48405).
LITERATURE CITED
- Alabert C, Bukowski-Wills J-C, Lee S-B, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, Groth A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nature Cell Biology. 2014;16:281–293. doi: 10.1038/ncb2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budhavarapu VN, Chavez M, Tyler JK. How is epigenetic information maintained through DNA replication? Epigenetics & Chromatin. 2013;6:1–1. doi: 10.1186/1756-8935-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramlich PME, Warncke S, Gierlich J, Carell T. Click–Click–Click: Single to Triple Modification of DNA. Angewandte Chemie International Edition. 2008;47:3442–3444. doi: 10.1002/anie.200705664. [DOI] [PubMed] [Google Scholar]
- Groth A. Replicating chromatin: a tale of histones. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2009;87:51–63. doi: 10.1139/O08-102. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Furuyama T, Ahmad K. Histone variants, nucleosome assembly and epigenetic inheritance. Trends in Genetics. 2004;20:320–326. doi: 10.1016/j.tig.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Kliszczak AEA, Rainey MDM, Harhen BB, Boisvert FMF, Santocanale CC. DNA mediated chromatin pull-down for the study of chromatin replication. Scientific Reports. 2011;1:95–95. doi: 10.1038/srep00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angewandte Chemie (International ed. in English) 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Marzluff WF, Wagner EJ, Duronio RJ. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nature Reviews Genetics. 2008;9:843–854. doi: 10.1038/nrg2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejlvang J, Feng Y, Alabert C, Neelsen KJ, Jasencakova Z, Zhao X, Lees M, Sandelin A, Pasero P, Lopes M, et al. New histone supply regulates replication fork speed and PCNA unloading. The Journal of Cell Biology. 2014;204:29–43. doi: 10.1083/jcb.201305017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes E, Das SR. Click Chemistry for Rapid Labeling and Ligation of RNA. ChemBioChem. 2010;12:125–131. doi: 10.1002/cbic.201000466. [DOI] [PubMed] [Google Scholar]
- Rando OJ. Global patterns of histone modifications. Current Opinion in Genetics & Development. 2007;17:94–99. doi: 10.1016/j.gde.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angewandte Chemie (International ed. in English) 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BMB, Couch FBF, Feigerle JTJ, Bhaskara SS, Hiebert SWS, Cortez DD. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes & development. 2011;25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Franco AA, Santos H, Nelson DM, Kaufman PD, Adams PD. Defective S Phase Chromatin Assembly Causes DNA Damage, Activation of the S Phase Checkpoint, and S Phase Arrest. Molecular Cell. 2003;11:341–351. doi: 10.1016/s1097-2765(03)00037-6. [DOI] [PubMed] [Google Scholar]