

ABSTRACT
Therapeutic antibodies continue to develop as an emerging drug class, with a need for preclinical tools to better predict in vivo characteristics. Transgenic mice expressing human neonatal Fc receptor (hFcRn) have potential as a preclinical pharmacokinetic (PK) model to project human PK of monoclonal antibodies (mAbs). Using a panel of 27 mAbs with a broad PK range, we sought to characterize and establish utility of this preclinical animal model and provide guidance for its application in drug development of mAbs. This set of mAbs was administered to both hemizygous and homozygous hFcRn transgenic mice (Tg32) at a single intravenous dose, and PK parameters were derived. Higher hFcRn protein tissue expression was confirmed by liquid chromatography-high resolution tandem mass spectrometry in Tg32 homozygous versus hemizygous mice. Clearance (CL) was calculated using non-compartmental analysis and correlations were assessed to historical data in wild-type mouse, non-human primate (NHP), and human. Results show that mAb CL in hFcRn Tg32 homozygous mouse correlate with human (r2 = 0.83, r = 0.91, p < 0.01) better than NHP (r2 = 0.67, r = 0.82, p < 0.01) for this dataset. Applying simple allometric scaling using an empirically derived best-fit exponent of 0.93 enabled the prediction of human CL from the Tg32 homozygous mouse within 2-fold error for 100% of mAbs tested. Implementing the Tg32 homozygous mouse model in discovery and preclinical drug development to predict human CL may result in an overall decreased usage of monkeys for PK studies, enhancement of the early selection of lead molecules, and ultimately a decrease in the time for a drug candidate to reach the clinic.
KEYWORDS: Allometric scaling, clearance, FcRn, hFcRn transgenic mice, Human PK prediction, IgG, monoclonal antibody, mAb, Neonatal Fc Receptor, pharmacokinetics, PK
Abbreviations
- mAb
Monoclonal antibody
- PK
Pharmacokinetic
- IV
Intravenous
- CL
Clearance
- FcRn
Neonatal Fc Receptor
- hFcRn
human FcRn
- mFcRn
mouse FcRn
- mAb-FcRn+
mAb engineered for enhanced binding to FcRn
- KO
knock out
- NHP
non-human primate
- FIH
First-in-human
- TMDD
Target-mediated drug disposition
- LBA
Ligand Binding Assay
- TCA
trichloroacetic acid
- LC-HRMS
Liquid chromatography-high resolution tandem mass spectrometry
- WT
wild type
- ADME
Absorption, distribution, metabolism and excretion
- PBPK
Physiologically based PK model
- ADC
Antibody-drug conjugate
- CCC
Concordance correlation coefficient
Introduction
Monoclonal antibodies (mAbs) continue to proliferate as a therapeutic drug class, with over 30 mAbs marketed for commercial use and over 300 estimated to be in development.1 Establishing preclinical tools to better profile these molecules could ultimately reduce the time needed for drug discovery and development by improving the lead molecule selection process.2 Obtaining preclinical pharmacokinetics (PK) is a critical component to predicting human PK and projecting first-in-human (FIH) doses. The current gold standard for preclinical PK assessment of mAbs is the non-human primate (NHP), commonly the cynomolgus monkey. PK studies in NHP provide a reasonable and early assessment of human PK;3-7 however, NHP studies are costly and challenging to implement in the discovery and early preclinical phases of drug development, and there are ethical issues with the use of the animals for medical research. The potential for a rodent model to accurately estimate the PK behavior of mAbs in higher species could dramatically improve the throughput for lead drug candidate selection, and potentially increase the overall success while decreasing the time for non-clinical development of mAbs.
Therapeutic mAbs are usually immunoglobulin G (IgG) subclasses, and the majority of drug candidates are of the IgG1 or IgG2 isotype. Multiple factors, such as charge, size, target binding affinity and interaction, pH dependent neonatal Fc receptor (FcRn) binding affinity, degree and type of glycosylation, are thought to affect the PK of these large macromolecular therapeutics, which in turn could influence their efficacy and safety.2 PK of mAbs can be described as a linear process in which clearance (CL) is constant, or a non-linear process in which the CL varies across the dose range. Non-linear PK can be due to target-mediated drug disposition (TMDD),8 where at low doses target-mediated CL is the dominating contributor to PK until the target is saturated at higher doses. Non-linear PK can also be observed when anti-drug antibodies (ADA) are induced by an immune response to the mAb. FcRn-mediated protection and recycling is well documented as one of the most important mechanisms in modulating mAb CL. FcRn is a major histocompatibility complex (MHC) class I like heterodimer, consisting of an Fc binding α-chain and a β2-microglobulin subunit, which is believed to play a central role in the persistence and disposition of IgG in a pH dependent manner.9-11 FcRn binds with high affinity to the CH2-CH3 interface of the IgG Fc domain in the acidic environment (pH ≤ 6.5) of endosomes following endocytosis, where non-FcRn-bound IgG can be processed for degradation by the lysosome, and FcRn-bound IgG can be recycled to the extracellular surface and then dissociate from FcRn at physiological pH (7.4).12-15 As a result of the important role of FcRn-mediated recycling of IgG and its relationship to PK, FcRn has become a common target of mAb engineering efforts aimed at improving mAb PK characteristics.16-19 FcRn is widely distributed among various tissues and FcRn mRNA and protein expression have been reported in endothelial, epithelial and circulating cells.20-24 To date, a comprehensive, quantitative understanding of the tissue-specific expression of FcRn, which is necessary to understand the effect FcRn has on mAb disposition in various organs or tissues, has not been well established in human. Early reports have demonstrated expression of FcRn protein in human fetal intestine using an immunoblotting approach,25 while later an ELISA was used to quantify FcRn protein in human and NHP intestinal tissues.26 It has been previously documented from mAb biodistribution studies in mice using a radiolabeled mAb that the liver and spleen are the primary sites for IgG catabolism.27,28 Measuring the concentration of FcRn protein in liver and spleen relative to other organs will help elucidate the role of FcRn in the distribution and catabolism of mAbs.
Studies that evaluated the potential for the human FcRn (hFcRn) expressing transgenic mouse to correlate to human PK have been reported.17,29-32 While human IgG is able to bind to both mouse FcRn (mFcRn) and hFcRn, species differences in the binding of FcRn have been demonstrated in vitro, and may contribute to the variability and an overall poor PK predictability of wild type (WT) mouse to human PK.33 The hFcRn transgenic mouse from The Jackson Laboratory32 is null for α-chain mFcRn and contains either one or 2 transgenes of hFcRn, hemizygous and homozygous, respectively. Two hFcRn transgenic mouse strains are currently available; the Tg32 strain (hFcRn promoter) and the Tg276 strain (chicken β-actin promoter).10 Since endogenous mouse IgG binds to hFcRn with weaker affinity relative to mFcRn and is not protected as efficiently, the Tg32 and Tg276 strains demonstrate decreased serum concentrations of endogenous mouse IgG compared to WT C57Bl/6 mouse.29 In addition to IgG binding, FcRn has a discrete albumin binding site. Endogenous serum albumin concentrations are similar in the Tg32 and Tg276 mouse compared to the WT C57Bl/6 strain, demonstrating that the functionality of the hFcRn heterodimer in the mouse model may hinder endogenous IgG recycling, but not albumin.29 Several studies have begun to characterize the PK of mAbs in hFcRn transgenic mice. Petkova et al.17 examined both the Tg32 and Tg276 strains and their ability to distinguish PK differences of a mAb and 3 different framework mutants engineered to have either diminished binding to hFcRn (decreased half-life) or enhanced binding to hFcRn (increased half-life). Interestingly, the Tg276 (hemizygous and homozygous) demonstrated a more rapid CL of the mAbs tested, while the Tg32 hemizygous showed a slower CL of each mAb and was the only mouse model tested that was able to differentiate the 4 mutant mAbs based on PK. Of note, Tg32 homozygous mouse was not examined in this study. Tam et al.29 and Roopenian et al.32 examined the PK of a single mAb in the different strains and similarly noted a decreased exposure in the Tg276 mouse compared to the Tg32 mouse, as well as a decreased exposure in the hemizygous compared to the homozygous mouse. In addition, Tam et al. examined a panel of 9 mAbs in the Tg32 hemizygous mouse having a human CL ranging from approximately 0.1-0.5 mL/hr/kg, and demonstrated a robust correlation of CL in the Tg32 hemizygous mouse to human CL. While the Tg32 homozygous mouse was still not examined for human CL correlation in this study, the authors did demonstrate that co-administration of IVIG with test mAb did not alter the resulting PK correlation.29
In this study we investigated the potential application of the hFcRn Tg32 transgenic mouse model to effectively predict mAb human PK, and set out to understand what role FcRn expression may play in interpreting the preclinical utility of this mouse model. Our approach was to utilize a broad range of 21 Pfizer mAbs and 6 marketed mAbs with varying CL values in human (0.06-0.45 mL/hr/kg) and in NHP (0.04-4.9 mL/hr/kg), including IgG1 and IgG2 isotypes, mAbs that bind different target types (e.g., soluble or cell membrane targets), and encompassing different framework classifications to examine the spectrum of utility and sensitivity of the hFcRn Tg32 hemizygous and homozygous strains to predict human PK. A subset of mAbs were also examined in the hFcRn Tg276 mouse, WT C57Bl/6 mouse and FcRn knock-out (KO) mouse for comparison of the PK characteristics with hFcRn Tg32 mice. In addition, a series of matched WT and Fc framework mutation mAbs engineered for enhanced FcRn binding were included to understand the degree of PK differentiation achievable with the hFcRn Tg32 mouse strain. This study considers hFcRn protein expression differences in tissues and the potential effect on PK characteristics of mAbs, as well as demonstrates the ability to use this mouse model to scale PK allometrically to predict mAb CL in human. With all of these factors considered, we sought to characterize the utility of the Tg32 mouse in the discovery and preclinical drug development of mAbs to predict human PK, enhance the ability to select lead molecules earlier in discovery, more accurately predict human PK and ultimately decrease the time needed for a drug candidate to reach the clinic.
Results
PK data from historical PK studies
For a retrospective analysis on the PK of mAbs across species, historical PK data in WT mouse, NHP and human were collected (supplemental Table 1). Of the 27 mAbs included in this study, 24 were IgG1 and 3 were IgG2; 16 bind to soluble ligand targets, 9 bind to membrane bound targets, and 2 bind to both soluble and membrane bound receptor targets; 12 were human, 13 were humanized, and 2 were chimeric. The linear CL values (apparent linear and definitive linear) of these mAbs ranged from 0.13-2.19 mL/hr/kg in WT mouse (n = 17), 0.04-1.16 mL/hr/kg in NHP (n = 25, excludes non-linear mab17) and 0.06-0.45 mL/hr/kg in human (n = 15) (Fig. 1A–C, respectively). The median mAb linear CL values in WT mouse, NHP and human were 0.36, 0.22 and 0.12 mL/hr/kg, respectively. CL values depicted in Fig. 1 represent the CL designation (definitive linear, apparent linear, or non-linear) for each mAb. As described in the methods section, historical mAb CL values in each species were assessed using strict criteria to define each CL value as definitive linear, apparent linear, or non-linear. Definitive linear CL was selected as a mean CL value over a linear dose range from a dose escalating study. Apparent linear CL values were determined from a dose escalation study where only the highest dose cohort is observed to approach linearity, or from a single dose study with a dose greater than 10 mg/kg, believed to saturate the target. Non-linear CL values (target-mediated CL, or CL affected by presumed ADA) were excluded from all PK correlative analyses.
Figure 1.
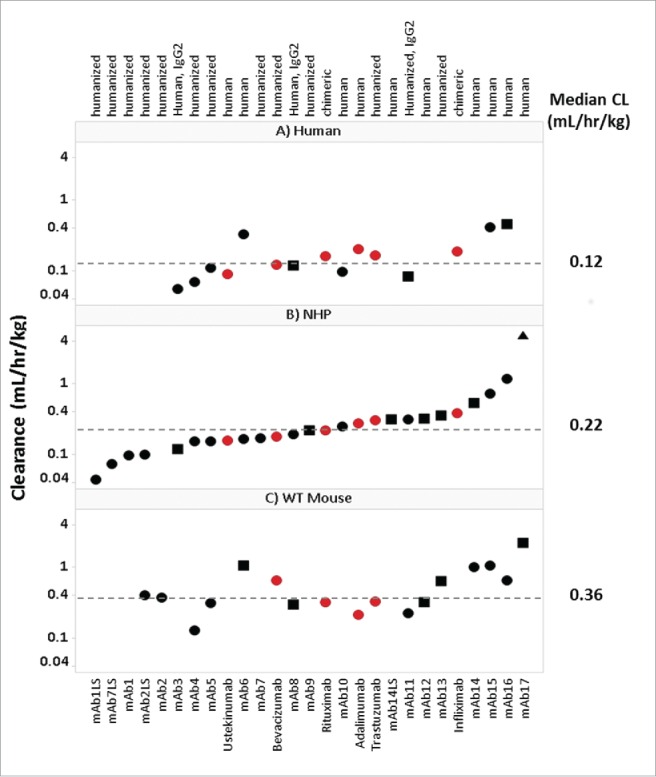
Panel of mAbs Used in Study. Interspecies CL for therapeutic mAbs. Panels A, B and C depict the 27 mAbs examined in this study, including 21 Pfizer mAbs (• ) and 6 marketed mAbs () and the CL values (mL/hr/kg) for human (15/27), NHP (26/27), and WT mouse (17/27), respectively. Mean CL value are depicted for each species. Symbols depict definitive linear CL values (•), apparent linear CL values (▪) and non-linear (▴). Of the 27 collected mAbs: 24 are IgG1 and 3 are IgG2; 16 have soluble ligand targets, 9 have membrane bound targets, and 2 target both soluble and membrane bound receptor targets; 12 are fully human, 13 are humanized, and 2 are chimeric.
) and 6 marketed mAbs () and the CL values (mL/hr/kg) for human (15/27), NHP (26/27), and WT mouse (17/27), respectively. Mean CL value are depicted for each species. Symbols depict definitive linear CL values (•), apparent linear CL values (▪) and non-linear (▴). Of the 27 collected mAbs: 24 are IgG1 and 3 are IgG2; 16 have soluble ligand targets, 9 have membrane bound targets, and 2 target both soluble and membrane bound receptor targets; 12 are fully human, 13 are humanized, and 2 are chimeric.
Initial PK data from hFcRn transgenic mouse strains – selection of Tg32 mouse model
To understand the role of human vs. mouse FcRn protection and assess the 2 hFcRn transgenic mouse strains (Tg32 and Tg276), PK studies were conducted and compared to WT mouse (C57Bl/6, mFcRn) and FcRn KO mice.
The first mAb evaluated was mAb14 with a known CL in NHP of 0.53 mL/hr/kg, and was administered to Tg32 hemizygous and homozygous, Tg276 homozygous and WT C57Bl/6 mice. Compared to the CL observed in WT C57Bl/6 mice, mAb14 showed an 8.9-fold faster CL in Tg276 homozygous, and 2.2-fold slower CL in Tg32 homozygous mice (Fig. 2A).
Figure 2.
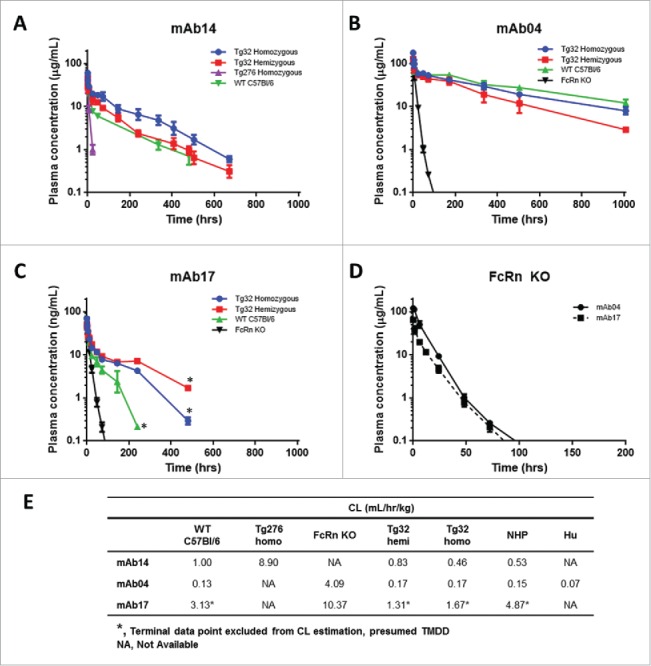
hFcRn Tg Mouse Strain Comparisons. PK profile results of Tg32 hemizygous and homozygous, Tg276 homozygous, WT C57Bl/6 and FcRn KO mice following administration of (A) mAb 14, (B) mAb 04 and (C) mAb17. (D) PK profile overlay in FcRn KO mice for mAb04 and mAb17. (E) PK parameters for mAb14, mAb04 and mAb17 in NHP and each mouse model. Data is depicted as the mean ± standard deviation for 3-6 animals/group for panels A-D.
Two mAbs with contrasting PK characteristics, mAb04 and mAb17 with observed CL in NHP of 0.15 (definitive linear) and 4.87 (non-linear) mL/hr/kg, respectively, were administered intravenously (IV) to Tg32 hemizygous and homozygous, WT C57Bl/6, and FcRn KO mice. MAb04 exhibited a comparable and slow elimination in the Tg32 hemizygous, Tg32 homozygous and WT C57Bl/6 mouse (Fig. 2B). MAb17 exhibited a 1.9-fold more rapid elimination in WT C57Bl/6 mice compared to Tg32 homozygous mice and a 2.4-fold more rapid elimination compared to Tg32 hemizygous mice (Fig. 2C). In both historical NHP data and PK studies conducted in mice, mAb17 appears to exhibit a TMDD effect at the terminal phase (i.e., non-linear CL) at low concentrations. It was therefore excluded from any subsequent correlative analyses in this study, and only the estimated linear portion of CL was used for the comparison of mouse strains. Both mAb04 and mAb17 demonstrated rapid elimination in FcRn KO mice with mAb CL of 4.09 and 10.37 mL/hr/kg, respectively, despite a > 30-fold CL difference observed for these mAbs in NHP (Fig. 2D). The mAb PK parameters in each mouse strain and NHP are summarized in Fig. 2E. The Tg32 mouse strain most closely paralleled the PK observed in NHP and contributed to the selection of the Tg32 mouse as the animal model to further characterize.
Total hFcRn protein expression in Tg32 hemizygous and homozygous mice
To interpret the role of genotype on the PK differences observed for mAbs administered to Tg32 hemizygous and homozygous, the total protein expression of hFcRn in each mouse genotype was quantified using an online Liquid chromatography-high resolution tandem mass spectrometry (LC-HRMS) immunoaffinity approach. Fig. 3 depicts the quantitative concentration of total hFcRn across a panel of tissue homogenates extracted from Tg32 hemizygous and homozygous mice. Harvested tissues were examined from n = 5 mice each where the inter-individual variability is depicted by the standard deviation error bars. As anticipated, increased expression of hFcRn protein was observed in the Tg32 homozygous compared to hemizygous mice for all tissues examined. The most significant increases (p < 0.05, Mann-Whitney test) were observed for lymph node, lung, skin, stomach, heart, kidney, skeletal muscle, and brain. A less significant increase (p < 0.1, Mann-Whitney test) was observed for large intestine. No significant difference was observed in liver, small intestine, spleen and fat when comparing Tg32 homozygous to Tg32 hemizygous mice. Interestingly, the tissues that did not show significant differences between the 2 genotypes of mice are the ones with the greatest intra-individual variability (CV range of 8.4-112.0% compared to 9.5-42.3% for those tissues with significant differences).
Figure 3.
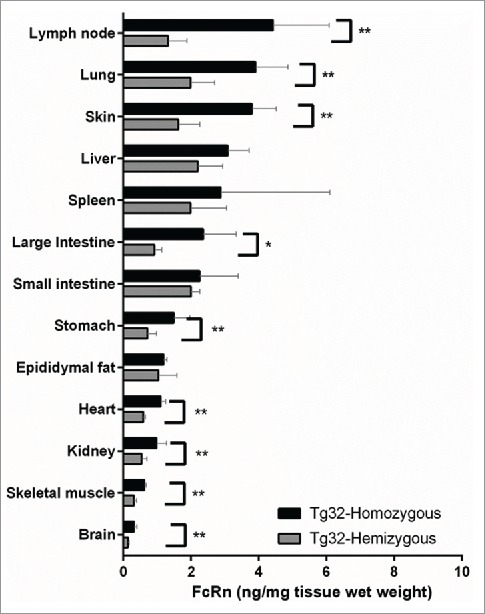
hFcRn Tissue Expression Profile in Tg32 Hemizygous and Homozygous Mice. Significant expression differences between homozygous and hemizygous genotypes were analyzed using an unpaired Mann-Whitney test where significance is indicated as single asterisk (*) for p < 0.1 and double asterisk (**) for p < 0.05.
PK studies conducted in Tg32 transgenic mouse
A panel of mAbs (Table S1) was administered to Tg32 hemizygous and homozygous mice for PK analysis and comparison. Two of 27 mAbs (mAb09 and mAb13, both with NHP CL < 0.4 mL/hr/kg) were not administered to Tg32 homozygous mice because they did not have corresponding human PK data, and sufficient data had been obtained from the 25/27 mAbs over the full range of PK to interpret the correlation of the Tg32 mouse to NHP.
Use of hFcRn Tg32 mouse model to distinguish PK of mAb framework variants
We sought to further characterize and elucidate the threshold of PK differentiation for each Tg32 mouse genotype, using 4 matched pairs of parent WT mAbs vs. mAbs engineered for enhanced binding to FcRn (mAb-FcRn+) administered to the Tg32 hemizygous and homozygous mice. Of note, only NHP PK was available for comparison (with the exception of mAb02). Fig. 4 depicts the relationship in CL values of each mAb between Tg32 hemizygous and NHP (Fig. 4A) or between Tg32 homozygous and NHP (Fig. 4B). The PK parameters are summarized in Fig. 4C. Similar to NHP, the Tg32 homozygous mouse was able to differentiate the WT mAb vs. mAb-FcRn+ mutation for 3/4 mAb pairs based on CL and 4/4 mAb pairs based on half-life, whereas the Tg32 hemizygous mouse was only able to differentiate 2/4 mAb pairs based on CL and 1/4 mAb pairs based on half-life, respectively.
Figure 4.
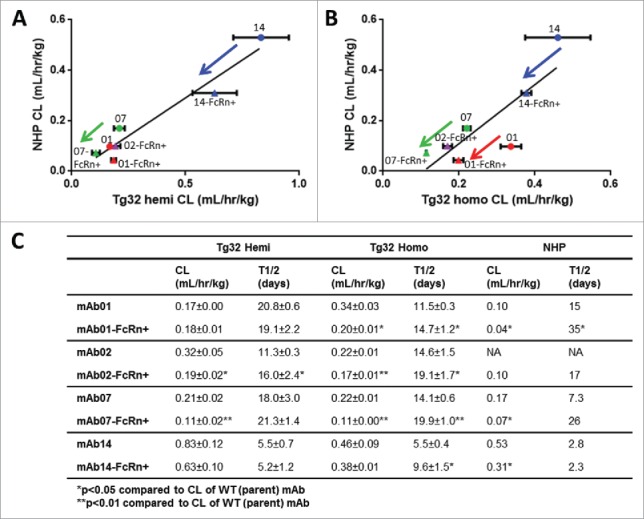
hFcRn Tg32 PK Differentiation in Framework Mutations. CL correlation between (A) Tg32 hemizygous and (B) homozygous mouse to NHP for 4 pairs of parent and FcRn+ mAbs (mAb01, mAb02, mAb07, mAb14) and (C) Summary of CL and T1/2 PK parameters for each parent and FcRn+ mAb in Tg32 hemizygous and homozygous mice and NHP. Statistical significance determined using a Student's paired t-test (*p < 0.05 and ** p < 0.01).
Correlation of mAb CL in rodents to NHP
The correlation of mAb CL in WT mouse, Tg32 hemizygous and Tg32 homozygous to NHP yield r2 values of 0.18, 0.82 and 0.75 (r = 0.43, 0.91, and 0.87), respectively, for the full data sets (Fig. 5). 25/27 mAbs have definitive linear or apparent linear CL in NHP for comparison, whereas only 15/27 mAbs have been administered to humans for comparison. mAb17, having non-linear CL in both mice and NHP, was excluded from all correlative analyses. The Pearson correlation test showed that the relationship between mAb CL in NHP and WT mouse was not significant (p = 0.11), but correlated similarly with Tg32 hemizygous and homozygous (p < 0.01) albeit better with Tg32 hemizygous mouse.
Figure 5.
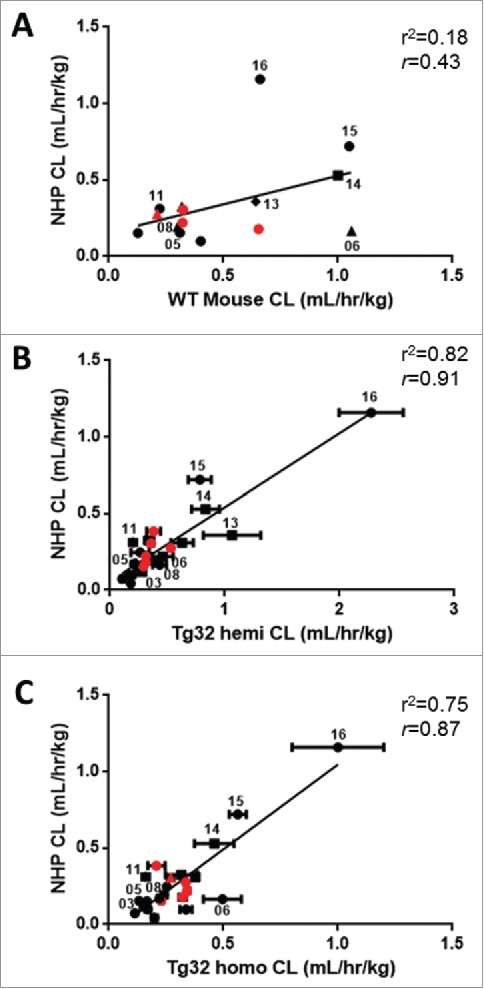
Correlation of mAb CL in Rodents to mAb CL in NHP. Linear correlation graphs of (A) WT mouse CL to NHP CL for 15/27 mAbs, (B) Tg32 hemizygous mouse CL to NHP CL for 25/27 mAbs and (C) Tg32 homozygous CL to NHP CL for 23/27 mAbs. Tg32 mouse CL results are shown as the mean ± standard deviation for 3-6 animals/group. Symbols: •, definitive linear CL values; ▪, apparent linear CL in NHP only; ▴, apparent linear CL in rodent only; ♦, apparent linear CL in both NHP and rodent. •, Pfizer mAbs , marketed therapeutic mAbs.
Correlation of mAb Cl in rodents vs human and NHP vs human
The relationship of the 15/27 mAbs having human CL values for comparison to WT mouse, NHP, Tg32 hemizygous and homozygous mice is illustrated in Fig. 6. Of note, only 11/15 mAbs had WT mouse PK data, while 15/15 mAbs had PK data for NHP, Tg32 hemizygous and homozygous mice. The correlation of mAb CL in each animal model to human CL yielded r2 of 0.61, 0.67, 0.87 and 0.83 (r = 0.78, 0.82, 0.79, 0.91) for WT mouse, NHP, Tg32 hemizygous and homozygous mice, respectively (p < 0.01), for the full data sets. Interestingly, Tg32 hemizygous mouse showed saturation at the higher range of CL, while all the other strains show a linear correlation to mAb CL in human. mAb16, with very high CL in Tg32 hemizygous mouse, heavily influenced the non-linear relationship observed to mAb CL in human, and removing it yielded a weaker linear relationship (r2 = 0.79, r = 0.89, and p < 0.01) (Fig. 6C inset). By contrast, removal of mAb16 from the CL correlation of Tg32 homozygous mouse compared to human did not affect the overall correlation and yielded the identical r2 of 0.83 (r = 0.91) (Fig. 6D inset). Taken together, these results revealed the Tg32 homozygous mouse as having the strongest correlation of mAb CL to human.
Figure 6.
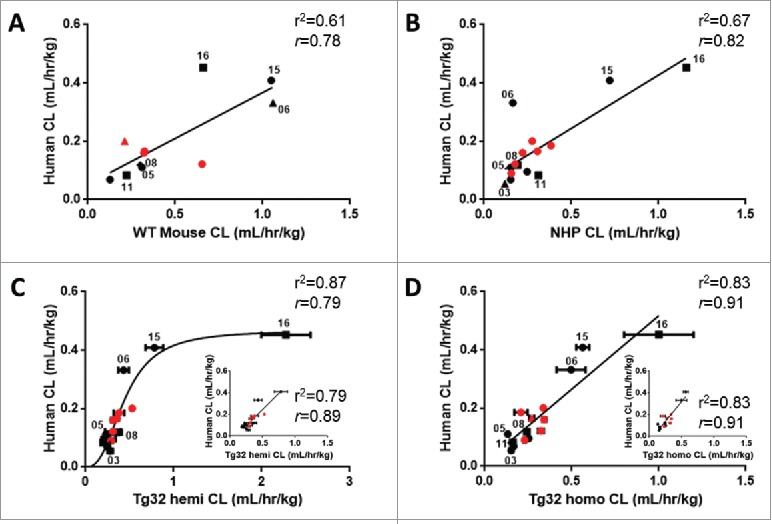
Correlation of mAb CL in Rodents vs Human and NHP vs Human. Linear correlation graphs of (A) WT mouse CL to human CL for 11/27 mAbs, (B) NHP CL to human CL for 15/27 mAbs, (C) Tg32 hemizygous CL to human CL for 15/27 mAbs and D) Tg32 homozygous CL to human CL for 15/27 mAbs. MAbs shown in panels B, C, and D represent the same 15 mAbs. Tg32 mouse CL results are shown as the mean ± standard deviation for 3-6 animals/group. Symbols: •, definitive linear CL values; ▴, apparent linear CL in rodent or NHP; ▪apparent linear CL in human; ♦, apparent linear CL in rodent or NHP and human. •, Pfizer mAbs, , marketed therapeutic mAbs.
Single species allometric scaling of mAb CL from WT mouse, NHP or hFcRn Tg32 mouse model to predict human CL
Allometric scaling methodology from a single species was used to assess prediction accuracy (percent of mAbs within 2-fold error from the line of unity) of human CL from each animal model. Fig. 7 depicts the allometric scaling results of human CL prediction for each mAb from preclinical species CL (definitive linear and apparent linear CL) observed in WT Mouse, NHP, Tg32 hemizygous and homozygous mice using either the standard exponent of 0.75 (top row) or an empirically derived best fit exponent applied to each animal model (middle row). Application of an exponent of 0.75 in this data set resulted in under prediction of human CL for all mouse models with a prediction accuracy of less than 15%. Consistent with literature,6 applying the exponent of 0.75 to NHP data resulted in reasonable prediction of human CL, for 93% of mAbs.
Figure 7.
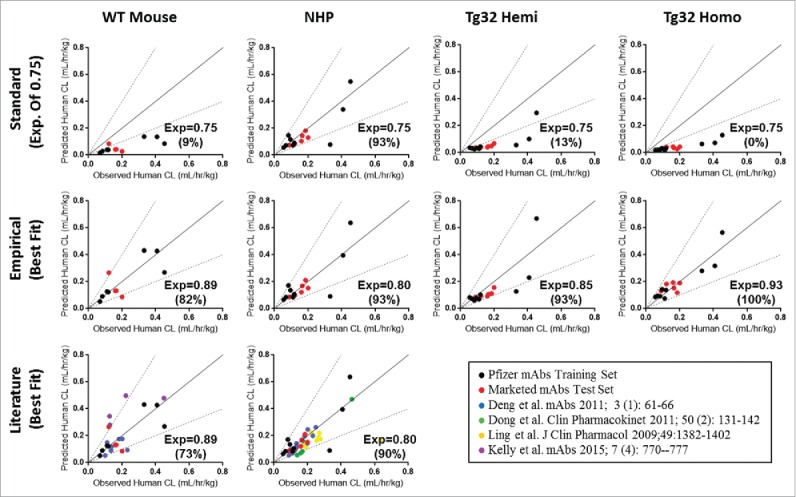
Single Species Allometric Scaling of CL for mAbs. Allometric Scaling for mAbs in NHP (15/27mAbs), WT mice (11/27mAbs), and hFcRn Tg32 hemizygous and homozygous mice (15/27 mAbs), comparing a standard scaling exponent of 0.75 (top row) to an empirically derived best fit exponent (middle row) per animal model. Results are plotted against a line of unity (solid black line) ± 2-fold error (dotted gray lines) where accuracy is described as the percentage (%) of mAbs predicted within 2-fold of the line of unity. Symbols: •, definitive linear CL values; ▴, apparent linear CL in rodent or NHP; ▪, apparent linear CL in human; ♦ apparent linear CL in rodent or NHP and human. •, training data set. , test set, marketed therapeutic mAbs.
Empirical exponents were then assessed from a training dataset of the 9 Pfizer mAbs with human PK available (Fig. 7, black circles) using a linear regression and statistical concordance correlation coefficient (ccc). The empirically derived exponent with the highest ccc values, i.e., with the best overall precision and accuracy relative to the line of unity, was determined as the best fit exponent. Resulting best fit exponents were 0.89, 0.80, 0.85 and 0.93 with ccc values of 0.86, 0.79, 0.76 and 0.93, for WT mouse, NHP, Tg32 hemizygous and homozygous mouse, respectively, and were then verified by application to a test set of mAbs, which consisted of the 6 marketed mAbs. For the test set of mAbs (Fig. 7, red circles), the same best fit exponents resulted with the highest ccc values of 0.78, 0.80, 0.75 and 0.91, respective to the same animal models listed above, confirming the best fit exponent selection and overall prediction and accuracy of the fit to the line of unity. Use of the best fit exponents for the combined data sets (training and test sets of mAbs) yielded prediction accuracies of 82%, 93%, 93% and 100% for WT Mouse, NHP, Tg32 hemizygous and homozygous mice, respectively. Further, addition of available literature data (Fig. 7, bottom row) for WT Mouse CL5,34 and NHP CL 4-7 and applying our best fit exponents of 0.89 (WT mouse) and 0.80 (NHP) could predict human CL with prediction accuracy of 73% and 90%, respectively, compared to 18% and 85% using the traditional exponent of 0.75 (not shown). Application of a best fit exponent 0.80 for scaling NHP CL provided similar results to the traditional exponent 0.75 for the training and test set of mAbs; however, addition of literary data further supported use of exponent 0.80 for improved CL projections from NHP PK.
Discussion
Prior to clinical testing, animal models have historically been a valuable tool in predicting human PK of drug candidates, where selection of the appropriate animal model is based on essential absorption, distribution, metabolism and elimination (ADME) properties of the drug that results in scalable parameters to accurately estimate human PK. Transgenic mouse models for human FcRn have been of recent interest for their potential to scale to human PK for mAbs, an established class of important biotherapeutics. We sought to characterize this preclinical model as a tool to provide guidance for its application in the preclinical development of mAbs. Using a study panel of 27 mAbs and a mouse serial sampling approach, we demonstrated a stronger correlation of mAb CL in the Tg32 homozygous than hemizygous mouse or wild type mouse to human CL. The serial sampling approach allows the opportunity to capture a single PK profile from one mouse as compared to composite sampling of a terminal time point for each mouse.35 In addition, we provided the first recommendation for use of this model to allometrically scale and predict human CL. Our results establish that the Tg32 homozygous mouse model can distinguish PK differences in mAbs containing Fc framework mutations to increase serum half-life from their WT mAb counterparts. NHP has proven to be a reliable model for the prediction of human PK; however, the early utility is often hindered by substantial expense, ethical concerns surrounding frequent use and added quantity of drug necessary to conduct a study. The Tg32 homozygous mouse model provides valuable utility in predicting mAb PK earlier in drug discovery, which may ultimately reduce the need for NHP PK studies and potentially be used as a selection tool, decreasing the time and costs needed to select an optimum lead mAb candidate with desired PK properties for success in the clinic.
Mouse models come in a wide range of background strains and genetic modifications, which results in biological differences36 that may in part contribute toward the large degree of variability noted when correlating mouse PK to human PK. Where FcRn has been widely identified as the predominant pathway for protection and regulating mAb CL, the species homology and physiological binding affinity of FcRn may play a significant role in the ability to scale animal to human PK. Human IgG, the backbone for mAb therapeutics, has been shown through in vitro binding studies to bind mFcRn with higher affinity than to hFcRn.33 This discrepancy in binding to FcRn in different species is likely variable across mAbs, and may result in large differences across animal models regarding the recycling efficiency and elimination rate of human IgG. Our results support this hypothesis where administration of mAb14 to WT (C57Bl/6) mice shown in Fig. 2A results in a decreased exposure compared to Tg32 mice, and administration of a different mAb with slower CL (mAb04), shown in Fig. 2B, results in an increased exposure in WT (C57Bl/6) compared to Tg32 mice, suggesting the 2 different mAbs have variable interactions with mFcRn compared to hFcRn. The in vitro binding affinity of mAbs was examined using a Biacore assay consisting of recombinant hFcRn-avitag-biotin conjugated to a streptavidin chip; however, a correlation was not observed to in vivo PK for human, NHP, or Tg32 mice (data not shown). There continues to be a need to understand the relationship between in vitro FcRn binding affinity and in vivo PK where the available literary data to date suggests mixed results toward a correlation.16,18,30,37,38 Of the 2 available hFcRn transgenic mouse strains (Tg32 and Tg276), there appear to be large differences in mAb PK from each mouse model, and the difference in the promoter-driven regulation of the hFcRn transgene likely affects protein expression or function. Our results are in line with literary observations where the Tg276 strain showed a more rapid mAb CL than the Tg32 strain,17,29,32,39 and that the Tg32 homozygous mice demonstrated a greater overall plasma exposure of mAb than the Tg32 hemizygous mice.29 Use of the Tg32 hemizygous mouse for PK analysis may be conducted in a shorter time course than the homozygous mouse due to decreased mAb exposure; however, the accuracy in CL prediction to human will be limited. Based on the range of CL values observed for the same set of mAbs in the Tg32 hemizygous mice compared to Tg32 homozygous mice (Figs. 5B, 5C vs and Figs. 6C inset versus 6D), the PK differences observed in the 2 Tg32 mice genotypes are likely attributed to the gene copy and observed protein expression differences of hFcRn (Fig. 3).
Unlike small molecules where passive diffusion and sometimes transporters are the major mechanisms of distribution, FcRn protection is critical in CL and distribution of IgGs in tissues throughout the body, including sites of distribution that may not be in rapid equilibrium with the plasma compartment. To date, most PK calculations of mAbs have employed non-compartmental analysis. These approaches assume that the site of drug elimination is in the central compartment. However, these approaches may not provide accurate assessment of the disposition of many mAbs in tissues. Appropriate assessment of mAb disposition ideally relies on a direct measure of the tissue concentrations, and it has been suggested that physiologically-based pharmacokinetic (PBPK) models may facilitate the appropriate characterization of mAb tissue disposition.40,41 FcRn protein concentrations (peripheral cells and tissue source) are an important parameter to consider in the construction of a mAb PBPK model. Shah and Bett's model incorporated FcRn–mAb interactions in all tissues considered in the model assuming that the concentration of FcRn in the vascular endothelial cells is conserved between different species.40 Quantitating individual tissue FcRn concentrations in different species would greatly improve the accuracy of a PBPK model; however, to date a quantitative analysis across a comprehensive tissue panel for FcRn expression has yet to be demonstrated, particularly for the hFcRn Tg32 mouse. A new mass spectrometry-based assay for the quantification of human FcRn in Tg32 tissues has been developed recently.42 Here, we demonstrated the utility of a quantitative FcRn tissue concentration profile in the characterization of the hFcRn Tg32 mouse model for early PK assessment of mAbs. Fig. 3 shows significantly increased protein expression of hFcRn in homozygous mice compared to hemizygous mice for several of the tissues studied, normalized by tissue weight. These results are in line with the double gene copy of hFcRn in the Tg32 homozygous mice compared to hemizygous. The observed high degree of inter-subject variability of hFcRn in several tissues can be related in part to the tissue cellular and vascular composition. The overall increased expression of hFcRn protein in homozygous vs. hemizygous Tg32 mice also supports the higher amount of circulating endogenous mouse IgG in homozygous vs. hemizygous (0.54 and 0.23 mg/mL 29), respectively.
Where the Tg32 hemizygous mouse has been more prevalent in the literature for conducting preclinical PK studies,29 17 the human CL range that was tested was > 2.5 mL/day/kg (0.10 mL/hr/kg). We examined mAbs CL values as low as 1 mL/day/kg (0.04 mL/hr/kg), and observed a saturating effect in the hemizygous mouse ability to differentiate very slow clearing mAbs compared to the Tg32 homozygous. Overall, superior correlations were noted for the Tg32 homozygous mouse CL to human CL (r2 = 0.83, r = 0.91, p < 0.01) compared to the Tg32 hemizygous mouse (Fig. 6). For the panel of 27 mAbs examined, there were no trends or correlations observed that suggest mAb isotype, soluble vs membrane-bound target ligand, or framework have an effect on the overall correlation results for this data set (data not shown). Existence of non-dose proportional PK can affect the analysis of CL correlation 6 because the non-linear PK can be due to TMDD or immunogenicity. In this study, we focused our analysis on the linear portion of CL by either selecting mAbs only exhibiting dose proportional PK or using CL at the highest dose level where non-linear PK should be saturated and does not contribute to the calculated CL value.
Predicting human PK from animal models serves as the first step toward dose selection for a FIH study, and it helps predict achievable pharmacodynamic ranges. Early allometric scaling efforts to predict human CL of small molecules were based on whole body metabolic rate differences between species, defined by an exponent of 0.75.43,44 For biotherapeutic mAbs with linear PK or saturated TMDD, it has recently been suggested that an exponent greater than 0.75 may provide more accurate estimates of human CL 4-7 from NHP PK. Results from our study corroborate this, with improved human CL predictions resulting from use of an exponent of 0.80 for NHP, allowing accurate prediction of human CL within 2-fold error for 93% of mAbs for this data set and 90% when also including literary mAb PK data (Fig. 7). Notably, the Tg32 homozygous mouse accurately (defined as within 2-fold of observed CL) predicted human CL for 100% of mAbs tested relative to 93%, 93% and 82% for NHP, Tg32 hemizygous mice, and WT mice, respectively. Moreover, applying the best fit exponent developed from this study to predict the human CL of 6 marketed biotherapeutic mAbs did not change the exponent selection or accuracy of human CL predictions. These results are in alignment with the premise that FcRn represents the predominant protective pathway contributing to CL for mAbs, and further supports the utility of the Tg32 homozygous model in the preclinical PK assessment of therapeutic mAbs in drug development.
In addition to the full-length standard mAbs included in this study, other drugs, such as Fc-fusion proteins and immunoconjugates, e.g., antibody-drug conjugates (ADCs), contain an Fc modality capable of binding to FcRn to undergo systemic recycling. While our investigation sought to characterize the potential of the Tg32 mouse model for the human PK prediction of mAbs that are commonly in development, there may be a broader application for this mouse model for additional classes of drugs that bind FcRn. Complexities are likely to arise in the interpretation of ADC PK in the hFcRn transgenic mouse model because the site and drug ratio of conjugation, antibody carrier, linker, and chemical payload may all have the possibility to influence PK.45-48 To date these drug classes have yet to be described in the hFcRn transgenic mouse model; however, they represent a complex next step of interest to continue to evaluate the capacity for PK interpretation of the Tg32 mouse and the relationship to human PK. In Fig. 8, we show a recommended approach for utility of the Tg32 hemizygous and homozygous mouse model to apply in preclinical drug development of mAbs. Fig. 8A describes the current approach for preclinical mAb PK assessment, where NHP is the gold standard for predicting mAb PK and allometric scaling to predict human PK and FIH dose. In Fig. 8B, we suggest an approach that uses the hFcRn Tg32 homozygous mouse model earlier in discovery (i.e., earlier than the common practice of using NHP PK studies during the preclinical phase) to select lead mAb therapeutics for ideal PK properties, and de-select compounds with non-ideal PK properties, and use the data to scale allometrically to predict human PK. While NHPs remain the appropriate animal model for evaluating preclinical toxicity of mAbs, the objective is to use the Tg32 homozygous mouse model earlier in drug development to provide an accurate PK assessment. Fig. 8C details the characteristics of the Tg32 hemizygous mouse model in comparison to the homozygous model for suggested applications of use in preclinical drug development of mAbs.
Figure 8.
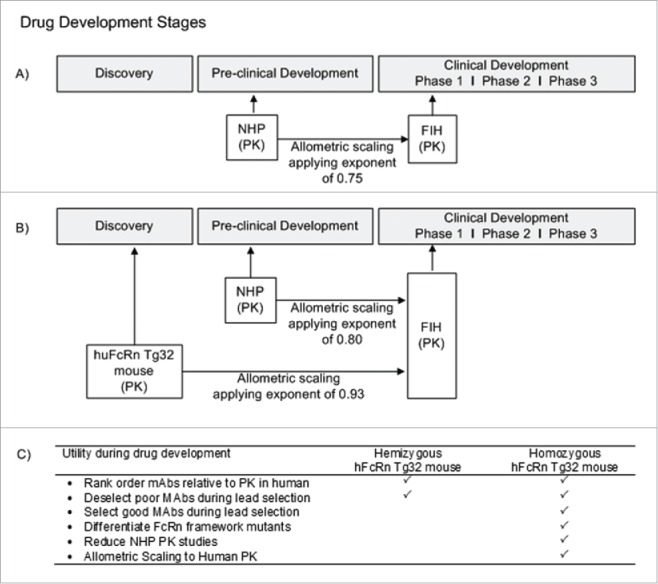
Utility of hFcRn Tg32 Mouse Model. (A) Current timeline for utility of NHP PK studies in preclinical development. (B) Suggested timeline for utility of hFcRn Tg32 PK studies, showing earlier use in discovery and application of allometric scaling using the exponent 0.93 to scale to human PK. (C) Use and selection of hemizygous vs homozygous Tg32 model.
Our work has expanded upon previous literature studies of mAb PK in the Tg32 mouse, further defining the mouse model genotypes and characterizing the utility of this mouse model to directly apply to early drug discovery and development processes. In preclinical drug development, researchers need to be able to select mAbs with favorable PK properties early in development, and to de-select earlier in development mAbs with PK properties that are unfavorable based on therapeutic need. This ability is particularly important to quantify PK with reasonable certainty when projecting human PK for potential lead drug candidates. Our results support the utility of the Tg32 homozygous mouse model in this regard. The Tg32 homozygous mouse is a particularly valuable tool to predict human PK alternatively to NHP for a number of reasons including: (1) economical and ethical concerns; (2) ease of handling; (3) lower costs and (4) lower drug material requirements. This mouse model therefore can be implemented in discovery and preclinical drug development areas to predict human PK, which can result in an overall decreased usage of monkeys for PK studies, enhance the ability to select lead molecules early in discovery, and ultimately decrease the development time needed for a mAb drug candidate to reach the clinic.
Materials and methods
Antibodies
In this study, a total of 27 mAbs were evaluated in the hFcRn transgenic mouse model, which included 21 Pfizer mAbs and 6 marketed mAbs (Rituximab-US, Infliximab-US, Trastuzumab-US, Adalimumab-US, Bevacizumab-US, Ustekinumab-US). The 21 Pfizer mAbs were generated as recombinant proteins using a Chinese hamster ovary cell platform. Test set mAbs are all commercially available compounds and were obtained as US pharmaceutical-grade drug product.
Historical PK data
For the 21 Pfizer mAbs evaluated in this study, a retrospective analysis of historical PK studies in WT mouse, NHP or human was conducted and CL determined (Supplemental Table 1). For the marketed mAbs CL data was obtained from literature sources.4-7,34,49
WT mouse
Historical WT mouse PK information was available for 13/21 Pfizer mAbs from single IV dose studies conducted in C57Bl/6, BALB/C, CD-1, DBA and target knockout mice for PK analysis.
NHP
Historical NHP PK information was available for 20/21 Pfizer mAbs. The PK studies were conducted in cynomolgus monkeys, administered as a single IV dose for PK analysis.
Human
Historical human PK information was available for 9/21 Pfizer mAbs. The single dose IV PK studies were conducted in healthy subjects or patients.
Quantitation of hFcRn in Tg32 mouse tissue via LC-HRMS
Five polyclonal antibodies against hFcRn immunogen peptides were generated in rabbits and ligand affinity purified to their respective individual peptide prior to use. A 2.1 × 30 mm PEEK cartridge was packed with 200 µl slurry of POROS protein G beads (Applied Biosystems, Life Technologies, Foster City, CA). The five independently ligand affinity-purified anti-peptide antibodies were mixed at 200 µg each, and loaded to the packed column in 100 mM sodium acetate, pH 5.7 in a closed loop. Antibodies were cross-linked to protein G with 7.78 mg/mL dimethyl pimelimidate (DMP) (Sigma, St. Louis, MO) in 100 mM triethanolamine, pH 8.2. This multiplex antibody column was used for hFcRn peptide immuno-affinity sample enrichment. Tissues from hemizygous and homozygous Tg32 mice were collected between weeks 15-18 at the end of their respective PK studies (n = 5 of each), including epididymal fat, spleen, brain, heart, stomach, small and large intestine, lymph node, kidney, liver, lung, skeletal muscle, pancreas, and skin.
Prior to tissue collection, whole body perfusion was conducted using heparinized phosphate-buffered saline (PBS). Tissues (50 mg) were bead homogenized in 1 mL of cold TER-I buffer (FNN0071, Invitrogen Life Technologies) with freshly added 1X Halt Protease Inhibitor Cocktail (78430, Thermo Scientific). Homogenates were clarified by centrifugation at 4°C, 10,000 × g for 15 min. Calibration standards were prepared by adding recombinant full-length human FcRn protein (OriGene) into control mouse liver lysate prepared from BALB/C mice (+mFcRn, -hFcRn) that served as calibration matrices for all tissues. Twelve calibration standards were prepared, ranging from 1.76–900 ng/ml.
Detergents were removed by protein precipitation with cold acetone (1:4, v/v) at −20°C for 1 hour. To solubilize precipitated protein pellets, 250 µg of TPCK-treated trypsin (T1426, Sigma) was added to each sample and incubated overnight at 37°C. After protein pellet solubilization, 500 fmol of stable isotope-labeled (SIL) peptides was added to the lysate, followed by 1 hour reduction with 5 mM dithiothreitol at 37°C, 1 hour alkylation with 10 mM iodoacetamide at room temperature (RT) in the dark and overnight digestion with 30 µg of fresh TPCK-treated trypsin. Digested peptides were diluted with 1 M urea in 25 mM ammonium formate (1:9, v/v), and loaded onto FcRn peptide immuno-affinity column. The antibody column was equilibrated with 25 mM ammonium formate, pH 7, followed by a wash with 500 mM ammonium formate. Bound peptides were eluted by 0.4% trifluoroacetic acid and trapped onto PepMap300 C18 trap column (5 × 0.3 mm, 5 µm, 300 Å, Thermo). The antibody column was then washed with 0.75% formic acid in 3% isopropanol, and re-equilibrated with 25 mM ammonium formate. Similar anti-peptide enrichment approach has been applied to soluble biomarker analysis in serum and tissue.50,51 Peptides on the C18 trap column were chromatographically separated by PepMap C18 (15 cm × 75 µm, 3 µm, 100 Å, Thermo). Analyte peptides were introduced into a Q Exactive quadrupole orbitrap mass spectrometer using an EasySpray Source (Thermo Scientific, USA). Targeted selected ion monitoring method was employed with a multiplexing degree of 4. Each target was monitored with a ± 1.5 min retention time window, 3-amu mass isolation window, AGC target value of 1 × 105, resolution of 140,000 FWHM at m/z = 200, and maximum ion injection time of 160 ms. Total hFcRn concentrations are determined via interpolation of a standard curve measured against purified recombinant hFcRn and normalized by tissue weight (mg).
Animal PK studies conducted
Supplementary Table 1 details all mouse PK studies conducted in this investigation: dose administered, strain of mice and method of mAb quantitative analysis. Mice used in this study included hFcRn Tg32 strain (cat#014565, hemizygous and homozygous), hFcRn Tg276 strain (cat#004919, homozygous), null for mouse FcRn (cat#003982), and WT C57Bl/6 (cat#000664), obtained from The Jackson Laboratory (Bar Harbor, ME). All mice were treatment-naïve male mice between the ages of 6 to 10 weeks at study start. PK studies were conducted at Pfizer Inc., and were designed and executed within accordance of the Animal Use Protocol (AUP) and adherence to the Pfizer institutional animal care and use committee (IACUC) regulations. MAbs were dosed at 5 mg/kg with a dose volume of 4 mL/kg (with the exception of Rituximab-US dosed at 10 mg/kg; mAb14 and mAb14-FcRn+ dosed at 2 mg/kg). A dose of 5 mg/kg was selected to saturate target binding in the event of cross reactivity to mouse target, thus mitigating any TMDD to obtain linear PK parameter estimates. For mAb14 and mAb14-FcRn+ dosed at 2 mg/kg, it had been previously determined that there was no cross reactivity in the mouse, and therefore results were considered to represent linear CL. A total of 4-6 animal replicates were evaluated for each mAb with study durations ranging from 4 to 12 weeks depending on the test mAb.
Selection of hFcRn transgenic mouse model
PK studies were conducted in Tg32 and Tg276 mouse strains and compared to WT mouse (C57Bl/6, mFcRn) and FcRn KO mice for mAb14, mAb04 and mAb17. The resulting mAb CL from the mouse strain that most closely correlates with NHP CL will be selected for further characterization and prediction of mAb human CL.
Use of hFcRn Tg32 mouse model to distinguish PK of mAb framework variants
To further understand the sensitivity of PK differentiation for each Tg32 mouse genotype, 4 matched pairs of WT mAb (mAb01, mAb02, mAb07, mAb14) compared to Fc framework mutation mAbs for enhanced FcRn binding (mAb-FcRn+) were administered to both the Tg32 hemizygous and homozygous mice. MAb CL was compared between the WT mAb and the FcRn+ mAb for each animal model.
Correlation of mAb CL to NHP and human
17/27, 25/27 and 15/27 mAbs had PK data for correlative analysis for WT mouse, NHP and human, respectively. 27/27 and 25/27 mAbs were administered to Tg32 hemizygous and homozygous mice, respectively, and the resulting mAb CL values were correlated to NHP and human CL. Each correlation was assessed using a regression fit and Pearson correlation coefficient as described in statistical analysis section.
Dose preparations
Dose preparations were made appropriately for 2 PK study and analytical approaches: 1) quantitative mAb concentration analysis using a ligand binding assay (LBA); or 2) administration of 125-iodine radiolabeled mAb (125I-mAb) and analysis measuring radioactivity counts. Study approach was determined based on available resources (Supplemental Table 1 details dose and method of analysis for each mAb administered). Previous reports have demonstrated the 125I-mAb vs. LBA method of analysis does not affect the resulting PK analysis.52
PK Studies using LBA analysis
22/27 mAbs were administered IV and analyzed by LBA. MAbs were prepared in 10 mM histidine, 5% sucrose, pH 6.0 buffer, to a final concentration of 1.25 mg/mL (2.5 mg/mL for Rituximab-US and 0.5 mg/mL for mAb14 and mAb14-FcRn+). Dose concentration was confirmed using a Nanodrop 8000 (Thermo Scientific, Waltham, MA) and standard IgG (150 KDa) molar extinction coefficient of 210,000 M−1cm−1. A previously described serial sampling approach35was used, where 10-12 serial tail stick bleeds were obtained per animal over a scheduled time course.
PK Studies using 125I-mAb
5/27 mAbs were administered as 125I-mAb. Radio-iodination (125I) of mAbs for PK studies was performed using the succinimidyl iodobenzoate (SIB) iodination method.52 Briefly, 2-3 mCi of Na125I (Perkin-Elmer, Billerica, MA) was reacted with 5-8 μg N-succinimidyl-3-(tri-n-butylstannyl) benzoate (American Advanced Scientific, College Station, TX) to generate [125I]SIB, which in turn was reacted with 1-2 mg of each test mAb, essentially as described.52 The labeled proteins were purified by gel filtration over PD-10 desalting columns (GE Healthcare, Piscataway, NJ) to remove unconjugated [125I]SIB and protein concentrations verified by UV spectroscopy. Dosing solutions were prepared by mixing unlabeled mAbs with the corresponding 125I-mAb to a final concentration of 1.25 mg/mL in buffer (PBS-CMF, 1% bovine serum albumin (BSA) or 10 mM histidine, 5% sucrose, 1% BSA, pH 6.0). BSA (US Biological, Salem, MA) was added to protect the test article against radiolysis due to free radicals generated by gamma radiation. The radioactive specific activity of the dosing solutions was 70-100 μCi/mg for 2 mAbs that utilized a composite plasma sampling approach (mAb04 and mAb17), and 400-500 μCi/mg for all other mAbs using a serial plasma sampling approach. Radiochemical purity of dosing solutions was characterized by trichloroacetic acid (TCA; Sigma-Aldrich, St. Louis, MO) precipitation52 and size-exclusion HPLC using an Agilent Bio SEC-3 column (Agilent Technologies, Santa Clara, CA). The percentage of free 125I was less than 1% in all dosing solution preparations. Animals receiving 125I -mAbs were pre-treated with 20 mM potassium iodine water for 3 d prior to study start to reduce the thyroid uptake of any unbound free 125I generated in vivo.
mAb quantification by radioactivity
For samples analyzed by serial sampling following 125I-mAb administration, 20 μL of whole blood was collected at each time point via a heparinized capillary tube and mixed with 80 μL of rabbit serum (Sigma Aldrich, St. Louis, MO), centrifuged at 1,100 × g for 10 minutes at RT and the diluted plasma supernatant aliquoted into a clean tube for analysis of radioactivity. For samples analyzed by composite sampling, whole blood was collected via sub-mandibular (non-terminal timepoint) or cardiac puncture (terminal) and processed directly to plasma following centrifugation at 1,100 × g for 10 minutes at RT and the plasma supernatant aliquoted into a clean tube for analysis of radioactivity.
Soluble 125I counts were determined by TCA precipitation to quantify the amount of free 125I (soluble) in the plasma samples and establish the integrity of mAb in vivo throughout the PK sampling time course compared to the precipitable (intact mAb) counts. An equal part of cold (4 °C) 4% TCA solution was added to each plasma sample, mixed, and centrifuged at 12,000 rpm for 15 minutes at 4 °C. The supernatant was transferred to a clean tube and analyzed for soluble 125I counts accounting for the time of analysis and for the 60.2 day half-life for the decay rate of 125I. Plasma concentrations were measured using a gamma counter (Wallac Wizard 1480, Perkin Elmer, Waltham, MA) and calculated accounting for hematocrit and sample dilution as appropriate.
mAb quantification by LBA
For samples collected via serial sampling for LBA analysis, 10 μL of whole blood was collected at each time point via a heparinized capillary tube and mixed with 90 μL of assay buffer [Rexxip A Buffer (Gyros US. Inc. Warren, NJ, USA) for mAb14 and mAb14-FcRn+, and 0.2 M Tricine, pH 8.5, 1% BSA, 0.5 M NaCl, 0.1% Zwittergent, 0.05% Proclin300 for all other mAbs] to obtain a minimum required dilution (MRD) of 1:10. The diluted blood sample was centrifuged at 1,100 × g for 10 minutes at RT and the supernatant aliquoted into a clean tube and stored at −80°C until analysis. To calculate plasma concentrations from the diluted whole blood matrix a dilution factor of 17.3635 for mAb14 and mAb14-FcRn+ and of 18.18 for all other mAbs were applied to correct for the hematocrit component and the lysis of red blood cells respectively. Quantitative bioanalysis was performed utilizing a Gyrolab (Gyrolab™ xP, Gyros US. Inc. Warren, NJ, USA) immunoassay platform. A generic human IgG assay format was utilized, each mAb assay was independently optimized and qualified to give the required sensitivity and dynamic range. mAb reference standards and quality controls were prepared in assay buffer. For each assay, inter-day accuracy, precision, selectivity, specificity and dilutional linearity were examined and precision and accuracy of spiked quality controls met pre-established acceptance criteria (%CV ≤ 20% and %bias ≤ 30% of nominal). The assay range of quantitation was 23.6 ng/mL –5760 ng/mL in 100% mouse plasma for all mAbs tested, with these exceptions: mAb06 and mAb16, which had an assay range of quantitation of 9.4 – 5760 ng/mL; and mAb14 and mAb14-FcRn+, which had an assay range of quantitation of 52.4–32,000 ng/mL. The qualification data was generated using Gyrolab Evaluator Software (Version 3.1.5.137).
A generic human IgG assay format utilized an anti-human IgG biotin reagent captured onto streptavidin coated beads on the affinity capture column of the Gyrolab Bioaffy microstructure (Gyrolab Bioaffy 1000 nL CD, Gyros US. Inc. Warren, NJ, USA). Quantification of all mAbs in this study used a capture reagent of mouse anti-human IgG biotin (Southern Biotech, Birmingham, AL, USA). Bound mAb was detected with Alexa 647-labeled donkey anti-human IgG (H+L) (Jackson Immunoresearch Labs, West Grove, PA, USA). Gyrolab Rexxip F Buffer was used for reagent dilution (Gyros US. Inc. Warren, NJ, USA). For mAb 14 and mAb14-FcRn+ only donkey anti-human IgG (H+L) biotin (Jackson ImmunoResearch Labs, West Grove, PA, USA) was used as capture reagent and Alexa 647-labeled mouse anti-human IgG (Southern Biotech, Birmingham, AL, USA) as detector reagent. The detected fluorescent signal on the column was quantified by response units and measured by the Gyrolab instrument using a 1% Photomultiplier tube (PMT) setting. The concentration of study samples from each mAb were determined by interpolation from a standard curve using a 5-parameter logistic curve fit with 1/y2 response weighting using Watson LIMS Software version 7.4 (Thermo Scientific Inc. Waltham, MA USA).
PK analysis
PK was determined from individual animal data using non-compartmental analysis in WinNonlin (Version 6.3.0.395, Pharsight, CA) using the Plasma Data Module. Concentration values below the limit of quantitation (BLQ) were set to 0 ng/mL for all PK calculations. PK profiles from an animal that showed a sharp drop in concentration, typical of anti-drug antibody (ADA) LBA interference, were excluded from PK calculations. Terminal data points in PK profiles that were presumed to be affected by TMDD were excluded from CL estimation (mAb17 only). Historical mAb CL values in each species were assessed using strict criteria to define each CL value as definitive linear, apparent linear, or non-linear. Definitive linear CL was selected as a mean CL value over a linear dose range from a dose escalating study. Apparent linear CL values were determined from a dose escalation study where only the highest dose cohort is observed to approach linearity, or from a single dose study with a dose greater than 10 mg/kg, believed to saturate the target. Non-linear CL values, presumed due to either a TMDD or ADA effect, were excluded from this study, with the exception of mAb17, where only non-linear CL was obtained, and was thus exempt from subsequent correlative or allometric scaling analyses.
Allometric scaling
A simple allometric scaling approach was used to calculate predictive human PK parameters:
| (1) |
where Y is the predicted parameter, BM (body mass) and X are the allometric coefficient and exponent, respectively. This approach was applied to CL estimates as follows:
| (2) |
where CLpred(human) is the human CL predicted from the observed animal CL (CLobs(animal)), BMhuman is the human body mass (70 kg), BManimal is the mouse body mass (0.02 kg) or cynomolgus monkey body mass (3.5 kg), and X is the allometric scaling exponent. CL predictions were calculated using the traditional Kleiber's law exponent of 0.75,43,44 and compared to CL predictions calculated using an empirical best fit approach.
Allometric scaling was performed for each animal model to predict human CL first using a training dataset of the 9 Pfizer mAbs (those with human PK data) and verified for each approach using a test set of 6 marketed mAbs. A traditional approach using an exponent of 0.75 was compared to empirically derived exponents for each animal model to predict human CL with greatest accuracy. Empirically derived best fit exponents derived from the training data set and verified by the test set of mAbs were determined for each animal model using a linear regression of observed human CL to predicted human CL to evaluate which exponent yielded greater accuracy and precision to the line of unity, determined by a concordance correlation coefficient (CCC) statistical analysis,53 detailed in the statistical analysis methods section. Percentage (%) accuracy in CL projections for each animal model at a given scaling exponent is determined as the number of predictions that fall within a 2-fold error from the line of unity relative to predictions that fall outside of the 2-fold error limits.
Statistical analysis
Analysis of CL correlations from each animal model to mAb CL in either NHP or human was performed using GraphPad Prism (version 6.03) to determine goodness of fit (r2), as well as Pearson correlation coefficient (r) where statistical significance level is set at 0.05. All correlations were fitted using a linear regression fit with the exception of the non-linear correlation of Tg32 hemizygous to human CL (Inset of Fig. 6C), which was fit using a one site specific binding regression with a Hill slope.
A Student's unpaired t-test analysis was performed to compare CL values for mAb-FcRn+ compared to parent mAb. Significant protein expression differences of hFcRn between Tg32 homozygous and hemizygous genotypes were analyzed using unpaired Mann-Whitney test. Differences were considered as statistically significant if p < 0.1 (labeled as *), and very statistically significant if p < 0.05 (labeled as **).
CCC is used to measure the agreement between the predicted human CL from each animal and the observed human CL. Like Pearson correlation coefficient (r), CCC also ranges from -1 to 1. However, these 2 coefficients are different correlation measures. Pearson correlation coefficient (r) measures how one variable is linearly related to the other variable. CCC measures not only the linear relationship between 2 variables, but also the closeness between the best-fit line and the 45 degree unity line. Therefore, CCC is affected by both the accuracy (reflected by the agreement) and the precision of the data. Higher accuracy and precision correspond to higher CCC. In this paper, CCC was calculated using the statistical package R i386 (version no. 3.1.2). The best exponent for each animal model was empirically chosen as the one that resulted in the highest CCC value. A Mann-Whitney test was performed to compare statistical difference of protein expression in Tg32 homozygous and hemizygous mice.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
All authors are employees of Pfizer Worldwide Research & Development. The authors are grateful to the following individuals for their contributions to this work: Steve Penn, Rohit Yadav, David Cirelli, Laird Bloom, Yulia Vugmeyster, Nicole Piche-Nicholas, Laura Danner, Karsten Fynboe, Angela Hernandez, Cynthia Filliettaz, Katie McGinn, Adam Root, Chris Lepsy, Shraddah Desal, Christine Ditondo, Terri Caiazzo, Jean Donley, Eugenia Kraynov, Rhys Jones, Bing Kuang, Haojing Rong, Scott Fountain, Tracey Clark, Allison Given-Chunyk, Joann Wentland, Kathleen Pelletier.
References
- 1.Reichert JM. Antibodies to watch in 2015. mAbs 2015; 7:1–8; PMID:25484055; http://dx.doi.org/ 10.4161/19420862.2015.988944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi S. Biologics: an update and challenge of their pharmacokinetics. Curr Drug Metab 2014; 15:271–90; PMID:24745789; http://dx.doi.org/ 10.2174/138920021503140412212905 [DOI] [PubMed] [Google Scholar]
- 3.Hotzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al.. A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012; 4:753–60; PMID:23778268; http://dx.doi.org/ 10.4161/mabs.22189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oitate M, Masubuchi N, Ito T, Yabe Y, Karibe T, Aoki T, Murayama N, Kurihara A, Okudaira N, Izumi T.. Prediction of human pharmacokinetics of therapeutic monoclonal antibodies from simple allometry of monkey data. Drug Metab Pharmacokinet 2011; 26:423–30; PMID:21606605; http://dx.doi.org/ 10.2133/dmpk.DMPK-11-RG-011 [DOI] [PubMed] [Google Scholar]
- 5.Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? mAbs 2011; 3:61–6; PMID:20962582; http://dx.doi.org/ 10.4161/mabs.3.1.13799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong JQ, Salinger DH, Endres CJ, Gibbs JP, Hsu CP, Stouch BJ, Hurh E, Gibbs MA.. Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first-in-human prediction. Clin Pharmacokinet 2011; 50:131–42; PMID:21241072; http://dx.doi.org/ 10.2165/11537430-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 7.Ling J, Zhou H, Jiao Q, Davis HM. Interspecies scaling of therapeutic monoclonal antibodies: initial look. J Clin Pharmacol 2009; 49:1382–402; PMID:19837907; http://dx.doi.org/ 10.1177/0091270009337134 [DOI] [PubMed] [Google Scholar]
- 8.Mager DE. Target-mediated drug disposition and dynamics. Biochem Pharmacol 2006; 72:1–10; PMID:16469301; http://dx.doi.org/ 10.1016/j.bcp.2005.12.041 [DOI] [PubMed] [Google Scholar]
- 9.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715–25; PMID:17703228; http://dx.doi.org/ 10.1038/nri2155 [DOI] [PubMed] [Google Scholar]
- 10.Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, et al.. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol 2003; 170:3528–33; PMID:12646614; http://dx.doi.org/ 10.4049/jimmunol.170.7.3528 [DOI] [PubMed] [Google Scholar]
- 11.Challa DK, Velmurugan R, Ober RJ, Sally Ward E. FcRn: from molecular interactions to regulation of IgG pharmacokinetics and functions. Curr Top Microbiol Immunol 2014; 382:249–72; PMID:25116104 [DOI] [PubMed] [Google Scholar]
- 12.Wines BD, Powell MS, Parren PW, Barnes N, Hogarth PM. The IgG Fc contains distinct Fc receptor (FcR) binding sites: the leukocyte receptors Fc gamma RI and Fc gamma RIIa bind to a region in the Fc distinct from that recognized by neonatal FcR and protein A. J Immunol 2000; 164:5313–8; PMID:10799893; http://dx.doi.org/ 10.4049/jimmunol.164.10.5313 [DOI] [PubMed] [Google Scholar]
- 13.Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, Dall'Acqua WF. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem 2014; 289:7812–24; PMID:24469444; http://dx.doi.org/ 10.1074/jbc.M113.537563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward ES, Zhou J, Ghetie V, Ober RJ. Evidence to support the cellular mechanism involved in serum IgG homeostasis in humans. Int Immunol 2003; 15:187–95; PMID:12578848; http://dx.doi.org/ 10.1093/intimm/dxg018 [DOI] [PubMed] [Google Scholar]
- 15.Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol 2004; 172:2021–9; PMID:14764666; http://dx.doi.org/ 10.4049/jimmunol.172.4.2021 [DOI] [PubMed] [Google Scholar]
- 16.Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006; 281:23514–24; PMID:16793771; http://dx.doi.org/ 10.1074/jbc.M604292200 [DOI] [PubMed] [Google Scholar]
- 17.Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC.. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol 2006; 18:1759–69; PMID:17077181; http://dx.doi.org/ 10.1093/intimm/dxl110 [DOI] [PubMed] [Google Scholar]
- 18.Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR.. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol 2010; 28:157–9; PMID:20081867; http://dx.doi.org/ 10.1038/nbt.1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robbie GJ, Criste R, Dall'acqua WF, Jensen K, Patel NK, Losonsky GA, Griffin MP. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother 2013; 57:6147–53; PMID:24080653; http://dx.doi.org/ 10.1128/AAC.01285-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu X, Meng G, Dickinson BL, Li X, Mizoguchi E, Miao L, Wang Y, Robert C, Wu B, Smith PD, et al.. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J Immunol 2001; 166:3266–76; PMID:11207281; http://dx.doi.org/ 10.4049/jimmunol.166.5.3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goebl NA, Babbey CM, Datta-Mannan A, Witcher DR, Wroblewski VJ, Dunn KW. Neonatal Fc receptor mediates internalization of Fc in transfected human endothelial cells. Mol Biol Cell 2008; 19:5490–505; PMID:18843053; http://dx.doi.org/ 10.1091/mbc.E07-02-0101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol 2007; 179:4580–8; PMID:17878355; http://dx.doi.org/ 10.4049/jimmunol.179.7.4580 [DOI] [PubMed] [Google Scholar]
- 23.Borvak J, Richardson J, Medesan C, Antohe F, Radu C, Simionescu M, Ghetie V, Ward ES.. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol 1998; 10:1289–98; PMID:9786428; http://dx.doi.org/ 10.1093/intimm/10.9.1289 [DOI] [PubMed] [Google Scholar]
- 24.Dickinson BL, Badizadegan K, Wu Z, Ahouse JC, Zhu X, Simister NE, Blumberg RS, Lencer WI.. Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J Clin Invest 1999; 104:903–11; PMID:10510331; http://dx.doi.org/ 10.1172/JCI6968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah U, Dickinson BL, Blumberg RS, Simister NE, Lencer WI, Walker WA. Distribution of the IgG Fc receptor, FcRn, in the human fetal intestine. Pediatr Res 2003; 53:295–301; PMID:12538789; http://dx.doi.org/ 10.1203/00006450-200302000-00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hornby PJ, Cooper PR, Kliwinski C, Ragwan E, Mabus JR, Harman B, Thompson S, Kauffman AL, Yan Z, Tam SH, et al.. Human and non-human primate intestinal FcRn expression and immunoglobulin G transcytosis. Pharma Res 2014; 31:908–22; PMID:24072267; http://dx.doi.org/ 10.1007/s11095-013-1212-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen N, Wang W, Fauty S, Fang Y, Hamuro L, Hussain A, Prueksaritanont T.. The effect of the neonatal Fc receptor on human IgG biodistribution in mice. mAbs 2014; 6:502–8; PMID:24492305; http://dx.doi.org/ 10.4161/mabs.27765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yip V, Palma E, Tesar DB, Mundo EE, Bumbaca D, Torres EK, Reyes NA, Shen BQ, Fielder PJ, Prabhu S,. Quantitative cumulative biodistribution of antibodies in mice: effect of modulating binding affinity to the neonatal Fc receptor. mAbs 2014; 6:689–96; PMID:24572100; http://dx.doi.org/ 10.4161/mabs.28254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tam SH, McCarthy SG, Brosnan K, Goldberg KM, Scallon BJ. Correlations between pharmacokinetics of IgG antibodies in primates vs. FcRn-transgenic mice reveal a rodent model with predictive capabilities. mAbs 2013; 5:397–405; PMID:23549129; http://dx.doi.org/ 10.4161/mabs.23836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T.. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos 2011; 39:1469–77; PMID:21610128; http://dx.doi.org/ 10.1124/dmd.111.039453 [DOI] [PubMed] [Google Scholar]
- 31.Haraya K, Tachibana T, Nanami M, Ishigai M. Application of human FcRn transgenic mice as a pharmacokinetic screening tool of monoclonal antibody. Xenobiotica 2014; 44:1127–34; PMID:25030041; http://dx.doi.org/ 10.3109/00498254.2014.941963 [DOI] [PubMed] [Google Scholar]
- 32.Roopenian DC, Christianson GJ, Sproule TJ. Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol Biol 2010; 602:93–104; PMID:20012394; http://dx.doi.org/ 10.1007/978-1-60761-058-8_6 [DOI] [PubMed] [Google Scholar]
- 33.Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol 2001; 13:1551–9; PMID:11717196; http://dx.doi.org/ 10.1093/intimm/13.12.1551 [DOI] [PubMed] [Google Scholar]
- 34.Kelly RL, Sun T, Jain T, Caffry I, Yu Y, Cao Y, Lynaugh H, Brown M, Vásquez M, Wittrup KD, et al.. High throughput cross-interaction measures for human IgG1 antibodies correlate with clearance rates in mice. mAbs 2015; 7:770–7; PMID:26047159; http://dx.doi.org/ 10.1080/19420862.2015.1043503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joyce AP, Wang M, Lawrence-Henderson R, Filliettaz C, Leung SS, Xu X, O'Hara DM. One mouse, one pharmacokinetic profile: quantitative whole blood serial sampling for biotherapeutics. Pharma Res 2014; 31:1823–33; PMID:24464271; http://dx.doi.org/ 10.1007/s11095-013-1286-y [DOI] [PubMed] [Google Scholar]
- 36.Van Oortmerssen GA. Biological Significance, Genetics and Evolutionary Origin of Variability in Behaviour Within and Between Inbred Strains of Mice (Mus Musculus). Behaviour 1971; 38:1–91; PMID:5100645; http://dx.doi.org/ 10.1163/156853971X00014 [DOI] [PubMed] [Google Scholar]
- 37.Borrok MJ, Wu Y, Beyaz N, Yu XQ, Oganesyan V, Dall'Acqua WF, Tsui P. pH-dependent binding engineering reveals an FcRn affinity threshold that governs IgG recycling. J Biol Chem 2015; 290:4282–90; PMID:25538249; http://dx.doi.org/ 10.1074/jbc.M114.603712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dall'Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S.. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol 2002; 169:5171–80; PMID:12391234; http://dx.doi.org/ 10.4049/jimmunol.169.9.5171 [DOI] [PubMed] [Google Scholar]
- 39.Proetzel G, Roopenian DC. Humanized FcRn mouse models for evaluating pharmacokinetics of human IgG antibodies. Methods 2014; 65:148–53; PMID:23867339; http://dx.doi.org/ 10.1016/j.ymeth.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn 2012; 39:67–86; PMID:22143261; http://dx.doi.org/ 10.1007/s10928-011-9232-2 [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, Balthasar JP. Evaluation of a catenary PBPK model for predicting the in vivo disposition of mAbs engineered for high-affinity binding to FcRn. AAPS J 2012; 14:850–9; PMID:22956476; http://dx.doi.org/ 10.1208/s12248-012-9395-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan Y-Y, Neubert H. Quantitative Analysis of Human Neonatal Fc Receptor (FcRn) Tissue Expression in Transgenic Mice by Online Peptide Immuno-Affinity LC-HRMS. Anal Chem 2016; 88:4239–47; PMID:27012525; http://dx.doi.org/ 10.1021/acs.analchem.5b03900 [DOI] [PubMed] [Google Scholar]
- 43.Adolph EF. Quantitative Relations in the Physiological Constitutions of Mammals. Science 1949; 109:579–85; PMID:17835379; http://dx.doi.org/ 10.1126/science.109.2841.579 [DOI] [PubMed] [Google Scholar]
- 44.Kleiber M. Body size and metabolic rate. Physiol Rev 1947; 27:511–41; PMID:20267758 [DOI] [PubMed] [Google Scholar]
- 45.Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, et al.. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 2012; 30:184–9; PMID:22267010; http://dx.doi.org/ 10.1038/nbt.2108 [DOI] [PubMed] [Google Scholar]
- 46.Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al.. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 2004; 10:7063–70; PMID:15501986; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-0789 [DOI] [PubMed] [Google Scholar]
- 47.Polson AG, Calemine-Fenaux J, Chan P, Chang W, Christensen E, Clark S, de Sauvage FJ, Eaton D, Elkins K, Elliott JM, et al.. Antibody-drug conjugates for the treatment of non-Hodgkin's lymphoma: target and linker-drug selection. Cancer Res 2009; 69:2358–64; PMID:19258515; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2250 [DOI] [PubMed] [Google Scholar]
- 48.Chapman AP, Antoniw P, Spitali M, West S, Stephens S, King DJ. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat Biotechnol 1999; 17:780–3; PMID:10429243; http://dx.doi.org/ 10.1038/11717 [DOI] [PubMed] [Google Scholar]
- 49.Dayde D, Ternant D, Ohresser M, Lerondel S, Pesnel S, Watier H, Le Pape A, Bardos P, Paintaud G, Cartron G. et al.. Tumor burden influences exposure and response to rituximab: pharmacokinetic-pharmacodynamic modeling using a syngeneic bioluminescent murine model expressing human CD20. Blood 2009; 113:3765–72; PMID:19029438; http://dx.doi.org/ 10.1182/blood-2008-08-175125 [DOI] [PubMed] [Google Scholar]
- 50.Palandra J, Finelli A, Zhu M, Masferrer J, Neubert H. Highly specific and sensitive measurements of human and monkey interleukin 21 using sequential protein and tryptic peptide immunoaffinity LC-MS/MS. Anal Chem 2013; 85:5522–9; PMID:23638938; http://dx.doi.org/ 10.1021/ac4006765 [DOI] [PubMed] [Google Scholar]
- 51.Neubert H, Muirhead D, Kabir M, Grace C, Cleton A, Arends R. Sequential protein and peptide immunoaffinity capture for mass spectrometry-based quantification of total human beta-nerve growth factor. Anal Chem 2013; 85:1719–26; PMID:23249404; http://dx.doi.org/ 10.1021/ac303031q [DOI] [PubMed] [Google Scholar]
- 52.Chen J, Wang M, Joyce A, DeFranco D, Kavosi M, Xu X, O'Hara DM. Comparison of succinimidyl [(125)I]iodobenzoate with iodogen iodination methods to study pharmacokinetics and ADME of biotherapeutics. Pharmaceutical research 2014; 31:2810–21; PMID:24844406; http://dx.doi.org/ 10.1007/s11095-014-1378-3 [DOI] [PubMed] [Google Scholar]
- 53.Lin LI. A concordance correlation coefficient to evaluate reproducibility. Biometrics 1989; 45:255–68; PMID:2720055; http://dx.doi.org/ 10.2307/2532051 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.