

Abstract
AIM: To clarify the relationship between autophagy and lipotoxicity-induced apoptosis, which is termed “lipoapoptosis,” in non-alcoholic steatohepatitis.
METHODS: Male C57BL/6J mice were fed a high-fat diet (HFD) for 12 wk, after which the liver histology and expression of proteins such as p62 or LC3 were evaluated. Alpha mouse liver 12 (AML12) cells treated with palmitate (PA) were used as an in vitro model.
RESULTS: LC3-II, p62, and Run domain Beclin-1 interacting and cysteine-rich containing (Rubicon) proteins increased in both the HFD mice and in AML12 cells in response to PA treatment. Rubicon expression was decreased upon c-Jun N-terminal kinase (JNK) inhibition at both the mRNA and the protein level in AML12 cells. Rubicon knockdown in AML12 cells with PA decreased the protein levels of both LC3-II and p62. Rubicon expression peaked at 4 h of PA treatment in AML12, and then decreased. Treatment with caspase-9 inhibitor ameliorated the decrease in Rubicon protein expression at 10 h of PA and resulted in enlarged AML12 cells under PA treatment. The enlargement of AML12 cells by PA with caspase-9 inhibition was canceled by Rubicon knockdown.
CONCLUSION: The JNK-Rubicon axis enhanced lipoapoptosis, and caspase-9 inhibition and Rubicon had effects that were cytologically similar to hepatocyte ballooning. As ballooned hepatocytes secrete fibrogenic signals and thus might promote fibrosis in the liver, the inhibition of hepatocyte ballooning might provide anti-fibrosis in the NASH liver.
Keywords: Ballooned hepatocyte, Caspase 9, c-Jun N-terminal kinase, Rubicon, SP600125
Core tip: Autophagy is interrupted in both in vivo and in vitro non-alcoholic steatohepatitis (NASH) models, and impaired autophagy is mediated by Run domain Beclin-1 interacting and cysteine-rich containing (Rubicon) protein expression via c-Jun N-terminal kinase phosphorylation. Rubicon expression appears prior to apoptosis and enhances palmitate toxicity in the hepatocytes, and caspase-9 decreases Rubicon at the protein level during lipoapoptosis. Caspase-9 inhibition with Rubicon expression induces both hepatocyte enlargement and endoplasmic reticulum stress accumulation. The present study extends our knowledge on the precise balance between lipoapoptosis and autophagy via Rubicon expression in NASH and reveals a possible pathophysiology of ballooned hepatocytes in NASH.
INTRODUCTION
The prevalence of non-alcoholic fatty liver disease (NAFLD) is drastically increasing in the Western countries[1]. In some NAFLD patients, persistent inflammation and progressive fibrosis develop in the liver, a condition termed non-alcoholic steatohepatitis (NASH)[2]. As NASH will progress to liver cirrhosis and end-stage liver disease, its pathogenesis needs to be clarified[3]. NASH is histologically characterized by lipid accumulation with inflammation in the liver, which is considered to be a result of lipotoxicity-induced hepatocyte apoptosis[2]. Infiltration of immune cells following hepatocyte death activates hepatic stellate cells, inducing the generation of collagen fiber and the development of liver fibrosis[2]. Therefore, hepatocyte apoptosis is considered to be an important step in development of NASH[4,5].
Lipid-induced apoptosis, which is termed “lipoapoptosis,” has been established to cause NASH[6,7]. Saturated free fatty acids (FFAs), such as palmitate (PA), induce hepatocyte lipoapoptosis via endoplasmic reticulum (ER) stress, c-Jun N-terminal kinase (JNK) phosphorylation, and mitochondrial dysfunction[8,9]. Recently, it has been reported that saturated FFAs can inhibit autophagy[10] which is a process of cellular self-digestion[11]. In addition, it has been demonstrated that autophagy is inhibited in the NASH liver[12]. As autophagy removes both aggregated proteins and damaged organelles, it maintains organelle quality and prevents apoptosis[11]. It has been reported that impaired autophagy is associated with lipoapoptosis and that chemical agent-induced apoptosis inhibits autophagy[10,13]. Accumulating evidence suggests that inhibition of autophagy is associated with hepatocyte apoptosis in the NASH liver. However, the underlying mechanism remains unclear, and gaining a clearer understanding of the inhibition of autophagy by lipoapoptosis may lead to the development of novel therapeutic strategies for NASH. In line with the above concept, we focused this study on a molecule associated with autophagy inhibition: Run domain Beclin-1 interacting and cysteine-rich containing (Rubicon)[14]. Rubicon has been found to inhibit the fusion of lysosomes to autophagosomes. However, whether Rubicon is expressed in the NASH liver and which role it would play in NASH has not been elucidated yet.
Another histopathological hallmark of NASH is the presence of ballooned hepatocytes. The number of ballooned hepatocytes generally correlates with the severity of liver inflammation and fibrosis in NASH[15-17]. Therefore, the prevalence of ballooned hepatocytes is considered to be a marker of NASH activity. Ballooned hepatocytes can be histologically identified on the basis of several features, such as swelling, central nuclei, rarefied cytoplasm, and Mallory-Denk bodies[18]. Mallory-Denk bodies contain several proteins, such as keratin, chaperones, kinases, and protein degradation machinery[19]. Although proteins in Mallory-Denk bodies should be decomposed via the protein degradation pathway, they are accumulated in ballooned hepatocytes. These findings indicate that the protein degradation pathway related to autophagy may be impaired in ballooned hepatocytes.
The aims of the present study were as follows: (1) to confirm the presence of an autophagic state in an in vivo NASH model using mice fed a high-fat diet (HFD); (2) to evaluate the intracellular signaling associated with both autophagy and lipoapoptosis; and (3) to clarify the relation between autophagy inhibition and ballooning of hepatocytes during lipoapoptosis.
MATERIALS AND METHODS
Animals
Male 5-wk-old C57BL/6J mice were obtained from Charles River Laboratories (Charles River, Yokohama, Japan) and were maintained on a 12-h light/12-dark cycle in humidity-controlled rooms at 22 °C with ad libitum access to drinking water. After 1 wk of habitation, 5 mice were assigned to each of normal chow and HFD (HFD-60, Oriental Yeast CO., Tokyo, Japan) groups and were fed their respective diets for 12 wk. All of the mice were sacrificed using isoflurane anesthesia after overnight fasting at 20 wk of age. All of the animal experiments were approved by the Animal Care and Use Committee of Iwate Medical University (Morioka, Japan; 25-025).
Cells
Alpha mouse liver 12 (AML 12) cells, a hepatocyte cell line from a mouse transgenic for human transforming growth factor α, were kindly supplied by Professor Itaru Kojima, Gunma University. Because autophagic status was confirmed in the NASH mouse model, we employed a cell line generated from the same species, AML 12, to investigate autophagic status in NASH in detail.
Antibodies and reagents: The antibodies used in this study were obtained from the following sources: anti-p62 (1:1000; #5114), anti-LC3 (1:1000; #4108), anti-Rubicon (1:1000; #8465), anti-cleaved caspase-3 (1:1000; #9661), and rabbit anti-phospho-JNK (1:1000; #9251) were obtained from Cell Signaling Technology, Tokyo, Japan; mouse anti-C/EBP homologous protein (CHOP) (1:500; sc-575), mouse anti-phospho-c-Jun (1:1000; sc-822), and goat anti-β-actin (1:1000; sc-1616) were obtained from Santa Cruz Biotechnology, Santa Cruz, CA, United States). Alexa Fluor 488-conjugated IgG was from Life Technologies (Tokyo, Japan). The JNK inhibitor SP600125 (#420119) was obtained from Calbiochem (San Diego, CA, United States). Caspase-9 inhibitor Z-LEHD-FMK (ab142026), and pan-caspase inhibitor QVD-OPh (ab141421) were obtained from Abcam Biochemicals (Tokyo, Japan).
Histological analysis: Liver tissues were collected from the mice, fixed in 10% neutral buffered formalin, paraffin-embedded, and sectioned. Liver specimens were stained with hematoxylin and eosin according to standard procedures. All samples were evaluated in a blinded manner by single pathologist, who scored inflammation, steatosis, and ballooning using non-alcoholic steatohepatitis activity scores[20].
Hepatocyte treatments: Palmitate (PA; #P5585, Sigma Aldrich, Tokyo, Japan), dissolved in isopropanol at a stock concentration of 160 mmol/L, was used to investigate signal transduction during lipoapoptosis. AML12 cells were treated for the indicated time periods or with the indicated concentrations of PA. For inhibition studies, AML12 cells were treated with PA (400 μmol/L or 800 μmol/L) for 4 h or 10 h in the presence or absence of 30 μmol/L SP600125, 20 mol/L z-LEHD-fmk, or 20 μmol/L QVD-OPh. All inhibitors simultaneously added with PA treatment.
Biochemical analysis of the in vivo NASH model: Blood samples were obtained from the mice by cardiac puncture. Serum was obtained from the blood by centrifugation at 3000 × g for 10 min. The serum levels of alanine transaminase (ALT) were analyzed using an autoanalyzer (JCA-BM2250; JEOL, Tokyo, Japan).
Cell proliferation assay
For AML12 proliferation studies, WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt] (Nacalai Tesque, Kyoto, Japan) incorporation experiments were performed using a microplate reader (Multiskan FC; Thermo Fisher Scientific, Yokohama, Japan). All proliferation assays were performed at least 4 times for each group. The results are presented as the ratio of incorporation in the cells that received the indicated treatment to that in vehicle-treated cells.
Immunocytochemistry for p62
AML12 cells seeded in 6-well plates were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.0125% (w/v) CHAPS in PBS. The primary antibody for anti-p62 was used at a dilution of 1:500. The secondary antibody was Alexa Fluor 488-conjugated IgG, and ProLong Antifade with DAPI (Molecular Probes, Eugene, OR, United States) was used as the mounting medium. Images were acquired using an EVOS microscope (AMF 4300; Life Technologies) with excitation and emission wavelengths of 488 nm and 507 nm, respectively. The experiments were repeated three times.
Measurement of cell size
AML12 cells seeded in 6-well plates received the indicated treatments. Images were acquired with the AMF 4300 microscope. Individual cells were identified and the sizes of 25 randomly selected cells were measured using the Image J software program ver. 1.47 (NIH, Bethesda, MD, United States). The results were presented as the ratio of the size of treated cells to that of control cells.
Quantitative real-time PCR
Total cellular RNA of AML 12 cells was extracted using an RNeasy Mini Kit (Qiagen, Tokyo, Japan) and was reverse-transcribed into complementary (c) DNA with Moloney murine leukemia virus reverse transcriptase (Invitrogen, Camarillo, CA, United States) and random primers (Invitrogen, Camarillo, CA, United States) as described previously[21]. Quantification of the cDNA template was conducted on a 7500 Real-Time PCR system (Applied Biosystems, Waltham, MA, United States) and analyzed using the 7500 Software program. The PCR primers were as follows: for mouse p62 (NM_011018): forward 5’-GAAGCTGCCCTATACCCACA-3’ and reverse 5’-TGGGAGAGGGACTCAATCAG-3’ (65 bp); for mouse Rubicon (NM_001200038): forward 5’-GATGGGGAGCGTCTGCTA-3’ and reverse 5’-TCCACAGTCGTCTTCAAATTACC-3’ (74 bp). Mouse Actin (NM_007393.4), amplified with the following primers: forward 5’-TAAGGCCAACCGTGAAAAG-3’ and reverse 5’-ACCAGAGGCATAGGGACA-3’ (104 bp), was used as an internal control. The target mRNA expression levels were expressed relative to Actin for each sample as described previously[21]. The experiment was repeated three times.
Small interfering RNA transfection
The small interfering RNAs (siRNAs) used for the knockdown of endogenous Rubicon protein and the negative control siRNA were purchased from Life Technologies. siRNA (5 pmol) was transfected into cells using Lipofectamine RNAiMAX (Life Technologies), according to manufacturer’s instructions. The examinations were performed after 30 h of transfection.
Immunoblotting analysis
Whole cell lysates were prepared as described previously[21]. Equal amounts of protein (10-50 μg) were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 4%-12% acrylamide gels, transferred to polyvinylidene difluoride membranes, and incubated with primary antibodies. Then, the membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (BioSource International, Camarillo, CA, United States). Bound antibody was visualized using a chemiluminescent substrate (ECL Prime; Amersham, Buckinghamshire, United Kingdom).
Statistical analysis
All data represent the results of at least three independent experiments and are expressed as the mean ± SD. The differences between the groups were compared using Student’s t-test and one-way analysis of variance with a post-hoc Dunnett’s test. P-values < 0.05 were considered to be statistically significant.
RESULTS
Autophagic process is inhibited in mice with steatohepatitis
The mice were divided into 2 groups: those fed a normal chow diet (CT, n = 5) and those fed an HFD diet (HFD, n = 5). After 12 wk, all mice were sacrificed, and liver histology, serum ALT levels, and protein expression in the liver were examined. Body weight, ALT measurement, and histological scores were evaluated in 5 mice per group. Protein expression was evaluated in 3 mice per group. The body weight of the HFD mice was significantly higher than that of the CT mice (Figure 1A). The ALT level was significantly higher in the HFD mice than in the CT mice (Figure 1A; 77 ± 11.0 IU/mL vs 25 ± 1.4 IU/mL). Histological analysis revealed the accumulation of lipid droplets in the liver (Figure 1B and C) and an increase in the number of ballooned hepatocytes (Figure 1C) in HFD mice as compared to CT mice. These findings confirmed that the HFD mice in the present study were compatible with a NASH model.
Figure 1.
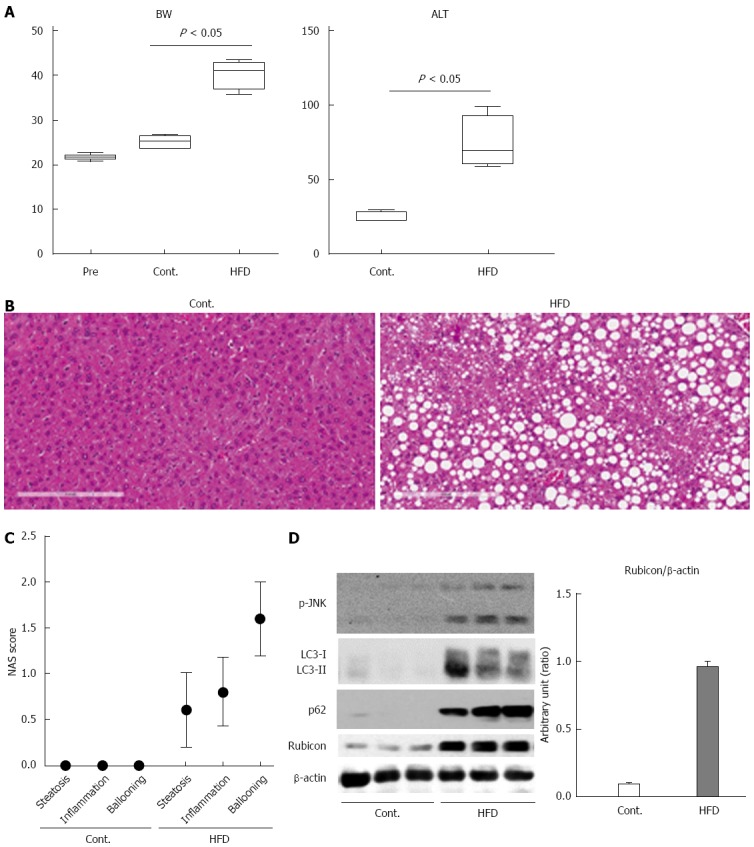
Mice fed high-fat diet show steatosis and inflammation of the liver, and impairment of the autophagic process. A: The body weights of mice at the start of feeding or at 12 wk after feeding with normal chow (Cont.) or high-fat diet (HFD) are presented in the left panel. The serum ALT levels after 12 wk of feeding are shown in the right panel; B: Histology of the liver of control and HFD mice is shown in the left and the right panel, respectively (Hematoxylin and Eosin staining); C: Non-alcoholic steatohepatitis activity scores (NAS) of control and HFD mice; D: Immunoblotting analyses of phosphorylated JNK, p62, LC3, Rubicon, and β-actin. Protein samples were prepared from the liver tissue of each of the control and HFD mice. All of the above experiments were repeated three times and representative results are shown. The quantitative data are presented as the means ± SD.
To confirm impairment of the autophagic process in this model, we evaluated autophagic marker protein expression in the liver by immunoblotting analysis. When protein was decomposed through autophagy, p62 was induced, and both p62 and LC3-II were subsequently degraded via the autophagic process. The expression of p62 in the livers of the HFD mice was higher than in those of CT mice (Figure 1D). However, the expression of both LC3-I and LC3-II was also increased in the HFD mice (Figure 1D). Expression of the autophagy inhibitor Rubicon was significantly higher in the HFD mice than in the CT mice (Figure 1D). These findings indicated that autophagy was impaired in the HFD mice. Additionally, we evaluated JNK phosphorylation, which is a key mediator of hepatocyte lipoapoptosis. Protein expression of phosphorylated JNK was increased in the HFD mice as compared to the CT mice.
PA induces cell death in a dose- and time-dependent manner
To investigate the detailed mechanism of the impaired autophagic process, we employed the mouse hepatocyte cell line AML12 in an in vitro study. A cell proliferation assay revealed that PA induced cell death in a dose- and a time-dependent manner, confirming PA cytotoxicity to AML12 cells (Figure 2A and C). JNK phosphorylation, ER stress, and cleaved caspase-3 were increased by PA in both a dose- and a time-dependent manner (Figure 2B and D). Therefore, we considered that PA induced apoptosis in AML12 cells.
Figure 2.
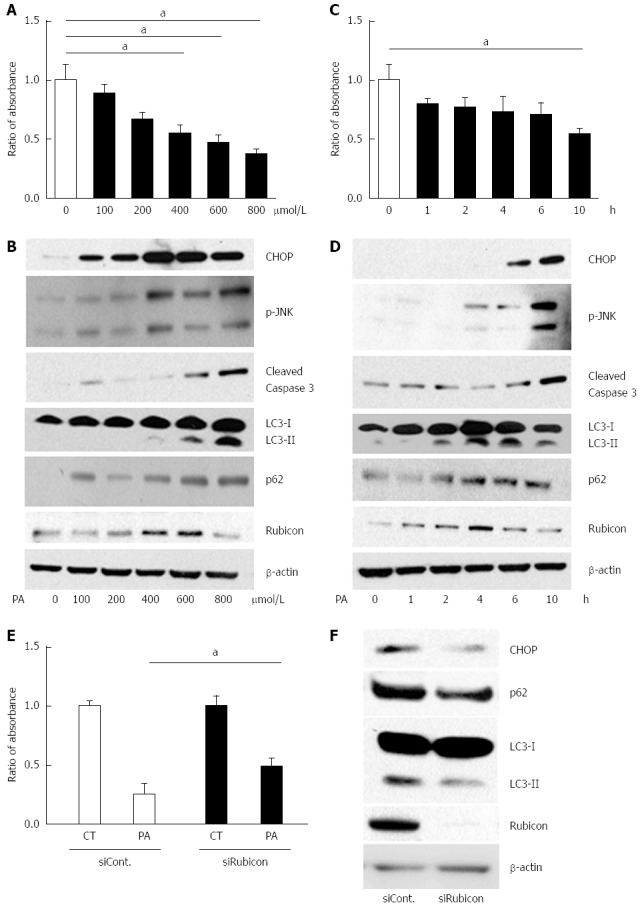
Treatment with palmitate induces apoptosis in a dose- and a time-dependent manner, and initiates but impairs the autophagic process in AML12 cells. A and B: AML12 cells were incubated with PA at the indicated concentrations for 10 h. Untreated AML12 cells were used as the control. A: PA cytotoxicity as evaluated by cell proliferation assay. Living cells are presented as the ratio of absorbance of cells treated with indicated conditions to that of untreated AML12 cells; B: Immunoblotting analyses of CHOP, phosphorylated JNK, cleaved Caspase-3, LC3, p62, Rubicon, and actin. Whole cell lysates were prepared from AML12 cells incubated at the indicated concentrations for 10 h; C and D: AML12 cells were incubated with 800 μmol/L of PA for the indicated periods. Untreated AML12 cells (0 h) were used as the control; C: PA cytotoxicity as evaluated by cell proliferation assay. Living cells are presented as the ratio of absorbance at the indicated incubation time to the absorbance at 0 h; D: Whole cell lysate was prepared from the AML12 cells with 800 μmol/L PA for the indicated incubation times; E and F: AML12 cells were incubated with 800 μmol/L PA for 4 h after transfection with the indicated siRNA: control siRNA (siCont) or Rubicon siRNA (siRubicon); E: PA cytotoxicity as evaluated by cell proliferation assay. Living cells are presented as the ratio of absorbance in AML12 cells with 800 μmol/L PA for 10 h (PA) to that in AML12 control (CT) cells; F: Whole cell lysate was prepared from AML12 cells treated with 800 μmol/L PA with the indicated siRNA. All of the above experiments were repeated three times and representative results are shown. The quantitative data are presented as the mean ± SD; aP < 0.05.
PA inhibits the autophagic process in AML12 cells and induces Rubicon expression
Next, we evaluated autophagy-related protein expression in PA-treated AML12 cells. PA induced p62 expression after 10 h of incubation with 800 μmol/L of PA in a dose- and a time-dependent manner (Figure 2B and D). The increased p62 level indicated induction of autophagy. If the autophagic process would progress normally, LC3-II would be expected to decrease with the decomposition of p62-associated protein aggregates. However, LC3-II expression peaked at 6 h of incubation with 800 μmol/L of PA. These findings indicated that PA induced autophagy, but that the autophagic process was interrupted around 6 h of incubation. Since LC3-II expression was further decreased at 10 h of incubation (Figure 2D), autophagy progressed with longer incubation times.
In the in vitro model, 600 μmol/L of PA and 4 h of incubation with PA increased Rubicon expression, while 10 h of incubation with 800 μmol/L of PA decreased Rubicon expression in AML12 cells (Figure 2B and D). Considering the progress of apoptosis at higher dose and longer incubation time of PA, Rubicon expression would be negatively related with apoptotic signals (Figure 2B and D).
PA-induced Rubicon expression inhibits autophagy, resulting in enhanced lipotoxicity in AML12 cells
To check whether PA-induced Rubicon expression was associated with lipoapoptosis, we knocked down Rubicon in AML12 cells using siRNA (siRubicon). After incubation with 800 μmol/L of PA for 4 h, the number of viable cells was significantly higher in siRubicon-treated than in control siRNA (siCont)-treated cells (Figure 2E). Expression of both p62 and LC3-II was lower in siRubicon- than in siCont-treated cells. Furthermore, PA-induced expression of the ER stress-related transcription factor CHOP was lower in Rubicon-silenced than in control cells (Figure 2F). These results indicated that PA-induced Rubicon expression inhibited autophagy, and that Rubicon enhanced lipotoxicity via the impairment of autophagy.
PA-induced Rubicon expression is mediated by JNK signaling
To evaluate the mechanism of PA-induced Rubicon expression, we used pharmacological inhibitors of lipoapoptosis-related molecules: JNK inhibitor (SP600125), caspase-9 inhibitor (Z-LEHD-FMK), and pan-caspase inhibitor (QVD-OPh). All of the inhibitors ameliorated lipotoxicity in the AML12 cells (Figure 3A). Interestingly, SP600125 decreased Rubicon expression in response to PA in comparison to the control (Figures 3B and 4A). The results confirmed that PA induced Rubicon expression and indicated that Rubicon expression was mediated by JNK signaling in both AML12 cells and the human hepatoma cell line HepG2 (Figures 3B, 4A, 4C and 5). JNK inhibition ameliorated PA-induced c-jun phosphorylation and decreased PA-induced protein expression of both p62 and LC3-II (Figure 4A). Immunohistochemical analysis revealed that p62 expression was diffuse in the cytoplasm of AML12 cells that were treated with PA, and was repressed by SP600125 inhibitor (Figure 4B). p62 mRNA expression in cells treated with PA and SP600125 was higher than that in cells treated with SP600125 alone, while it was lower than in cells treated with PA alone, as indicated by RT-qPCR (Figure 4C). These data indicated that p62 expression was mediated by both JNK-independent and JNK-dependent signals. When PA-induced Rubicon expression was inhibited by siRNA or JNK inhibitor, both PA-induced LC3-II expression and p62 expression decreased (Figures 2F and 4A).
Figure 3.
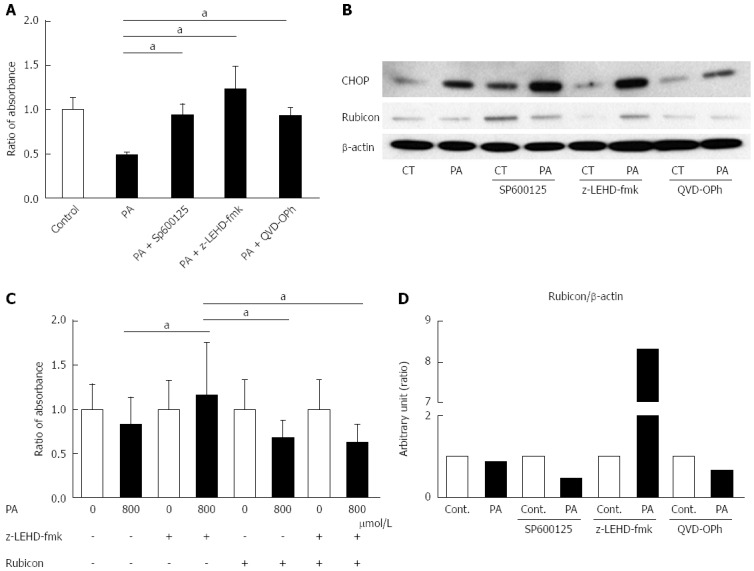
c-Jun N-terminal kinase inhibitor, Caspase-9 inhibitor, and Pan-Caspase inhibitor ameliorate palmitate-induced cell death, and Caspase-9 inhibition attenuates the decrease of Rubicon protein. A: AML12 cells were incubated with 800 μmol/L PA for 10 h in the presence or absence of either 30 μmol/L SP600125, 20 μmol/L LEHD-fmk, or 20 μmol/L QVD-OPh. Untreated AML12 cells were used as the control. PA cytotoxicity was evaluated by cell proliferation assay. Living cells are presented as the ratio of the absorbance at the indicated conditions to that of AML12 without PA treatment; B: Whole cell lysates were prepared from AML12 cells treated with PA (800 μmol/L) for 10 h in the presence or absence of either 30 μmol/L SP600125, 20 μmol/L z-LEHD-fmk, or 20 μmol/L QVD-OPh. Immunoblot analysis of CHOP and Rubicon. β-actin was used as the loading control; C: The size of the AML12 cells after PA treatment (800 μmol/L) for 10 h was compared to that of untreated AML12 cells using the Image J software program. z-LEHD-fmk and/or siRubicon were used for Caspase-9 inhibition or Rubicon knockdown. The difference in cell size under each treatment condition is presented as the ratio to the cell size of the respective controls. All of the above experiments were repeated three times and representative results are shown. The quantitative data are presented as the mean ± SD; aP < 0.05.
Figure 4.
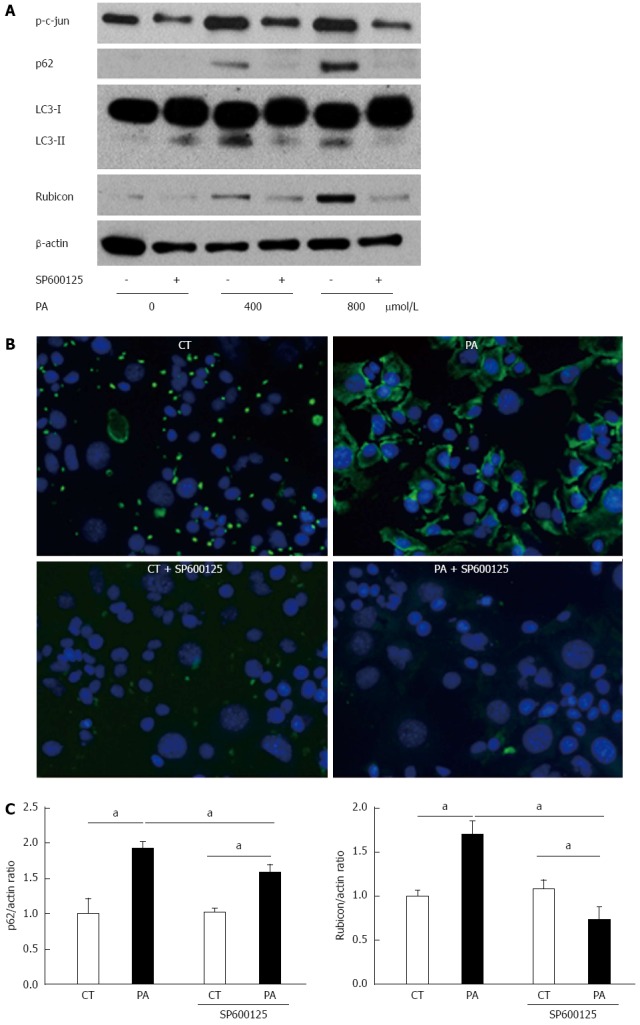
Palmitate induces expression of both p62 and Rubicon, and PA-induced Rubicon expression is mediated by c-Jun N-terminal kinase phosphorylation. A: Whole cell lysates were prepared from AML12 cells treated with PA (400 or 800 μmol/L) for 4 h in the presence or absence of SP600125. Immunoblotting analyses were performed for p62, LC3, and Rubicon. β-actin was used as the loading control; B: AML12 cells were incubated with 800 μmol/L PA for 4 h in the presence or absence of 30 μmol/L SP600125. p62 antibody was used as the primary antibody, and Alexa Fluor® 488-labeled secondary antibody was used for detecting p62 antibody. Samples were visualized using an EVOS microscope; C: Total RNA was prepared from AML12 cells treated with PA (800 μmol/L) for 4 h in the presence or absence of 30 μmol/L SP600125. Vehicle-treated cells were used as the control (CT). Both p62 and Rubicon mRNA were quantified by RT-qPCR, normalized to Actin, and expressed as the fold-change vs control cells without SP600125. All of the above experiments were repeated three times and representative results are shown. The quantitative data are presented as the mean ± SD; aP < 0.05 vs control.
PA-induced Rubicon expression decreases with progression of lipoapoptosis, which might be affected by caspase-9 activation
We observed that Rubicon expression decreased at the protein level under the condition of lipotoxicity (Figure 2B and D). Because apoptosis was correlated with autophagy, we hypothesized that the lipoapoptosis signaling would affect Rubicon expression. When inhibitors of lipoapoptosis were used in AML12 cells that were treated with PA, Z-LEHD-fmk maintained a greater amount of Rubicon protein than both AML12 with PA alone and AML12 with other inhibitors (Figure 3B). Because the pan-caspase inhibitor QVD-OPh did not ameliorate the decreased Rubicon expression during lipoapoptosis, the cause of the decreased Rubicon expression was upstream of the executioner caspases (caspase-3 and -7). We considered that caspase-9 activation was associated with inhibition of Rubicon expression during lipoapoptosis.
Caspase-9 inhibition induces ER stress accumulation and hepatocyte enlargement, which is required for Rubicon expression
Both caspase-9 deletion and JNK phosphorylation have been previously reported to be associated with hepatocyte ballooning[22]. Therefore, we suspected that Rubicon also affects hepatocyte morphology. To test this hypothesis, the effect of lipotoxicity on cell size was evaluated under the conditions of caspase-9 inhibition and/or Rubicon knockdown. The size of PA-treated AML12 cells was significantly larger in the z-LEHD-fmk group than in the PA-alone group (Figure 3C). In contrast, Rubicon knockdown ameliorated hepatocyte enlargement in the PA-treated z-LEHD-fmk group (Figure 3C). In addition, caspase-9 inhibition induced accumulation of CHOP protein (Figure 3B).
DISCUSSION
NASH is characterized by both inflammation and fibrosis in the liver. Lipotoxic insults lead to persistent hepatocyte apoptosis, resulting in fibrosis in the liver. During these processes, ER stresses, such as the unfolding of proteins, accumulate in the hepatocytes as a result of apoptotic insults[23,24]. Autophagy can inhibit the accumulation of these insults by decomposing the aggregated proteins and removing the damaged organelles[11,25]. Therefore, autophagy can be considered not only a process of energy supply but also a survival mechanism that protects against lipoapoptosis. When the autophagic process is interrupted, insults accumulate and damaged cells are led to lipoapoptosis (Figure 2B, D and F).
JNK phosphorylation is a key signal of hepatocyte lipoapoptosis[5,9,21]. Previous studies have shown that a JNK-dependent pathway leads to inflammation and fibrosis in the NASH liver[26], and that JNK phosphorylation is associated with hepatocyte ballooning, which is a hallmark of NASH[22]. The present study demonstrated that JNK phosphorylation was involved in the induction of lipoapoptosis as well as in the inhibition of autophagy via Rubicon expression, revealing a new role of JNK signaling in hepatocyte lipoapoptosis. When Rubicon was knocked down, ER stress-induced lipoapoptosis was decreased. Impaired autophagy via Rubicon induced the accumulation of ER stress, consequently enhancing lipotoxicity. Indeed, Rubicon expression appeared before apoptosis (Figure 2B, D and F). These data suggest that impairment of autophagy occurs before lipoapoptosis. Thus, decreasing the expression of Rubicon could be an important early intervention to prevent NASH development.
As lipoapoptosis progressed, Rubicon expression decreased (Figure 2B and D). The pan-caspase inhibitor did not ameliorate the decrease of Rubicon protein (Figure 3B). In contrast, caspase-9 inhibitor Z-LEHD-fmk attenuated the decrease of Rubicon expression (Figure 3B). Thus, we speculate that caspase-9 might decompose Rubicon. Interestingly, caspase-9 has been suspected to be a key molecule in hepatocyte ballooning. Levels of caspase-9 were found to be lower in ballooned than in normal hepatocytes in the NASH liver[22]. Furthermore, caspase-9-knockdown Huh-7 cells showed biological similarities with ballooned hepatocytes, such as intracellular lipid accumulation, accumulation of ER stress, and lipotoxic insult-induced sonic hedgehog signaling via JNK phosphorylation[22]. PA treatment upon caspase-9 inhibition in AML12 induced the accumulation of CHOP protein (Figure 3B). In contrast, a recent report demonstrated that pan-caspase inhibitor improved inflammation and fibrosis in the liver, and decreased the number of ballooned hepatocytes in HFD mice[27]. These data indicate that caspase-9 might play an important role in the formation of ballooned hepatocytes. According to the present study, caspase-9 inhibition has cytological effects reminiscent of hepatocyte ballooning, such as hepatocyte enlargement and accumulation of ER stress. Interestingly, knockdown of Rubicon counteracted the caspase-9 inhibition-induced hepatocyte enlargement. These data indicate that Rubicon expression and caspase-9 inhibition are required for the enlargement of hepatocytes. We hypothesize that caspase-9, JNK, and Rubicon are key molecules for hepatocyte ballooning in the NASH liver.
The present study revealed a number of significant findings: (1) autophagy is interrupted in both in vivo and in vitro NASH models; (2) PA-induced lipoapoptosis inhibits autophagy by Rubicon expression via JNK phosphorylation; (3) Rubicon expression appears prior to apoptosis and enhances PA toxicity in the hepatocytes; (4) Caspase-9 decreases Rubicon at the protein level during lipoapoptosis; and (5) Caspase-9 inhibition with Rubicon expression induces hepatocyte enlargement. We conclude that PA-induced JNK phosphorylation directly induced apoptosis and indirectly enhanced apoptosis via the inhibition of autophagy by Rubicon expression, and that Rubicon, caspase-9, and JNK are key molecules for lipotoxic insult-induced hepatocyte ballooning.
ACKNOWLEDGMENTS
We thank Asako Watanabe for western blotting and Koko Motodate for providing excellent secretarial support.
COMMENTS
Background
Lipoapoptosis in hepatocytes leads to infiltration of immune cells, activation of hepatic stellate cells, and generation of collagen fibers in the liver. Thus, prevention of hepatocyte apoptosis is considered to be a therapeutic strategy for non-alcoholic steatohepatitis (NASH). As apoptosis and autophagy negatively interact with each other, we focused this study on the Rubicon protein, a negative regulator of autophagy, in the NASH liver.
Background
Ballooned hepatocytes, which are a well-known hallmark of NASH disease severity, accumulate toxic insult. Hepatocytes in which caspase-9 was knocked down with lipotoxicity demonstrated a number of similarities with ballooned hepatocytes, such as accumulation of protein degradation machinery or secretion of fibrogenic signal. The interaction between exceeded apoptosis, impaired autophagy, and ballooning of hepatocytes has never been elucidated. The detailed interaction between these signals may be a therapeutic target of the NASH liver.
Innovations and breakthroughs
This is the first report to show that Rubicon is overexpressed in hepatocyte lipoapoptosis, Rubicon enhances lipotoxicity, caspase-9 decomposes Rubicon protein, and caspase-9 inhibition leads to ballooning of hepatocytes. Although interactions between apoptosis and autophagy have been previously reported elsewhere, the present study demonstrates the role of each of Rubicon and caspase-9 during hepatocyte lipoapoptosis.
Applications
Rubicon is induced during lipoapoptosis by c-Jun N-terminal kinase phosphorylation and enhances apoptosis via autophagy inhibition. Since caspase-9 inhibition and Rubicon overexpression led to hepatocyte ballooning, Rubicon may be associated with ballooning in hepatocytes. As the prevalence of ballooned hepatocytes correlates with disease severity in the NASH liver, control of Rubicon has potential for NASH therapy.
Terminology
Lipoapoptosis is a term for lipotoxicity-induced apoptosis. Lipoapoptosis in hepatocytes induces the infiltration of immune cells into the NASH liver, which leads to the generation of collagen fibers. Thus, lipoapoptosis is considered a leading cause of NASH development.
Peer-review
This manuscript contains fascinating and novel data for understanding of pathophysiology of NASH.
Footnotes
Manuscript source: Invited manuscript
Specialty Type: Gastroenterology and Hepatology
Country of Origin: Japan
Peer-Review Report Classification
Grade A (Excellent): A, A
Grade B (Very good): 0
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional animal care and use committee statement: All of the animal experiments were approved by the Animal Care and Use Committee of Iwate Medical University (Morioka, Japan; 25-025).
Conflict-of-interest statement: There is no conflict-of-interest.
Data sharing statement: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 6, 2016
First decision: May 12, 2016
Article in press: June 13, 2016
P- Reviewer: Kucera O S- Editor: Qi Y L- Editor: A E- Editor: Ma S
References
- 1.Rinella ME, Sanyal AJ. NAFLD in 2014: Genetics, diagnostics and therapeutic advances in NAFLD. Nat Rev Gastroenterol Hepatol. 2015;12:65–66. doi: 10.1038/nrgastro.2014.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yeh MM, Brunt EM. Pathological features of fatty liver disease. Gastroenterology. 2014;147:754–764. doi: 10.1053/j.gastro.2014.07.056. [DOI] [PubMed] [Google Scholar]
- 3.Wong RJ, Cheung R, Ahmed A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology. 2014;59:2188–2195. doi: 10.1002/hep.26986. [DOI] [PubMed] [Google Scholar]
- 4.Guicciardi ME, Malhi H, Mott JL, Gores GJ. Apoptosis and necrosis in the liver. Compr Physiol. 2013;3:977–1010. doi: 10.1002/cphy.c120020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 6.Cazanave SC, Gores GJ. Mechanisms and clinical implications of hepatocyte lipoapoptosis. Clin Lipidol. 2010;5:71–85. doi: 10.2217/clp.09.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Unger RH, Orci L. Lipoapoptosis: its mechanism and its diseases. Biochim Biophys Acta. 2002;1585:202–212. doi: 10.1016/s1388-1981(02)00342-6. [DOI] [PubMed] [Google Scholar]
- 8.Ibrahim SH, Akazawa Y, Cazanave SC, Bronk SF, Elmi NA, Werneburg NW, Billadeau DD, Gores GJ. Glycogen synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte lipoapoptosis. J Hepatol. 2011;54:765–772. doi: 10.1016/j.jhep.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cazanave SC, Elmi NA, Akazawa Y, Bronk SF, Mott JL, Gores GJ. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G236–G243. doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.González-Rodríguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillón J, Lo Iacono O, Corazzari M, Fimia GM, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5:e1179. doi: 10.1038/cddis.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. doi: 10.1038/cr.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Del Bello B, Toscano M, Moretti D, Maellaro E. Cisplatin-induced apoptosis inhibits autophagy, which acts as a pro-survival mechanism in human melanoma cells. PLoS One. 2013;8:e57236. doi: 10.1371/journal.pone.0057236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 15.Caldwell S, Ikura Y, Dias D, Isomoto K, Yabu A, Moskaluk C, Pramoonjago P, Simmons W, Scruggs H, Rosenbaum N, et al. Hepatocellular ballooning in NASH. J Hepatol. 2010;53:719–723. doi: 10.1016/j.jhep.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 17.Guy CD, Suzuki A, Abdelmalek MF, Burchette JL, Diehl AM. Treatment response in the PIVENS trial is associated with decreased Hedgehog pathway activity. Hepatology. 2015;61:98–107. doi: 10.1002/hep.27235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brunt EM, Tiniakos DG. Histopathology of nonalcoholic fatty liver disease. World J Gastroenterol. 2010;16:5286–5296. doi: 10.3748/wjg.v16.i42.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM, Cadrin M, Omary MB. From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res. 2007;313:2033–2049. doi: 10.1016/j.yexcr.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 20.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 21.Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, Mott JL, Gores GJ. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2012;302:G77–G84. doi: 10.1152/ajpgi.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kakisaka K, Cazanave SC, Werneburg NW, Razumilava N, Mertens JC, Bronk SF, Gores GJ. A hedgehog survival pathway in ‘undead’ lipotoxic hepatocytes. J Hepatol. 2012;57:844–851. doi: 10.1016/j.jhep.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akazawa Y, Cazanave S, Mott JL, Elmi N, Bronk SF, Kohno S, Charlton MR, Gores GJ. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J Hepatol. 2010;52:586–593. doi: 10.1016/j.jhep.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, Kahraman A, Garrison SP, Zambetti GP, Charlton MR, et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009;284:26591–26602. doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshii SR, Mizushima N. Autophagy machinery in the context of mammalian mitophagy. Biochim Biophys Acta. 2015;1853:2797–2801. doi: 10.1016/j.bbamcr.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 26.Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–320. doi: 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barreyro FJ, Holod S, Finocchietto PV, Camino AM, Aquino JB, Avagnina A, Carreras MC, Poderoso JJ, Gores GJ. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015;35:953–966. doi: 10.1111/liv.12570. [DOI] [PubMed] [Google Scholar]