

ABSTRACT
Fusion proteins combining oligomeric assemblies of a genetically obtained single-chain (sc) variant of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) with antibodies directed against tumor-associated antigens represent a promising strategy to overcome the limited therapeutic activity of conventional soluble TRAIL. To further improve the scTRAIL module in order to obtain a robust, thermostable molecule of high activity, we performed a comprehensive analysis of the minimal TNF homology domain (THD) and optimized linkers between the 3 TRAIL subunits constituting a scTRAIL. Through a stepwise mutagenesis of the N- and C-terminal region and the joining linker sequences, we generated bioactive scTRAIL molecules comprising a covalent linkage of the C-terminal Val280 and the N-terminal position 122 by only 2 amino acid residues in combination with conservative exchanges at positions 122 and 279. The increased thermal stability and solubility of such optimized scTRAIL molecules translated into increased bioactivity in the diabody-scTRAIL (Db-scTRAIL) format, exemplified here for an epidermal growth factor receptor-specific Db-scTRAIL. Additional modifications within the diabody linkers resulted in a fusion protein exerting high, target-dependent apoptosis induction in tumor cell lines in vitro and potent antitumor activity in vivo. Our results illustrate that protein engineering of scTRAIL and associated peptide linkers provides a promising strategy to develop antibody-scTRAIL fusion proteins as effective antitumor therapeutics.
KEYWORDS: Apo2L, apoptosis, diabody, EGFR targeting, mouse xenograft, thermal stability, TRAIL, TNF homology domain
Abbreviations
- aa
amino acid
- ALT
alanine aminotransferase
- AUC
area under the curve
- CD
cluster of differentiation
- CI
confidence interval
- Db
diabody
- DR
death receptor
- EC50
half maximal effective concentration
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- Fc
fragment crystallizable
- Ig
immunoglobulin
- i.p.
intraperitoneal(ly)
- i.v.
intravenous(ly)
- PE
phycoerythrin
- PK
pharmacokinetics
- PNGase F
protein N-glycosidase F
- s.c.
subcutaneous(ly)
- scFv
single-chain Fv fragment
- scTRAIL
single-chain TRAIL
- SD
standard deviation
- SEC
size-exclusion chromatography
- THD
TNF homology domain
- TMB
3,3′,5,5′-tetramethylbenzidine
- TNF
tumor necrosis factor
- TRAIL
TNF-related apoptosis-inducing ligand
- VH
variable domain of immunoglobulin heavy chain
- VL
variable domain of immunoglobulin light chain
Introduction
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is capable of specifically inducing apoptosis in tumor cells without affecting non-transformed cells. In contrast to other cell death-triggering tumor necrosis factor (TNF) superfamily members, e.g., TNF or CD95L (FasL), soluble, homotrimeric TRAIL is well tolerated upon systemic application in humans.1-3 TRAIL activates the extrinsic apoptotic pathway via binding to death receptors (DR) 4 and 5, which leads to apoptosis of tumor cells. Nevertheless, in several clinical trials, conventional TRAIL such as dulanermin4,5 and a circularly permuted TRAIL (CPT)6,7 proved to be largely ineffective. This clinical failure has been subsequently attributed to the low in vivo bioactivity of the TRAIL molecules administered, and their rather short terminal plasma half-life (∼1 hour).4,6 Consequently, alternative therapeutic strategies targeting TRAIL death receptors have been developed, such as agonistic monoclonal antibodies directed against either DR4, e.g., mapatumumab,8,9 or DR5, e.g., conatumumab10 and lexatumumab.11 However, Phase 1 and 2 clinical studies revealed ambiguous results (for review see refs. 12, 13). Trials investigating combination treatments of recombinant TRAIL with chemotherapeutics, e.g., paclitaxel and carboplatin, that might break potential resistance toward TRAIL-induced apoptotic pathways did not substantially improve clinical responses.2
Mechanistically, apoptosis induction by TRAIL, as for other ligands of the TNF family, occurs via receptor clustering.14 Under physiologic conditions, TRAIL is expressed as a non-covalently associated homotrimeric membrane protein, from which it is proteolytically cleaved into a soluble, homotrimeric form. In vitro, both the membrane and the soluble forms are, in principle, able to induce apoptosis, albeit with different efficiency and, depending on cell type, via distinct receptors. The soluble form can induce apoptosis in some tumor cells via triggering DR4, whereas for many types of solid tumors DR5 plays a dominant role in TRAIL-mediated apoptosis induction.15,16 DR5, however, is poorly activated by soluble TRAIL, but efficiently triggered by the membrane form of natural TRAIL or membrane-mimetic agonists causing DR5 clustering in the membrane, such as genetically engineering controlled oligomeric forms of soluble TRAIL.17 A suitable molecular basis for generation of oligomeric TRAIL variants is the single-chain TRAIL format, in which the 3 extracellular domains of TRAIL are covalently linked by 2 short peptide sequences.18 Thus, unlike the naturally processed soluble TRAIL and recombinant therapeutics derived thereof,19 a scTRAIL lacks inactivating dissociation at dilute concentrations, i.e., is stable as a trimer at physiologic temperatures.18,20 ScTRAIL molecules have been successfully used to generate antibody TRAIL fusion proteins to achieve specific targeting and to improve pharmacokinetic properties with the aim to enrich the therapeutic protein at the tumor site.18,20,21 Moreover, depending on the antibody format used as fusion partner for scTRAIL, we recently showed that a controlled oligomerization of the scTRAIL module can be achieved, for example, through fusion of a diabody or a dimerization module such as the heavy chain domain 2 of IgE, resulting in substantially increased biological activity compared to sTRAIL, scTRAIL, and targeted scTRAIL in a monomeric scFv fusion protein.20,21
Because we noted a rather low thermal stability of scTRAIL with a melting temperature of approximately 46°C, as determined by dynamic light scattering, we set out to develop derivatives of scTRAIL with increased biophysical and biochemical properties as a prerequisite for clinical development of tumor-targeted TRAIL-based therapeutics. Here, we describe the results of a rational design of single-chain TRAIL variants comprising TNF homology domains (THD) of minimal length. The use of N- or C-terminal deletions in combination with distinct mutations at the termini and appropriate linker sequences led to fully bioactive scTRAIL molecules with increased thermal stability, solubility and production rate.22-24 Most importantly, the use of the evolved scTRAIL molecules as components in the diabody (Db)-scTRAIL format, additionally modified in linker peptides between the VH, VL and scTRAIL moieties, led to a further improvement of product quality of this second generation of Db-scTRAIL, and resulted in potent antitumor activities in a xenograft mouse model.
Results
Stepwise evolution of minimal scTRAIL variants
We started with a scTRAIL variant composed of 3 identical TRAIL subunits (scTRAIL) comprising residues 95 to 281 (95L8), i.e., having the following composition: subunit-1 (aa 95–281)-(GGGS)2-subunit 2 (aa 95–281)-(GGGS)2-subunit 3 (aa 95–281), with the aim of generating a panel of new scTRAIL molecules comprising THDs with reduced length. In a first set, 4 new scTRAIL molecules with individual subunits starting either with amino acid (aa) residue Val114 or Gln120, respectively, were generated. Two different Gly/Ser polypeptides with a length of 4 (GGGS) or 8 residues (GGGS)2 were used in these molecules, leading to the variants scTRAIL-FLVGGGGSVRERGPQRVA, scTRAIL-FLVGGGGSGGGSVRERGPQRVA, scTRAIL-FLVGGGGSQRVA and scTRAIL-FLVGGGGSGGGSQRVA (wild-type TRAIL sequences are underlined, additional linker aa residues are in italics, see Fig. 1 and Table 1). All scTRAIL variants were expressed in soluble form upon transient or stable transfection of HEK293 cells, and could be purified by anti-FLAG affinity chromatography. A predominantly monomeric composition for the scTRAIL molecules was observed in size-exclusion chromatography (SEC), although main peaks were accompanied by minor amounts of low-order oligomers in some cases (Fig. S1). The melting points of these scTRAIL variants determined by dynamic light scattering were between 46 to 50°C (Table 1, Fig. S3). For comparison, homotrimeric soluble TRAIL (sTRAIL115–281) exhibited a melting point of 46°C. The apoptosis-inducing activity of the new scTRAIL variants was tested in a cytotoxicity assay using Colo205 colon carcinoma cells sensitized with 250 ng/ml bortezomib.20 All scTRAIL variants were bioactive with EC50 values in the range of 77 to 199 pM (Table 1, Fig. S2).
Figure 1.
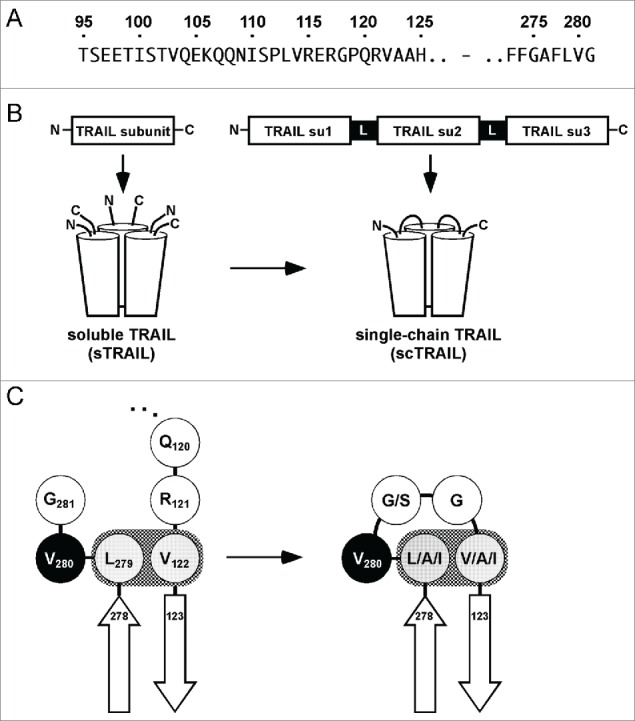
A single-chain format for human TRAIL. (A) Amino acid sequences in the extracellular domain of human TRAIL in which the modifications described in this work are located. For the previous generation of scTRAIL, we used the N-terminal position 95 as a start, involving 26 aa residues of the stalk region, preceding the receptor binding domain. Each individual TRAIL subunit of the scTRAIL molecules described in this work begins in close proximity of the N-terminus of the receptor binding domain (122–281), as realized for the aa positions 118, 120, 121, 122 and 123. (B) Scheme depicting the generation of single-chain TRAIL from soluble TRAIL. Su, subunit. (C) Val122 and Leu279 of human TRAIL are involved in a hydrophobic interaction, thereby defining beginning and end of the TNF homology domain. Both residues were connected by Val280 and 2 linking residues as demonstrated for scTRAIL-FLVGGVA and derived variants scTRAIL-F(L/A/I)V(G/S)G(V/A/I)A in order to form scTRAIL molecules of minimal size.
Table 1.
Protein solubility, denaturation temperatures and in vitro bioactivity of scTRAIL variants.
| Molecule | Solubility | Tm (°C) | EC50 (pM) of bioactivity1 |
|---|---|---|---|
| sTRAIL | yes | 46 | |
| scTRAIL-95L8 | yes | 47 | 219 ± 17 |
| scTRAIL-FLVGGGGSGGGSVRERGPQRVA | yes | 46 | 199 ± 110 |
| scTRAIL-FLVGGGGSVRERGPQRVA | yes | 47 | 135 ± 35 |
| scTRAIL-FLVGGGGSGGGSQRVA | yes | 49 | 123 ± 21 |
| scTRAIL-FLVGGGGSQRVA | yes | 50 | 77 ± 16 |
| scTRAIL-FLVGGGGPQRVA | yes | 52 | 65 ± 28 |
| scTRAIL-FLVGGGPQRVA | yes | 52 | 56 ± 13 |
| scTRAIL-FLVGGPQRVA | yes | 53 | 69 ± 9 |
| scTRAIL-FLVGGQRVA | yes | nd | nd |
| scTRAIL-FLVGGRVA | yes | 54 | 39 ± 3 * |
| scTRAIL-FLVGGVA | yes | 57 | 41 ± 6 * |
| scTRAIL-FLVGGGA | no | — | — |
| scTRAIL-FGVGGGA | no | — | — |
| scTRAIL-FLGGGGA | no | — | — |
| scTRAIL-FLGGGA | no | — | — |
| scTRAIL-FLVGGLA | no | — | — |
| scTRAIL-FLVGGMA | no | — | — |
| scTRAIL-FVVGGLA | no | — | — |
| scTRAIL-FLVGGIA | yes | 54 | 47 ± 19 * |
| scTRAIL-FLVGGAA | yes | 53.5 | 96 ± 43 |
| scTRAIL-FAVGGAA | yes | 53 | 129 ± 34 |
| scTRAIL-FLVSGIA | yes | 51.5 | 261 ± 68 |
| scTRAIL-FIVGGIA | yes | 52 | 45 ± 0.6 * |
| scTRAIL-FIVSGIA | yes | 51 | 229 ± 52 |
| scTRAIL-FAVGGIA | yes | 53 | 53 ± 11 * |
| scTRAIL-FAVSGIA | yes | 50.5 | 274 ± 55 |
| scTRAIL-FAVSGAA | yes | 54 | 80 ± 8 * |
analyzed on Colo 205 cells in presence of 250 ng/ml bortezomib, mean ± SD (n = 3).
percentage of oligomers >15% of the total protein.
Next, we analyzed if the TRAIL subunits can also be connected directly or with a short linker sequence without affecting production and bioactivity. Glycine 118 was chosen as the N-terminal residue (see Fig. 1). ScTRAIL molecules starting from Gly118 were generated, whereby those molecules only comprised wild-type TRAIL sequences (scTRAIL-FLVGGPQRVA) or comprised a single or 2 glycine residues for connecting the modules (scTRAIL-FLVGGGPQRVA, scTRAIL-FLVGGGGPQRVA). These scTRAIL variants were expressed in soluble form upon transient or stable transfection of HEK293 cells, with melting points increased to 52 or 53°C, respectively (Table 1, Fig. 2A, Fig. S3). The bioactivity of these 3 scTRAIL molecules was slightly increased compared to the scTRAIL 114/120 variants (EC50 values between 56–69 pM) (Table 1, Fig. S2).
Figure 2.

Biochemical properties of new scTRAIL molecules. (A) Melting point curves measured by dynamic light scattering of sTRAIL (115–281), scTRAIL-95L8 (left), scTRAIL-FLVGGGPQRVA, scTRAIL-FLVGGRVA (middle) and scTRAIL-FLVGGVA, scTRAIL-FAVSGAA (right) are shown (symbols represent mean of 2 measurements). (B) Size exclusion chromatograms of scTRAIL-FAVSGAA, in comparison to the old variant scTRAIL-95L8 (filled graph), and scTRAIL-FAVSGIA.
Based on these findings, we further reduced the N-terminal position to residues Gln120 (scTRAIL-FLVGGQRVA), Arg121 (scTRAIL-FLVGGRVA), or Val122 (scTRAIL-FLVGGVA), connecting the shortened subunits with a single glycine residue (Fig. 1, Table 1). All three scTRAIL variants could be purified in soluble form from cell culture supernatants of transfected HEK293 cells. Melting points were determined for scTRAIL-FLVGGRVA and scTRAIL-FLVGGVA. Both variants exhibited a further increased stability, evident from a melting point of 54° C (scTRAIL-FLVGGRVA) and 57°C (scTRAIL-FLVGGVA) (Fig. 2A, Fig S3). EC50 values in the range of 40 pM were determined for cell death induction in Colo205 cells in the presence of bortezomib (Table 1, Fig. S2). However, the substantially higher thermal stabilities and bioactivities of scTRAIL-FLVGGRVA and scTRAIL-FLVGGVA correlated with an increase in multimeric species compared to scTRAIL 118 molecules (Fig. S1).
Influence of amino acid residues at N- and C-terminus of the TRAIL THD on protein solubility
The structure of soluble TRAIL bound to DR525 indicates that Val122, preceding the first β-strand of soluble TRAIL, forms a hydrophobic bond with Leu279 of the C-terminus (Fig. 1C). The importance of Val122 and Leu279 for the formation of active scTRAIL molecules was analyzed by substituting these residues by glycine, alanine or other hydrophobic residues. Substitution of Val122 in scTRAIL-FLVGGVA by glycine or methionine, as well as substitution of Leu279 and Val122 by glycine, resulted in insoluble products as shown by immunoblotting analysis (Fig. S4). Of note, the hydrophobic interaction of Leu279 and Val122 in scTRAIL-FLVGGVA, determined by soluble expression, could not be retained by switching of these residues (L279V/V122L) (Fig. S4). In contrast, substitution of Val122 by isoleucine or alanine, as well as double substitutions Leu279A/Val122A, yielded soluble protein upon expression in HEK293 cells.
To further shed light on the importance of N-terminal aa residues at positions 120 to 123, mutants of homotrimeric sTRAIL with glycine substitutions at the respective positions were stably expressed in HEK293 cells. Glycine substitutions at positions 120 or 120 and 121 (Q120G/Q120G/R121G) yielded soluble proteins, whereas proteins carrying the substitutions at positions 120–122 (Q120G/R121G/V122G) or 120–123 (Q120G/R121G/V122G/A123G) were insoluble (Fig. S5). A further mutant with glycine substitutions at positions 121 and 123 (R121G/A123G), thus maintaining V122, was expressed in soluble form, further supporting the crucial role of Val122.
In a final round of optimization, positions 279, 281 and 122 were varied with distinct combinations of isoleucine, alanine or serine residues (Table 1). The molecules scTRAIL-FAVSGAA (L279A/G281S/V123A), scTRAIL-FAVGGIA (L279A/V123I), scTRAIL-FIVGGIA (L279I/V123I), scTRAIL-FLVSGIA (G281S/V123I), scTRAIL-FIVSGIA (L279I/ G281S/V123I) and scTRAIL-FAVSGIA (L279A/G281S/V123I) could be expressed in a soluble form. The integrity of the proteins was confirmed by SDS-PAGE and SEC. Most intriguingly, a high grade of sequence reduction, as realized for instance in scTRAIL-FAVSGAA, directly correlated with more compact molecules showing reduced hydrodynamic radii (Fig. 2B), corroborated also by a Stoke's radius of 3.2 nm versus 3.8 nm for scTRAIL-95L8. In particular, scTRAIL variants comprising substitutions at aa positions 281, 121 and 122 (e.g., scTRAIL-FLVSGIA, scTRAIL-FIVSGIA and scTRAIL-FAVSGIA) were found to have remarkably low oligomer content of 5% or less of the total protein (Fig. 2B, Fig. S1). ScTRAIL-FAVSGAA showed the best stability with a melting point of 54°C (Fig. 2A, Fig. S3). This variant was further analyzed via substitution of the linker glycine residue with arginine, originally present at this position (aa 121), or with lysine. Interestingly, both variants exhibited a rather low melting point of 47°C and 48°C, respectively, indicating the beneficial effect of substitution by the small and aliphatic glycine residue at this position.
In summary, all scTRAIL variants comprising shortened N-terminal sequences and combined with short intervening peptide sequences (0–2 aa residues) exhibit higher thermal stability compared to previously described scTRAIL molecules, e.g., scTRAIL-95L8 and sTRAIL. Importantly, all soluble scTRAIL molecules were shown to be bioactive with EC50 values in the sub-nanomolar range. Possible differences among the bioactivities of individual scTRAIL variants may correlate with the amount of spontaneously formed oligomers. This was further analyzed for scTRAIL-FLVGGVA exhibiting ∼24% oligomeric molecules (Fig. S1). Equal concentrations of SEC-purified oligomers and monomers were analyzed for their apoptotic activity on the cancer cell lines Colo205, NCI-H460 and HT1080 (Fig. S6). The oligomeric fraction of scTRAIL-FLVGGVA exerted considerably higher bioactivity than the monomeric fraction on all tested cell lines. Monomers of scTRAIL-FLVGGVA were either nearly inactive (Colo205) or were 50-fold (HT1080), and 13-fold (NCI-H460), respectively, less bioactive than the oligomers.
Epidermal growth factor receptor-targeted Db-scTRAIL proteins based on new scTRAIL variants
The variants scTRAIL-FAVSGAA, representing a molecule of high thermal stability and good expression, and scTRAIL-FAVSGIA, representing a molecular variant with superior monomeric property, were used to construct dimeric Db-scTRAIL molecules targeting epidermal growth factor receptor (EGFR) (Fig. 3A, B).20 For both variants, an increased portion of the dimeric, full-length form was obtained, compared with Db-scTRAIL-95L8 used in previous studies (Fig. 3D, E; 72% for Db-Glyco-scTRAIL-FAVSGAA and 82% for Db-Glyco-scTRAIL-FAVSGIA vs. 54% for the reference Db-scTRAIL-95L8). Db-scTRAIL variants utilizing scTRAIL-FAVSGAA and scTRAIL-FAVSGIA were characterized by 4 to 6°C higher thermal stability compared to the reference protein Db-scTRAIL-95L8 corresponding to the values obtained for the respective scTRAIL variants (Table 2, Fig. S7). Protein quality (full-length protein, low aggregate content) could be further enhanced by introduction of a modified connecting linker (see Fig. 3B) between diabody and scTRAIL moieties composed of 16 aa residues and comprising 2 N-glycosylation sites. Incubation of the purified protein with peptide N-glycosidase F (PNGase F) confirmed functional N-glycosylation (Fig. 3C). In addition, a diabody linker (Db linker, see Fig. 3B) of 10 aa residues length (Db10-Glyco-scTRAIL-FAVSGAA), which still allowed quantitative dimerization of the fusion protein, reduced the amounts of higher order complexes (below 12%, Fig. 3E) without affecting thermal stability (Table 2).
Figure 3.
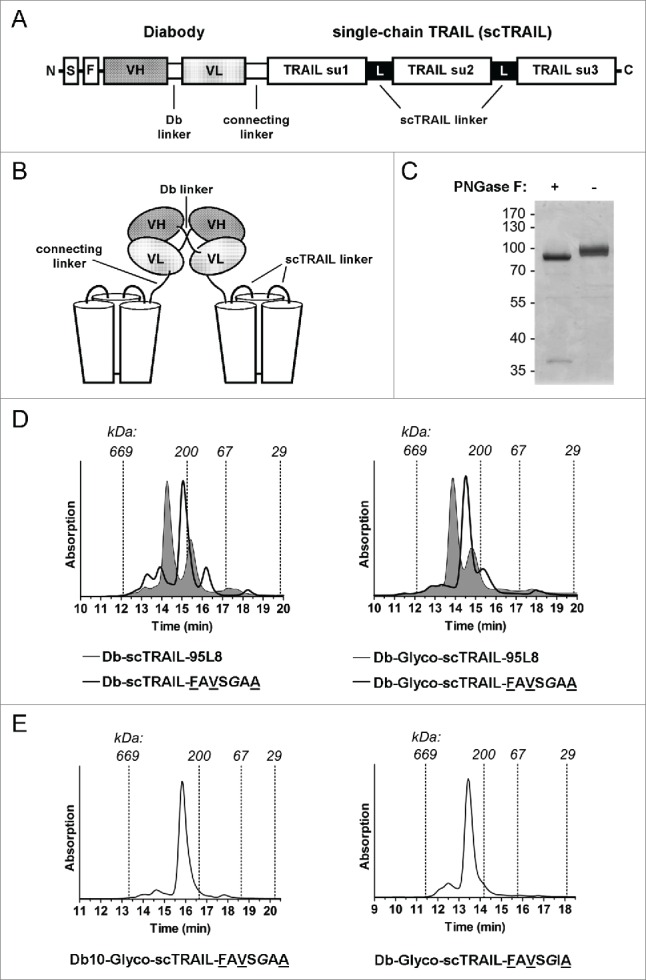
Characteristics of the diabody-scTRAIL format. (A) Scheme of the polypeptide chain of the diabody scTRAIL fusion proteins described in this work. The diabody linker has in standard the sequence GGGGS. The connecting linker was either AAAEFGG or AAAGNGTSNGTSEFGG, the latter comprises 2 N-glycosylation sites. S, signal peptide; F, FLAG tag; su, subunit. (B) Schematic illustration of the non-covalently dimerized Db-scTRAIL molecule. (C) Functional N-glycosylation of the connecting linker in the Db-Glyco-scTRAIL formats was exemplified for Db-Glyco-scTRAIL-FAVSGAA by PNGase F digestion and subsequent SDS-PAGE/ Coomassie staining of the reactions. (D) Size-exclusion chromatograms of non-glycosylated (left) and glycosylated forms (right) of Db-scTRAIL-FAVSGAA in comparison with Db-scTRAIL-95L8 (filled). (E) Size-exclusion chromatograms of Db10-Glyco-scTRAIL-FAVSGAA and Db-Glyco-scTRAIL-FAVSGIA.
Table 2.
Denaturation temperatures of EGFR-specific Db-scTRAIL variants.
| Molecule | Tm (°C) |
|---|---|
| Db-scTRAIL-95L8 | 50 |
| Db-Glyco-scTRAIL-95L8 | 50 |
| Db-scTRAIL-FAVSGAA | 55 |
| Db-Glyco-scTRAIL-FAVSGAA | 56 |
| Db10-scTRAIL-FAVSGAA | 56 |
| Db10-Glyco-scTRAIL-FAVSGAA | 56 |
| Db-Glyco-scTRAIL-FAVSGIA | 54 |
As shown by flow cytometry, the new Db-scTRAIL variants displayed similar specific binding as the reference Db-scTRAIL molecules on EGFR+ HCT116 (data not shown), Colo205 and HT1080 target cells (Fig. 4A). The EGFR-specific binding of Db10-Glyco-scTRAIL-FAVSGAA was also proven by competition with cetuximab on tumor cells and purified EGFR-Fc, revealing a sub-nanomolar EC50 for EGFR (Fig. 4B, C). We exemplified also the binding to death receptors for Db10-Glyco-scTRAIL-FAVSGAA (Fig. 4D). The protein bound significantly better to DR5-Fc compared with Db-Glyco-scTRAIL-95L8 (0.22 ± 0.05 nM vs. 0.37 ± 0.01 nM, p = 0.013). However, both proteins bound with generally higher EC50 values to DR4-Fc (5.28 ± 1.36 nM for Db10-Glyco-scTRAIL-FAVSGAA, 3.05 ± 0.44 nM for Db-Glyco-scTRAIL-95L8, p = 0.091, n.s.), suggesting a preference for DR5 binding associated with the scTRAIL format.
Figure 4.

Binding of EGFR-targeted Db-scTRAIL proteins. (A) The binding of Db-Glyco-scTRAIL-FAVSGAA (left), Db10-Glyco-scTRAIL-FAVSGAA (middle) and Db-Glyco-scTRAIL-FAVSGIA (right, squares) to EGFR+ HT1080 fibrosarcoma (upper) and Colo205 colon carcinoma cells (lower) was measured by flow cytometry in reference to Db-scTRAIL-95L8 (circles) and Db-Glyco-scTRAIL-95L8 (triangles). Mean ± SD (n = 3). (B) Db10-Glyco-scTRAIL-FAVSGAA was tested in 3 concentrations for combined binding to the EGFR+ tumor cell lines Colo205 and HCT116 by flow cytometry. In addition, a 200-fold molar excess of cetuximab was used for competition with the scTRAIL fusion protein, demonstrating residual binding to TRAIL receptors mediated by scTRAIL in its dimeric configuration. Mean ± SD (n = 3). (C) The binding (ELISA) of Db10-Glyco-scTRAIL-FAVSGAA (filled circles) and Db10-Glyco-scTRAIL-FAVSGAA competed with 10 µM cetuximab (open circles) was tested on purified EGFR-Fc. In addition, a FLAG-tagged, fully humanized anti-EGFR IgG derived from cetuximab (M. Siegemund, R. Kontermann, unpublished data) was tested with (open squares) and without (filled squares) competition by cetuximab. Db10-Glyco-scTRAIL-FAVSGAA and αEGFR-IgG bound to EGFR-Fc with EC50 values of 284 ± 34 pM and 77 ± 8 pM, respectively. Mean ± SD (n = 3). (D) The binding (ELISA) of Db10-Glyco-scTRAIL-FAVSGAA (filled symbols) was tested on purified DR4-Fc and DR5-Fc in comparison to Db-Glyco-scTRAIL-95L8 (open symbols). Mean ± SD (n = 3).
Furthermore, enhanced bioactivities were observed in vitro with 3 different target cell lines, especially for Db10-Glyco-scTRAIL-FAVSGAA and Db-Glyco-scTRAIL-FAVSGIA, with EC50 values in the low picomolar or even sub-picomolar range, depending on the tumor cell line (Fig. 5, Table 3). As exemplified for Db10-Glyco-scTRAIL-FAVSGAA, high bioactivities were also observed without addition of bortezomib (Fig. S8). Importantly, bioactivity was reduced in the presence of excess amounts of cetuximab, which blocks binding of the Db-scTRAIL molecules to EGFR (Fig. 5, Fig. 4B, C), indicating that targeting via EGFR contributes to enhanced cell death induction of these scTRAIL fusion proteins on appropriate target cells.
Figure 5.

Cell death induction of Db-scTRAIL proteins in vitro. The bioactivity of Db-Glyco-scTRAIL-FAVSGAA (left), Db10-Glyco-scTRAIL-FAVSGAA (middle) and Db-Glyco-scTRAIL-FAVSGIA (right, filled circles) was assayed in vitro in presence of the apoptosis sensitizer bortezomib (Brt) on Colo205 (upper, 250 ng/ml Brt), HT1080 (middle, 10 ng/ml Brt) and HCT116 (lower, 5 ng/ml Brt) tumor cells. Db-scTRAIL-95L8 served as a reference (filled squares). After 16 hours of incubation, crystal violet staining was performed. In order to demonstrate EGFR-dependent increase of the bioactivity, the binding of the fusion proteins was competed by blocking with 70 nM cetuximab (open symbols), resulting in abolished EGFR targeting and thus lower bioactivity. Mean ± SD (n = 3).
Table 3.
In vitro bioactivity of EGFR-specific Db-scTRAIL variants with or without EGFR blocking by cetuximab, in presence of bortezomib (mean ± SD, n = 3).
| EC50 (pM) |
||||
|---|---|---|---|---|
| Molecule | Cetuximab | Colo205 | HT1080 | HCT116 |
| Db-scTRAIL-95L8 | − | 24 ± 4 | 3 ± 0.7 | 127 ± 2 |
| + | 48 ± 5 | 15 ± 4 | 381 ± 16 | |
| Db-Glyco-scTRAIL-FAVSGAA | − | 33 ± 6 | 1 ± 0.6 | 66 ± 10 |
| + | 74 ± 15 | 10 ± 4 | 450 ± 2 | |
| Db10-Glyco-scTRAIL-FAVSGAA | − | 13 ± 2 | 0.7 ± 0.2 | 39 ± 4 |
| + | 66 ± 17 | 8 ± 2 | 298 ± 3 | |
| Db-Glyco-scTRAIL-FAVSGIA | − | 19 ± 3 | 0.6 ± 0.2 | 43 ± 2 |
| + | 32 ± 5 | 5 ± 1 | 161 ± 3 | |
In vivo study of Db10-Glyco-scTRAIL-FAVSGAA
The antitumor bioactivity of Db10-Glyco-scTRAIL-FAVSGAA was investigated using the established mouse xenograft model of the human colon carcinoma cell line Colo205. Db-scTRAIL fusion was intravenously administered when tumors reached a volume of ∼100 mm3. Three different doses of 0.1 nmol, 0.3 nmol and 1 nmol protein were applied in a daily regimen for 8 d in combination with intraperitoneal injection of clinical grade bortezomib. Whereas treatment with 0.1 nmol did not induce measurable antitumor effects, a transient, partial inhibition of tumor growth was observed for the 0.3 nmol group, which was, however, statistically not significant (Fig. 6A). In contrast, a strong and rapid reduction of tumor volumes with macroscopically undetectable tumors in 9/12 cases at day 20 was observed for the 1 nmol dose. A re-growth of tumors was observed for a subpopulation around day 25 (subgroup I) and for a second subpopulation around day 42 (subgroup II). Both subgroups received a second, identical treatment cycle starting at day 28 (subgroup I) or day 43 (subgroup II) (Fig. 6A). An antitumor response was again observed for both groups. At the end of the observation period (d109), 4/12 tumors stayed in complete macroscopic remission and 3 other tumors were in a stable, not actively growing state with volumes below 100 mm3.
Figure 6.
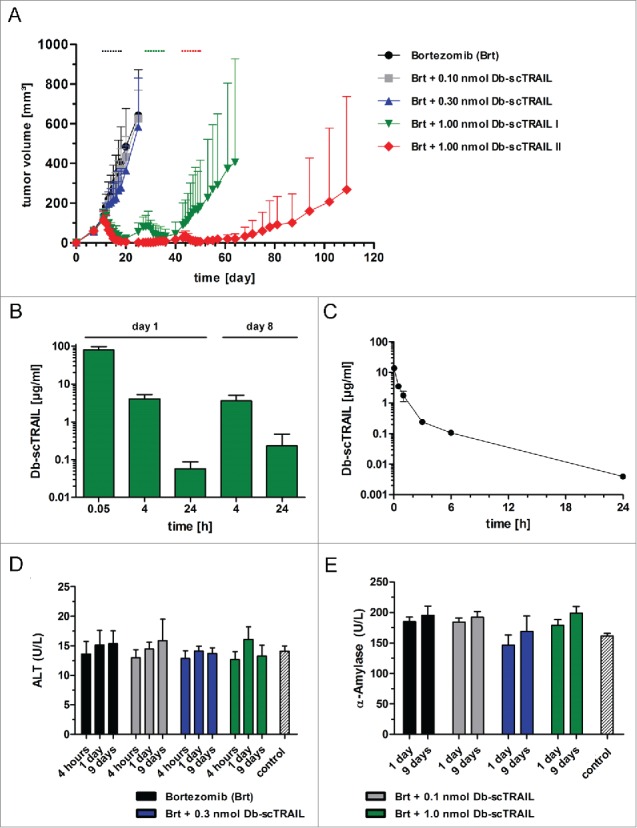
In vivo antitumor activity, pharmacokinetics and safety of Db10-Glyco-scTRAIL-FAVSGAA. (A) Colo205-bearing nude mice received 8 i.v. injections of the Db10-Glyco-scTRAIL-FAVSGAA fusion protein (0.1 nmol, 0.3 nmol, or 1.0 nmol) in combination with 8 intraperitoneal (i.p.) injections of bortezomib (Brt; 5µg per injection) every day indicated by dots. Mean ± 95% CI (n = 12 tumors per group). The 1.0 nmol-treated group received an identical regime of Db10-Glyco-scTRAIL-FAVSGAA and bortezomib after regrowth of tumors (volume ∼100 mm3). Therefore, animals were divided into 2 subgroups of fast (I) and slow (II) regrowth. (B, C) Db10-Glyco-scTRAIL-FAVSGAA (168 µg per animal in B / 25 µg per animal in C) was injected i.v. into Colo205-bearing nude mice (B) or CD-1 mice (C). The serum concentrations of the fusion protein were analyzed via ELISA. Mean ± SD (n = 3). (D, E) Activity of alanine aminotransferase (ALT, D) and α-amylase (E) was measured after 4 hours (only for D), 1 day, and 9 d after the first of total 8 injections (every day) of Db10-Glyco-scTRAIL-FAVSGAA (0.1 nmol, 0.3 nmol, or 1.0 nmol; i.v.) and/or bortezomib (Brt, 5 µg; i.p.). Control mice were non-treated. Mean ± SD (n = 3).
Serum concentrations of Db10-Glyco-scTRAIL-FAVSGAA were determined by ELISA 0.05 h, 4 h, and 24 h after the first injection, as well as 4 h and 24 after the last injection of the first cycle, showing that similar serum concentrations were reached after the first and last injection (Fig. 6B). Additionally, the pharmacokinetics of Db10-Glyco-scTRAIL-FAVSGAA were studied in immunocompetent CD-1 mice, revealing a terminal half-life of 3.6 ± 0.1 h and an AUC of 8.8 ± 1.4 (µg/ml)*h (Fig. 6C).
The safety of the applied doses of Db10-Glyco-scTRAIL-FAVSGAA was monitored by measuring serum activities of alanine aminotransferase (ALT) and α-amylase (Fig. 6D, E). Serum samples were taken from all groups 4 hours and 24 hours after the first injection, as well as 24 h after the last injection of the first treatment cycle, and compared to untreated animals. In both assays, no statistically significant increase of ALT and α-amylase levels were observed for all treatment groups compared to the untreated animals, except for the α-amylase value of the 1 nmol group at day 9 (24 h after last injection, 199 U/L vs. 161 U/L, p < 0.05).
Discussion
To explore the therapeutic potential of ligands of the TNF family, single-chain variants that are superior to naturally occurring or recombinant homotrimeric ligands, and represent a versatile core module that allows engineering of bi- or multifunctional reagents, e.g., with target-specific activity, have recently been developed.18,20,21,26,27 In addition to a high specific activity, clinically useful recombinant protein therapeutics must also have suitable protein stability and pharmacokinetic properties.24,28,29 Toward this end, we here describe single-chain variants of the apoptosis-inducing TRAIL with improved stability and superior product quality in the form of a tumor-targeted fusion protein, resulting in high anti-tumor activity in a xenograft tumor model in vivo.
We achieved our goal by combining 3 approaches, 1) reducing the TRAIL subunit to its minimal THD domain (aa 122–281); 2) reducing the scTRAIL linkers connecting the individual subunits to a minimal length (0–2 aa residues); and 3) optimizing the flanking TRAIL sequences (aa 122, 279, 281) through site-directed mutagenesis, resulting in a scTRAIL molecule with an increased thermal stability of ≥ 10°C compared to parental homotrimeric sTRAIL and the first-generation scTRAIL molecules.18
The minimal THD was defined here to start with aa residue 122, which is in line with results from sequence alignments of TNF family members.30 In further support of this, crystallographic studies showed that Val122 is the first residue of the first β-strand A.25,31,32 The structure of the TRAIL THD further suggests a hydrophobic interaction of Val122 with Leu279. With respect to this, only certain combinations of double mutations, e.g., A-A, L-I, A-I, I-I, can be introduced without affecting functional expression, i.e., correct folding and secretion of a soluble protein, whereas other mutations like L-G, L-L, L-M, V-L and G-G failed in that relationship.
Two other approaches for the generation of scTRAIL molecules have recently been described. In one approach, a scTRAIL molecule was obtained by connecting a first TRAIL subunit (aa 91–281) with 2 shorter TRAIL subunits (aa 108–281), i.e., using part of the native extracellular stalk region of TRAIL (aa 108–120) to connect the subunits (TR3).33 This scTRAIL molecule (TR3) displayed increased stability, confirming earlier results for a single-chain TNF molecule showing that physical connection of the 3 subunits increases stability and bioactivity in vitro and in vivo.26 In another study, 3 TRAIL subunits comprising aa 121–281 were connected with glycine/serine linkers of 8 aa length comprising one N-glycosylation site (GSGSGNGS).34 Both studies did not report on the thermal stability of these scTRAIL molecules. By defining Val122 as the N-terminus of the TRAIL THD, the molecules described by Spitzer et al.33 and Gieffers et al.34 comprise 14 and 9 aa residues, respectively, that functionally serve as linker. Our results suggest that glycine/serine linkers of 10 or 12 residues have no or only marginal positive effects on the thermal stability of a scTRAIL, whereas a linker of 6 or fewer residues is required to significantly increase thermal stability, demonstrated most obviously for scTRAIL-FLVGGVA. Furthermore, we identified aa substitutions at positions 122, 279 and 281 in this minimal scTRAIL, which strongly reduced the spontaneous formation of oligomers.
The optimized scTRAIL modules can be favorably used as building blocks to generate highly potent scTRAIL fusion proteins for therapeutic applications. This was exemplified here with construction of a strictly dimeric Db-scTRAIL, capable of hexavalent TRAIL receptor interaction and bivalent targeting of EGFR, a receptor over-expressed in various types of cancer.35 An optimization strategy was applied to the linker connecting the diabody with the scTRAIL moiety (connecting linker), as well as the linker connecting the VH and VL domain of the diabody (Db linker), resulting in a Db10-Glyco-scTRAIL molecule with an improved expression profile, biochemical properties and an in vitro tumor cell killing activity with EC50 values in the low picomolar range. Specifically, the observed low content of higher molecular weight oligomers and aggregates (< 12%) in this product is of particular relevance because dose-limiting toxicity has been ascribed to high molecular weight species of TRAIL,36,37 as well as to antibody-based multivalent TRAILR agonists.38 Importantly, an improved thermal stability was retained in this Db-scTRAIL molecule. Furthermore, these optimized molecules had a drastically reduced Stoke's radius compared to the parental form (5.8 nm vs. 6.4 nm for the glycosylated forms) used in a previous study, very probably due to the strongly reduced Stoke's radius of the scTRAIL moiety. A smaller size might facilitate extravasation and tumor penetration, thus increasing anti-tumor activity.39-41 In support of this reasoning and in accordance with high specific bioactivity in in vitro assays, a first animal study using a xenograft Colo205 tumor model showed a potent and long-lasting tumor response without dose-limiting toxicity, though a direct comparison with pharmacodynamics data from Db-scTRAIL-95L8 20 is actually hampered due to differences in protein administration (doses, intraperitoneal vs. intravenous injection). To explore the therapeutic potential of fusion proteins comprising engineered scTRAIL variants in a broader way, the modification of pharmacokinetic properties by fusing half-life elongation moieties like Fc might be revealing.42 Accordingly, we propose that dimeric, highly bioactive fusion protein formats like scTRAIL-Fc/ Fc-scTRAIL are promising candidates for difficult to treat cancers, in particular when combined with antibody-mediated targeting of relevant tumor-associated antigens, for example in the scFv-Fc-scTRAIL format. The structural optimization described here could potentially also be applied for death receptor (DR4/DR5) selective TRAIL variants in order to avoid competition with decoy receptors that could affect the efficacy of TRAIL-based therapies.43 A possible approach combining such receptor-selective mutations with our refined concept for single-chain TRAIL described here could lead to the development of novel TRAIL-based therapies in oncology.
In summary, we engineered an improved scTRAIL, which represents an amenable structure for a second generation of TRAIL-based cancer therapeutics displaying target-selective activity. Moreover, given the strong conservation of tertiary structure of all members of the TNF ligand family, the strategy of rational molecular evolution of a scTRAIL and scTRAIL-based fusion proteins used here may serve as a paradigm for other members of the TNF family, e.g., for immunostimulatory members of this family to be applied in cancer immunotherapeutic approaches.44,45
Materials and methods
Cell lines and materials
The pIRESpuro3-scTRAIL-95L8 and pCR3-Db-scTRAIL-95L8 expression plasmids were previously published.20 HEK293, HT1080, HCT116, NCI-H460 and Colo205 cells were purchased from ATCC. Cells were cultured in RPMI 1640 medium (Thermo Fisher, 21875–034) supplemented with 10% fetal bovine serum (FBS Premium, PAN Biotech, P30-3302), respectively 5% FBS for HEK293. Anti-FLAG M2 antibody (F1804) was purchased from Sigma-Aldrich and bortezomib was from UBPBio (F1200). Velcade (clinical grade bortezomib) and cetuximab were kindly provided by Dr. J. Schmid (Institute of Clinical Pharmacology, Margarete Fischer-Bosch Foundation, Stuttgart, Germany). Soluble TRAIL (sTRAIL, aa 115–281, 310–04) was purchased from Peprotech.
Generation of expression plasmids
For generation of scTRAIL variants, PCR products of individual TRAIL modules were obtained with the oligonucleotide pairs fw_TRAIL 1_EcoRI/ rv_TRAIL 1, fw_TRAIL 2/ rv_TRAIL 2 and fw_TRAIL 3/ rv_TRAIL 3_NotI. After agarose gel purification, 10% aliquots of both PCR products coding for TRAIL modules 1 and 2 were annealed at the introduced overlapping regions and 3′ strand elongated in a 30 µl reaction using Pfu DNA polymerase (Thermo Fisher, EP0501) for 5 cycles. Then, 20 µl of this reaction was used in another 5 cycles of annealing/ elongation with a 10% aliquot of the PCR product for TRAIL module 3 in a volume of 80 µl. Finally, the reaction was made up to 100 µl by addition of dNTPs, 10 × Pfu buffer and 50 pmol of oligonucleotides comprising EcoRI and NotI restriction sites, followed by PCR amplification of the fully assembled scTRAIL DNA construct and EcoRI/ NotI cloning into pIRESpuro3-scTRAIL-95L8.
All EGFR-specific pCR3-Db-Glyco-scTRAIL constructs were derived from the vector pCR3-scFv-scTRAIL-95L8.20 The scFv sequence from this vector was amplified by PCRs to insert the sequence coding for 2 N-glycosylation sites by XhoI/ EcoRI cloning into the construct. PCR3-Db-Glyco-scTRAIL-95L8 was then obtained from pCR3-scFv-Glyco-scTRAIL-95L8 by XhoI/ NotI cloning of the diabody sequence from pCR3-Db-scTRAIL-95L8. The scTRAIL sequences were amplified from pIRESpuro3-scTRAIL-FAVSGAA and pIRESpuro3-scTRAIL-FAVSGIA followed by EcoRI/ XbaI cloning into pCR3-Db-scTRAIL-95L8 or pCR3-Db-Glyco-scTRAIL-95L8 to yield pCR3-Db-scTRAIL-FAVSGAA, pCR3-Db-Glyco-scTRAIL-FAVSGAA and pCR3-Db-Glyco-scTRAIL-FAVSGIA, respectively. PCR3-Db10-scTRAIL-FAVSGAA was generated by PCR of VH and VL from pCR3-scFv-scTRAIL-95L8 using oligonucleotides comprising complementary regions. Next, the gel-purified DNA strands were assembled, amplified with terminal oligonucleotides and cloned via XhoI/ EcoRI into pCR3-Db-scTRAIL-FAVSGAA. PCR3-Db10-Glyco-scTRAIL-FAVSGAA was obtained in a similar way from pCR3-Db-Glyco-scTRAIL-FAVSGAA using the PCR template pCR3-scFv-Glyco-scTRAIL-95L8.
Production and purification of recombinant proteins
ScTRAIL proteins were produced in HEK293 cells and purified from supernatants by anti-FLAG affinity chromatography as described elsewhere.46 In brief, stably transfected cell pools were cultivated for 3 d at 37°C/ 5% CO2 with OptiMEM I medium (Thermo Fisher, 31985–070) supplemented with 50 µM ZnCl2. Cell-free supernatants were then incubated with anti-FLAG M2 Affinity Gel (3 ml bead volume/ l supernatant, Sigma-Aldrich, A2220) for at least 2 h or alternatively overnight at 4°C on a roller mixer, prior to collecting of beads in an empty column, washing with 1 × TBS and eluting with 100 µg/ml FLAG peptide/ 1 × TBS (peptides&elephants). After dialysis in 1 × PBS, eluates were concentrated with Vivaspin 20 devices (10 or 50 kDa, Sartorius, VS2001/ VS2031). Protein concentrations were determined spectrophotometrically using the calculated extinction coefficients. Aliquots were stored at −80°C. DR4-Fc, DR5-Fc and EGFR-Fc fusion proteins were produced and purified as previously described.21
Biochemical and biophysical protein analysis
Affinity-purified scTRAIL proteins were analyzed by SDS-PAGE under reducing conditions and stained with InstantBlue (Expedeon, ISB1L). For Western blotting, monoclonal anti-FLAG M2 antibody (1 µg/ml in PBS) was used, followed by anti-mouse IgG alkaline phosphatase-coupled secondary antibody (1:15,000 in PBS with 0.05% Tween 20, Sigma-Aldrich, A3562) for detection. For SEC, proteins were applied to a Yarra 3 µm SEC-2000 or Yarra 3 µm SEC-3000 (300 × 7.8 mm) HPLC column (Phenomenex, 00H-4512-K0/ 00H-4513-K0) equilibrated in 0.1 M Na2HPO4/ NaH2PO4, 0.1 M Na2SO4, pH 6.7 and eluted at a flow rate of 0.5 ml/min. The following reference molecules were used: thyroglobulin, β-amylase, BSA, carbonic anhydrase and FLAG peptide. For enzymatic removal of N-glycosylation, 5 µg of protein was denatured and treated with PNGase F (New England BioLabs, P0704S) according to manufacturer's protocol. The thermal stability of the proteins was determined by measuring denaturation temperatures with dynamic light scattering using a ZetaSizer Nano ZS instrument (Malvern) as published.21
ELISA
ELISA plates were coated with TRAIL-R1-Fc, TRAIL-R2-Fc or EGFR-Fc 21 (300 ng/well in 0.1 M sodium carbonate buffer, pH 9.5) overnight at 4°C. The plates were blocked with 3% (w/v) skim milk powder in PBS (MPBS). The scTRAIL fusion proteins were diluted in MPBS, titrated 1:3 in duplicates starting from 500 nM and incubated for 2 h at room temperature on a shaker. For competition experiments, wells were incubated with 10 µM cetuximab in MPBS for 30 min before adding the scTRAIL fusion proteins. Bound proteins were detected by anti-FLAG M2 horseradish peroxidase conjugate (1:15,000 in MPBS, Sigma-Aldrich, A8592) using 3,3′,5,5′-tetramethylbenzidine (TMB) as substrate (0.1 mg/ml TMB, 100 mM sodium acetate buffer, pH 6.0, 0.006% H2O2). After stopping the reaction with 50 µl of 2 M sulfuric acid, plates were measured in an ELISA reader at 450 nm. Data were fitted with Prism (GraphPad Software) in order to calculate EC50 values ± SD from 3 independent experiments. The t-test was applied for statistical analysis of the data.
Flow cytometry
Cells were trypsinized, washed once with FACS buffer (PBS, 2% FBS, 0.05% NaN3) and re-suspended in the same buffer. 1.5 × 105 cells were incubated for 2 h on ice in presence of serially diluted scTRAIL fusion proteins in FACS buffer starting from 500 nM or without (background control). For competition experiments, cells were incubated with a 200-fold molar excess of cetuximab for 30 min on ice before adding the scTRAIL fusion proteins. After washing the cells 2 times with ice-cold FACS buffer, bound proteins were detected by anti-DYKDDDDK-PE labeled antibody (Miltenyi Biotec, 130-101-576) diluted 1:200 in FACS buffer. After two washing steps, cells were analyzed using a MACSQuant Analyzer 10 equipped with a 585/40 nm filter or a MACSQuant VYB with 586/15 nm filter (Miltenyi Biotec). Data were fitted with Prism from 3 independent binding curves.
Cell death assays
Colo205 (4 × 104/ well), HT1080 (2 × 104/ well) or HCT116 cells (2 × 104/ well) were grown in 100 µl culture medium in 96-well plates for 24 h, followed by treatment with serial dilutions (1:3) of scTRAIL or Db-scTRAIL fusion proteins in triplicates. Molar concentrations were normalized to one scTRAIL unit. For positive control, cells were lysed with 0.5% Triton X-100. Cell death assays were performed in the presence of bortezomib (250 ng/ml for Colo205, 10 ng/ml for HT1080, 5 ng/ml for HCT116; UBPBio, F1200). Bortezomib was added 30 min prior incubation with the proapoptotic ligands to sensitize carcinoma cells for cell death induction. After 16 h of incubation, cell viability was determined by crystal violet staining.47 To demonstrate target antigen-dependent induction of cell death, cells were preincubated for 30 min with 70 nM of the competing antibody cetuximab. Data were fitted with Prism in order to calculate EC50 values ± SD from 3 independent experiments.
Pharmacokinetic studies in CD-1 mice
Female CD-1 (Charles River, 8 weeks old, 3 animals) received an i.v. injection of 25 µg of Db10-Glyco-scTRAIL-FAVSGAA in a total volume of 100 µl. Blood samples (50 µl) were taken at 0.05, 0.5, 1, 3, 6, and 24 hours and incubated on ice for 10 minutes. Clotted blood was centrifuged at 16,100 × g for 30 minutes, 4°C and serum samples were stored at −20°C. The serum concentration of Db10-Glyco-scTRAIL-FAVSGAA was analyzed with BD OptEIA Human TRAIL ELISA Set (BD, 550948) according to the manufacturer's instructions. The first value (0.05 h) was set to 100%. The initial (t1/2α) and terminal (t1/2β) half-lives and the bioavailability (area under the curve, AUC) were calculated with MS Excel.
Xenograft mouse tumor model
Female NMRI nu/nu mice (Charles River, 8 weeks old) were injected s.c. with 3 × 106 Colo205 cells in 100 µl PBS at the left and right dorsal sides. Treatment started 11 d after tumor cell inoculation when tumors reached an average volume of 100 mm3. Mice received 8 daily i.v. injections of 0.1 nmol, 0.3 nmol or 1.0 nmol of purified Db10-Glyco-scTRAIL-FAVSGAA in 100 µl PBS and additionally 8 daily intraperitoneal (i.p.) injections of 5 µg bortezomib one hour before protein application. The control groups received only 8 daily i.p. injections of 5 µg bortezomib. This amount of bortezomib was shown in previous studies to have no effects on tumor growth.20 For PK studies and toxicity assays, blood samples were taken as indicated and incubated on ice for 10 minutes. Clotted blood was centrifuged at 16,100 × g for 30 minutes at 4°C, and serum samples were stored at −20°C. The activity of alanine aminotransferase (ALT) and α-amylase were measured using an enzymatic assay kit (abcam) according to the manufacturer's instructions. A second treatment cycle (identical regimen) was applied for the 1.0 nmol treated group when tumor regrowth was observed. Tumor volume was monitored as described.18 One-way ANOVA and Tukey post hoc tests were performed for statistical analysis.
Supplementary Material
Disclosure of potential conflicts of interest
MS, MH, OS, RK, KP are named inventors on patent applications covering the scTRAIL technology.
Acknowledgments
We thank Jens Schmid from IKP Stuttgart for providing clinical grade bortezomib and cetuximab. This study was supported by grants from BioNTech AG and BMBF (PREDICT Project No. 0316185A).
References
- 1.Yee L, Fanale M, Dimick K, Calvert S, Robins C, Ing J, Ling J, Novotny W, Ashkenazi A, Burris H III. A phase IB safety and pharmacokinetic (PK) study of recombinant human Apo2L/TRAIL in combination with rituximab in patients with low-grade non-Hodgkin lymphoma. J Clin Oncol 2007; 25: abstract 8078; http://meeting.ascopubs.org/cgi/content/short/25/18_suppl/8078 [Google Scholar]
- 2.Soria JC, Mark Z, Zatloukal P, Szima B, Albert I, Juhasz E, Pujol JL, Kozielski J, Baker N, Smethurst D, et al.. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J Clin Oncol 2011; 29: 4442-51; PMID:22010015; http://dx.doi.org/ 10.1200/JCO.2011.37.2623 [DOI] [PubMed] [Google Scholar]
- 3.Wainberg ZA, Messersmith WA, Peddi PF, Kapp AV, Ashkenazi A, Royer-Joo S, Portera CC, Kozloff MF. A phase 1B study of dulanermin in combination with modified FOLFOX6 plus bevacizumab in patients with metastatic colorectal cancer. Clin Colorectal Cancer 2013; 12: 248-54; PMID:24075777; http://dx.doi.org/ 10.1016/j.clcc.2013.06.002 [DOI] [PubMed] [Google Scholar]
- 4.Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O'Dwyer PJ, Gordon MS, Novotny W, Goldwasser MA, Tohnya TM, Lum BL, et al.. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol 2010; 28: 2839-46; PMID:20458040; http://dx.doi.org/ 10.1200/JCO.2009.25.1991 [DOI] [PubMed] [Google Scholar]
- 5.Soria JC, Smit E, Khayat D, Besse B, Yang X, Hsu CP, Reese D, Wiezorek J, Blackhall F. Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J Clin Oncol 2010; 28: 1527-33; PMID:20159815; http://dx.doi.org/ 10.1200/JCO.2009.25.4847 [DOI] [PubMed] [Google Scholar]
- 6.Chen W, Qiu L, Hou J, Zhang X, Ke X, Wang Z, Zhou F, Yang S, Zhao Y, Leng Y, et al.. Phase Ib study of recombinant circularly permuted TRAIL (CPT) in relapsed or refractory multiple myeloma patients. 54th ASH annual meeting abstr 2012a; 2012: 1857; https://ash.confex.com/ash/2012/webprogram/Paper47989.html [Google Scholar]
- 7.Chen W, Hou J, Zhao Y, Qiu L, Ke X, Wang Z, Leng Y, Jing H, Xi H, Zheng X, et al.. Circularly permuted TRAIL (CPT) combined with thalidomide for the treatment of relapsed or refractory multiple myeloma: an open-label, multicenter phase II clinical trial. 54th ASH annual meeting abstr 2012: 2958; https://ash.confex.com/ash/2012/webprogram/Paper48023.html [Google Scholar]
- 8.Tolcher AW, Mita M, Meropol NJ, von Mehren M, Patnaik A, Padavic K, Hill M, Mays T, McCoy T, Fox NL, et al.. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. J Clin Oncol 2007; 25: 1390-5. Erratum in: J Clin Oncol 2007; 25: 4701; PMID:17416859; http://dx.doi.org/ 10.1200/JCO.2006.08.8898 [DOI] [PubMed] [Google Scholar]
- 9.Trarbach T, Moehler M, Heinemann V, Köhne CH, Przyborek M, Schulz C, Sneller V, Gallant G, Kanzler S. Phase II trial of mapatumumab, a fully human agonistic monoclonal antibody that targets and activates the tumour necrosis factor apoptosis-inducing ligand receptor-1 (TRAIL-R1), in patients with refractory colorectal cancer. Br J Cancer 2010; 102: 506-12; PMID:20068564; http://dx.doi.org/ 10.1038/sj.bjc.6605507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herbst RS, Kurzrock R, Hong DS, Valdivieso M, Hsu CP, Goyal L, Juan G, Hwang YC, Wong S, Hill JS, et al.. A first-in-human study of conatumumab in adult patients with advanced solid tumors. Clin Cancer Res 2010; 16: 5883-91; PMID:20947515; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-0631 [DOI] [PubMed] [Google Scholar]
- 11.Plummer R, Attard G, Pacey S, Li L, Razak A, Perrett R, Barrett M, Judson I, Kaye S, Fox NL, et al.. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin Cancer Res 2007; 13: 6187-94; PMID:17947486; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-0950 [DOI] [PubMed] [Google Scholar]
- 12.Holland PM. Death receptor agonist therapies for cancer, which is the right TRAIL? Cytokine Growth Factor Rev 2014; 25: 185-93; PMID:24418173; http://dx.doi.org/ 10.1016/j.cytogfr.2013.12.009 [DOI] [PubMed] [Google Scholar]
- 13.Micheau O, Shirley S, Dufour F. Death receptors as targets in cancer. Br J Pharmacol 2013; 169: 1723-44; PMID:23638798; http://dx.doi.org/ 10.1111/bph.12238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalves F, Ashenazi A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 2010; 29: 4752-65; PMID:20531300; http://dx.doi.org/ 10.1038/onc.2010.221 [DOI] [PubMed] [Google Scholar]
- 15.Wajant H, Pfizenmaier K, Scheurich P. TNF-related apoptosis inducing ligand (TRAIL) and its receptors in tumor surveillance and cancer therapy. Apoptosis 2002; 7: 449-59; PMID:12207178; http://dx.doi.org/ 10.1023/A:1020039225764 [DOI] [PubMed] [Google Scholar]
- 16.van Roosmalen IA, Quax WJ, Kruyt FA. Two death-inducing human TRAIL recpetors to target in cancer: similar or distinct regulation and function? Biochem Pharmacol 2014; 91: 447-56; PMID:25150214; http://dx.doi.org/ 10.1016/j.bcp.2014.08.010 [DOI] [PubMed] [Google Scholar]
- 17.Wajant H, Moosmayer D, Wuest T, Bartke T, Gerlach E, Schönherr U, Peters N, Scheurich P, Pfizenmaier K. Differential activation of TRAIL-R1 and −2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRIAL-R2 by a soluble TRAIL derivative. Oncogene 2001; 20: 4101-16; PMID:11494138; http://dx.doi.org/ 10.1038/sj.onc.1204558 [DOI] [PubMed] [Google Scholar]
- 18.Schneider B, Münkel S, Krippner-Heidenreich A, Grunwald I, Wels WS, Wajant H, Pfizenmaier K, Gerspach J. Potent antitumoral activity of TRAIL through generation of tumor-targeted single-chain fusion proteins. Cell Death Dis 2010; 1: e68; PMID:21364672; http://dx.doi.org/ 10.1038/cddis.2010.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Bruyn M, Bremer E, Helfrich W. Antibody-based fusion proteins to target death receptors in cancer. Cancer Lett 2013; 332: 175-83; PMID:21215513; http://dx.doi.org/ 10.1016/j.canlet.2010.11.006 [DOI] [PubMed] [Google Scholar]
- 20.Siegemund M, Pollak N, Seifert O, Wahl K, Hanak K, Vogel A, Nussler AK, Göttsch D, Münkel S, Bantel H, et al.. Superior antitumoral activity of dimerized targeted single-chain TRAIL fusion proteins under retention of tumor selectivity. Cell Death Dis 2012; 3: e295; PMID:22495350; http://dx.doi.org/ 10.1038/cddis.2012.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seifert O, Plappert A, Fellermeier S, Siegemund M, Pfizenmaier K, Kontermann RE. Tetravalent antibody-scTRAIL fusion proteins with improved properties. Mol Cancer Ther 2014; 13: 101-11; PMID:24092811; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0396 [DOI] [PubMed] [Google Scholar]
- 22.Kowalski JM, Parekh RN, Mao J, Wittrup KD. Protein folding stability can determine the efficiency of escape from endoplasmic reticulum quality control. J Biol Chem 1998; 273: 19453-8; PMID:9677365; http://dx.doi.org/ 10.1074/jbc.273.31.19453 [DOI] [PubMed] [Google Scholar]
- 23.Shusta EV, Holler PD, Kieke MC, Kranz DM, Wittrup KD. Directed evolution of a stable scaffold for T-cell receptor engineering. Nat Biotechnol 2000; 18: 754-9; PMID:10888844; http://dx.doi.org/ 10.1038/77325 [DOI] [PubMed] [Google Scholar]
- 24.Kim DY, Hussack G, Kandalaft H, Tanha J. Mutational approaches to improve the biophysical properties of human single-domain antibodies. Biochim Biophys Acta 2014; 1844: 1983-2001; PMID:25065345; http://dx.doi.org/ 10.1016/j.bbapap.2014.07.008 [DOI] [PubMed] [Google Scholar]
- 25.Hymowitz SG, Christinger HW, Fuh G, Ultsch M, O'Connell M, Kelley RF, Ashkenazi A, de Vos AM. Triggering cell death: the crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol Cell 1999; 4: 563-71; PMID:10549288; http://dx.doi.org/ 10.1016/S1097-2765(00)80207-5 [DOI] [PubMed] [Google Scholar]
- 26.Krippner-Heidenreich A, Grunwald I, Zimmermann G, Kühnle M, Gerspach J, Sterns T, Shnyder SD, Gill JH, Männel DN, Pfizenmaier K, et al.. Single-chain TNF, a TNF derivative with enhanced stability and antitumoral activity. J Immunol 2008; 180: 8176-83; PMID:18523283; http://dx.doi.org/ 10.4049/jimmunol.180.12.8176 [DOI] [PubMed] [Google Scholar]
- 27.Fischer R, Maier O, Siegemund M, Wajant H, Scheurich P, Pfizenmaier K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS One 2011; 6: e27621; PMID:22110694; http://dx.doi.org/ 10.1371/journal.pone.0027621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szlachcic A, Zakrzewska M, Otlewski J. Longer action means better drugs: tuning up protein therapeutics. Biotechnol Adv 2011; 29: 436-41; PMID:21443940; http://dx.doi.org/ 10.1016/j.biotechadv.2011.03.005 [DOI] [PubMed] [Google Scholar]
- 29.Marshall SA, Lazar GA, Chirino AJ, Desjarlais JR. Rational design and engineering of therapeutic proteins. Drug Discov Today 2003; 8: 212-21; PMID:12634013; http://dx.doi.org/ 10.1016/S1359-6446(03)02610-2 [DOI] [PubMed] [Google Scholar]
- 30.Bodmer JL, Schneider P, Tschopp J. The molecular architecture of the TNF superfamily. Trends Biochem Sci 2002; 27: 19-26; PMID:11796220; http://dx.doi.org/ 10.1016/S0968-0004(01)01995-8 [DOI] [PubMed] [Google Scholar]
- 31.Cha SS, Kim MS, Choi YH, Sung BJ, Shin NK, Shin HC, Sung YC, Oh BH. 2.8 A resolution crystal structure of human TRAIL, a cytokine with selective antitumor activity. Immunity 1999; 11: 253-61; PMID:10485660; http://dx.doi.org/ 10.1016/S1074-7613(00)80100-4 [DOI] [PubMed] [Google Scholar]
- 32.Hymowitz SG, O'Connell MP, Ultsch MH, Hurst A, Totpal K, Ashkenazi A, de Vos AM, Kelley RF. A unique zinc-binding site revealed by a high-resolution X-ray structure of homotrimeric Apo2L/TRAIL. Biochemistry 2000; 39: 633-40; PMID:10651627; http://dx.doi.org/ 10.1021/bi992242l [DOI] [PubMed] [Google Scholar]
- 33.Spitzer D, McDunn JE, Plambeck-Suess S, Goedegebuure PS, Hotchkiss RS, Kawkins WG. A genetically encoded multifunctional TRAIL trimer facilitates cell-specific targeting and tumor cell killing. Mol Cancer Ther 2010; 9: 2142-51; PMID:20571073; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-0225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gieffers C, Klug M, Merz C, Sykora J, Thiemann M, Schaal R, Fischer C, Branschädel M, Hohenberger P, Fulda S, et al.. APG350 induces superior clustering of TRAIL receptors and shows therapeutic antitumor efficacy independent of cross-linking via Fcγ receptors. Mol Cancer Ther 2013; 12: 2735-47; PMID:24101228; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0323 [DOI] [PubMed] [Google Scholar]
- 35.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2: 127-37; PMID:11252954; http://dx.doi.org/ 10.1038/35052073 [DOI] [PubMed] [Google Scholar]
- 36.Lawrence D, Shahrokh Z, Marsters S, Achilles K, Shih D, Mounho B, Hillan K, Totpal K, DeForge L, Schow P, et al.. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat Med 2001; 7: 383-5; PMID:11283636; http://dx.doi.org/ 10.1038/86397 [DOI] [PubMed] [Google Scholar]
- 37.Ganten TM, Koschny R, Sykora J, Schulze-Bergkamen H, Büchler P, Haas TL, Schader MB, Untergasser A, Stremmel W, Walczak H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin Cancer Res 2006; 12: 2640-6; PMID:16638878; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-2635 [DOI] [PubMed] [Google Scholar]
- 38.Papadopoulos KP, Isaacs R, Bilic S, Kentsch K, Huet HA, Hofmann M, Rasco D, Kundamal N, Tang Z, Cooksey J, et al.. Unexpected hepatotoxicity in a phase I study of TAS266, a novel tetravalent agonistic Nanobody® targeting the DR5 receptor. Cancer Chemother Pharmacol 2015; 75: 887-95; PMID:25721064; http://dx.doi.org/ 10.1007/s00280-015-2712-0 [DOI] [PubMed] [Google Scholar]
- 39.Beckman RA, Weiner LM, Davis HM. Antibody constructs in cancer therapy: protein engineering strategies to improve exposure in solid tumors. Cancer 2006; 109: 170-9; PMID:17154393; http://dx.doi.org/ 10.1002/cncr.22402 [DOI] [PubMed] [Google Scholar]
- 40.Thurber GM, Schmidt MM, Wittrup KD. Factors determining antibody distribution in tumors. Trends Pharmacol Sci 2007; 29: 57-61; http://www.sciencedirect.com/science/article/pii/S0165614707002854; PMID:187982824151537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muchekehu R, Lliu D, Horn M, Campbell L, Rosario JD, Bacica M, Moskowitz H, Osothprarop T, Dirksen A, Doppelapudi V, et al.. The effect of molecular weight, PK and valency on tumor biodistribution and efficacy of antibody-based drugs. Transl Oncol 2013; 6: 562-72; PMID:24151537; http://dx.doi.org/ 10.1593/tlo.13409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7: 715-25; PMID:17703228; http://dx.doi.org/ 10.1038/nri2155 [DOI] [PubMed] [Google Scholar]
- 43.O'Leary L, van der Sloot AM, Reis CR, Deegan S, Ryan AE, Dhami SP, Murillo LS, Cool RH, de Sampaio PC, Thompson K, Murphy G, Quax WJ, Serrano L, Samali A, Szegezdi E. Decoy receptors block TRAIL sensitivity at a supracellular level: the role of stromal cells in controlling tumour TRAIL sensitivity. Oncogene 2015; PMID:26050621; http://dx.doi.org/ 10.1038/onc.2015.180 [DOI] [PubMed] [Google Scholar]
- 44.Kontermann RE. Antibody-cytokine fusion proteins. Arch Biophys Biochem 2012; 526: 194-205; PMID:22445675; http://dx.doi.org/ 10.1016/j.abb.2012.03.001 [DOI] [PubMed] [Google Scholar]
- 45.Bremer E. Targeting of the tumornecrosis factor receptor superfamily for cancer immunotherapy. ISRN Oncol 2013: 371854; PMID:23840967; http://dx.doi.org/10.1155/2013/371854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siegemund M, Richter F, Seifert O, Unverdorben F, Kontermann RE. Expression and purification of recombinant antibody formats and antibody fusion proteins. Methods Mol Biol 2014; 1131: 273-95; PMID:24515473; http://dx.doi.org/ 10.1007/978-1-62703-992-5_18 [DOI] [PubMed] [Google Scholar]
- 47.Wüest T, Gerlach E, Banerjee D, Gerspach J, Moosmayer D, Pfizenmaier K. TNF-Selectokine: a novel prodrug generated for tumor targeting and site-specific activation of tumor necrosis factor. Oncogene 2002; 21: 4257-65; PMID:12082613; http://dx.doi.org/ 10.1038/sj.onc.1205193 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.