

Abstract
A long-standing paradox in the pathophysiology of metabolic diseases is the selective insulin resistance of the liver. It is characterized by a blunted action of insulin to reduce glucose production, contributing to hyperglycemia, while de novo lipogenesis remains insulin sensitive, participating in turn to hepatic steatosis onset. The underlying molecular bases of this conundrum are not yet fully understood. Here, we established a model of selective insulin resistance in mice by silencing an inhibitor of insulin receptor catalytic activity, the growth factor receptor binding protein 14 (Grb14) in liver. Indeed, Grb14 knockdown enhanced hepatic insulin signaling but also dramatically inhibited de novo fatty acid synthesis. In the liver of obese and insulin-resistant mice, downregulation of Grb14 markedly decreased blood glucose and improved liver steatosis. Mechanistic analyses showed that upon Grb14 knockdown, the release of p62/sqstm1, a partner of Grb14, activated the transcription factor nuclear factor erythroid-2-related factor 2 (Nrf2), which in turn repressed the lipogenic nuclear liver X receptor (LXR). Our study reveals that Grb14 acts as a new signaling node that regulates lipogenesis and modulates insulin sensitivity in the liver by acting at a crossroad between the insulin receptor and the p62-Nrf2-LXR signaling pathways.
INTRODUCTION
The prevalence of metabolic diseases, including obesity and type 2 diabetes, is expanding worldwide, in close association with nonalcoholic fatty liver diseases (NAFLD). These pathologies are characterized by a decreased action of insulin on its target tissues. However, in the liver, insulin resistance leads to a blunted inhibitory action on hepatic glucose production, inducing hyperglycemia, whereas de novo lipogenesis, which is positively regulated by insulin, is paradoxically exacerbated, contributing to hepatic steatosis and hypertriglyceridemia. This phenomenon is known as the paradox of liver selective insulin resistance (1).
Insulin acts by binding to its membrane receptor to stimulate the receptor tyrosine kinase activity and consequently the Erk1/2 and phosphoinositol 3-kinase (PI3K)–Akt pathways. Insulin signaling bifurcates below Akt into two distinct pathways, one of which inhibits hepatic glucose production through inactivation of the transcription factor FoxO1 while the other stimulates de novo lipogenesis through the activation of sterol regulatory element binding protein 1c (SREBP-1c) (2). In insulin-resistant fatty liver, the activation of the PI3K-Akt pathway is strongly blunted, leading to a lack of FoxO1 inhibition and preserving an active gluconeogenesis, whereas SREBP-1c remains functional, stimulating the lipogenic pathway (3). The molecular mechanisms involved in this exacerbated SREBP-1c expression and activity remain to be fully clarified (4). In this context, the molecular adapter Grb14, an inhibitor of insulin signaling highly expressed in the liver, appears as an interesting candidate (5). Genome-wide association studies indeed recently revealed that the human GRB14 gene locus is associated with type 2 diabetes and insulin sensitivity (6–8). Moreover, GRB14 expression is enhanced in adipose tissue of type 2 diabetic patients and in skeletal muscle from morbidly obese women (9, 10), and its expression is restored to normal value following gastric surgery, which improves insulin sensitivity (10). In contrast, liver expression of Grb14 is not altered in physiopathological states, but it is decreased by insulin-sensitizing treatment in ob/ob mice (9). The Grb14 expression level is thus inversely correlated with insulin sensitivity in human and animal models of insulin resistance. We previously reported that Grb14 is recruited to the activated insulin receptor and inhibits its catalytic activity and downstream insulin signaling (5, 11, 12). Furthermore, Grb14 expression is stimulated by insulin, suggesting that it might be involved in a negative feedback loop of insulin signaling and action (9). Of note, we showed that downregulation of Grb14 expression in cultured hepatocytes improved insulin signaling but led to an unexpected decrease in SREBP-1c activation and lipogenic gene expression (13). Grb14 can thus regulate insulin signaling through mechanisms that are independent of the receptor kinase activity (14), suggesting that partners other than the insulin receptor are likely to be implicated in the Grb14-mediated modulation of lipogenesis.
In the present work, we addressed the molecular mechanisms involved in the regulation of hepatic lipogenesis by Grb14. Liver-specific Grb14 knockdown improves insulin signaling and simultaneously inhibits de novo fatty acid synthesis. Interestingly, the reduction of Grb14 expression in liver of ob/ob insulin-resistant mice ameliorates both glycemia and hepatic steatosis and consequently improves their metabolic profile. Further investigation of the molecular mechanism that drives lipogenesis inhibition after Grb14 downregulation revealed that the release of p62/sqstm1, a partner of Grb14 (15), triggers a signaling pathway leading to inhibition of the lipogenic nuclear receptor liver X receptor (LXR) activity and to subsequent decrease in fatty acid synthesis. Our study thus provides the first evidence that p62, a multitask adapter exerting a central role in cellular homeostasis through the regulation of nutrient sensing, autophagy, oxidative stress, and genomic stability (16), is involved in the regulation of liver metabolic homeostasis. Together, our data further identify Grb14 as a novel signaling node in the control of liver lipogenesis and selective insulin resistance.
MATERIALS AND METHODS
Animals and treatments.
Nine-week-old male mice were purchased from Harlan Laboratories (C57BL/6J) and Elevage Janvier (ob/ob) and were adapted to the environment for 1 week before the study. Nrf2−/− mice were previously described (17) and were studied at 15 weeks of age. Mice had free access to water and a regular diet (65% carbohydrate, 11% fat, and 24% protein). Mice were anesthetized with isoflurane before injection through the penis vein of a final volume of 150 μl sterile physiological serum containing 2 × 109 PFU of sh-scramble (USi) or shGrb14 (Grb14i) adenovirus (13). Experiments and analyses were performed 4 to 7 days after the adenoviral injection. For LXR activation studies, a single oral gavage of T0901317 (50 mg/kg body weight in 0.5% carboxymethylcellulose solution) was administered to mice 18 h before their sacrifice (18). All mice were housed in colony cages with a 12-h light/dark cycle in a temperature-controlled environment (the dark cycle going from 3:00 a.m. to 3:00 p.m.). For “fed” conditions, mice were sacrificed between 9:00 and 10:00 a.m. (during the dark cycle), for “fasting” conditions, mice were fasted overnight, and for “refed” conditions, mice starved for 24 h were fed a regular diet and 20% glucose water for 18 h. All procedures were carried out according to the French guidelines for the care and use of experimental animals. All animal studies were approved by the Direction départementale des services vétérinaires de Paris.
Glucose tolerance tests.
Glucose tolerance tests were performed by glucose gavage (1 g d-glucose/kg body weight) after an overnight fast. Blood glucose concentration was determined using the One-Touch AccuCheck glucometer (Roche).
Primary culture of hepatocytes and luciferase reporter assays.
Hepatocytes were isolated from livers of 8- to 10-week-old fed male C57BL/6J mice by an in situ collagenase method as described previously (19). Primary cultured hepatocytes were infected with USi, Grb14i, green fluorescent protein (GFP) (13), p62, p62i (Vector BioLabs), or p62 S351E mutant (20) recombinant adenovirus (1 to 3 PFU/cell) or transfected with plasmid vectors expressing wild-type (WT), S351A, or S351E p62 constructs or a control empty vector. For luciferase reporter assays, the reporter construct pGL-3xLXRE-luc or pGL-8xARE-luc (a kind gift from Roland Wolf [Dundee, United Kingdom] [21]) were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Luciferase activity was measured at room temperature 64 h after reporter transfection, and values were normalized for transfection efficiency using a Rous sarcoma virus β-galactosidase reporter (RSV β-Gal) as an internal control. For rapamycin experiments, hepatocytes were incubated in the presence of 0.5 μM rapamycin (Sigma) for the last 22 h. Experimental data are means from at least 3 independent experiments conducted in triplicate, with luciferase activity normalized to β-galactosidase activity.
Ex vivo measurement of liver lipogenic flux.
Mouse liver pieces (100 to 150 mg of liver tissue per flask, chopped into 20 to 30 pieces) were incubated in duplicate at 37°C in 25-ml conical flasks sealed with rubber caps in a final volume of 3 ml Krebs-Henseleit bicarbonate buffer (pH 7.4) containing 2% (wt/vol) defatted bovine serum albumin (BSA) and 5 mM [1-14C]acetate (0.4 Ci/mol; PerkinElmer). The flasks were gassed with O2-CO2 (19:1, wt/vol) prior to sealing and incubation. After 2 h, incubations were ended by centrifugation at 3,000 × g for 10 min at 4°C, and labeled lipids were extracted as described previously (22). Briefly, liver samples were saponified by heating in 0.5 ml of 30% (wt/vol) KOH at 70°C for 15 min, followed by the addition of 0.5 ml of 95% (vol/vol) ethanol and continued heating at 70°C for 1 h. After cooling and acidification with 0.5 ml of H2SO4 (8 M), lipids were extracted by shaking 3 times with 4 ml of petroleum ether. The petroleum ether fractions containing lipids were combined, washed 3 times with 5 ml of H2O, and then evaporated to dryness at room temperature with an overdraft of air. Labeled lipids were then quantified by scintillation counting.
Analytical procedures.
Liver triglycerides (TGs) and cholesterol (Diasys), glutathione (GSH/GSSG-Glo assay; Promega), and lactate (BioVision) were measured according to the manufacturers' kit instructions.
Isolation of total mRNA and analysis of mRNA expression by quantitative PCR.
Extraction and reverse transcription-quantitative PCR (qRT-PCR) analysis of RNAs from whole liver or from primary cultured hepatocytes were performed as described previously (13). Primer sequences are available upon request.
Western blot analysis and EMSA.
Protein extracts from cultured cells or mouse livers were prepared and analyzed by Western blotting as previously described (13). Liver cytoplasmic and nuclear extracts were prepared as described previously (23). Protein lysates were immunoblotted with the following antibodies: anti-p-4EBP1 (Cell Signaling number 9451), anti-4EBP1 (Cell Signaling number 9644), anti-pAKT (pS473; Cell Signaling number 9271), anti-AKT (Cell Signaling number 9272), anti-phosphorylated extracellular signal-regulated kinase 1/2 (anti-pERK1/2; Cell Signaling number 9101), anti-ERK1/2 (Cell Signaling number 9102), anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH; Santa Cruz Biotechnology sc-25778), anti-Grb14 (5), anti-lamin A/C (Cell Signaling number 2032), anti-Nrf2 (gift from Ken Itoh, Japan), anti-p62 (15), anti-pS351-p62 (20), anti-pS6K (Cell Signaling number 9234), anti-S6K (Cell Signaling number 9202), anti-SREBP-1c (Santa Cruz Biotechnology sc-8984). Electromobility shift assays (EMSA) were performed as described previously (23). The supershift was performed using LXR antibodies from abcam (ab41902).
Immunohistochemistry.
For histology studies, livers were fixed in 4% neutral buffered formalin and embedded in paraffin. Then, 5-μm sections were cut and stained with hematoxylin and eosin.
Transcriptomic analyses.
Gene expression profiles for liver from C57BL/6 mice injected with recombinant adenovirus expressing scramble (USi, n = 6) or Grb14 (Grb14i, n = 6) short hairpin RNA (shRNA) were analyzed using Affymetrix Mouse Gene 1.0 ST arrays (Gene Expression Omnibus [GEO] data set GSE53521). Samples were normalized using the RMA algorithm (Bioconductor affy package), and probe set intensities were then averaged per gene symbol. Differential expression was measured with the moderated t test (limma R package).
Statistical analysis.
Results are reported as means ± standard errors of the means (SEM). Comparison between two groups was carried out using a Student t test, and multiple-group comparisons were performed by one-way analysis of variance (ANOVA) followed by the Bonferroni posttest. Differences were considered statistically significant at P values of <0.05. Gene set enrichment analyses (GSEA) (24) were performed with a data set from mice with LXR knockout in liver (GEO no. GSE38083 [25]) and with a data set from mice with a genetic activation of Nrf2 in liver (GEO no. GSE15633 [26]).
RESULTS
Liver-specific Grb14 knockdown improves insulin signaling but represses de novo lipogenesis in lean and obese mice.
To investigate the metabolic consequences of an acute depletion of Grb14 expression in adult mouse liver, C57BL/6 mice were injected with recombinant adenovirus expressing scramble (USi) or Grb14 (Grb14i) shRNA and analyzed 7 days later. Grb14 expression was specifically blunted in the liver and not modified in other tissues (data not shown). As expected, hepatic Grb14 silencing induced a significant improvement in insulin signaling, as illustrated by the increased phosphorylation state of Akt, ERK1/2, and Akt targets (FoxO1, S6K, and 4EBP1) in liver of fed mice (Fig. 1A). In correlation with the enhanced insulin signal, the expression of gluconeogenic genes was significantly decreased in liver from Grb14i fasted mice, resulting in reduced blood glucose level and improved glucose tolerance, associated with reduced plasma insulin concentration (Fig. 1B to D). Despite enhanced insulin signaling, Grb14 silencing induced a paradoxical decrease in the expression of insulin-regulated glycolytic and lipogenic genes in the refed state (Fig. 1E and F), associated with a reduction in triglyceride liver content (Fig. 1G). The expressions of genes involved in lipid uptake, lipid oxidation, or lipid export were either unchanged or decreased (data not shown), suggesting that the lower liver lipid content was attributable mostly to the inhibition of the de novo lipid synthesis pathway. This hypothesis was confirmed by ex vivo measurement of the lipogenic flux, showing that Grb14 depletion led to a 40% decrease in de novo fatty acid synthesis and esterification (Fig. 1H). Gene expression of the transcription factor SREBP-1c (Srebf1), which mediates the effect of insulin on lipogenic gene expression, was blunted by Grb14 silencing (Fig. 1F). Insulin activates SREBP-1c by acting at both transcriptional and posttranslational levels, stimulating the proteolytic cleavage of the precursor anchored in the endoplasmic reticulum (27, 28). As shown in Fig. 1I, both precursor and nuclear forms of SREBP-1c were decreased in the absence of Grb14. As a result, these data suggest that the paradoxical decrease in the hepatic lipogenic pathway induced by Grb14 knockdown is linked to an impaired activation of SREBP-1c by insulin.
FIG 1.

Liver-specific silencing of Grb14 improves insulin signaling but inhibits de novo lipogenesis. C57BL/6J mice were injected with unspecific (USi) or Grb14 sh-adenovirus (Grb14i) and studied after 4 days (D) or 7 days (A to C and E to I). (A) Western blot analysis of Grb14 expression and insulin signaling pathways in liver of fed mice. (B) qRT-PCR analysis of gluconeogenic gene expression in liver from 24-h-starved mice. Expression is relative to that of 18S. (C) Blood glucose and plasma insulin in fed mice. (D) Oral glucose tolerance tests. (E, F) qRT-PCR analysis of glycolytic (E) and lipogenic (F) gene expression in liver from refed mice. (G) Liver triglyceride contents measured in liver lysates from fed mice. (H) Lipogenic flux measured ex vivo on liver pieces. (I) Precursor and mature SREBP-1c protein content in cytosolic and nuclear liver extracts. GAPDH and lamin A/C were used as loading controls for cytosolic and nuclear extracts, respectively. Results are the means ± SEM (n = 5 to 7 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.005 for Grb14i compared to USi mice. For Western blot analysis, representative blots of 3 samples from the 5 to 7 samples per group are shown.
The dual effect of Grb14 inhibition on insulin-regulated glucose metabolism prompted us to investigate the metabolic consequences of liver Grb14 silencing in obese and insulin-resistant mice, which exhibit hyperglycemia and liver steatosis. Grb14i treatment of ob/ob mice enhanced insulin signaling, improved glucose tolerance, and decreased gluconeogenic gene expression, lowering glycemia to a level below the value measured in lean mice with no change in plasma insulin level (Fig. 2A to D). Hepatic Grb14 silencing also induced a 60% decrease in liver lipid content, restoring it to the level observed in WT mice (Fig. 2G). As shown in lean mice, the reduction in lipid stores was associated with a decrease in glycolytic and lipogenic gene expression (Fig. 2E and F) and with a reduction in SREBP-1c protein expression (Fig. 2H). Inhibition of hepatic Grb14 expression thus results in a striking amelioration of both glucose and lipid homeostasis in the ob/ob diabetic mouse model.
FIG 2.

Targeting hepatic Grb14 content improves metabolic homeostasis in ob/ob mice. ob/ob mice were injected with USi- or Grb14i-expressing adenovirus and studied after 4 days (D) or 7 days (A to C and E to H). (A) Western blot analysis of Grb14 expression and insulin signaling pathways in liver of fed mice. (B) Gluconeogenic gene expression in liver from fasted mice. (C) Blood glucose and plasma insulin of fed mice. In panels C and G, the dashed line refers to the level measured in USi-treated C57BL/6 mice. (D) Oral glucose tolerance tests. Relative expression of glycolytic (E) and lipogenic (F) gene expression measured by qRT-PCR in liver from refed mice. (G) Quantification of liver triglyceride content in fed mice. (H) Western blot analysis of precursor and mature SREBP-1c protein content in cytosolic and nuclear liver extracts. Results are the means ± SEM (n = 6 or 7 per group); *, P < 0.05; **, P < 0.01; ***, P < 0.005 for Grb14i compared to USi. For Western blot analysis, representative blots of 3 samples from the 5 to 7 samples per group are shown.
Altogether these data show that liver Grb14 depletion increases insulin signaling and improves glucose metabolism but leads to a paradoxical inhibition of SREBP-1c expression, decreasing fatty acid synthesis and liver lipid content. The combination of these two effects restores both glucose and lipid metabolic defects to physiological conditions.
Liver Grb14 depletion abrogates LXR activity.
Since the nuclear receptor LXR is key for the insulin-stimulated transcriptional regulation of SREBP-1c (29), we examined its activity in liver of Grb14i mice. The significant overlap of genes dysregulated by Grb14 silencing with genes controlled by LXR (determined by microarrays from LXRα-LXRβ double knockout mice [LXR DKO] [25]) suggested that Grb14 silencing in liver induced an LXR DKO-like phenotype (Fig. 3A). When focusing on lipid metabolism, the comparative liver transcriptomic analysis showed that 34% of the genes that were diminished in LXR DKO mice were also downregulated in Grb14i mice (Fig. 3B; see also Table S1 in the supplemental material). LXRα and LXRβ mRNA levels were not altered (Fig. 3C), but LXR DNA-binding and transcriptional activity were decreased as shown by the electromobility shift experiments (EMSA) using a canonical LXRE sequence and the qRT-PCR analysis of LXR target gene expression, respectively (Fig. 3D and E). In accordance with LXR inhibition by Grb14 knockdown, circulating and liver cholesterol contents were enhanced after hepatic Grb14 silencing (Fig. 3F and G). To validate that Grb14i-induced lipogenesis inhibition was mediated by a decreased LXR activity, we measured the expression of LXR targets after an oral gavage with the synthetic LXR agonist T0901317. In control animals, a single high dose of T0901317 significantly induced mRNA expression of LXR target genes involved in cholesterol metabolism and lipid synthesis (Fig. 4). However, the induction of these genes was severely blunted in Grb14i mice, suggesting that Grb14 is required for ligand-induced LXR activation (Fig. 4).
FIG 3.

Grb14 expression is required to maintain liver LXR activity. (A) Gene set enrichment analysis (GSEA) showed a significant enrichment of genes up- and downregulated by LXR knockout in liver (GEO no. GSE38083 [25]) with gene expression profile induced in liver from Grb14i mice. NES, normalized enrichment score. (B) Venn diagrams of genes involved in lipid metabolism that are downregulated in liver of LXR DKO mice and Grb14i mice. See Table S1 in the supplemental material for the list of the 67 genes involved in lipid metabolism that are downregulated in both LXR DKO and Grb14i mice. (C) Liver gene expression of LXRα and LXRβ. Expression is relative to that of 18S. (D) Electrophoretic mobility shift assay using an LXRE probe and liver nuclear extracts from Grb14i and USi mice (upper blot). Supershift was performed using anti-LXR antibodies (lanes 4 and 8). The LXR-LXRE complexes are shown by an arrow, and the LXR supershift is shown by a star. The black line indicates a cropped lane of the original blot. The lower blot shows results of similar experiments performed with a CCAAT probe to assess the quality of protein extracts. The NFY-CCAAT complex is shown by an arrowhead. (E) qRT-PCR analysis of LXR target genes. (F and G) Liver cholesterol content (F) and concentration of cholesterol in plasma, triglycerides, and nonesterified fatty acids (G) in Grb14i or USi mice. Results are means ± SEM (n = 5 to 7 per group); *, P < 0.05; **, P < 0.01 for Grb14i compared to USi mice.
FIG 4.

Loss of Grb14 impedes the transcriptional activation of LXR by its agonist T0901317. Mice injected with Grb14i or USi were subjected to oral gavage with T0901317 (T0; 50 mg/kg) or with vehicle (Vhc; 0.5% methylcellulose) 18 h before being sacrificed at day 7 postinfection. Liver gene expression was measured by qRT-PCR. Results are means ± SEM (n = 5 or 6 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.005.
LXR inhibition consecutive to hepatic Grb14 silencing is mediated by the activation of Nrf2.
The recently reported inhibition of LXR-dependent lipogenesis by the transcription factor Nrf2 (30) prompted us to investigate Nrf2 activation upon liver Grb14 silencing. We first confirmed that the activation of Nrf2 by sulforaphane treatment blocked T0901317-induced expression of lipogenic genes in mouse primary hepatocytes (Fig. 5A), as well as the expression of an LXRE reporter gene (Fig. 5B). The significant overlap of genes dysregulated by Grb14 silencing with genes controlled by Nrf2 (determined by microarrays from a model of genetic activation of Nrf2 in the liver [26]) suggested that the liver transcriptomic profile induced by the inhibition of Grb14 exhibits an activated Nrf2 signature (Fig. 6A). Quantification of Nrf2 target gene expression by qRT-PCR confirmed the activation of the Nrf2 antioxidant pathway upon Grb14 downregulation (Fig. 6B). The expression of an Nrf2 reporter gene (antioxidant response element [ARE]-luciferase) was enhanced 2-fold in cultured hepatocytes depleted for Grb14 (Fig. 6C), providing direct evidence for the activation of Nrf2 by Grb14 silencing. Furthermore, the Nrf2 protein level, the expression of glutathione-synthesizing genes, and the ratio of reduced to oxidized glutathione were significantly increased in liver of Grb14i mice (Fig. 6B, D, and E). To evaluate in vivo the role of Nrf2 in the blockade of LXR-induced lipogenesis upon Grb14 depletion, Nrf2−/− mice were then injected with USi or Grb14i and subjected to T0901317 gavage to stimulate LXR. As shown in Fig. 7A, the repression of lipogenic gene expression induced by Grb14 knockdown was blunted in Nrf2−/− mice. Of note, the increase in circulating concentrations of alanine aminotransferase (ALAT) and aspartate aminotransferase (ASAT) observed upon Grb14i treatment in WT mice was blocked in Nrf2−/− mice (Fig. 7B), and no major change in hepatic histology was observed in control and Nrf2−/− mice treated with USi or Grb14i (Fig. 7C). These data suggest that the harmful effect of Grb14i observed in C57 mice was dependent on Nrf2 activation, possibly through the inhibition of LXR anti-inflammatory action (31).
FIG 5.
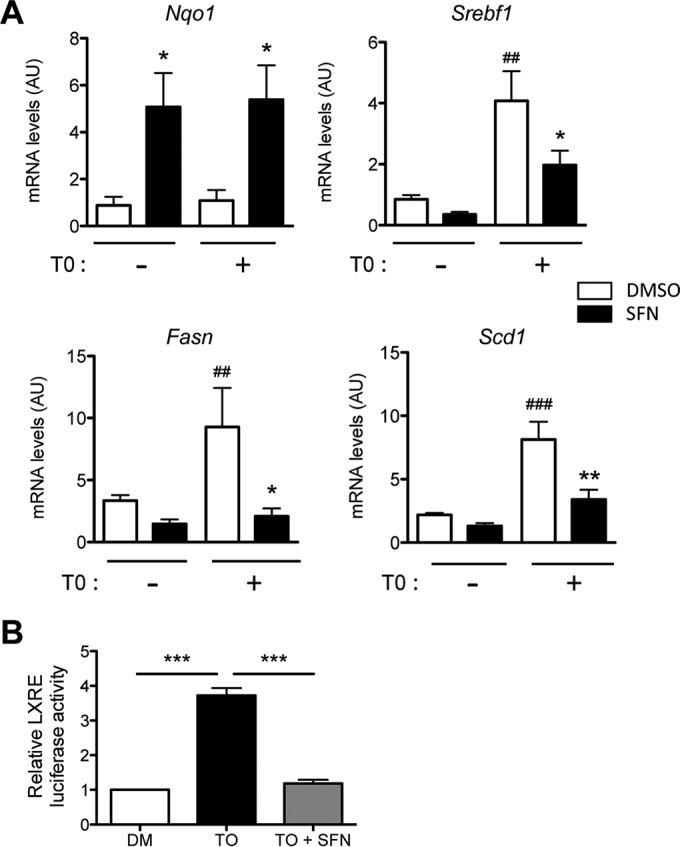
Activation of Nrf2 inhibits the LXR-induced lipogenic pathway. Primary mouse hepatocytes were cultivated in the presence of 25 mM glucose with 100 nM insulin and supplemented or not for 24 h with 10 μM T0901317 (T0) and 20 μM sulforaphane (SFN) as indicated. (A) Gene expression was analyzed by qRT-PCR and is given relative to that of 18S. Nqo1 is a transcriptional target of Nrf2. Results are the means ± SEM from three independent experiments. *, P < 0.05 for the effect of sulforaphane compared to control dimethyl sulfoxide (DMSO); ##, P < 0.01; ###, P < 0.005 for the effect of the T0901317 treatment. (B) Activity of a 3× LXRE-luc reporter gene transfected in primary mouse hepatocytes treated or not with 10 μM T0901317 and 20 μM sulforaphane as indicated. ***, P < 0.005.
FIG 6.

Grb14 silencing activates the Nrf2 pathway in the liver. (A) Gene set enrichment analysis (GSEA) showed a significant enrichment of genes up- and downregulated by Nrf2 activation in liver (GEO no. GSE15633 [26]) with gene expression profile in liver from Grb14i mice. NES, normalized enrichment score. (B) Quantification by qRT-PCR of Nrf2 target genes in liver from USi and Grb14i mice. Gene expression is relative to that of 18S. (C) Activity of an 8x-ARE-luciferase Nrf2 reporter gene in primary mouse hepatocytes treated with USi or Grb14i. Results are means ± SEM (n = 3). (D) Western blot analysis of Nrf2 expression in liver lysates from USi and Grb14i mice. Liver samples are from the same experiment as the one whose results are shown in Fig. 1A. Representative blots of 3 samples from the 5 to 7 samples per group are shown. (E) Ratio of reduced to oxidized glutathione (GSH/GSSG) in liver extracts. Results in panels B, C, and E are means ± SEM (n = 5 or 6 per group). **, P < 0.01; ***, P < 0.005 for Grb14i compared to USi mice.
FIG 7.

Grb14 silencing is unable to decrease LXR-induced gene expression in liver of Nrf2−/− (KO) mice. C57BL/6J or Nrf2−/− mice injected with Grb14i or USi were submitted to an oral gavage of T0901317 for 18 h and analyzed 7 days after adenoviral injection. (A) Liver gene expression was measured by qRT-PCR. (B) Concentrations of alanine aminotransferase (ALAT) and aspartate aminotransferase (ASAT) in plasma of C57BL/6J and Nrf2−/− mice infected with USi or Grb14i. Results are means ± SEM (n = 5 or 6 per group); *, P < 0.05; **, P < 0.01; ***, P < 0.005 for Grb14i compared to USi mice. (C) Hematoxylin and eosin histological analysis of livers from C57BL/6J and Nrf2−/− mice.
Taken together, these results demonstrate that Nrf2 is required for the paradoxical inhibition of LXR-dependent lipogenesis after Grb14 downregulation.
Nrf2 activation in liver of Grb14i mice is mediated by p62.
The next step was to elucidate the molecular mechanism(s) implicated in Nrf2 activation upon Grb14 downregulation. We previously showed that p62/sqstm1 was a binding partner of Grb14, both proteins being highly expressed in the liver and interacting constitutively (15). p62 is an activator of Nrf2, which acts by competitively blocking the interaction of Nrf2 with its inhibitor Keap1, resulting in the stabilization of Nrf2 and expression of its molecular targets (32, 33). We first confirmed in mouse primary hepatocytes that the forced expression of p62 upregulated Nrf2 transactivation activity, as illustrated by the ARE-luciferase reporter assay and by the expression of Nrf2 target genes (Fig. 8A and B). We thus hypothesized that depleting liver Grb14 content could induce the release of a p62 pool and consequently stimulate Nrf2. As shown in Fig. 8C, the increased Nrf2 transcriptional activity induced by Grb14i treatment was abolished by simultaneous depletion of p62, indicating that the activation of Nrf2 following Grb14 silencing was indeed mediated by p62. Interestingly, coimmunoprecipitation experiments provided evidence that a greater amount of Keap1 was associated with p62 in Grb14i compared to USi liver extracts from refed mice (Fig. 8D). The activation of Nrf2 in Grb14i mice liver is probably linked to the release of its inhibitor Keap1, which is trapped by the p62-Keap1 complexes.
FIG 8.

p62 mediates Nrf2 activation in Grb14i mice. (A) Activity of the 8x-ARE-luciferase Nrf2 reporter gene transfected in primary mouse hepatocytes infected with control GFP- or p62-expressing adenovirus. (B) qRT-PCR analysis of Nrf2 target genes in primary mouse hepatocytes infected with adenovirus expressing control GFP or p62. (C) Activity of the 8x-ARE-luciferase Nrf2 reporter gene in primary mouse hepatocytes infected with control USi, p62i, and Grb14i as indicated (lower panel); Western blot analysis of p62 expression in these hepatocytes (upper panel). (D) Anti-p62 immunoprecipitates and liver lysates from USi and Grb14i refed mice were immunodetected using the indicated antibodies. Representative blots of 2 samples from the 4 samples per group are shown. (E) Activity of the 8x-ARE-luciferase Nrf2 reporter gene in primary mouse hepatocytes transfected with plasmid vectors expressing the indicated p62 constructs or with a control empty vector (EV). (F) Effect of a rapamycin treatment on the 8x-ARE-luciferase Nrf2 reporter gene activity in primary mouse hepatocytes infected with Grb14i. (G) qRT-PCR analysis of Nqo1 and lipogenic genes (Fasn, Acaca, and Scd1) in primary mouse hepatocytes infected with adenovirus expressing control GFP or the indicated p62 constructs. Hepatocytes were cultivated in 5 mM glucose (G5) or 25 mM glucose in the presence of 100 nM insulin (G25i) and 10 μM T0901317 (T0), as indicated. (H) Western blot analysis of p62 expression in the experiment whose results are shown in panel G. The arrowhead indicates p62 and p62 S351E overexpression, and the lower band corresponds to endogenous p62. Results are means ± SEM (n = 3 for panels A, B, and E to G; n = 4 for panel C); *, P < 0.05; **P < 0.001; ***, P < 0.005; ###, P < 0.005 for rapamycin effect compared to Grb14i DMSO control in panel F.
It was recently shown that p62 is a target of mammalian target of rapamycin complex 1 (mTORC1) and that mTORC1-induced S351-p62 phosphorylation enhances Nrf2 activation (20). We showed that, as expected, Nrf2 transcriptional activity analyzed by the ARE-luciferase reporter assay was activated by overexpression of p62 or S351A (phosphorylation defective) and that a higher increase was observed with S351E (phosphorylation mimic) (Fig. 8E). In addition, rapamycin treatment blunted the induction of Nrf2 transcriptional activity induced by Grb14 depletion (Fig. 8F), suggesting that mTORC1 may play a role in the p62-dependent activation of Nrf2. To investigate the effect of S351 phosphorylation of p62 on the inhibition of lipogenic gene expression, primary cultured hepatocytes were infected with adenoviral p62 constructs overexpressing similar amounts of WT and S351E p62 isoforms (Fig. 8H). Following adenoviral treatment, cells were incubated in the presence of high concentrations of glucose and insulin with T0901317 to stimulate the lipogenic pathway. Under these conditions, Nrf2 target gene expression was further enhanced by S351E p62 compared to WT p62, as expected (20) (Fig. 8G). Importantly, lipogenic gene expression was significantly decreased by 35 to 45% in the presence of S351E p62, while it was not altered by WT p62 (Fig. 8G). These results indicate that p62-mediated inhibition of the lipogenic pathway is dependent on its S351 phosphorylation.
p62 is a receptor for selective autophagy, and its protein expression is enhanced when autophagy is inhibited (34). Since mTORC1 is the main physiological inhibitor of autophagy, it was then important to determine upon Grb14 downregulation whether p62 was increased as a result of mTORC1 stimulation or whether it was released from preexisting Grb14-p62 complexes (15). To discriminate between these two mechanisms, p62 expression and phosphorylation levels were examined in liver of fasted (low mTORC1 activity/active autophagy condition) or refed (high mTORC1 activity/inactive autophagy condition) mice (35) treated with USi or Grb14i. Refeeding in USi mice increased the p62 protein level, as a consequence of mTORC1 activation (illustrated by S6K phosphorylation) (Fig. 9A). However, p62 was not phosphorylated in liver of USi refed mice, suggesting that p62 that accumulates during refeeding is a poor substrate for mTORC1 and is unable to activate Nrf2 and target genes (Fig. 9A). In Grb14i mice, p62 accumulated in fasted liver and was even more expressed in the refed state. Interestingly, p62 was phosphorylated under both nutritional states. In correlation with p62 phosphorylation, the expression of Nrf2 and Nqo1 was increased in Grb14i mice liver, hence inferring that p62 accumulated upon Grb14 downregulation was a potent activator of Nrf2 (Fig. 9A). The increased mTORC1 activity and p62 expression in Grb14i livers were concomitant to enhanced Keap1 and LC3-II expression, suggesting that autophagy was inhibited upon Grb14 downregulation. Finally, we observed that p62 phosphorylation and Nrf2 expression were also increased by Grb14 inhibition in liver of ob/ob mice, suggesting that a similar molecular mechanism was involved in the restoration of hepatic lipid homeostasis in this mouse model of liver metabolic disease (Fig. 9B).
FIG 9.

Grb14i activation of Nrf2 is mediated by an increase in p62 protein availability. (A) Western blot analysis of liver extracts from mice injected with USi or Grb14i and studied 7 days after the infection in the fasted or refed state as indicated. (B) Western blot analysis of liver extracts from ob/ob mice injected with USi or Grb14i and studied in the refed state 7 days after the infection. Western blots shown are representative blots of 3 samples from the 6 samples per group.
DISCUSSION
A role for Grb14 as a physiological modulator of insulin action was first suggested by its inhibitory effect on the catalytic activity of the insulin receptor and by the metabolic phenotype of Grb14−/− mice, which displayed improved glucose tolerance and insulin signaling in the liver and skeletal muscle (11, 36, 37). However, the regulation by Grb14 of hepatic metabolism likely involves additional molecular mechanisms besides its role as a pseudo-substrate inhibitor of the insulin receptor. Indeed, we reported that the downregulation of Grb14 in primary hepatocytes enhanced insulin signaling and potentiated insulin-induced inhibition of hepatic glucose production but simultaneously blocked the induction by insulin of lipogenic gene expression (13). To elucidate the physiological role of Grb14 in mouse liver, we thus investigated the metabolic modifications induced by an acute liver-specific knockdown of Grb14 expression. In the present study, we unravel that hepatic Grb14 inhibition stimulates insulin signaling, improves hepatic glucose production, and, in parallel, enhances Nrf2 activity via p62, thereby counteracting de novo lipogenesis through LXR inhibition. Grb14 appears then as a critical regulator of hepatic metabolic homeostasis, mediating a cross talk between the insulin receptor-dependent prolipogenic and p62-Nrf2-LXR-dependent antilipogenic pathways (Fig. 10). Importantly, inhibition of hepatic Grb14 expression counteracts the metabolic defects of obese and insulin-resistant mice, leading to decreased hepatic glucose production and steatosis. Our data suggest that Grb14, by acting through p62 binding and by modulating Nrf2 activation, plays a central role in hepatic insulin resistance selectivity.
FIG 10.
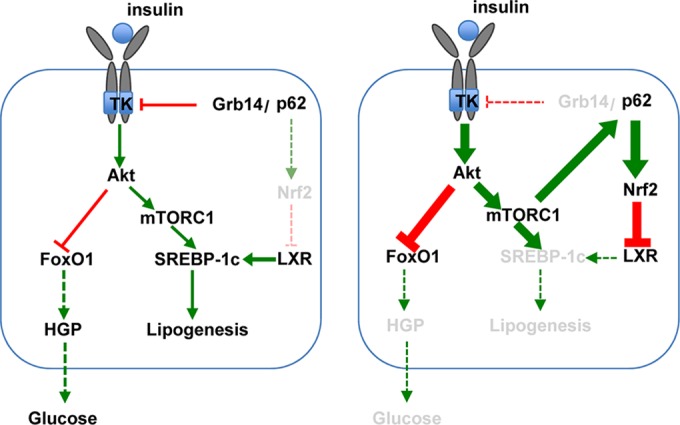
Model for the regulation of liver glucose and lipid homeostasis in the presence (left) or the absence (right) of Grb14. (Left) Grb14 binds to the activated insulin receptor, inhibiting its catalytic activity, and to p62. Hepatic glucose production (HGP) is regulated by the transcription factor FoxO1, which is phosphorylated and inactivated by Akt in an insulin-dependent manner. Lipogenesis is controlled by the transcription factor SREBP-1c, which is regulated at the transcriptional level by LXR, and at the transcriptional and the posttranslational levels by insulin through the Akt-mTORC1 pathway. Under basal physiological conditions, Nrf2 is degraded by the proteasome and is expressed at a low level. (Right) Grb14 depletion enhances insulin signaling, improving insulin-dependent inhibition of hepatic glucose production. The increased p62 availability upon liver Grb14 depletion combined with the phosphorylation of p62 by the activated mTORC1 stabilized Nrf2, leading to the repression of LXR activity and blocking SREBP-1c and lipogenic gene expression.
After Grb14 downregulation, SREBP-1c expression and the lipogenic program are dramatically decreased despite the presence of an activated PI3K/Akt/mTORC1 pathway. Our data demonstrate that the leading cause for lipogenesis inhibition upon Grb14 knockdown is a defect in LXR transcriptional activity. This paradoxical blockage of insulin-induced lipogenesis in liver from Grb14i mice is a cell-autonomous phenomenon, as supported by data obtained in vitro in cultured hepatocytes (13). LXRs are important regulators of cholesterol homeostasis, acting in a coordinate manner on macrophages, intestine, and liver to stimulate reverse cholesterol transport, high-density lipoprotein (HDL) circulating levels, and cholesterol conversion into bile acids (38). However, besides their beneficial effects on HDL and cholesterol homeostasis, treatments with synthetic LXR agonists stimulate liver lipogenesis and increase triglyceride levels, thus preventing their utilization to treat atherosclerosis and cardiovascular disease (39, 40). Our study suggests that targeting hepatic LXR through Grb14 inhibition could be efficient in reducing the prolipogenic effects of LXR agonists while preserving their protective effect against cardiovascular diseases, which is primarily driven by intestine LXR activation (41).
Grb14 silencing in Nrf2−/− mice provided a direct demonstration of the role of Nrf2 in Grb14-induced LXR inhibition. Nrf2 is the master regulator of the adaptive response to exogenous and endogenous oxidative and electrophilic stress, playing a crucial role in cellular homeostasis (42). Nrf2 recently emerged as a potent inhibitor of liver de novo lipogenesis (43, 44). Consistent with these findings, pharmacological activation of Nrf2 inhibited the insulin- and T0901317-induced activation of SREBP-1c in hepatocytes, underlying the fact that acute stimulation of Nrf2 can overcome the positive insulin-LXR signal on the lipogenic pathway (45). The nuclear receptors FXR and SHP are inhibitors of LXR activity. It has been proposed that Nrf2 represses LXR through the activation of FXR, leading to SHP induction and the formation of inactive LXR-SHP dimers (30, 46). The fact that the expression of both FXR and SHP was not increased supports the lack of their involvement in the inhibition of liver lipogenesis induced by Grb14 silencing (unpublished data). Considering that Nrf2 activation can inhibit liver gluconeogenesis (47), the decreased gluconeogenic gene expression and the associated reduction in blood glucose levels that we observed upon Grb14 depletion in liver could be also driven by Nrf2 activation, in parallel to hyperactivation of the Akt signaling pathway. It has been recently reported that an active PI3K-Akt pathway augments the nuclear accumulation of Nrf2 and enables Nrf2 to promote metabolic reprogramming (48). Upon Grb14 downregulation, insulin signaling activation may thus participate in the induction of the Nrf2 pathway and help the expression of enzymes involved in the pentose phosphate and glutathione synthesis pathways (48).
Our data indicate that Grb14i-mediated Nrf2 activation is linked to an increased availability of p62. The pool of p62 that is released after Grb14 downregulation is phosphorylated in an mTORC1-dependent manner and is therefore able to activate Nrf2 (20). Since Nrf2 is a transcriptional activator of p62 (49), this could initiate a positive feedback loop ultimately leading to LXR inactivation and lipogenic gene inhibition. Interestingly, amino acids are required for the activation of mTORC1, and their effect is mediated by p62 (50). The increased mTORC1 activity in Grb14i mice can therefore be also driven by p62 in addition to insulin receptor stimulation, contributing to the positive feedback loop. After a nutritional challenge, insulin-dependent de novo fatty acid synthesis and liver lipid accumulation are mediated by the activation of SREBP-1c in an mTORC1-dependent manner (51, 52). The observation that p62 is not phosphorylated by mTORC1 in liver of refed control mice is thus consistent with the activation of the lipogenic pathway in this situation. In addition, this dissociation between p62 accumulation and phosphorylation following mTORC1 activation in refed control mice could be a way to protect cells from unwanted cytoprotective responses after common nutritional challenges. p62 is involved in an increasing number of cellular processes (16), but this is, to our knowledge, the first study reporting its role in the regulation of liver energy homeostasis.
The relationships between oxidative stress and insulin sensitivity are complex. It is established that chronic reactive oxygen species (ROS) production contributes to the development of insulin resistance, but insulin signaling is also improved by transient and discrete generation of ROS following physiological stimuli (53). The level and duration of the oxidative stress are thus determinants for its beneficial or detrimental consequences (54). Nrf2 is activated in human fatty liver disease, and impairment of Nrf2 activity represents a major risk factor for the evolution of NAFLD to nonalcoholic steatohepatitis (NASH) (55, 56). Nrf2 therefore arises as a critical regulator of liver metabolic homeostasis, regulating fatty acid synthesis and the progression from steatosis to more-severe liver pathologies (57, 58). Since Grb14 regulates both insulin signaling and Nrf2 activity in the liver, it will be interesting to investigate to what extent it is involved in the pathophysiological evolution of hepatic metabolic diseases.
Supplementary Material
ACKNOWLEDGMENTS
We thank Florent Dumont and Franck Letourneur from the Plateform Génomique (Institut Cochin, Inserm U1016, Paris, France) for microarray experiments and analysis, Maryline Favier and the HISTIM facility for histochemistry analysis, Isabelle Lagoutte and the staff of the Cochin's animal facility, the Plateforme Biochimie from the Institut Claude Bernard, Paris, France, for plasma analysis, the Laboratoire de Thérapie Génique (Inserm UMR1089, Nantes, France) for the production of the p62 adenovirus, Ken Itoh (Japan) for anti-Nrf2 antibodies, and Roland Wolf (University of Dundee, United Kingdom) for the 8x-ARE reporter plasmid. C. Perret, R. Dentin, A. Leturque, S. Vaulont, M. Moldes, S. Lotersztajn, B. Cariou, and A. Iroz are gratefully acknowledged for helpful discussions and critical reading of the manuscript. Mice used in this study were housed in an animal facility equipped with the help of the Région Ile de France.
The work performed within the Département Hospitalo-Universitaire (DHU) Autoimmune and Hormonal Diseases (AUTHORS) was supported by grants from the Agence Nationale de la Recherche (ANR-06-Grb14, ANR-10-Wnt-Metaboliv, and ANR-09-JCJC-0057) and by the Fondation pour la Recherche Médicale (Labélisation Equipe). L.P. and L.M. are supported by a Ph.D. grant from the French Ministry of Research, and L.P. has a fellowship from the Fondation pour la Recherche Médicale.
We declare that we have no conflict of interest.
Funding Statement
Renaud Dentin was the recipient of Agence Nationale de la Recherche (ANR) grant ANR-09-JCJC-0057.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00170-16.
REFERENCES
- 1.Brown MS, Goldstein JL. 2008. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 7:95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Taniguchi CM, Emanuelli B, Kahn CR. 2006. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96. [DOI] [PubMed] [Google Scholar]
- 3.Smith BW, Adams LA. 2011. Nonalcoholic fatty liver disease and diabetes mellitus: pathogenesis and treatment. Nat Rev Endocrinol 7:456–465. doi: 10.1038/nrendo.2011.72. [DOI] [PubMed] [Google Scholar]
- 4.Jeon TI, Osborne TF. 2012. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab 23:65–72. doi: 10.1016/j.tem.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasus-Jacobi A, Perdereau D, Auzan C, Clauser E, Van Obberghen E, Mauvais-Jarvis F, Girard J, Burnol A-F. 1998. Identification of the rat adapter Grb14 as an inhibitor of insulin actions. J Biol Chem 273:26026–26035. doi: 10.1074/jbc.273.40.26026. [DOI] [PubMed] [Google Scholar]
- 6.Kooner JS, Saleheen D, Sim X, Sehmi J, Zhang W, Frossard P, Been LF, Chia KS, Dimas AS, Hassanali N, Jafar T, Jowett JB, Li X, Radha V, Rees SD, Takeuchi F, Young R, Aung T, Basit A, Chidambaram M, Das D, Grundberg E, Hedman AK, Hydrie ZI, Islam M, Khor CC, Kowlessur S, Kristensen MM, Liju S, Lim WY, Matthews DR, Liu J, Morris AP, Nica AC, Pinidiyapathirage JM, Prokopenko I, Rasheed A, Samuel M, Shah N, Shera AS, Small KS, Suo C, Wickremasinghe AR, Wong TY, Yang M, Zhang F, Abecasis GR, Barnett AH, Caulfield M, Deloukas P, Frayling TM, Froguel P, Kato N, Katulanda P, Kelly MA, Liang J, Mohan V, Sanghera DK, Scott J, Seielstad M, Zimmet PZ, Elliott P, Teo YY, McCarthy MI, Danesh J, Tai ES, Chambers JC. 2011. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet 43:984–989. doi: 10.1038/ng.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, Rybin D, Liu CT, Bielak LF, Prokopenko I, Amin N, Barnes D, Cadby G, Hottenga JJ, Ingelsson E, Jackson AU, Johnson T, Kanoni S, Ladenvall C, Lagou V, Lahti J, Lecoeur C, Liu Y, Martinez-Larrad MT, Montasser ME, Navarro P, Perry JR, Rasmussen-Torvik LJ, Salo P, Sattar N, Shungin D, Strawbridge RJ, Tanaka T, van Duijn CM, An P, de Andrade M, Andrews JS, Aspelund T, Atalay M, Aulchenko Y, Balkau B, Bandinelli S, Beckmann JS, Beilby JP, Bellis C, Bergman RN, Blangero J, Boban M, Boehnke M, Boerwinkle E, et al. 2012. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 44:659–669. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, Prokopenko I, Kang HM, Dina C, Esko T, Fraser RM, Kanoni S, Kumar A, Lagou V, Langenberg C, Luan J, Lindgren CM, Muller-Nurasyid M, Pechlivanis S, Rayner NW, Scott LJ, Wiltshire S, Yengo L, Kinnunen L, Rossin EJ, Raychaudhuri S, Johnson AD, Dimas AS, Loos RJ, Vedantam S, Chen H, Florez JC, Fox C, Liu CT, Rybin D, Couper DJ, Kao WH, Li M, Cornelis MC, Kraft P, Sun Q, van Dam RM, Stringham HM, Chines PS, Fischer K, Fontanillas P, et al. 2012. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cariou B, Capitaine N, Le Marcis V, Vega N, Bereziat V, Kergoat M, Laville M, Girard J, Vidal H, Burnol AF. 2004. Increased adipose tissue expression of Grb14 in several models of insulin resistance. FASEB J 18:965–967. [DOI] [PubMed] [Google Scholar]
- 10.Park JJ, Berggren JR, Hulver MW, Houmard JA, Hoffman EP. 2006. GRB14, GPD1, and GDF8 as potential network collaborators in weight loss-induced improvements in insulin action in human skeletal muscle. Physiol Genomics 27:114–121. doi: 10.1152/physiolgenomics.00045.2006. [DOI] [PubMed] [Google Scholar]
- 11.Bereziat V, Kasus-Jacobi A, Perdereau D, Cariou B, Girard J, Burnol A-F. 2002. Inhibition of insulin receptor catalytic activity by the molecular adapter Grb14. J Biol Chem 277:4845–4852. doi: 10.1074/jbc.M106574200. [DOI] [PubMed] [Google Scholar]
- 12.Desbuquois B, Bereziat V, Authier F, Girard J, Burnol AF. 2008. Compartmentalization and in vivo insulin-induced translocation of the insulin-signaling inhibitor Grb14 in rat liver. FEBS J 275:4363–4377. doi: 10.1111/j.1742-4658.2008.06583.x. [DOI] [PubMed] [Google Scholar]
- 13.Carré N, Caüzac M, Girard J, Burnol A-F. 2008. Dual effect of the adapter Grb14 on insulin action in primary hepatocytes. Endocrinology 149:3109–3117. doi: 10.1210/en.2007-1196. [DOI] [PubMed] [Google Scholar]
- 14.Goenaga D, Hampe C, Carre N, Cailliau K, Browaeys-Poly E, Perdereau D, Holt LJ, Daly RJ, Girard J, Broutin I, Issad T, Burnol AF. 2009. Molecular determinants of Grb14-mediated inhibition of insulin signaling. Mol Endocrinol 23:1043–1051. doi: 10.1210/me.2008-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cariou B, Perdereau D, Cailliau K, Browaeys-Poly E, Béréziat V, Vasseur-Cognet M, Girard J, Burnol A-F. 2002. The adapter protein ZIP binds Grb14 and regulates its inhibitory action on insulin signaling by recruiting protein kinase Cz. Mol Cell Biol 22:6959–6970. doi: 10.1128/MCB.22.20.6959-6970.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moscat J, Diaz-Meco MT. 2012. p62: a versatile multitasker takes on cancer. Trends Biochem Sci 37:230–236. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. 1997. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. 2007. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 19.Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, Girard J, Postic C. 2006. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 55:2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 20.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, Hoshii T, Hirao A, Takagi K, Mizushima T, Motohashi H, Lee MS, Yoshimori T, Tanaka K, Yamamoto M, Komatsu M. 2013. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 21.Wang XJ, Hayes JD, Wolf CR. 2006. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer Res 66:10983–10994. doi: 10.1158/0008-5472.CAN-06-2298. [DOI] [PubMed] [Google Scholar]
- 22.Stansbie D, Brownsey RW, Crettaz M, Denton RM. 1976. Acute effects in vivo of anti-insulin serum on rates of fatty acid synthesis and activities of acetyl-coenzyme A carboxylase and pyruvate dehydrogenase in liver and epididymal adipose tissue of fed rats. Biochem J 160:413–416. doi: 10.1042/bj1600413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bobard A, Hainault I, Ferre P, Foufelle F, Bossard P. 2005. Differential regulation of sterol regulatory element-binding protein 1c transcriptional activity by insulin and liver X receptor during liver development. J Biol Chem 280:199–206. doi: 10.1074/jbc.M406522200. [DOI] [PubMed] [Google Scholar]
- 24.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ducheix S, Podechard N, Lasserre F, Polizzi A, Pommier A, Murzilli S, Di Lisio C, D'Amore S, Bertrand-Michel J, Montagner A, Pineau T, Loiseau N, Lobaccaro JM, Martin PG, Guillou H. 2013. A systems biology approach to the hepatic role of the oxysterol receptor LXR in the regulation of lipogenesis highlights a cross-talk with PPARalpha. Biochimie 95:556–567. doi: 10.1016/j.biochi.2012.09.028. [DOI] [PubMed] [Google Scholar]
- 26.Yates MS, Tran QT, Dolan PM, Osburn WO, Shin S, McCulloch CC, Silkworth JB, Taguchi K, Yamamoto M, Williams CR, Liby KT, Sporn MB, Sutter TR, Kensler TW. 2009. Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 30:1024–1031. doi: 10.1093/carcin/bgp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hegarty BD, Bobard A, Hainault I, Ferre P, Bossard P, Foufelle F. 2005. Distinct roles of insulin and liver X receptor in the induction and cleavage of sterol regulatory element-binding protein-1c. Proc Natl Acad Sci U S A 102:791–796. doi: 10.1073/pnas.0405067102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldstein JL, DeBose-Boyd RA, Brown MS. 2006. Protein sensors for membrane sterols. Cell 124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 29.Chen G, Liang G, Ou J, Goldstein JL, Brown MS. 2004. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc Natl Acad Sci U S A 101:11245–11250. doi: 10.1073/pnas.0404297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kay HY, Kim WD, Hwang SJ, Choi HS, Gilroy RK, Wan YJ, Kim SG. 2011. Nrf2 inhibits LXRalpha-dependent hepatic lipogenesis by competing with FXR for acetylase binding. Antioxid Redox Signal 15:2135–2146. doi: 10.1089/ars.2010.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steffensen KR, Jakobsson T, Gustafsson JA. 2013. Targeting liver X receptors in inflammation. Expert Opin Ther Targets 17:977–990. doi: 10.1517/14728222.2013.806490. [DOI] [PubMed] [Google Scholar]
- 32.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. 2010. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 33.Bryan HK, Olayanju A, Goldring CE, Park BK. 2013. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol 85:705–717. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 34.Komatsu M. 2012. Liver autophagy: physiology and pathology. J Biochem 152:5–15. doi: 10.1093/jb/mvs059. [DOI] [PubMed] [Google Scholar]
- 35.Rabinowitz JD, White E. 2010. Autophagy and metabolism. Science 330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Depetris RS, Hu J, Gimpelevich I, Holt LJ, Daly RJ, Hubbard SR. 2005. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Mol Cell 20:325–333. doi: 10.1016/j.molcel.2005.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooney GJ, Lyons RJ, Crew AJ, Jensen TE, Molero JC, Mitchell CJ, Biden TJ, Ormandy CJ, James DE, Daly RJ. 2004. Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J 23:582–593. doi: 10.1038/sj.emboj.7600082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calkin AC, Tontonoz P. 2012. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol 13:213–224. doi: 10.1038/nrm3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jakobsson T, Treuter E, Gustafsson JA, Steffensen KR. 2012. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol Sci 33:394–404. doi: 10.1016/j.tips.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 40.Hong C, Tontonoz P. 2014. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov 13:433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Breevoort SR, Angdisen J, Fu M, Schmidt DR, Holmstrom SR, Kliewer SA, Mangelsdorf DJ, Schulman IG. 2012. Liver LXRalpha expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J Clin Invest 122:1688–1699. doi: 10.1172/JCI59817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baird L, Dinkova-Kostova AT. 2011. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol 85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 43.Kitteringham NR, Abdullah A, Walsh J, Randle L, Jenkins RE, Sison R, Goldring CE, Powell H, Sanderson C, Williams S, Higgins L, Yamamoto M, Hayes J, Park BK. 2010. Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J Proteomics 73:1612–1631. doi: 10.1016/j.jprot.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka Y, Aleksunes LM, Yeager RL, Gyamfi MA, Esterly N, Guo GL, Klaassen CD. 2008. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J Pharmacol Exp Ther 325:655–664. doi: 10.1124/jpet.107.135822. [DOI] [PubMed] [Google Scholar]
- 45.Hwahng SH, Ki SH, Bae EJ, Kim HE, Kim SG. 2009. Role of adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway in repression of liver X receptor-alpha-dependent lipogenic gene induction and hepatic steatosis by a novel class of dithiolethiones. Hepatology 49:1913–1925. doi: 10.1002/hep.22887. [DOI] [PubMed] [Google Scholar]
- 46.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. 2004. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest 113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, Sugawara A, Kensler TW, Yamamoto M. 2013. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol 33:2996–3010. doi: 10.1128/MCB.00225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. 2012. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 49.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. 2010. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. 2011. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 44:134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li S, Brown MS, Goldstein JL. 2010. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 107:3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS, Lee CH, Manning BD. 2011. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab 14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, Bruce C, Shields BJ, Skiba B, Ooms LM, Stepto N, Wu B, Mitchell CA, Tonks NK, Watt MJ, Febbraio MA, Crack PJ, Andrikopoulos S, Tiganis T. 2009. Reactive oxygen species enhance insulin sensitivity. Cell Metab 10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tiganis T. 2011. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol Sci 32:82–89. doi: 10.1016/j.tips.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 55.Hardwick RN, Fisher CD, Canet MJ, Lake AD, Cherrington NJ. 2010. Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos 38:2293–2301. doi: 10.1124/dmd.110.035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chowdhry S, Nazmy MH, Meakin PJ, Dinkova-Kostova AT, Walsh SV, Tsujita T, Dillon JF, Ashford ML, Hayes JD. 2010. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic Biol Med 48:357–371. doi: 10.1016/j.freeradbiomed.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 57.Meakin PJ, Chowdhry S, Sharma RS, Ashford FB, Walsh SV, McCrimmon RJ, Dinkova-Kostova AT, Dillon JF, Hayes JD, Ashford ML. 2014. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol Cell Biol 34:3305–3320. doi: 10.1128/MCB.00677-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uruno A, Yagishita Y, Yamamoto M. 2015. The Keap1-Nrf2 system and diabetes mellitus. Arch Biochem Biophys 566:76–84. doi: 10.1016/j.abb.2014.12.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.