

ABSTRACT
Proper regulation of mitophagy for mitochondrial homeostasis is important in various inflammatory diseases. However, the precise mechanisms by which mitophagy is activated to regulate inflammatory responses remain largely unknown. The NLRP3 (NLR family, pyrin domain containing 3) inflammasome serves as a platform that triggers the activation of CASP1 (caspase 1) and secretion of proinflammatory cytokines. Here, we demonstrate that SESN2 (sestrin 2), known as stress-inducible protein, suppresses prolonged NLRP3 inflammasome activation by clearance of damaged mitochondria through inducing mitophagy in macrophages. SESN2 plays a dual role in inducing mitophagy in response to inflammasome activation. First, SESN2 induces “mitochondrial priming” by marking mitochondria for recognition by the autophagic machinery. For mitochondrial preparing, SESN2 facilitates the perinuclear-clustering of mitochondria by mediating aggregation of SQSTM1 (sequestosome 1) and its binding to lysine 63 (Lys63)-linked ubiquitins on the mitochondrial surface. Second, SESN2 activates the specific autophagic machinery for degradation of primed mitochondria via an increase of ULK1 (unc-51 like kinase 1) protein levels. Moreover, increased SESN2 expression by extended LPS (lipopolysaccharide) stimulation is mediated by NOS2 (nitric oxide synthase 2, inducible)-mediated NO (nitric oxide) in macrophages. Thus, Sesn2-deficient mice displayed defective mitophagy, which resulted in hyperactivation of inflammasomes and increased mortality in 2 different sepsis models. Our findings define a unique regulatory mechanism of mitophagy activation for immunological homeostasis that protects the host from sepsis.
KEYWORDS: autophagy, mitochondrial priming, mitophagy, NLRP3 inflammasome, sepsis, SESN2
Introduction
Mitophagy, a selective autophagic process that specifically removes damaged or excess mitochondria, is critical for maintaining the mitochondrial population and cellular homeostasis.1-4 Failure of mitophagy regulation results in abnormal cellular function caused by the accumulation of damaged mitochondria, leading to many pathophysiological states.5-8 Accumulating data suggests that mitophagy has an essential role in the regulation of the innate immune response.9-12 When mitophagy is impaired, the increase of damaged mitochondria caused by immune stimulators results in the generation of mitochondrial ROS (reactive oxygen species) and release of mitochondrial DNA, which induces hyperactivation of the NLRP3 inflammasome, and in turn leads to over-inflammation, tissue injury and increased mortality in the host.9,10,12,13
Although a number of mitophagy-related factors have been identified, detailed mechanisms by which they take action are still largely unknown. Recent studies on mitophagy in mammalian cell studies reveal that signal-dependent removal of damaged mitochondria by mitophagy requires 2 steps. The first is preparing damaged mitochondria and the second is activating specific autophagy machinery for the degradation of primed mitochondria.5 Mitochondrial priming is initiated by PINK1 (PTEN induced putative kinase 1) stabilization and the E3 ubiquitin ligase PARK2/PARKIN (Parkinson disease [autosomal recessive, juvenile] 2, parkin) recruitment to damaged mitochondria.7,14,15 Activated PARK2 promotes ubiquitination of outer membrane proteins on the mitochondria, which in turn triggers translocation of the ubiquitin-binding receptor SQSTM1 or NBR1 (neighbor of Brca1 gene 1) to mitochondria, thus completing mitochondrial priming.6,16-19 Among the components of the autophagy machinery required for mitophagy, ULK1 and BNIP3L (BCL2/adenovirus E1B 19kDa interacting protein 3-like), are critical for clearance of mitochondria during erythroid cell maturation.2,20,21 However, it remains unclear how these 2 steps are connected so that the mitophagy process can be completed under inflammatory conditions.
SESNs (sestrins) are highly conserved proteins that protect cells exposed to a variety of environmental stresses, including oxidative stress and DNA damage.22,23 Apart from their antioxidant function, SESNs maintain metabolic homeostasis through regulation of AMPK (AMP-activated protein kinase) and MTOR (mechanistic target of rapamycin [serine/threonine kinase]) signaling.23-26 SESN2 is also able to induce autophagy through activation of AMPK and inhibition of MTOR under conditions of genotoxic stress and in cancer cell lines.24,27,28 In addition, SESN2 can induce autophagic degradation of KEAP1 (kelch-like ECH-associated protein 1) through association with SQSTM1 triggered by the acute lipogenic stimulus.29 Furthermore, despite significant progress in understanding the function of SESN2 in metabolic pathways and diseases, the regulatory roles of SESN2 in immune responses and the mechanisms involved therein have not yet been revealed. Here, we demonstrate that bone marrow-derived macrophages (BMDMs) from sesn2−/− mice displayed defective mitophagy upon immune stimulation, which resulted in hyperactivation of the NLRP3 inflammasome and increased mortality from sepsis. We showed that in order to induce mitophagy, SESN2 facilitates mitochondrial priming by mediating the aggregation of SQSTM1 and its binding to ubiquitinated mitochondria, and also activates specific autophagy machinery for degradation of primed mitochondria via increase of ULK1 protein levels. Our results highlight previously unknown mechanisms of 2 different phases of mitophagy activation regulated by SESN2, leading to the suppression of hyperactivation of the NLRP3 inflammasome.
Results
sesn2−/− macrophages display increased CASP1 activation in response to LPS and ATP, after extended (12 h) LPS priming
To investigate the involvement of SESN2 in inflammasome activation in macrophages, we isolated BMDMs from Sesn2+/+ and sesn2−/− mice, primed the cells with LPS for 0, 6, and 12 hours (h), and then stimulated them with ATP for 30 minutes (min). ATP-driven activation of CASP1 in LPS-primed macrophages is a well-established model for NLRP3 inflammasome-mediated activation of CASP1 in vitro, which involves signaling pathways mediated by TLR4 (toll-like receptor 4) and P2RX7 (purinergic receptor P2X, ligand-gated channel, 7).30,31 First, we examined the effect of SESN2 on the activation of CASP1 in BMDMs. The level of the active form of CASP1, indicated by the appearance of the cleaved form of CASP1 (p10), increased in Sesn2+/+ BMDMs, reaching its peak when primed with LPS for 6 h, and then dramatically decreased at 12 h (Fig. 1A). However, in sesn2−/− BMDMs, significant amounts of cleaved CASP1 remained at 12 h, similar to amounts of cleaved CASP1 detected in Sesn2+/+ BMDMs at 6 h after LPS treatment (Fig. 1A). We also examined cleavage of CASP1 in the supernatants of stimulated BMDMs by western blot. The expression patterns of cleaved CASP1 in the supernatants were identical to those in the lysates of stimulated BMDMs (Fig. S1). However, activation of the NFKB (nuclear factor of kappa light polypeptide gene enhancer in B cells) signaling pathway by LPS was not affected by the ablation of Sesn2 (Fig. S2). Moreover, protein levels of pro-IL1B (interleukin 1 β), NLRP3 and PYCARD (PYD and CARD domain containing), components of the NLRP3 complex, and P2RX7 did not change in sesn2−/− macrophages in response to LPS and ATP (Fig. 1A).
Figure 1.
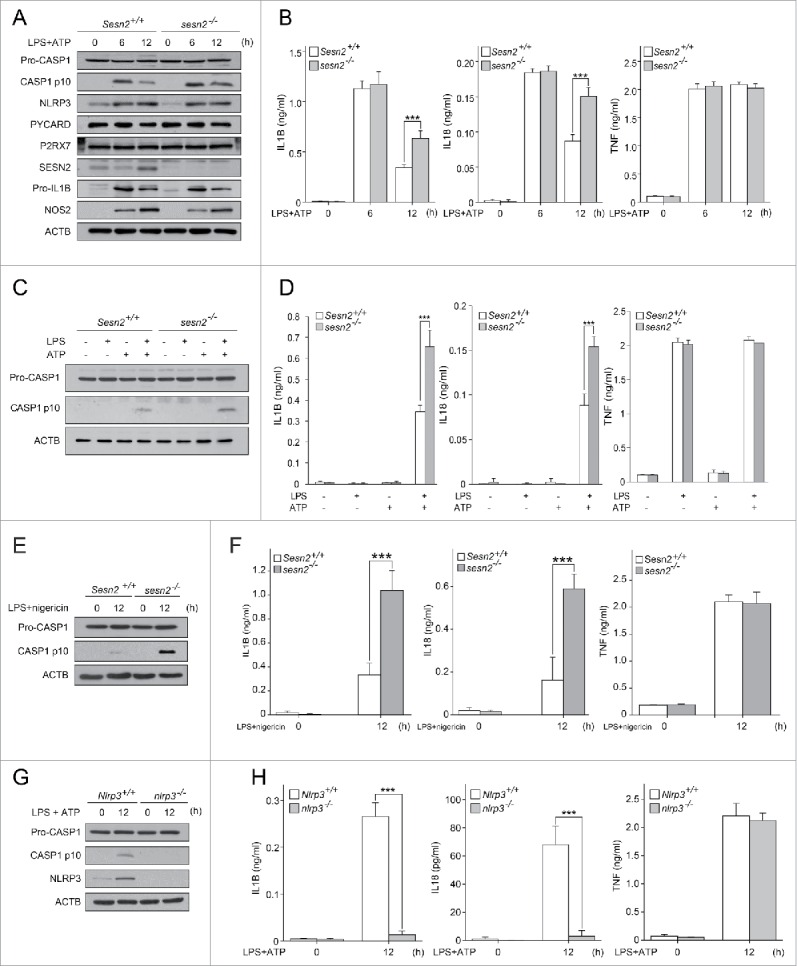
SESN2 suppresses NLRP3-dependent CASP1 activation in response to extended (12 h) LPS priming followed by ATP treatment. (A and B) sesn2−/− mice and the corresponding wild-type littermate mice (Sesn2+/+) bone marrow-derived macrophages were primed with LPS (0, 6, and 12 h) followed by 30 min of ATP treatment. (A) Immunoblot analysis of the indicated proteins and (B) production of IL1B, IL18 and TNF as measured by ELISA. (C and D) Sesn2+/+ and sesn2−/− BMDMs were treated with LPS and/or ATP. (C) Immunoblot analysis of the indicated proteins and (D) production of IL1B, IL18 and TNF as measured by ELISA. (E and F) Sesn2+/+ and sesn2−/− BMDMs were primed with LPS followed by nigericin treatment. (E) Immunoblot analysis of the indicated proteins and (F) production of IL1B, IL18 and TNF as measured by ELISA. (G and H) Nlrp3+/+ and nlrp3−/− BMDMs were primed with LPS followed by ATP treatment. (G) Immunoblot analysis of the indicated proteins and (H) production of IL1B, IL18 and TNF as measured by ELISA. Data shown are representative of 3 independent experiments and are the mean ± s .e.m. ***, p < 0.005 from an ANOVA followed by Tukey's post hoc test.
We next examined the secretion level of the CASP1-dependent cytokines, IL1B and IL18 (interleukin 18), in the supernatant of the treated BMDMs. Although the cytokine levels were comparable at 6 h of priming with LPS, much higher secretion levels of IL1B and IL18 were detected in sesn2−/− BMDMs compared to those in Sesn2+/+ BMDMs at 12 h (Fig. 1B), which is in accord with the effect of Sesn2 deficiency on cleaved CASP1 detection. Unlike IL1B and IL18 secretion, the secretion of TNF (tumor necrosis factor) was unchanged by the absence of Sesn2 (Fig. 1B), supporting our data that activation of NFKB signaling was not affected in sesn2−/− cells. In addition, CASP1 activation and secretion of IL1B and IL18 at 12 h treatment with LPS or ATP alone did not change in sesn2−/− BMDMs (Fig. 1C, D, Fig. S3). Since stimulation with nigericin induces NLRP3 activation in LPS-primed macrophages,11,32,33 we examined whether nigericin had the same effect as ATP on inflammasome activation at 12 h in sesn2−/− BMDMs. Similar to ATP, treatment with LPS and nigericin increased CASP1 activation and secretion of IL1B and IL18 in sesn2−/− BMDMs (Fig. 1E, F, Fig. S4). Stimulation with flagellin receptor NLRC4 (NLR family, CARD domain containing 4) activator or double-stranded (dA:dT) DNA (an AIM2 inflammasome activator) in LPS-primed BMDMs, did not influence secretion of IL1B or IL18 in sesn2−/− BMDMs (Fig. S5A, B). In contrast, CASP1 activation and secretion of IL1B and IL18 at 12 h after treatment with LPS and ATP were significantly decreased in nlrp3−/− BMDMs, compared to Nlrp3+/+ BMDMs (Fig. 1G, H, Fig. S6).
To investigate the possible role for SESN2 in CASP11 activation in our experimental condition, we examined the expression levels of cleaved CASP11 p26 and CASP11-mediated cytokines IL1A (interleukin 1 α) and HMGB1 (high mobility group box 1) in the supernatants of Sesn2+/+ or sesn2−/− BMDMs treated with LPS and ATP. Increases in cleaved CASP11 and secretion of IL1A and HMGB1 upon stimulation were not affected in sesn2−/− BMDMs (Fig. S7A, B), suggesting that SESN2 is not involved in CASP11 activation in BMDMs stimulated with LPS and ATP. Taken together, SESN2 suppresses NLRP3-dependent CASP1 activation and the secretion of IL1B and IL18 in macrophages primed with LPS for 12 h, rather than 6 h, followed by stimulation with ATP.
SESN2 expression increased by extended LPS priming is mediated by NOS2-generated NO
As shown in Figure 1A, SESN2 protein expression increased at 12 h, but not 6 h, of priming with LPS when followed by stimulation with ATP, while the transcription level of Sesn2 was not increased at any time points within 12 h by the same stimulation (data not shown), suggesting that the LPS-dependent SESN2 increase is a result of post-transcriptional regulation of SESN2 expression. The increase of NOS2-derived NO in macrophages primed with LPS for 12 h results in the suppression of the ATP-induced NLRP3 inflammasome activation.34,35 To investigate the effect of NO on SESN2 levels, we primed wild-type BMDMs with LPS for 6 or 12 h in the presence or absence of a NOS inhibitor, L-NAME (NG-nitro-L-arginine methyl ester), followed by treatment with or without ATP. Interestingly, the increases in SESN2 caused by stimulation with LPS alone, or LPS plus ATP at 12 h were decreased by L-NAME treatment, whereas increases in NOS2 expression by the same stimulation were not affected by L-NAME, although increased NO was decreased by L-NAME (Fig. 2A, B). Consistent with the effect of Sesn2 deficiency, L-NAME also significantly increased both ATP-induced CASP1 activation (Fig. 2B, Fig. S8) and secretion of IL1B and IL18 after 12 h, but not 6 h, of priming with LPS (Fig. 2C).
Figure 2.
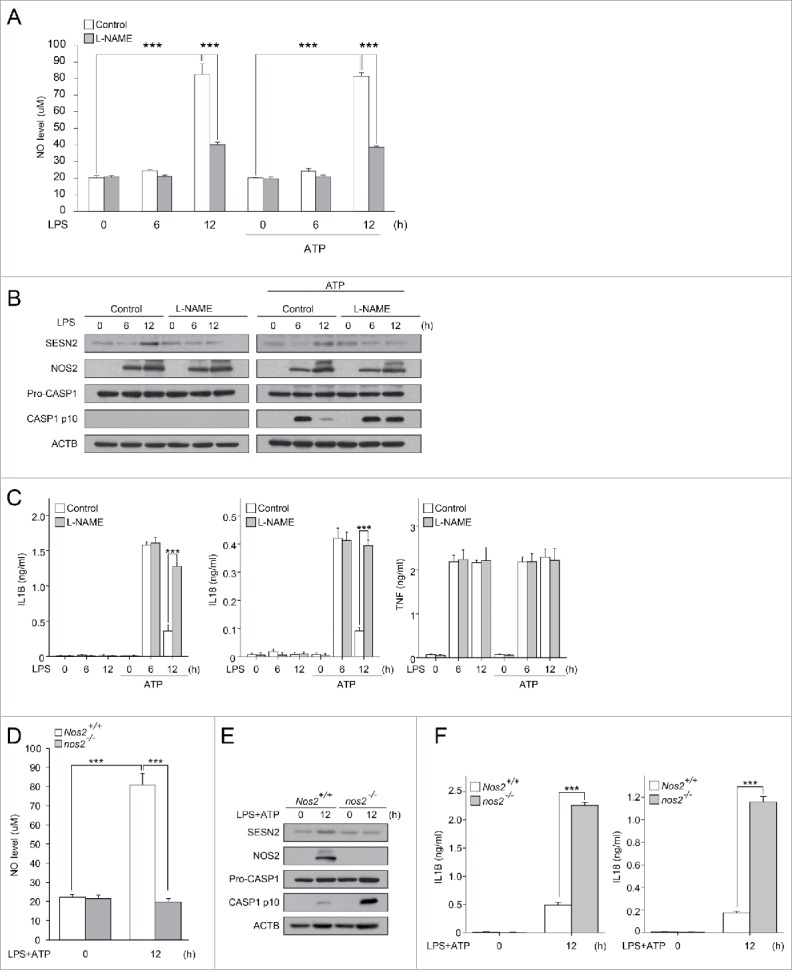
Inhibition of NOS2-mediated NO production decreases SESN2 expression, and increased CASP1 activation. (A-C) BMDMs were primed with LPS (0, 6, 12 h) in the presence or absence of L-NAME followed by the presence or absence of ATP treatment. (A) NO production, (B) immunoblot analysis of the indicated proteins and (C) production of IL1B, IL18 and TNF as measured by ELISA. (D-F) Nos2+/+ and nos2−/− BMDMs were primed with LPS (12 h) followed by ATP treatment. (D) NO production, (E) immunoblot analysis of the indicated proteins and (F) production of IL1B and IL18 as measured by ELISA. Data shown are representative of 3 independent experiments and are the mean ± s .e.m. ***, p < 0.005 from an ANOVA followed by Tukey's post hoc test.
To examine whether SESN2 expression and inflammasome suppression induced by NO production upon stimulation at 12 h is dependent on expression of NOS2, we used BMDMs isolated from Nos2+/+ and nos2−/− mice. Similar to the effect of L-NAME, Nos2 deficiency decreased SESN2 expression and NO production, and reversed the decrease in CASP1 activation (Fig. 2D, E, Fig. S9) and secretion of IL1B and IL18 (Fig. 2F). However, Sesn2 deficiency did not affect the NOS2 increase observed upon stimulation (Fig. 1A). To investigate whether autophagic or proteasomal activity is responsible for SESN2 degradation upon treatment with L-NAME, we examined changes in SESN2 expression levels in BMDMs in the presence of L-NAME when pretreated with bafilomycin A1 (an inhibitor of autophagosome fusion with the lysosome) or MG132 (a proteasome inhibitor). Treatment with bafilomycin A1, rather than MG132, rescued SESN2 expression decreased by L-NAME treatment (Fig. S10), indicating that SESN2 degradation in the presence of L-NAME was due to autophagic degradation. Collectively, these results indicate that SESN2 expression increased by extended LPS priming is mediated by NOS2-mediated NO, and NOS2-generated NO production contributes to SESN2-mediated suppression of NLRP3 inflammasome activation.
SESN2 is required for maintenance of mitochondrial homeostasis in response to LPS and ATP stimulation
Treatment with LPS and ATP produces damaged mitochondria, followed by mitochondrial ROS generation, leading to NLRP3 inflammasome activation in macrophages.9 We thus investigated whether SESN2 can suppress NLRP3 activation through regulation of mitochondrial homeostasis. We first examined mitochondrial superoxide production using MitoSOX (a mitochondrial superoxide indicator) at 12 h after priming with LPS. Stimulation with LPS and ATP increased the production of mitochondrial superoxide in sesn2−/− BMDMs to a greater extent than in Sesn2+/+ BMDMs, although there were no differences in the basal levels of mitochondrial superoxide production between the 2 groups (Fig. 3A).
Figure 3.
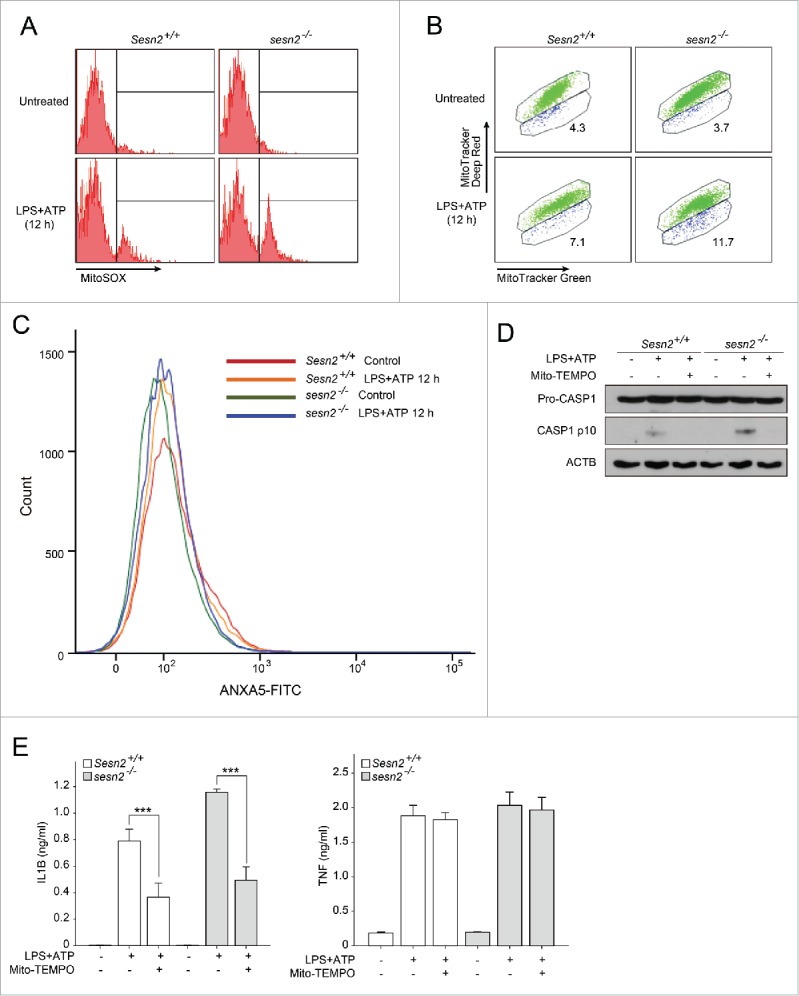
SESN2 is required for maintenance of mitochondrial homeostasis in response to LPS and ATP stimulation. (A-C) Sesn2+/+ or sesn2−/− BMDMs were primed with LPS (12 h) followed by ATP treatment. Flow cytometry of BMDMs with (A) MitoSOX staining for 15 min, (B) MitoTracker Deep Red and MitoTracker Green staining for 15 min and (C) ANXA5-FITC for 5 min. (D and E) Sesn2+/+ or sesn2−/− BMDMs incubated with Mito-TEMPO (500 µM) for 1 h followed by LPS and ATP treatment. (D) Immunoblot analysis of the indicated proteins and (E) production of IL1B and TNF as measured by ELISA. Data shown are representative of 3 or more independent experiments and are the mean ± s .e.m. ***, p < 0.005 from an ANOVA followed by Tukey's post hoc test.
We further analyzed the functional mitochondrial pool in sesn2−/− BMDMs using MitoTracker Deep Red, a probe sensitive to the mitochondrial inner transmembrane potential, and MitoTracker Green, a probe for mitochondrial membrane lipids which is independent of membrane potential. The percentages of damaged mitochondria (positive for MitoTracker Green and negative for MitoTracker Deep Red) following stimulation were 11.7% and 7.1% in sesn2−/− and Sesn2+/+ BMDMs, respectively (Fig. 3B). These results revealed an approximately 1.6-fold greater mitochondrial damage index in sesn2−/− than in Sesn2+/+ cells. Although increases in mitochondrial damage were noted in sesn2−/− BMDMs, the cells did not show increased apoptotic cell death upon stimulation with LPS and ATP (Fig. 3C), suggesting that stimulation-dependent mitochondria damage in sesn2−/− BMDMs was not enough to induce apoptotic cell death. We next investigated the effect of mitochondrial ROS on CASP1 activation in sesn2−/− BMDMs. We observed that treatment with Mito-TEMPO ([2-{2, 2, 6, 6-tetramethylpeperidin-1-oxyl-4-ylamino}-2-oxoethyl]triphenylphosphonium chloride), a mitochondria-targeted ROS scavenger, abrogated the increase in CASP1 activation (Fig. 3D, Fig. S11) and secretion of IL1B, but not of TNF (Fig. 3E), in sesn2−/− cells. These data indicate that SESN2 suppresses prolonged activation of inflammasomes in response to LPS and ATP through maintenance of mitochondrial integrity.
SESN2 induces mitochondrial priming by facilitating perinuclear clustering of damaged mitochondria
We sought to uncover the regulatory mechanism whereby SESN2 prevents the over-production of mitochondrial ROS and the aggravation of mitochondrial permeability transition caused by damaged mitochondria. We found that SESN2 protein increased and associated with mitochondria after 12 h of priming with LPS and subsequent ATP treatment (Fig. 4A). Similar to SESN2, SQSTM1 protein increased and was translocated to mitochondria at 12 h following stimulation (Fig. 4A). To examine whether recruited SESN2 and SQSTM1 at mitochondria can interact with each other upon stimulation, we performed mitochondrial fractionation, followed by a co-immunoprecipitation (co-IP) with SESN2 antibody and western blot analysis. SESN2 interacted with SQSTM1 in both the cytoplasm and mitochondria after stimulation, whereas this interaction did not occur in the absence of stimulation (Fig. 4B). To confirm the specific interactions between SESN2 and SQSTM1, we performed immunoprecipitation experiments using the lysates of sesn2−/− BMDMs. Endogenous SQSTM1 immunoprecipitation by SESN2 antibody was detected in only Sesn2+/+ BMDMs stimulated with LPS and ATP, not in sesn2−/− BMDMs (Fig. S12).
Figure 4.

SESN2 induces mitochondrial preparing by facilitating perinuclear clustering of damaged mitochondria. (A-C) BMDMs were primed with LPS (12 h) followed by ATP treatment. (A) Immunoblot analysis of the indicated proteins in cytosolic (C), mitochondrial (M) protein fractions, and whole cell lysates (Input) (B) immunoblot analysis of SQSTM1 in SESN2 immunoprecipitates (IP) in cytosolic and mitochondrial protein fractions and input, and (C) representative confocal images of the PLA (proximity-ligation assay) using SESN2 and SQSTM1, and their colocalization with MitoTracker Deep Red. Scale bar: 25 μm. (D-J) Sesn2+/+ or sesn2−/− BMDMs were primed with LPS (12 h) followed by ATP treatment. (D) Representative confocal immunofluorescence images of SQSTM1 colocalizing with TOMM20 (mitochondria surface membrane protein), Scale bar: 25 μm, (E) quantification of the cells that have SQSTM1 among total cells and (F) quantification of the cells that have SQSTM1 colocalized with perinuclear-clustered mitochondria among total cells (>150 cells per group in 2 independent experiments), and (G) immunoblot analysis of the indicated proteins in cytosolic and mitochondrial protein fractions in the presence of bafilomycin A1 and (H) PLA of SQSTM1 and Lys63-ubiquitin (UB) and their colocalization with MitoTracker Deep Red. Scale bar: 25 μm, and (I) quantification of cells that have SQSTM1 and Lys63-ubiquitin among total cells and (J) quantification of the cells that have SQSTM1 and Lys63-ubiquitin colocalized with perinuclear-clustered mitochondria among total cells (>150 cells per group in 2 independent experiments). Purity of the fractions was assessed by immunoblotting for VDAC1 (voltage dependent anion channel 1) and CASP3 (cytosolic protein). Data shown are representative of 3 independent experiments. *, p < 0.05; ***, p < 0.005 from an ANOVA followed by Tukey's post hoc test. NS, not significant.
We also investigated the spatial proximity of SESN2 to SQSTM1 on the mitochondrial surface following stimulation using an in situ proximity-ligation assay, which is a method that permits visualization of the spatial proximity of 2 proteins. Interestingly, we observed that the perinuclear aggregates of mitochondria were detected by stimulation with LPS and ATP, and that SESN2 and SQSTM1 colocalized on the perinuclear-aggregated mitochondria (Fig. 4C). In accordance with the co-IP result, some of the SESN2 and SQSTM1 also were colocalized in the cytoplasm (Fig. 4C). In this context, we asked whether SESN2 was responsible for both perinuclear clustering of the mitochondria and SQSTM1 recruitment onto the clustered mitochondria upon stimulation. As shown in Fig. 4D, SQSTM1 aggregation and perinuclear aggregates of mitochondria were induced in Sesn2+/+ BMDMs upon stimulation. Meanwhile, however, the intensity of SQSTM1 aggregation decreased in sesn2−/− BMDMs upon stimulation, compared to those in Sesn2+/+ BMDMs, although the number of cells containing SQSTM1 was not different between Sesn2+/+ BMDMs and sesn2−/− BMDMs (Fig. 4D, E). Furthermore, stimulation-induced perinuclear aggregates of mitochondria in Sesn2+/+ BMDMs were not observed in sesn2−/− BMDMs (Fig. 4D). Thus, SQSTM1 proteins colocalized to perinuclear-clustered mitochondria were rarely detected in Sesn2−/− BMDMs (Fig. 4D, F).
Given these findings, we wondered how SESN2 induces SQSTM1 aggregation toward the perinuclear-clustered mitochondria. Several studies in neuronal cells have shown that perinuclear clustering of mitochondria is initiated when mitochondria are damaged by various cellular stimuli, and is a prerequisite for the specific removal of impaired mitochondria through mitophagy.1,36 In neuronal cells, SQSTM1 is required for ubiquitination-dependent clustering of damaged mitochondria caused by treatment with a mitochondrial uncoupler, carbonyl cyanide m-chlorophenylhydrazone.6,18,37 Thus, we hypothesized that SESN2 may induce perinuclear clustering of mitochondria by mediating the aggregation of SQSTM1 via SESN2-SQSTM1 interaction and by linking SQSTM1 to ubiquitins on the surface of damaged mitochondria. As shown in Fig. 4G, both ubiquitins and SQSTM1 were increased in mitochondrial fractions upon stimulation in Sesn2+/+ BMDMs in the presence of bafilomycin A1. Interestingly, the increased level of ubiquitins and SQSTM1 observed in mitochondria upon stimulation were greatly decreased in sesn2−/− BMDMs (Fig. 4G), indicating that SESN2 was required for the recruitment of both ubiquitins and SQSTM1 to damaged mitochondria destined for autolysomal degradation. We also examined the spatial proximity of SQSTM1 to Lys63- or Lys48-linked ubiquitins on mitochondria following stimulation using the in situ proximity-ligation assay. Both the number of cells showing SQSTM1 colocalized with Lys63-linked ubiquitin-chains and the number of cells with SQSTM1-Lys63-linked ubiquitin-chains colocalized with perinuclear-clustered mitochondria were increased to a greater extent in Sesn2+/+ BMDMs, compared to those in sesn2−/− BMDMs upon stimulation (Fig. 4H–J). The cells showing SQSTM1 colocalized with Lys48-linked ubiquitin-chains were rarely detected in Sesn2+/+ BMDMs (Fig. S13).
SESN2 has already been shown to be associated with SQSTM1 through in vitro binding assays.29,38 Several studies have revealed that signal-dependent phosphorylation of SQSTM1 at specific sites (Ser349, Ser351, Ser403) is critical to degradation of ubiquitinated proteins targeted by SQSTM1 by reinforcing interactions between them.29,39,40 Therefore, we first examined the extent of phosphorylation of SQSTM1 at Ser349, Ser351, and Ser403 in Sesn2+/+ BMDMs in the absence or presence of stimulation. Therein, phosphorylation of SQSTM1 at Ser349 and Ser351, but not Ser403, increased upon stimulation (Fig. S14). Nevertheless, the amounts of increased phosphorylation of SQSTM1 in Sesn2+/+ BMDMs were almost the same as those in sesn2−/− BMDMs (Fig. S14), suggesting that SESN2 is not involved in phosphorylation of SQSTM1 in BMDMs treated with LPS and ATP. Collectively, SESN2 induces mitochondrial priming through facilitating perinuclear clustering of damaged mitochondria by mediating SQSTM1 aggregation and its recruitment to Lys63-linked ubiquitins on the mitochondrial surface.
SESN2 induces autophagosome formation and increases mitophagic activity
In addition to the critical role of SESN2 in mitochondrial priming, we hypothesized that SESN2 may also activate specific autophagy machinery for the degradation of damaged mitochondria upon stimulation. We first examined the level of autophagosome formation by counting the green fluorescent protein-microtubule-associated protein 1 light chain 3 (GFP-MAP1LC3) puncta in transgenic mice with either Sesn2+/+ (Sesn2+/+ GFP-MAP1LC3) or sesn2−/− (sesn2−/− GFP-MAP1LC3) genetic backgrounds. When primed with LPS for 6 h, a higher number of GFP-MAP1LC3 puncta (Fig. 5A), and cells with greater than 3 strong GFP-MAP1LC3 puncta among the total cells (Fig. 5B) were observed in both Sesn2+/+ GFP-MAP1LC3 and sesn2−/− GFP-MAP1LC3 BMDMs, compared to those in the absence of stimulation. However, there were no differences between Sesn2+/+ GFP-MAP1LC3 and sesn2−/− GFP-MAP1LC3 BMDMs (Fig. 5A, B). Conversely, after 12 h of priming with LPS, the number of GFP-MAP1LC3 puncta in Sesn2+/+ cells was almost similar to the number observed at 6 h, whereas they were significantly decreased in sesn2−/− cells (Fig. 5A, B).
Figure 5.

Sesn2 induces autophagosome formation and increases mitophagic activity. (A-C) Sesn2+/+ GFP-MAP1LC3 and sesn2−/− GFP-MAP1LC3 BMDMs were primed with LPS (0, 6, 12 h) followed by ATP treatment. (A) Representative confocal immunofluorescence images of GFP-MAP1LC3 and TOMM20. Scale bar: 25 μm. (B) Quantification of cells with more than 3 strong GFP-MAP1LC3 puncta from among total cells and (C) cells that have GFP-MAP1LC3 puncta colocalized with perinuclear clustered mitochondria from among cells with GFP-MAP1LC3 puncta (>150 cells per group in 2 independent experiments for B, C). (D and E) Sesn2+/+ and sesn2−/− BMDMs were primed with LPS (12 h) followed by ATP treatment in the presence or absence of bafilomycin A1. (D) Immunoblot analysis for MAP1LC3B-II and (E) quantification of MAP1LC3B-II immunoblot intensity. (F-H) Sesn2+/+ and sesn2−/− BMDMs were primed with LPS (12 h) followed by ATP treatment. (F) Representative confocal immunofluorescence images of mt-mKeima. Scale bar: 25 μm. (G) Quantification of the ratio of high (550ex:438ex) signal area (red) to total mitochondrial area (>30 cells per group in 2 independent experiments) and (H) transmission electron microscopy of morphological changes in mitochondria (arrow, mitochondria within autophagosomes; arrowhead, damaged mitochondria; red ‘N’, nucleus). NS, not significant. Scale bar: 1 µm. Data shown are representative of 3 independent experiments and are the mean ± s .e.m. *, p < 0.05; ***, p < 0.005 from an ANOVA followed by Tukey's post hoc test.
To verify whether suppression of autophagosome formation in sesn2−/− GFP-MAP1LC3 cells upon stimulation correlated with autophagic activity, we employed in vitro autophagic flux assays in the absence or presence of bafilomycin A1. In the presence of bafilomycin A1, endogenous MAP1LC3-II was detected in the absence of stimulation, and increased 3.1-fold upon stimulation in Sesn2+/+ BMDMs (Fig. 5D, E). However, in the absence of bafilomycin A1, MAP1LC3-II was not detected in Sesn2+/+ BMDMs even when stimulated with LPS and ATP (Fig. 5D, E). In the presence of bafilomycin A1 and stimulation, levels of endogenous MAP1LC3-II in sesn2−/− BMDMs was much lower than in Sesn2+/+ BMDMs (about 32.3 %), (Fig. 5D, E). These data indicate that SESN2 was critical not only for autophagosome formation, but also for autophagic activity after long-term (12 h) priming with LPS and subsequent ATP treatment.
We next investigated whether SESN2 is involved in selective autophagy of damaged mitochondria by analyzing the number of cells with GFP-MAP1LC3 puncta colocalized with perinuclear-clustered mitochondria from among cells with GFP-MAP1LC3 puncta. Colocalization was apparent in Sesn2+/+ GFP-MAP1LC3 BMDMs at 12 h, but not 6 h (Fig. 5A, C). However, in sesn2−/− GFP-MAP1LC3 BMDMs, GFP-MAP1LC3 puncta colocalized with perinuclear-clustered mitochondria were barely detected 12 h after stimulation (Fig. 5A, C). For quantification of mitophagic activity in BMDMs, we employed the mitochondria-targeted monomeric Keima (mt-mKeima) probe to identify functional mitophagy41 because mt-mKeima undergoes a reversible change in color in response to acidic pH (<pH 6.0) which occurs during autolysosome maturation. Thus, increases in the 550:438 nm excitation ratio of mt-mKeima-transfected BMDMs, represented by a pseudo-red color, indicate the presence of damaged mitochondria in maturing autolysosomes. Using this assay, we found that the frequency of red mt-mKeima puncta (Fig. 5F), and high (550ex:438ex) signal area (Fig. 5G) increased approximately 3.2-fold in Sesn2+/+ BMDMs 12 h after stimulation, relative to those observed in the untreated BMDMs. The number of red puncta and high (550ex:438ex) signals in sesn2−/− BMDMs were much lower than those in Sesn2+/+ BMDMs (about 42.2 %) (Fig. 5F, G) Furthermore, transmission electron microscopy analysis showed that damaged mitochondria surrounded by autolysosomes were detected in the perinuclear region of Sesn2+/+ BMDMs following stimulation, whereas a greater abundance of swollen mitochondria with severely disrupted cristae were observed throughout the cytoplasm of sesn2−/− BMDMs (Fig. 5H).
We also examined the basal rate of autophagy and mitophagy in Sesn2+/+ BMDMs and sesn2−/− BMDMs without stimulation. The number of cells with GFP-MAP1LC3 puncta slightly increased in Sesn2+/+ BMDMs upon treatment with bafilomycin A1, while the number of cells with GFP-MAP1LC3 puncta colocalized with mitochondria did not increase upon bafilomycin A1 treatment (Fig. S15A-C). Both the number of cells with GFP-MAP1LC3 puncta and the number of cells with GFP-MAP1LC3 puncta colocalized with mitochondria in sesn2−/− BMDMs were no different from those in Sesn2+/+ BMDMs (Fig. S15A-C). When we transfected mt-mKeima into Sesn2+/+ BMDMs and sesn2−/− BMDMs, we failed to observe red mt-mKeima puncta and high (550ex:438ex) signal areas in either of them (Fig. S15D, E). Taken together, these findings suggest that SESN2 is required for mitophagic activity via the induction of autophagosome formation and autophagic activity in a stimulation-dependent manner.
SESN2 induces autophagic activity for mitophagy by increase of ULK1 protein level
We wondered how SESN2 can initiate autophagosome formation in response to LPS and ATP. We first examined whether SESN2 regulates the activity of AMPK and MTOR signaling in BMDMs upon stimulation, since SESN2 regulates the activity of AMPK and MTOR in response to genotoxic stress,24 and AMPK and MTOR are closely involved in the initiation of autophagy in response to nutrient depletion.42-44 The extent of phosphorylation of AMPK, RPS6 (ribosomal protein S6), and ACACA (acetyl-Coenzyme A carboxylase α) were not affected by the deletion of Sesn2 (Fig. S16), indicating that SESN2 is not involved in the activation of the AMPK or MTOR signaling pathways in response to LPS and ATP in macrophages. We next investigated whether SESN2 regulates the expression of various autophagy-related (ATG) proteins that initiate or activate autophagy. Surprisingly, increases in ULK1 after 12 h of LPS priming and subsequent ATP treatment were significantly lower in sesn2−/− BMDMs, whereas the expression levels of other ATG proteins including ULK2, BECN1 (Beclin 1, autophagy related), ATG5, ATG7, and ATG13, in sesn2−/− BMDMs were similar to levels in wild-type cells (Fig. 6A). Similar to ULK1 itself, the increased expression levels of 2 phosphorylated versions of ULK1 (S555 and S757) observed in the wild-type cells at 12 h could hardly be detected in sesn2−/− BMDMs (Fig. 6A). As was observed for SESN2, ULK1 protein increased after 12 h, but not 6 h, of priming with LPS and subsequent ATP stimulation (Fig. 6A), whereas the transcription level of ULK1 was not increased by the same stimulation at any time point within 12 h (data not shown). ULK1 also associated with mitochondria after 12 h of LPS priming and subsequent ATP treatment (Fig. 6B). To verify that defective autophagosome formation in sesn2−/− BMDMs was attributable to a decrease in ULK1, we examined the possible rescue effect by injecting human ULK1-expressing virus particles into sesn2−/− BMDMs, after which ULK1 expression was confirmed (Fig. 6C). Rescue with ULK1 increased the number of cells with GFP-MAP1LC3 puncta but did not rescue the perinuclear clustering of mitochondria (Fig. 6D, E). Both mitochondrial ROS and damaged mitochondria were decreased by rescue with ULK1 (Fig. 6F, G). In accordance with the effects on mitochondrial homeostasis, cleaved CASP1 and secretion of IL1B and IL18 were also decreased by rescue with ULK1 (Fig. 6H, I, Fig. S17). These results indicate that SESN2 induces autophagic activity for mitophagy by increase of ULK1 protein levels.
Figure 6.
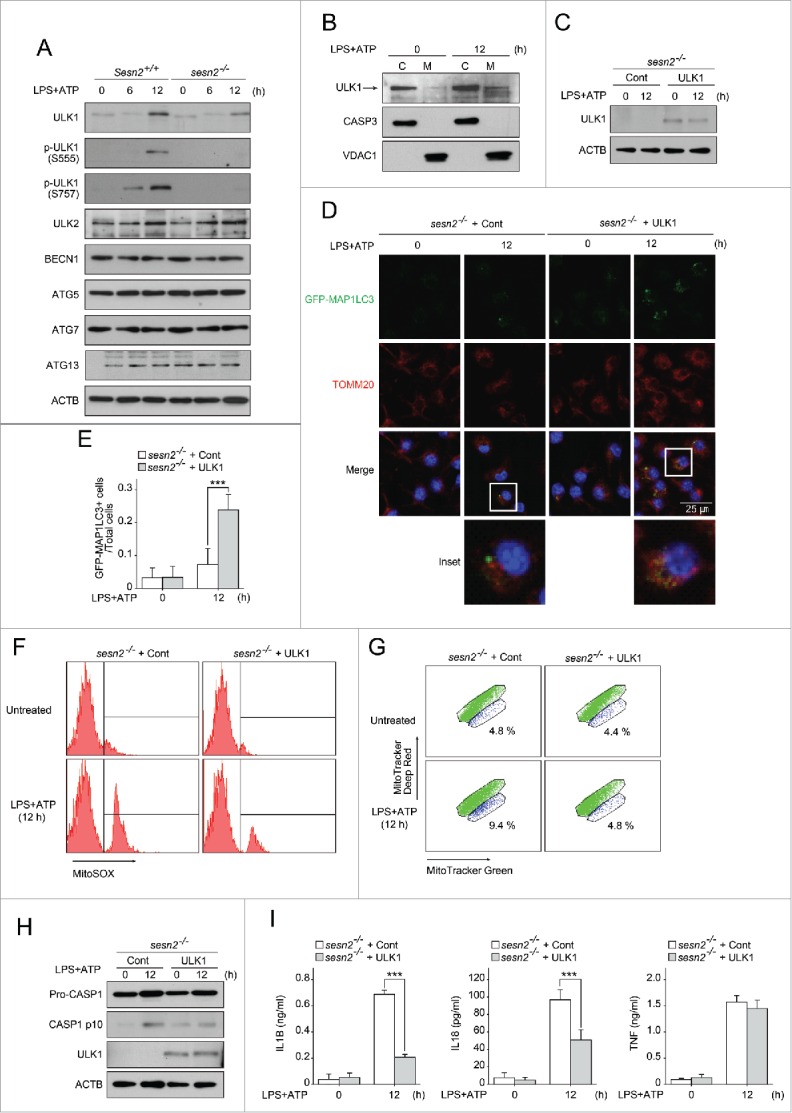
SESN2 induces autophagic activity for mitophagy by increase of ULK1 protein levels. (A) Immunoblot analysis of the indicated proteins from Sesn2+/+ and sesn2−/− BMDMs primed with LPS (0, 6, and 12 h) followed by ATP treatment. (B) Immunoblot analysis for human ULK1 in cytosolic (C) and mitochondrial (M) protein fractions from BMDMs primed with LPS (12 h) followed by ATP treatment. (C-I) sesn2−/− BMDMs infected with retrovirus containing control vector, or human ULK1 expression vector (sesn2−/− + Cont, sesn2−/− + ULK1, respectively), were primed with LPS (12 h) followed by ATP treatment. (C) Immunoblot analysis for human ULK1 using anti-human ULK1 antibody, (D) representative confocal immunofluorescence images of GFP-MAP1LC3 colocalizing with TOMM20, (E) quantification of number of cells with more than 3 strong GFP-MAP1LC3 puncta from among the total cells (>150 cells per group in 2 independent experiments), flow cytometry of BMDMs with (F) MitoSOX staining for 15 min and (G) MitoTracker Deep Red and MitoTracker Green staining for 15 min, (H) immunoblot analysis of the indicated proteins and (I) production of IL1B, IL18 and TNF as measured by ELISA. Data shown are representative of 3 independent experiments and are the mean ± s .e.m. ***, p < 0.005 from an ANOVA followed by Tukey's post hoc test.
SESN2 plays a protective role in 2 different sepsis mouse models
To examine the physiological role of SESN2 in the inflammatory response to septic shock, we employed the CLP (cecal ligation and puncture) technique, a clinically relevant murine model of polymicrobial sepsis. The serum levels of IL1B and IL18 were markedly elevated in Sesn2+/+ mice after CLP compared with those in sham-operated control mice (Fig. 7A). However, even higher levels of IL1B and IL18 were observed in sesn2−/− mice after CLP, (Fig. 7A). Similar to their serum levels, the levels of IL1B and IL18 in lung tissues after CLP were significantly increased in sesn2−/− mice compared with those in Sesn2+/+ mice (Fig. 7B). We also examined organ dysfunction by measuring biochemical indicators of organ function in the serum. Much higher levels of creatinine, GOT1/AST (glutamic-oxaloacetic transaminase 1, soluble), and GPT/ALT (glutamic pyruvic transaminase, soluble) were observed in sesn2−/− mice after CLP, compared with those in Sesn2+/+ mice (Fig. 7C). Strikingly, sesn2−/− mice had a significantly higher mortality rate (Fig. 7D). To confirm that these findings in CLP sesn2−/− mice were attributable to the deficiency of Sesn2 itself, we examined the rescue effect of SESN2 through the intravenous injection of an adenoviral vector expressing human SESN2 (Ad-SESN2) into sesn2−/− mice. We verified that SESN2 expression in the liver was detectable following the injection with Ad-SESN2, but not with Ad-control (Ad-Cont) (Fig. 7E). This rescue with SESN2 significantly decreased the serum concentrations of IL1B and IL18 (Fig. 7F). The survival rate was improved by the rescue with Ad-SESN2 (Fig. 7G). Similar to the results obtained with the CLP model, sesn2−/− mice had higher serum concentrations of IL1B and IL18 (Fig. 7H), and higher mortality in endotoxemia-induced sepsis (Fig. 7I). As with the CLP model, rescue with SESN2 decreased the serum concentrations of IL1B and IL18 (Fig. 7J), and improved the survival rate in endotoxemia-induced sesn2−/− mice (Fig. 7K). These data indicate that SESN2 is an essential factor in protection against septic shock, as shown in 2 different mouse models.
Figure 7.

Sesn2 plays a protective role against septic shock in mouse sepsis models. (A-D) Sepsis was induced by CLP in Sesn2+/+ and sesn2−/− mice. (A) Serum IL1B and IL18 as measured by ELISA (n = 5), (B) IL1B and IL18 in homogenized lung tissues as measured by ELISA (n = 5), (C) serum creatinine, GOT1 and GPT (n = 5) 24 h after CLP surgery, and (D) survival of Sesn2+/+(n = 11) mice or sesn2−/−(n = 12) mice. (E-G) CLP was performed in Sesn2+/+ and sesn2−/− mice. (E) Immunoblot analysis for SESN2 in liver tissue lysates from sesn2−/−+ Ad-Cont and sesn2−/−+ Ad-SESN2; ‘ns’ indicates a nonspecific band. (F) Serum IL1B and IL18 as measured by ELISA (n = 5) 24 h after CLP surgery, and (G) survival of sesn2−/−+ Ad-Cont (n = 13) or sesn2−/−+ Ad-SESN2 (n = 13). (H and I) Sesn2+/+ and sesn2−/− mice were challenged with LPS. (H) Serum IL1B and IL18 as measured by ELISA 24 h after LPS challenge (12 mg/kg, i.p.) and (I) Survival of Sesn2+/+(n = 13) mice or sesn2−/−(n = 13) mice after LPS challenge (25 mg/kg, i.p.). ((J)and K) sesn2−/−+ Ad-Cont and sesn2−/−+ Ad-SESN2 mice were challenged with LPS. (J) Serum IL1B and IL18 as measured by ELISA 24 h after LPS challenge (12 mg/kg, i.p.) and (K) survival of sesn2−/−+ Ad-Cont (n = 13) or sesn2−/−+ Ad-SESN2 (n = 13) mice after LPS challenge (25 mg/kg, i.p.). Data shown are the mean ± s .e.m. The survival rates were analyzed by Kaplan-Meier log-rank test. *, p < 0.05; **, p < 0.01; ***, p < 0.005 from an ANOVA followed by Tukey's post hoc test.
Protein levels of SESN2 are increased in monocytes from the sepsis mouse model
Since signal-dependent SESN2 protein, which was increased at specific time points suppressed inflammasome activation in macrophages, and the presence of SESN2 played a protective role in 2 septic shock mouse models, we wondered if the SESN2 protein levels were regulated in blood monocytes under septic conditions. SESN2 protein levels in monocytes isolated from mouse blood increased and peaked 48 h after intraperitoneal injection with LPS into the mice, and significantly decreased at 96 h (Fig. 8A). In accordance with the fluctuation of SESN2 protein levels in monocytes according to LPS injection time points, serum concentrations of IL1B and IL18 were most increased at 24 h and decreased at 48 h after LPS injection, which was when the SESN2 expression level was the highest (Fig. 8B). Interestingly, serum concentrations of IL1B and IL18 were radically decreased at 96 h at which time SESN2 expression was also decreased (Fig. 8B). These results indicate that SESN2 is increased when sepsis is exacerbated, and decreased when sepsis is recovered, suggesting that SESN2 plays a protective role in sepsis through the regulation of its expression level.
Figure 8.
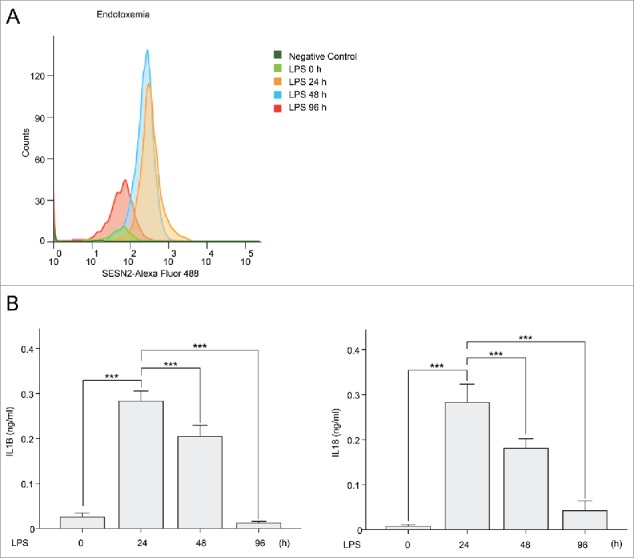
SESN2 is increased at the time when sepsis is exacerbated, and decreased at the time when sepsis is recovered. (A and B) Mice were injected with LPS (0, 24, 48, and 96 h, n = 9 per group). (A) Flow cytometry of SESN2 in mouse monocytes from blood stained with anti-SESN2-Alexa Flour 488 antibody and (B) serum IL1B and IL18 as measured by ELISA. Data shown are representative of 3 independent experiments and are the mean ± s .e.m. ***, p < 0.005 from an ANOVA followed by Tukey's post hoc test.
Human SESN2 is highly expressed in monocytes of septic shock patients
To investigate whether our observations from the sepsis mice models were relevant to human patients with septic shock syndrome, we first measured the secretion levels of IL1B and IL18 in the sera of 8 septic patients in the medical intensive care unit. Similar to the mouse sepsis model, higher levels of IL1B and IL18 were observed in the sera of septic shock patients compared to healthy volunteers (Fig. 9A). More damaged mitochondria were also detected in monocytes isolated from the blood of septic patients (Fig. 9B, C). In this context, we investigated the protein level of human SESN2 to determine if it was altered in monocytes of septic shock patients, as in the sepsis mouse model. Interestingly, the SESN2 protein level was significantly increased in monocytes of nearly all the septic shock patients, compared to the healthy volunteers in our study (Fig. 9D). Although we were not able to follow up on the serum SESN2 when the same sepsis patients recovered from their septic shock condition, we think that SESN2 plays a protective role against systemic inflammatory conditions such as sepsis via increasing its expression in monocytes.
Figure 9.
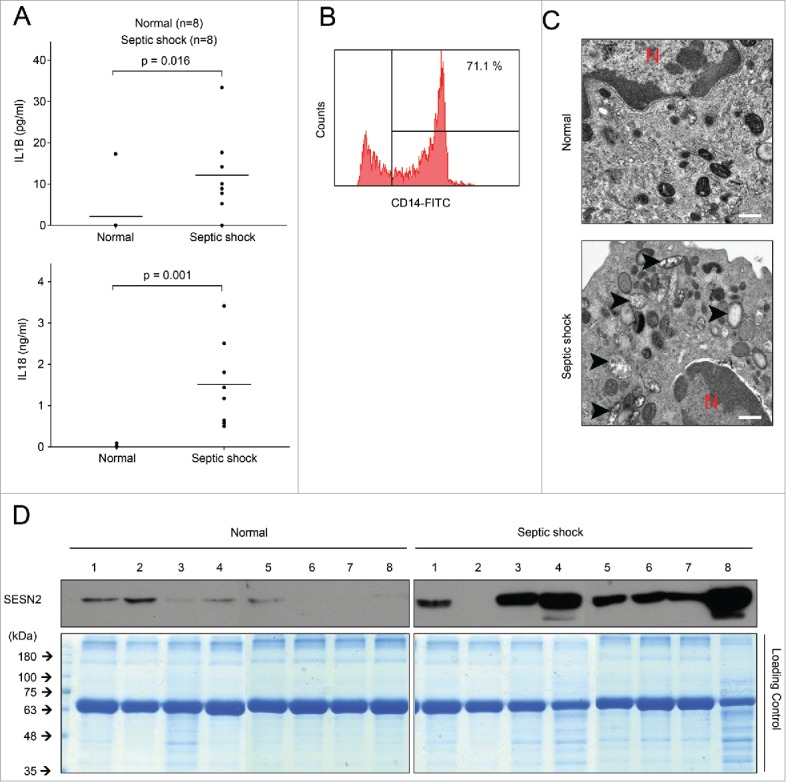
Human SESN2 is highly expressed in monocytes of septic shock patients. (A-D) Human blood samples were collected from normal subjects (n = 8) and septic shock patients (n = 8). (A) Plasma IL1B and IL18 as measured by ELISA, (B) flow cytometry of isolated human monocytes from blood stained with anti-CD14-FITC antibody to evaluate purity, (C) transmission electron microscopy of morphological changes in mitochondria in isolated human peripheral blood monocytes (arrowhead, damaged mitochondria; red ‘N’, nucleus). Scale bar, 1µm, and (D) immunoblot analysis for SESN2 and Coomassie Blue staining as a control for equal loading of isolated human monocytes. Each symbol represents an individual subject. Data shown are representative of 2 independent experiments and p values from an unpaired Student t test.
Discussion
In this study, SESN2 protein was increased by NO generated by increased NOS2 12 h after stimulation with LPS and ATP in macrophages. This increased SESN2 induced mitophagy activation through regulation of 2 synchronized procedures in a cooperative manner. First, SESN2 induced mitochondrial priming by mediating the aggregation of SQSTM1 and its binding to Lys63-ubiquitinated mitochondria. Second, SESN2 activated specific autophagic machinery for the degradation of primed mitochondria via maintenance of ULK1 protein levels. Altogether, we found that mitophagy was accomplished by SESN2- and ULK1-mediated selective autophagy of perinuclear-clustered mitochondria primed by SESN2-SQSTM1, leading to the suppression of prolonged NLRP3 inflammasome activation (Fig. 10).
Figure 10.
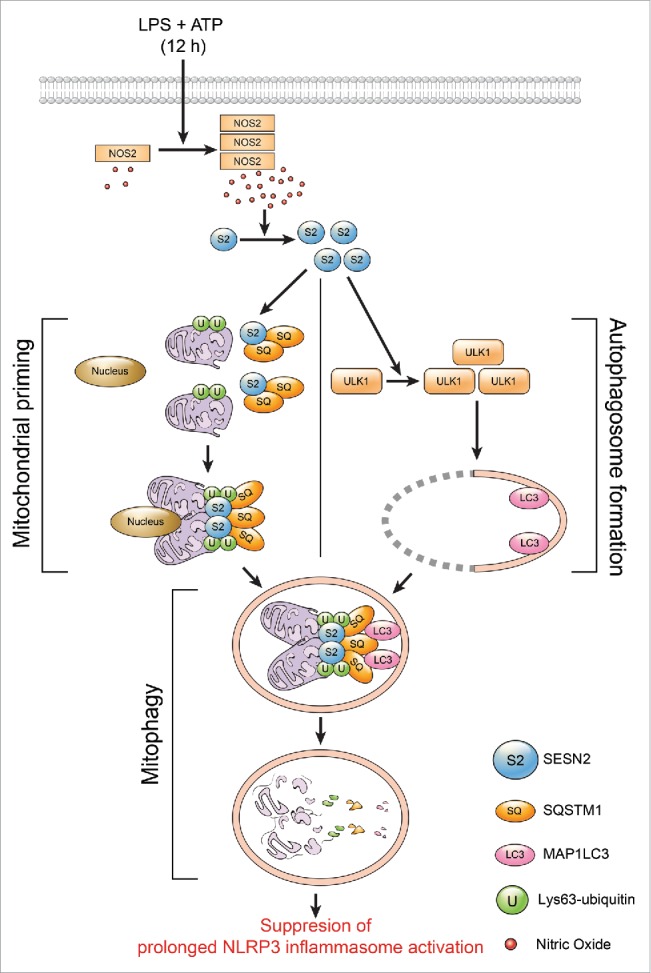
SESN2 suppresses prolonged NLRP3 activation by preserving mitochondrial homeostasis through mitophagy induction. In macrophages, SESN2 (S2) protein is increased by NO generated by increased NOS2 12 h after stimulation with LPS and ATP. Increased SESN2 induces mitophagy activation through regulation of 2 synchronized events. First, SESN2 induces mitochondrial priming by mediating the aggregation of SQSTM1 (SQ) and its binding to Lys63-ubiquitinated (U) mitochondria. Second, SESN2 activates specific autophagic machinery for the degradation of primed mitochondria via maintenance of the ULK1 protein level. Altogether, mitophagy is accomplished by SESN2- and ULK1-mediated selective autophagy of perinuclearly-clustered mitochondria primed by SESN2-SQSTM1, leading to the suppression of prolonged inflammasome activation.
Despite apparent lack of intrinsic catalytic antioxidant activity of SESN2, it protects cells from oxidative stress by lowering intracellular ROS levels.45 Two distinct pathways are known for the antioxidant function of SESN2. One is by upregulating NFE2L2 (nuclear factor, erythroid derived 2, like 2) signaling and thereby promoting the expression of genes for antioxidant enzymes.29 The other is by blocking MTOR activation, which results in ROS accumulation.23,24,46 The SESN2-promoted mitophagy in our study provides the third pathway for SESN2 to suppress ROS accumulation. Under oxidative stress, SESN2 serves as an adapter protein that mediates the interaction between SQSTM1 and KEAP1, which results in the autophagic degradation of KEAP1.29 In a recent study, SESN2 was shown to also induce autophagic degradation of SQSTM1 by promoting ULK1-mediated SQSTM1 phosphorylation via its interaction with ULK1 and SQSTM1.38 Based on these data and our result that SESN2 induces mitophagy by mediating SQSTM1 binding to ubiquitinated mitochondria via its interaction with SQSTM1, we speculate that SESN2 may be involved in SQSTM1-mediated autophagic degradation through its binding to SQSTM1 and/or SQSTM1 target proteins in response to various cellular stresses. Although we did not identify any Lys63 ubiquitinated proteins on the mitochondrial surface which interacted with SESN2 or SQSTM1, we suggest that SESN2 functions as a scaffold protein that strengthens the otherwise weak association of SQSTM1 with ubiquitinated proteins on mitochondria, resulting in mitophagy.
To date, it has been shown that SESN2 is responsible for autophagy activation through the regulation of AMPK activation and MTOR suppression under conditions of genotoxic stress and in cancer cell lines.24,27 Although phosphorylations of RPS6 and ACACA were increased in wild-type BMDMs upon stimulation with LPS and ATP, they were not affected in sesn2−/− BMDMs, suggesting that molecules other than SESN2 may be involved in the activation of AMPK and/or the MTOR signaling pathway in macrophages.
ULK1 was first identified as an autophagy initiator,47 and has recently been proposed to be involved in mitophagy during hematopoietic development or viral infection through its phosphorylation.10,48 In this study, SESN2 induced autophagosome formation and mitophagy activation through the conservation of ULK1 protein levels, rather than ULK1 phosphorylation upon stimulation, supporting a novel mechanism whereby ULK1-dependent mitophagy activation can be regulated by ULK1 protein stability. Upon stimulation, endogenous interaction with SESN2 and ULK1 was not observed when evaluated by co-IP with SESN2 antibody and western blot analysis (data not shown), suggesting that stimulation-dependent ULK1 translocation to mitochondria and maintenance of its protein levels may not be attributable to its association with SESN2. Further research will be needed to identify the exact mechanism whereby SESN2 regulates ULK1 protein levels in a signal-dependent manner. Given that reduced autophagosome formation in sesn2−/− BMDMs upon stimulation with LPS and ATP was rescued by addition of human ULK1, while perinuclear clustering of mitochondria was not, it can be deduced that ULK1 is involved in autophagosome formation, but not in perinuclear clustering of mitochondria in response to the same stimuli. Interestingly, in spite of the inability of ULK1 to rescue perinuclear clustering of mitochondria in sesn2−/− BMDMs, mitochondrial integrity was enhanced by rescue with ULK1, suggesting that perinuclear clustering of damaged mitochondria may not always be required for mitophagy induction in BMDMs in response to LPS and ATP.
It has been reported that NOS2-generated NO can suppress the activation of the NLRP3 inflammasome by maintaining mitochondrial homeostasis in macrophages,35 though the mechanism by which NO regulates mitochondrial homeostasis has not been reported. Decreases in the SESN2 protein levels and hyperactivation of CASP1 in nos2−/− BMDMs or L-NAME-treated BMDMs upon stimulation indicate that Sesn2 is increased by NOS2-generated NO, and suggest that NOS2-mediated regulation of mitochondrial homeostasis may be accomplished through the regulation of SESN2 protein levels. Until now, the accumulating data has only shown that SESN2 expression can be upregulated in response to oxidative stress at the transcriptional level.24,46,49,50 However, for the first time, we were able to demonstrate that SESN2 protein levels were translationally upregulated by NOS2-generated NO.
In addition to the importance of the signal-dependent SESN2 increase at the cellular level, SESN2 expression in blood monocytes may be associated with protection from sepsis, since much higher levels of SESN2 protein are observed in sepsis mouse models and in septic shock patients, compared to normal subjects. It has long been thought that sepsis is closely related to mitochondrial dysfunction, and a recent proteomic and metabolomic analysis in septic patients showed that proteins derived from mitochondrial deregulation may represent candidate biomarkers of severe sepsis.45 Thus, the promotion of mitophagy by increasing SESN2 expression may protect the host from sepsis. Furthermore, our novel findings regarding SESN2-regulated mitophagy in sepsis may possibly explain the cause of other diseases associated with deregulation of mitophagy, including neurodegenerative disorders, metabolic diseases, cardiovascular diseases, and cancer.
Materials and methods
Mice
sesn2−/− and sesn2−/− GFP-MAP1LC3 mice in the CL57BL/6 background were kindly provided by Dr. Seo Goo Rhee (Yonsei University, Seoul, Korea), nos2−/− and nlrp3−/− mice in the CL57BL/6 background were kindly gifted by Dr. Su-Cheong Yeom (Seoul National University, Seoul, Korea) and Dr. Je-Wook Yu (Yonsei University, Seoul, Korea) respectively. All mice were 12-16 wk of age at use. Mice were maintained in specific pathogen-free conditions and male mice were used for experiments.
Cell culture and treatment
Bone marrow-derived macrophages were cultured in DMEM (Lonza, 12-604F) supplemented with 10% fetal bovine serum (FBS; Gibco/Life Technologies, 16000-044), 1% penicillin/streptomycin (Gibco/Life Technologies, 15140-122) and 25% L929 mouse fibroblast supernatant (DMEM cultured with L929 mouse fibroblasts) or MEM-α (Gibco/Life Technologies, 12571) supplemented with 10% FBS, 1% penicillin/streptomycin and 20 ng/ml CSF1/M-CSF (R&D Systems, 416-ML). Cells were primed with LPS (200ng/ml; Sigma-Aldrich, L8274) for 6 h or 12 h and treated with ATP (Sigma-Aldrich, A2383) (2 mM) for 30 min. To inhibit NO synthesis, cells were incubated with 100 µM L-NAME (Sigma-Aldrich, N5751) with LPS. 100 nM bafilomycin A1 (Sigma-Aldrich, B1793) was used to inhibit autophagic flux and 10 µM MG132 (Sigma-Aldrich, C2211) was used to inhibit the proteasome. To inhibit mitochondrial ROS, Mito-TEMPO (Enzo Life Sciences, ALX-430-150) was administered and cells were pre-incubated for 1 h prior to LPS treatment. Cells were treated for 45 min with 5 µM nigericin (Sigma-Aldrich, N7143) or for 2 h with 0.2 µg/ml flagellin (Enzo Life Sciences, ALX-522-058) 12 h after LPS priming. One µg/ml (dA:dT) (Sigma-Aldrich, P0883) was transfected with LPS for 12 h.
Flow cytometry
Mitochondrial ROS were measured by staining cells with MitoSOX (Invitrogen, M36008) at 5 µM for 15 min at 37°C. To measure mitochondrial mass, cells were stained with 25 nM MitoTracker Green FM (Invitrogen, M7514) and MitoTracker Deep Red FM (Invitrogen, M22426) for 15 min at 37°C. Cells were then washed with phosphate-buffered saline (PBS; Lonza, 17-516F), trypsinized by trypsin-EDTA (Gibco/Life Technologies, 25200), and resuspended in cold PBS containing 1% FBS. Apoptosis was determined by flow cytometric analysis following staining with ANXA5 (annexin A5)-FITC (Biovision, K101) for 5 min at room temperature. To measure SESN2 expression in mouse monocytes from blood, buffy coats containing PBMCs (peripheral blood mononuclear cells) were stained with anti-SESN2 antibody (abclon by custom antibody service), anti-ITGAM (integrin α M) eFluor®450 (eBioscience, 48-0112) and Ly6g (lymphocyte antigen 6 complex, locus G) PerCP-Cy5.5 (eBioscience, 45-5931) antibodies for 20 min at 4°C, washed, and stained with Alexa Flour 488-conjugated secondary antibody for anti-SESN2 antibody. ITGAM+ Ly6g− cells were counted as monocytes and SESN2 expression was measured. To check the purity of isolated monocytes from human blood, CD14-FITC (Miltenyi Biotec, 130-050-201) was used. Data were acquired and analyzed with a BD LSR II flow cytometer (BD Biosciences, San Jose, CA, USA).
Transmission electron microscopy
BMDMs were fixed with glutaraldehyde and analyzed by electron microscopy as described previously.51
ELISA
Cell culture supernatants, serum and homogenized mouse lung tissues in PBS after centrifugation at 16,000 x g for 10 min were assayed by ELISA for human IL1B (R&D Systems, DY201), mouse IL1B (R&D Systems, DY401), human IL18 (MBL, 7620), mouse IL18 (MBL, 7625), mouse TNF (R&D Systems, DY410), mouse IL1A (R&D Systems, DY400) and HMGB1 (IBL international, ST51011).
Immunoblotting and immunoprecipitation
The following primary antibodies were used: SESN2 (1:2000; ProteinTech, 10795-1-AP), SQSTM1 (1:2000; Abnova, H00008878-M01), phospho-SQSTM1 (Ser349, 1:2000; Cell Signaling Technology, 95697), phospho-SQSTM1 (Ser403, 1:2000; Millipore, MABC186), NOS2 (1:2000; Millipore, 06-573), CASP1 (1:1000; Santa Cruz Biotechnology, sc-514), CASP11 (1:2000; Novus Biologicals, NB120-10454), NLRP3 (1:2000; Adipogen, AG-20B-0014-C100), P2X7P2RX7 (1:2000; Alomone Labs, APR-004-AO), ASC (1:2000; Santa Cruz Biotechnology, sc-22514-R), GFP (1:2000; Santa Cruz Biotechnology, sc-8334), AMPK (1:2000; Cell Signaling Technology, 2532), phospho-AMPK (1:2000; Cell Signaling Technology, 2535), RPS6/S6 (1:2000; Cell Signaling Technology, 2317), phopho- RPS6/S6 (1:2000; Cell Signaling Technology, 2211), MAP1LC3B (1:2000; Sigma-Aldrich, L7543), CASP3 (1:2000; Cell Technology, 9662), VDAC1 (1:2000; Abcam, ab14734), BECN1 (1:2000; Cell Signaling Technology, 3495), ATG5 (1:2000; Cell Signaling Technology, 12994), ATG7 (1:2000; Cell Signaling Technology, 8558), ATG13 (1:2000; Sigma-Aldrich, SAB4200100), mouse ULK1 (1:2000; Sigma-Aldrich, A7481), human ULK1 (1:5000; Santa Cruz Biotechnology, sc-33182), phospho-ULK1 S555 (1:2000; Cell Signaling Technology, 5869), phospho-ULK1 S757 (1:2000; Cell Signaling Technology, 6888), ULK2 (1:2000; AbD Serotec, AAM80), total NFKBIA (1:2000; Cell Signaling Technology, 9242), UBIQUITIN (1:2000; Santa Cruz Biotechnology, sc-8017) and phospho-NFKBIA (1:2000; Cell Signaling Technology, 9246). ACTB/β-actin (1:2000; Santa Cruz Biotechnology, sc-47778) was used as a loading control. Phospho-SQSTM1 (S351) was kindly provided by Dr. Seo Goo Rhee (Yonsei University, Seoul, Korea). Cells were lysed with lysis buffer (Invitrogen, FNN0011) and cell culture supernatants were concentrated by a methanol/chloroform precipitation method described previously.52 Lysates were quantified and equal amounts were loaded onto and separated by SDS-PAGE, transferred to nitrocellulose membranes (Merck Millipore, HATF09025), and incubated with primary antibodies followed by HRP-conjugated anti-rabbit secondary antibody (Santa Cruz Biotechnology, sc-2004) and HRP-conjugated anti-mouse secondary antibody (Santa Cruz Biotechnology, sc-2005). The blots were visualized using the ECL system (Animal Genetics, LR 01-01). For immunoprecipitation, 200 µg of whole cell lysates, cytosolic and mitochondrial fractions were incubated with the appropriate 2 µg of primary antibodies overnight and then incubated with protein A-agarose (Sigma-Aldrich, P9424) for 2 h with gentle rotation at 4°C. Normal rabbit immunoglobulin G (IgG) (Santa Cruz Biotechnology, sc-2027) was used as a negative control. The immunoprecipitates were washed 3 times with lysis buffer and boiled in 2x SDS-loading buffer (4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, 0.125 M Tris-HCl, pH 6.8), then separated by SDS-PAGE and analyzed by immunoblotting.
Mitochondria-cytosol fractionation
Mitochondrial fractions were obtained from BMDM following the manufacturer's instructions using a Qproteome Mitochondria Isolation Kit (QIAGEN, 37612) with minor modifications. Briefly, 5 × 106 cells were suspended in lysis buffer, and incubated on an end-over-end shaker for 10 min at 4°C, and centrifuged at 1,000 x g for 10 min at 4°C. The pellet fraction was resuspended in Disruption Buffer, passed through a 26 gauge needle (Korea Vaccine, ND.SY1030-001) 10 times to ensure complete cell disruption, and centrifuged at 1,000 x g for 10 min at 4°C. The supernatant fraction was centrifuged at 6,000 x g for 10 min at 4°C to pellet mitochondria. The mitochondria were washed with Mitochondria Storage Buffer and centrifuged at 6,000 x g for 20 min at 4°C. The pellet fraction was resuspended in an appropriate amount of Mitochondria Storage Buffer or lysis buffer and the concentration was determined with a BCA protein assay kit (Pierce, 23227). Five to 10 µg of cytosolic or mitochondrial protein extracts were used for immunoblotting.
Measurement of NO production
Production of NO was measured in cell supernatants of BMDMs using a kit following the instruction of the kit (R&D Systems, KGE001). Briefly, cell supernatants were filtered using 3,000 molecular weight cut-off filters (Merck Millipore, UFC800324); 50 µl of the filtered cell supernatants were diluted with 50 µl of reaction diluent and mixed with 25 µl each of nitrate reductase and NADH and then incubated at 37°C for 30 min to convert nitrate to nitrite. After incubation, total nitrite was measured at 540 nm with Griess reagents (R&D systems, 892878 and 892879).
Animal experiments
CLP was performed as described53 with minor modifications. Briefly, the mouse cecum was exposed through a 1.5-cm incision and the cecum was ligated with 4-0 silk without causing bowel obstruction and then perforated with a 22-gauge needle (Korea Vaccine, ND.SY1030-010) (1 hole injury). A small amount of stool was extruded to ensure wound patency. Sham-operated mice underwent the same procedure without ligation and puncture of the cecum (n = 5 per group for harvesting serum and tissue and Sesn2+/+: n = 11; sesn2−/−: n = 12 for survival rates). For the LPS endotoxemia model, mice were intraperitoneally injected with 12 mg/kg LPS or its solvent (PBS) before serum cytokines and biochemical indicators of organ function were measured (n = 5 per group for harvesting serum and Sesn2+/+: n = 13; sesn2−/−: n = 13 for survival rates). Blood samples were collected from mice via cardiac puncture. Aliquots of serum were stored at −80°C. Buffy coats containing PBMCs were separated by Ficoll (GE Healthcare Life Sciences, 17-1440-02) density gradient centrifugation.54 Serum creatinine (BioAssaySystems, DICT-500), GOT1/AST (glutamic-oxaloacetic transaminase 1, soluble) (BioAssaySystems, EASTR-100) and GPT/ALT (glutamic pyruvic transaminase, soluble) (BioAssaySystems, EALT-100) were measured using a kit. The concentrations were determined spectrophotometrically according to the manufacturer's instructions. For endotoxic shock experiments, 25 mg/kg LPS was injected intraperitoneally and survival rates were monitored for 1 wk. For ‘add-back’ rescue, adenoviruses (Ad) encoding human SESN2 (Ad-SESN2) and Ad Control (Ad-Cont) were obtained from Genenmed. Adenoviruses (5 × 109 viral particles) were injected intravenously 48 h before LPS challenge or CLP surgery (n = 5 per group for harvesting serum and n = 13 per group for survival rates).
Human study
Buffy coats containing PBMCs were separated from blood by Ficoll density gradient centrifugation. Aliquots of plasma were stored at −80°C and monocytes were isolated from buffy coats and enriched by depletion using a Monocyte Isolation Kit II (Miltenyi Biotec, 130-091-153).
Immunofluorescence staining and confocal microscopy
Cells seeded on glass coverslips were primed with LPS followed by ATP treatment and then fixed and permeabilized with methanol for 10 min at room temperature. After 3 PBS washes, cells were blocked with 1% BSA (Sigma-Aldrich, A7030) in antibody diluent (Dako, S0809) for 1 h at room temperature. They were then incubated overnight with primary antibodies in antibody diluent at 4°C. After 3 PBS washes, cells were incubated with secondary antibodies for 30 min at room temperature. Finally, cells were washed with PBS 3 times and stained with 4', 6'-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, D9542) for 2 min at room temperature. Cells were then washed with PBS, mounted onto slides with mounting medium and observed on an LSM700 confocal microscope (Carl Zeiss, Jena, Germany) at 800x magnification. The following primary antibodies were used: SESN2 (1:500; abclon by custom antibody service), SQSTM1 (1:500; Abnova, H00008878-M01), TOMM20 (1:500; Santa Cruz Biotechnology, sc-11415), Lys63-ubiquitin (1:500; Merck Millipore, 05-1308), and Lys48-ubiquitin (1:500; Merck Millipore, 05-1307). Secondary antibodies used are from Invitrogen: Alexa Fluor 488 donkey anti-mouse IgG (A21202), Alexa Fluor 488 donkey anti-rabbit IgG (A21206), Alexa Fluor 568 donkey anti-mouse IgG (A10037) and Alexa Fluor 568 donkey anti-rabbit IgG (A10042). To perform the proximity ligation assay (PLA), Duolink II PLA probes (Olink Bioscience, DUO92001 and DUO92005) and Detection Reagents (Olink Bioscience, DUO92014) were used following the manufacturer's instructions. The nuclei were counterstained with DAPI and PLA signals were visualized on a confocal microscope at 800x magnification. Cells with punctate GFP-MAP1LC3 were counted manually following the method described previously.55 mt-mKeima was used as a sensitive and quantitative assessment of mitophagy as described.56 BMDMs were infected with mt-mKeima retroviral particles for 2 d just before use, primed with LPS for 12 h followed by ATP treatment, fixed with 4% paraformaldehyde for 15 min at room temperature, and analyzed by confocal microscopy using 2 excitation filters (438 nm and 550 nm) and a 610LP emission filter at 800 x magnification. Ratio (550ex:438ex) images of mt-mKeima were created and analyzed using MetaMorph software. High (550ex:438ex) signal areas and mitochondrial areas were calculated, and the ratio (high [550ex:438ex] signal area:mitochondrial area) was used as an index of mitophagic activity.
Retroviral overexpression of GFP-MAP1LC3, human ULK1 and mKeima
GFP-MAP1LC3 (Addgene, 21073) and HA-tagged human ULK1 (Addgene, 31963) were originally deposited by Tamotsu Yoshimori (The National Institute of Genetics, Japan), and Dr. Do-Hyung Kim (University of Minnesota, USA), respectively. mt-mKeima plasmids were kindly provided by H. Katayama and A. Miyawaki (RIKEN, Tokyo, Japan). The GFP-MAP1LC3, ULK1 and mt-mKeima insert were cloned into the LZR retroviral vector and retroviral particles were produced in HEK293 GPG packaging cells. The LZR retroviral vector and HEK293 GPG packaging cells were kindly gifted by Dr. Jaesang Kim (Ewha Womans University, Korea). Target cells were infected with retroviral particles 2 d before use.
Study approval
All animal experiments and protocols were approved by the Yonsei University College of Medicine Institutional Animal Care and Use Committee. Human blood samples from healthy volunteers and septic shock patients were obtained in Severance Hospital (Seoul, Korea). This study was approved by the Institutional Review Board of Severance Hospital, Yonsei University College of Medicine (4-2013-0585). Informed consent was obtained directly from each subject and documented in writing before the start of study-related procedures.
Statistical analysis
All results are presented as mean ± s.e.m. An unpaired Student t test was performed for the comparison of 2 samples, and ANOVA followed by Tukey's post hoc test was performed for the comparison of multiple samples. The Kaplan-Meier log-rank test was used for the statistical analysis of survival experiments. p values < 0.05 were considered statistically significant. In each figure legend, s.e.m. means between-subjects standard error of the mean. IBM, SPSS software was used for all.
Supplementary Material
Abbreviations
- Ad-SESN2
adenoviral vector containing the human SESN2 gene
- AMPK
AMP-activated protein kinase
- ATG
autophagy related
- BMDMs
bone marrow-derived macrophages
- CASP
caspase
- CLP
cecal ligation puncture
- co-IP
co-immunoprecipitation
- GFP-MAP1LC3
green fluorescent protein-microtubule-associated protein 1 light chain 3
- IL1B
interleukin 1 β
- IL18
interleukin 18
- KEAP1
kelch-like ECH-associated protein 1
- L-NAME
NG-nitro-L-arginine methyl ester
- LPS
lipopolysaccharide
- Lys63
lysine 63
- Mito-TEMPO
(2-[2, 2, 6, 6-tetramethylpeperidin-1-oxyl-4-ylamino]-2-oxoethyl)triphenylphosphonium chloride
- mt-mKeima
mitochondria-targeted monomeric Keima
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NLRP3
NLR family, pyrin domain containing 3
- NO
nitric oxide
- NOS2
nitric oxide synthase 2, inducible
- PLA
proximity-ligation assay
- ROS
reactive oxygen species
- SESN2
sestrin 2
- SQSTM1
sequestosome 1
- TNF
tumor necrosis factor
- ULK1
unc-51 like kinase 1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Yonsei-Carl Zeiss Advanced Imaging Center, Yonsei University College of Medicine, for technical assistance. We also thank D.S. Jang for his excellent support with medical illustration.
Funding
This work was supported by the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (2014R1A2A1A01003385). This research was supported by the Basic Science Research Program through the NRF funded by the Ministry of Education (2013R1A1A2008511), by a NRF grant funded by the Korean government (MSIP) (No. 2007-0056092), and a NRF grant funded by the Ministry of Science, ICT & Future Planning (2012M3A9C5048709), (2013M3A9D5072551), (2013M3A9D5072550).
References
- [1].Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12(1):9-14; PMID:21179058; http://dx.doi.org/ 10.1038/nrm3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454(7201):232-5; PMID:18454133; http://dx.doi.org/ 10.1038/nature07006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Geisler S, Holmstrom KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010; 6(7):871-8; PMID:20798600; http://dx.doi.org/ 10.4161/auto.6.7.13286 [DOI] [PubMed] [Google Scholar]
- [4].Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 2009; 16(7):939-46; PMID:19229244; http://dx.doi.org/ 10.1038/cdd.2009.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 2012; 393(7):547-64; PMID:22944659; http://dx.doi.org/ 10.1515/hsz-2012-0119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12(2):119-31; PMID:20098416; http://dx.doi.org/ 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- [7].Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183(5):795-803; PMID:19029340; http://dx.doi.org/ 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jin S. Autophagy, mitochondrial quality control, and oncogenesis. Autophagy 2006; 2(2):80-4; PMID:16874075; http://dx.doi.org/ 10.4161/auto.2.2.2460 [DOI] [PubMed] [Google Scholar]
- [9].Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al.. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12(3):222-30; PMID:21151103; http://dx.doi.org/ 10.1038/ni.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, et al.. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 2013; 14(5):480-8; PMID:23525089; http://dx.doi.org/ 10.1038/ni.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469(7329):221-5; PMID:21124315; http://dx.doi.org/ 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
- [12].Zhang Q, Kuang H, Chen C, Yan J, Do-Umehara HC, Liu XY, Dada L, Ridge KM, Chandel NS, Liu J. The kinase Jnk2 promotes stress-induced mitophagy by targeting the small mitochondrial form of the tumor suppressor ARF for degradation. Nat Immunol 2015; 16(5):458-66; PMID:25799126; http://dx.doi.org/ 10.1038/ni.3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang Z, Xu X, Ma J, Wu J, Wang Y, Zhou R, Han J. Gene deletion of Gabarap enhances Nlrp3 inflammasome-dependent inflammatory responses. J Immunol 2013; 190(7):3517-24; PMID:23427251; http://dx.doi.org/ 10.4049/jimmunol.1202628 [DOI] [PubMed] [Google Scholar]
- [14].Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 2010; 191(5):933-42; PMID:21115803; http://dx.doi.org/ 10.1083/jcb.201008084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A 2008; 105(33):12022-7; PMID:18687899; http://dx.doi.org/ 10.1073/pnas.0802814105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hollville E, Carroll RG, Cullen SP, Martin SJ. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol Cell 2014; 55(3):451-66; PMID:24999239; http://dx.doi.org/ 10.1016/j.molcel.2014.06.001 [DOI] [PubMed] [Google Scholar]
- [17].Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137(6):1001-4; PMID:19524504; http://dx.doi.org/ 10.1016/j.cell.2009.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, et al.. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010; 15(8):887-900; PMID:20604804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 2010; 285(36):27879-90; PMID:20573959; http://dx.doi.org/ 10.1074/jbc.M110.119537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, et al.. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A 2007; 104(49):19500-5; PMID:18048346; http://dx.doi.org/ 10.1073/pnas.0708818104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008; 112(4):1493-502; PMID:18539900; http://dx.doi.org/ 10.1182/blood-2008-02-137398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004; 304(5670):596-600; PMID:15105503; http://dx.doi.org/ 10.1126/science.1095569 [DOI] [PubMed] [Google Scholar]
- [23].Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, et al.. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010; 327(5970):1223-8; PMID:20203043; http://dx.doi.org/ 10.1126/science.1182228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008; 134(3):451-60; PMID:18692468; http://dx.doi.org/ 10.1016/j.cell.2008.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, et al.. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab 2012; 16(3):311-21; PMID:22958918; http://dx.doi.org/ 10.1016/j.cmet.2012.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen CC, Jeon SM, Bhaskar PT, Nogueira V, Sundararajan D, Tonic I, Park Y, Hay N. FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev Cell 2010; 18(4):592-604; PMID:20412774; http://dx.doi.org/ 10.1016/j.devcel.2010.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sanli T, Linher-Melville K, Tsakiridis T, Singh G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One 2012; 7(2):e32035; PMID:22363791; http://dx.doi.org/ 10.1371/journal.pone.0032035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 2009; 8(10):1571-6; PMID:19377293; http://dx.doi.org/ 10.4161/cc.8.10.8498 [DOI] [PubMed] [Google Scholar]
- [29].Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, Lee HE, Kang D, Rhee SG. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab 2013; 17(1):73-84; PMID:23274085; http://dx.doi.org/ 10.1016/j.cmet.2012.12.002 [DOI] [PubMed] [Google Scholar]
- [30].Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al.. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 1995; 80(3):401-11; PMID:7859282; http://dx.doi.org/ 10.1016/0092-8674(95)90490-5 [DOI] [PubMed] [Google Scholar]
- [31].Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW, et al.. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 2006; 24(3):317-27; PMID:16546100; http://dx.doi.org/ 10.1016/j.immuni.2006.02.004 [DOI] [PubMed] [Google Scholar]
- [32].Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013; 153(2):348-61; PMID:23582325; http://dx.doi.org/ 10.1016/j.cell.2013.02.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 2013; 14(5):454-60; PMID:23502856; http://dx.doi.org/ 10.1038/ni.2550 [DOI] [PubMed] [Google Scholar]
- [34].Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I, Akira S, Mitsuyama M. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol 2012; 189(11):5113-7; PMID:23100513; http://dx.doi.org/ 10.4049/jimmunol.1202479 [DOI] [PubMed] [Google Scholar]
- [35].Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, He Z, Zeng Y, Hu Y, Sun S, et al.. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res 2013; 23(2):201-12; PMID:23318584; http://dx.doi.org/ 10.1038/cr.2013.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Springer W, Kahle PJ. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 2011; 7(3):266-78; PMID:21187721; http://dx.doi.org/ 10.4161/auto.7.3.14348 [DOI] [PubMed] [Google Scholar]
- [37].Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010; 6(8):1090-106; PMID:20890124; http://dx.doi.org/ 10.4161/auto.6.8.13426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ro SH, Semple IA, Park H, Park H, Park HW, Kim M, Kim JS, Lee JH. Sestrin2 Promotes Unc-51-like Kinase 1 (ULK1)-Mediated Phosphorylation of p62/sequestosome-1. FEBS J 2014; 281(17):3816-27; PMID:25040165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al.. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 2013; 51(5):618-31; PMID:24011591; http://dx.doi.org/ 10.1016/j.molcel.2013.08.003 [DOI] [PubMed] [Google Scholar]
- [40].Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011; 44(2):279-89; PMID:22017874; http://dx.doi.org/ 10.1016/j.molcel.2011.07.039 [DOI] [PubMed] [Google Scholar]
- [41].Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 2011; 18(8):1042-52; PMID:21867919; http://dx.doi.org/ 10.1016/j.chembiol.2011.05.013 [DOI] [PubMed] [Google Scholar]
- [42].Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32(1):2-11; PMID:22025673; http://dx.doi.org/ 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shang L, Wang X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 2011; 7(8):924-6; PMID:21521945; http://dx.doi.org/ 10.4161/auto.7.8.15860 [DOI] [PubMed] [Google Scholar]
- [44].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13(2):132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Woo HA, Bae SH, Park S, Rhee SG. Sestrin 2 is not a reductase for cysteine sulfinic acid of peroxiredoxins. Antioxid Redox Signal 2009; 11(4):739-45; PMID:19113821; http://dx.doi.org/ 10.1089/ars.2008.2360 [DOI] [PubMed] [Google Scholar]
- [46].Rhee SG, Bae SH. The antioxidant function of sestrins is mediated by promotion of autophagic degradation of Keap1 and Nrf2 activation and by inhibition of mTORC1. Free Radic Biol Med 2015; 88(Pt B):205-11 [DOI] [PubMed] [Google Scholar]
- [47].Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 1993; 333(1-2):169-74; PMID:8224160; http://dx.doi.org/ 10.1016/0014-5793(93)80398-E [DOI] [PubMed] [Google Scholar]
- [48].Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, Zhang J, Iyengar R, Jung CH, Suen DF, et al.. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell 2011; 43(4):572-85; PMID:21855797; http://dx.doi.org/ 10.1016/j.molcel.2011.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Olson N, Hristova M, Heintz NH, Lounsbury KM, van der Vliet A. Activation of hypoxia-inducible factor-1 protects airway epithelium against oxidant-induced barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 2011; 301(6):L993-L1002; PMID:21926263; http://dx.doi.org/ 10.1152/ajplung.00250.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Horino T, Fujieda M, et al.. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol 2013; 305(4):F495-509; PMID:23698117; http://dx.doi.org/ 10.1152/ajprenal.00642.2012 [DOI] [PubMed] [Google Scholar]
- [51].Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, et al.. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 2008; 3(10):e3316; PMID:18830406; http://dx.doi.org/ 10.1371/journal.pone.0003316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gross O. Measuring the inflammasome. Methods Mol Biol 2012; 844:199-222; PMID:22262445; http://dx.doi.org/ 10.1007/978-1-61779-527-5_15 [DOI] [PubMed] [Google Scholar]
- [53].Chung SW, Liu X, Macias AA, Baron RM, Perrella MA. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J Clin Invest 2008; 118(1):239-47; PMID:18060048; http://dx.doi.org/ 10.1172/JCI32730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mendez-David I, El-Ali Z, Hen R, Falissard B, Corruble E, Gardier AM, Kerdine-Romer S, David DJ. A method for biomarker measurements in peripheral blood mononuclear cells isolated from anxious and depressed mice: beta-arrestin 1 protein levels in depression and treatment. Front Pharmacol 2013; 4:124; PMID:24133448; http://dx.doi.org/ 10.3389/fphar.2013.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140(3):313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, et al.. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 2014; 124(9):3987-4003; PMID:25083992; http://dx.doi.org/ 10.1172/JCI74985 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.