

Abstract
ONC201 (also called TIC10) is a small molecule that inactivates the cell proliferation- and cell survival-promoting kinases AKT and ERK and induces cell death through the pro-apoptotic protein TRAIL. ONC201 is currently in early phase clinical testing for various malignancies. Here, we found through gene expression and protein analyses that ONC201 triggered an increase in TRAIL abundance and cell death through an integrated stress response (ISR) involving the transcription factor ATF4, the transactivator CHOP, and the TRAIL receptor DR5. ATF4 was not activated in ONC201-resistant cancer cells, and in ONC201-sensitive cells, knockdown of ATF4 or CHOP partially abrogated ONC201-induced cytotoxicity and diminished the ONC201-stimulated increase in DR5 abundance. The activation of ATF4 in response to ONC201 required the kinases HRI and PKR, which phosphorylate and activate the translation initiation factor eIF2α. ONC201 rapidly triggered cell cycle arrest, which was associated with decreased abundance of cyclin D1, decreased activity of the kinase complex mTORC1, and dephosphorylation of the retinoblastoma (Rb) protein. The abundance of X-linked inhibitor of apoptosis protein (XIAP) negatively correlated with the extent of apoptosis in response to ONC201. These effects of ONC201 were independent of whether cancer cells had normal or mutant p53. Thus, ONC201 induces cell death through the coordinated induction of TRAIL by an ISR pathway.
Introduction
The discovery of the pro-apoptotic protein tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and its receptors in the mid-to-late 1990’s generated much excitement because of TRAIL’s targeted apoptotic activity in cancer cells but not normal tissue (1). Unfortunately, the anti-tumor efficacy of recombinant TRAIL or TRAIL receptor antibodies [against TRAIL receptors 1 or 2 (TRAIL-R1 or TRAIL-R2), also known as death receptors 4 or 5, respectively (DR4 or DR5)] have not yet been convincingly demonstrated in clinical trials (2). While the underlying mechanism for poor clinical efficacy of pro-apoptotic TRAIL or TRAIL receptor agonist antibodies is unclear, investigators have several theories, including increased abundance of anti-apoptotic proteins such as Fas-associated death domain-like interleukin-1beta-converting enzyme (FLICE) inhibitory protein (FLIP), myeloid cell leukemia-1 (Mcl-1), inhibitor of apoptosis protein-2 (IAP-2) or TRAIL decoy receptors, or decreased abundance of target pro-apoptotic proteins DR4 or DR5 (2, 3), or insufficient receptor clustering and activation (4). Nevertheless, much effort has been put forth in identifying therapeutics that can be combined with TRAIL with the intent of downregulating these inhibitory proteins or upregulating the TRAIL receptors (2). On the other hand, DR5 also possesses ligand-independent pro-apoptotic capability (5, 6) that can be exploited therapeutically (7).
We previously identified a small molecule, currently in early phase clinical trials, called ONC201 (also known as TIC10) that not only induces TRAIL production but also increases DR5 abundance (8). Both TRAIL abundance and the TRAIL-DR5 interaction are required for ONC201 pro-apoptotic activity. Although ample evidence has been presented that TRAIL induction results from ONC201-induced activation of the transcription factor Forkhead box O3a (FOXO3a), the mechanism of DR5 upregulation in response to ONC201 was not directly evaluated nor were the early events that facilitate the anti-cancer effects of ONC201. To address these questions, we analyzed the gene expression profile of cancer cells that were treated with ONC201 at time points that precede the dual inactivation of protein kinase B (also known as Akt) and extracellular signaling-regulated kinase (ERK) and the upregulation of TRAIL abundance. Herein we report the early engagement of stress response pathway mediated by the α-subunit of the eukaryotic initiation factor 2 (eIF2α) and activating transcription factor 4 (ATF4) in ONC201-treated cancer cells and detail how this pathway mediates ONC201-induced cyclin D1 downregulation, proliferation inhibition and apoptosis.
Results
The broad spectrum anti-cancer activity of ONC201 can be attributed to its anti-proliferative and pro-apoptotic effects
ONC201 is a novel anti-cancer drug that acts in part through TRAIL pathway activation. To further understand the efficacy and mechanism of action of ONC201, we treated 23 cancer cell lines, representing 9 tumor types, with ONC201, determined EC50 values (Fig. 1A, fig. S1A, and table S1) and assessed the effect of the respective EC50 dose on cell viability (fig. S1B). KMS18 and MM1S multiple myeloma cells were especially sensitive to ONC201, with an EC50 in the nanomolar range. Because ONC201 decreases the phosphorylation of Akt, ERK, and Foxo3a in various cell lines, we assessed the phosphorylation status of these proteins in a subset of the cell lines used in this study, including lymphoma and multiple myeloma cells. We confirmed the dual inhibition of Akt and ERK pathway in many cell lines. On the other hand, there several cell types in which Akt and ERK phosphorylation was not decreased under the conditions used in the study (fig. S1C). We found that ONC201 not only induced apoptosis in many cell lines as measured by sub-G1 fraction and caspase activation (Fig. 1B, and fig. S1, D to F) but also induced cell cycle arrest in the cell lines tested as early as 24 hours after ONC201 treatment (Fig. 1C), before apoptosis was observed (fig. S1, E and F). In addition, ONC201 impeded cell cycle progression regardless of whether cancer cells also underwent apoptosis in response to the drug or harbored a mutant p53 protein (Fig. 1C and fig. S1G). Bromodeoxyuridine (BrdU) labeling experiments confirmed that cell proliferation was inhibited by ONC201 (Fig. 1D). The early cell cycle arrest in response to ONC201 treatment caused a significant decrease in the number of viable cells within 48 hours (Fig. 1E), even in cells that did not undergo apoptosis (fig. S1, D to F). Moreover, ONC201 slowed down the increase in cell number even of cells that did not eventually succumb to apoptosis. This could explain at least in part how ONC201 exhibited broad spectrum and potent anti-cancer effects, as indicated by the near-complete absence of colony formation in the clonogenic assays (Fig. 1F).
Fig. 1. ONC201’s broad-spectrum activity can be attributed to its anti-proliferative and pro-apoptotic effects.

(A) EC50 of ONC201 was assessed by Cell Titer Glo assay in 23 cell lines treated with ONC201 for 72 hours. (B) Apoptosis measured by Sub-G1 analysis in cells treated with ONC201 for 72 hours. (C) Cell cycle profile analyses in HCT 116 and A549 cells treated with ONC201 for 24 hours. Data is representative of two biological replicates. (D) BrdU incorporation assays in HCT116 and A549 cells treated with ONC201 for 48 hours. (E) Viable cell counts assessed by trypan blue exclusion in cells treated with ONC201 for up to 72 hours. *p < 0.05, Student’s t-test with Holm-Sidak correction. (F) Proliferative capacity of cells after 72 hours in the presence of ONC201, assessed by clonogenic assays. Bars that are not visible indicate a proliferative fraction of 0. Concentrations of ONC201 used for each cell line are listed in table S1. Data in (A), (B), (D), (E) and (F) are means ± SEM from three biological replicates.
The anti-cancer effects of ONC201 are dependent on its engagement of the ATF4 pathway
The observation that ONC201 exerted anti-cancer effects, even without eventually inducing apoptosis, suggested that ONC201 perturbs pathways in addition to those previously reported (8). To study the possible pathways regulated by ONC201, gene expression profiles were assessed in HCT116 colon cancer cells treated with ONC201 for 18 and 48 hours, and in RKO colon cancer cells treated for 48 hours (table S4). Pathway analyses of microarray results suggested that ONC201 engaged the activating transcription factor 4 (ATF4) pathway: Many of the genes that showed increased expression with ONC201 are regulated by ATF4 (9) and possess binding sites for ATF4 and the protein encoded by the ATF4 transcriptional target gene CCAAT/Enhancer Binding Protein Homology Protein (CHOP) (10) (table S2). Western blotting analyses results confirmed that ONC201 treatment increased the abundance of ATF4 in a dose-and time-dependent manner (Fig. 2A) and in different cell types (fig. S2).
Fig. 2. ONC201 activates the integrated stress response.

(A)Western blotting analysis for ATF4, CHOP, ATF3, and TRB3 on lysates from HCT116 cells cultured with ONC201 (0–10 μM) for 3 to 24 hours. Brefeldin A (BFA, an ER stressor) served as a positive control. Blots are representative of 2 experiments. (B) qRT-PCR for the expression of CHOP and DR5 relative to GAPDH in HCT116 cells treated with ONC201 (O; 10 μM) or vehicle (V) for 24 or 48 hours. (C) Immunohistochemical analysis for CHOP in HCT116-derived subcutaneous xenografts from mice that received either vehicle (−) or ONC201 (+; 25 mg/kg). Results are representative of tissues from two vehicle-treated mice and six mice intraperitoneally or intravenously injected with ONC201 (3 mice each). Scale bar = 10 μm (D) Western blotting analysis for CHOP in lysates from colorectal cancer stem cell-like cell-derived xenografts (45) in athymic nude mice extracted 72 hours after treatment [vehicle (V) or ONC201 (O) (50 mg/kg, i.p.)]. Tumor lysates from untreated mice (−) served as controls. (E) Densitometric analyses of the bands in the Western blots in (D). (F) qRT-PCR for the fold change in expression of CHOP and DR5 relative to GAPDH in different cell types cultured with ONC201 (compared to vehicle) for 72 hours. Data in (B), (E) and (F) are means ± SEM of three biological replicates. *p < 0.05, Student’s t-test with Holm-Sidak correction, comparing ONC201-treated versus vehicle-treated cells.
ATF4 promoted apoptosis, in part, by regulating the expression of two genes encoding the pro-apoptotic proteins CHOP (11) and DR5. CHOP and DR5 expression increased in response to exposure to ONC201 in cultured HCT116 cells (Fig. 2B). We have previously reported that DR5 protein abundance is increased in tumor xenograft tissue from mice treated with ONC201 (8). In this study, we performed immunohistochemical and Western blot analyses on colorectal cancer cell line-derived subcutaneous xenograft tumors from ONC201-treated mice, and we confirmed that ONC201 increased CHOP protein in vivo (Fig. 2, C to E). Similar to what was observed with ATF4, the increase in CHOP and DR5 abundance was also cell type-independent. Furthermore, CHOP and DR5 expression was increased even in cell types that did not undergo ONC201-induced apoptosis despite exhibiting decreased cell proliferation (Fig. 2F). ATF4 has been suggested to induce cell death by promoting persistent protein synthesis, even when proteostasis has not been restored (10). ATF4 accomplishes this, at least in part, by increasing the expression of genes encoding protein synthesis factors, particularly amino acyl transfer RNA (tRNA) synthetases. Our microarray analyses indicated that ONC201 induced the expression of genes encoding amino acyl tRNA synthetases (Table 1).
Table 1. ONC201 induces the expression of mRNAs encoding amino acyl tRNA synthetases.
Fold changes in expression of mRNAs encoding aminoacyl tRNA synthetases in HCT116 and RKO cells in response to 18 and 48 hours treatment of ONC201, as determined by microarray analyses (table S4).
| Aminoacyl tRNA synthetase | Fold change vs untreated |
|---|---|
| 18 hours ONC201 incubation | |
| HCT116 | |
| Cysteinyl-tRNA synthetase (CARS) | 3.4 |
| CARS | 2.5 |
| Seryl-tRNA synthetase (SARS) | 2.4 |
| Alanyl-tRNA synthetase (AARS) | 2.2 |
| Glycyl-tRNA synthetase (GARS) | 2.2 |
| Tryptophanyl-tRNA synthetase (WARS) | 2 |
| Tyrosyl-tRNA synthetase (YARS) | 2 |
| Leucyl-tRNA synthetase (LARS) | 1.8 |
| Isoleucyl-tRNA synthetase (IARS) | 1.7 |
| 48 hours ONC201 incubation | |
| HCT116 | |
| GARS | 2.2 |
| RKO | |
| SARS | 1.9 |
| WARS | 1.8 |
| WARS | 1.6 |
We examined more closely whether ATF4 activation is critical for ONC201’s anti-cancer effects. Cancer cells that acquired resistance to ONC201 (fig. S3, A and B) did not engage the ATF4 pathway when treated with ONC201 (Fig. 3, A and B, and table S3). Because ATF4 can be activated by pharmacological inducers of endoplasmic reticulum (ER) stress (12), we treated ONC201-resistant cells with ER stress inducers to verify whether the ATF4 pathway had become dysfunctional in these cells. Treatment of ONC201-resistant cells with pharmacological ER stressors brefeldin A (BFA) or bortezomib resulted in increased ATF4 and CHOP abundance (Fig. 3A and fig. S3C), indicating that the ATF pathway was intact.
Fig. 3. ATF4 is required for ONC201-induced cytotoxicity.

(A) Western blotting analysis for the indicated proteins in wild-type RKO (RKOp) and ONC201-resistant RKO (RKOr1) cells treated with 10 μM brefeldin A (an ER stressor, positive control) or 5 μM ONC201. (B) qRT-PCR for the fold-change in expression of CHOP relative to GAPDH in RKOp and RKOr1 cells treated with ONC201 (5 μM, 24–48 hours) compared with the 0 time point (H, hours). (C) Viability of HCT116 cells assessed by trypan blue exclusion assay after ATF4 and CHOP knockdown and treatment with vehicle (V) or ONC201 (O) (10μM, 24 and 48 hours). (D) Western blotting analysis for ATF4, CHOP, and uncleaved (unclvd) and cleaved (clvd) PARP in HCT116 cells transfected with ATF4 or CHOP siRNA. (E) qPCR of DR5 expression relative to GAPDH in HCT116 cells transfected with ATF4 or CHOP siRNA and subsequently treated with vehicle (V) or ONC201 (O) (10 μM, 48 hours). (F) Western blotting for the indicated proteins in HCT116 cells transfected with ATF4 or TRB3 siRNA and subsequently treated with ONC201 (10 μM, 72 hours upper set, 48 hours lower set). Blots in (A), (D), and (F) are representative of 2 experiments. Data in (B), (C) and (E) are means ± SEM of 3 biological replicates. *p < 0.05, Student’s t-test with Holm-Sidak correction.
Knocking down ATF4 or CHOP impeded the ability of ONC201 to reduce cell viability (Fig. 3C). However, unlike the impact of ATF4 knockdown on the efficacy of ONC201, CHOP reduction alone did not diminish the effect of ONC201 in inducing Poly Adenosine diphosphate-ribose polymerase (PARP) cleavage (Fig. 3D). Because ATF4 and CHOP are transcriptional activators of DR5, knockdown of ATF4 and CHOP was performed and DR5 expression was assessed. The reduction in ATF4 or CHOP amounts was sufficient to ablate ONC201-induced DR5 (Fig. 3E).
ATF4 may also play a role in the ONC201-induced inactivation of AKT, through its transcriptional target, tribbles-like protein 3 (TRB3) (13). TRB3 inhibits the kinase activity of AKT (14) and regulate mitogen-activated protein kinase (MAPK) family members (15). Knocking down TRB3 by siRNA abrogated ONC201-induced Akt inactivation (Fig. 3F). TRB3, therefore, could provide a link between early engagement of the ISR and late suppression of the Akt pathway in ONC201-treated cells.
The ATF4 pathway is engaged as part of the ISR to enable cells to survive conditions that threaten proteostasis, such as nutrient starvation and accumulation of misfolded proteins (a condition that causes ER stress) (12). ISR engagement acutely suspends protein synthesis to prevent further ER stress and the translation inhibition, in part, results in cell cycle arrest (16). We demonstrated that ONC201 inhibited cell cycle progression (Fig. 1C–D and figs. S1-E). The cell cycle delay caused by ONC201 correlated with a reduction in mammalian target of rapamycin complex 1 (mTORC1) activity and cyclin D1 abundance (Fig. 4 and fig. S4A) but not with a decrease in AKT1 and ERK phosphorylation (fig. S4B). The decrease in cyclin D1 abundance was associated with a partial reduction in the phosphorylation of Rb at Ser795 (Fig. 4 and fig. S4A).
Fig. 4. ONC201 induces a decrease in cyclin D1 abundance.
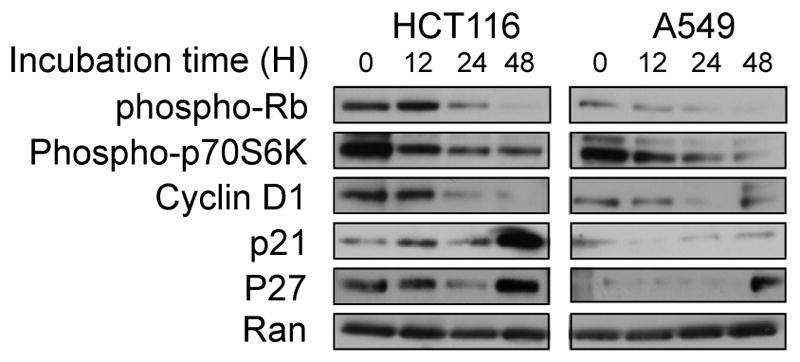
Western blotting analyses for the indicated proteins were performed in HCT116 or A549 cells treated with ONC201 (10 or 5 μM, respectively) for up to 48 hours. Blots are representative of two experiments.
Given the critical role that ATF4 plays in ONC201’s mechanism of cytotoxicity, we determined how ONC201 engages the ATF4 pathway. ATF4 protein abundance was generally increased after 12 hours of treatment with ONC201 (Figs. 2A and 3A, and fig. S2). An increase in ATF4 protein abundance can result from the combination of increased transcription and preferential translation, the latter of which occurs because of phosphorylation of the α subunit of the eukaryotic translation initiation factor 2 (eIF2α) (17). Indeed, ONC201 treatment induced eIF2α phosphorylation (Fig. 5A). Increased phosphorylation of eIF2α can result from the action of eIF2α kinases – general control non-derepressible-2 (GCN2), heme-regulated inhibitor (HRI), RNA-dependent protein kinase (PKR) and PKR-like endoplasmic reticulum-resident protein kinase (PERK), or from the inhibition of the eIF2α phosphatase, serine/threonine protein phosphatase 1 (PP1) (18). Phosphorylation of histone H3 (another PP1 substrate) and eIF2α was increased when cells were treated with the PP1 inhibitor calyculin A. In contrast, ONC201 induced only the phosphorylation of eIF2α, thus suggesting that ONC201 did not inhibit PP1 (fig. S5A). To identify the eIF2α kinase(s) involved in ONC201 action and to determine whether eIF2α phosphorylation was critical for the ONC201-induced increase of ATF4, we decreased the abundance of the different eIF2α kinases using siRNA. Although decreased GCN2 and PERK amounts did not abrogate the increase in ATF4, knockdown of HRI and PKR partially ablated ONC201-induced ATF4 abundance (Fig. 5B and fig. S5B). ONC201 still increased ATF4 abundance in GCN−/− and PERK−/− mouse embryonic fibroblasts (MEFs) (fig. S5C), thus providing further evidence that GCN2 and PERK may not mediate ONC201-induced eIF2 α phosphorylation, at least in the cell types used in this study. Double knockdown experiments confirmed the relevance of HRI and PKR to ATF4 activation in ONC201-treated cells: Decreased ATF4 abundance was seen only in PERK- or GCN2-deficient cells when either HRI or PKR was also knocked down (Fig. 5C and fig. S5D). Next, we treated cells with plasmid constructs encoding wild-type or non-phosphorylatable (S51A) mutant eIF2α and treated them with ONC201. ATF4 abundance was largely abrogated in cells expressing non-phosphorylatable mutant eIF2α (Fig. 5D). These results, therefore, demonstrated that ONC201 activated the integrated stress response by acting through HRI and PKR, to induce the phosphorylation of eIF2α and upregulate ATF4. ONC201, however, did not appear to increase PKR kinase activity, as indicated by an absence of a substantial increase in PKR autophosphorylation (fig. S5E). Phosphorylation of eIF2α caused cell cycle arrest, in part by downregulating cyclin D (19, 20). Given that ONC201 downregulated cyclin D1 (Fig. 4 and fig. S4), we determined whether HRI and PKR were involved in the ONC201-induced cyclin D1 decrease. The ONC201-induced decrease in cyclin D1 abundance was partially abrogated when PKR was knocked down, either alone or in conjunction with that of HRI (Fig. 5E). These results suggest that PKR likely plays a role in ONC201-induced decrease of cyclin D1.
Fig. 5. ONC201 activates the ATF4 pathway through the eIF2α kinases HRI and PKR.

(A) Western blotting analysis for phosphorylated and total eIF2α was performed in the indicated cell types treated with ONC201. (B to D) Western blotting analysis for the indicated proteins in lysates from HCT116 cells that were transfected with: siRNA against one (B) or two eIF2α kinases (C) as indicated for 24 hours, and subsequently treated with ONC201 (10 μM) for 12 hours. (D) Western blotting analysis for indicated proteins in lysates from HCT116 cells that were transfected with plasmid constructs for wild-type (wt) and a nonphosphorylatable eIF2α mutant (S51A) for 48 hours, and subsequently treated with ONC201 for 12 hours. (E) Western blotting analysis for cyclin D1 in HCT116 cells transfected with the indicated siRNA(s) and treated with ONC201 for 24 hours. Blots in (A) to (E) are representative of at least two independent experiments.
XIAP abundance correlates with sensitivity to ONC201-induced apoptosis
The ability of ONC201 to not only induce apoptosis but also inhibit cell proliferation may underlie the broad spectrum anti-cancer activity of ONC201. The amount of X-linked inhibitor of apoptosis (XIAP) can prevent apoptosis in response to TRAIL induction (21, 22). Here, XIAP abundance appeared to correlate with the apoptotic response of cancer cells to ONC201. In cells that undergo ONC201-induced apoptosis, XIAP amount was reduced after treatment with ONC201. In contrast, XIAP abundance persisted – although it decreased somewhat – in cells that did not undergo apoptosis (Fig. 6). By contrast, the abundance of other inhibitors of apoptosis proteins (IAPs) and B cell lymphoma 2 (BCL-2) family members was decreased by ONC201 in different cell types regardless of whether or not they underwent ONC201-induced apoptosis (fig. S6). Thus, ONC201-induced suppression of cellular inhibitor of apoptosis 2 (cIAP2) and anti-apoptotic BCL-2 family members did not appear to be sufficient to push cells to undergo cell death in response to ONC201 treatment. By contrast, the extent of decrease in XIAP amounts correlated with the degree of apoptosis in response to ONC201. To determine whether altering the amount of XIAP in cells changed their susceptibility to ONC201-induced apoptosis, we overexpressed XIAP in HCT116 cells (a cell type that underwent apoptosis, Fig. 1B) and knocked down XIAP in A549 cells (a cell type that did not undergo apoptosis, Fig. 1B) (Fig. S7). The extent of ONC201-induced PARP cleavage was not affected by changing XIAP abundance, indicating that XIAP by itself did not determine cell fate in response to ONC201 treatment.
Fig. 6. The effect of ONC201 on XIAP amount correlates with fate of cells treated with ONC201.
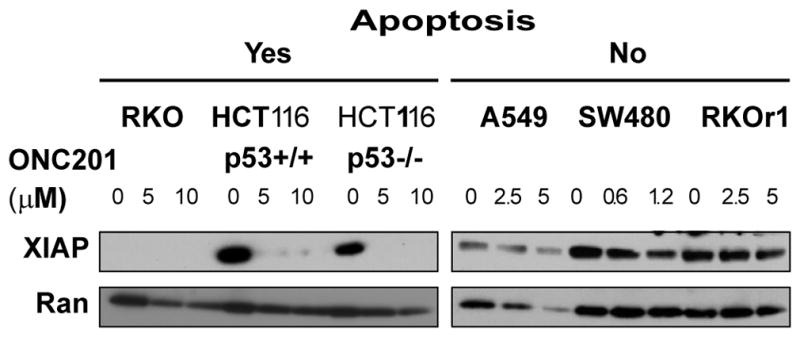
Western blotting analysis for XIAP in cells treated with the indicated concentrations of ONC201 for 72 hours. Blots are representative of at least two independent experiments.
Discussion
We uncovered a molecular mechanism that was involved in the anti-cancer effects of ONC201. ONC201 elicited the integrated stress response (ISR) through the eIF2α kinases HRI and PKR, resulting in activation of the eIF2α-ATF4 pathway. PKR played a role in ONC201-induced decrease in cyclin D1 abundance and increased ATF4 abundance was required for ONC201-induced increases in DR5 abundance. Thus, we implicated the eIF2α-ATF4 pathway in the ability of ONC201 to not only induce apoptosis but also impede cell cycle progression. We confirmed the broad spectrum anti-cancer activity of ONC201 and characterized the early molecular events that are critical for its efficacy.
ONC201 activated the ATF4 pathway within the first 24 hours of treatment, preceding changes in AKT, ERK and FOXO3a phosphorylation and caspase-3 activity. The ATF4 increase in ONC201-treated solid tumor cells appeared to be involved in the anti-cancer effects of ONC201, namely a decrease in cell proliferation (19), and apoptosis that partly resulted from ATF4-mediated increases in DR5 and TRAIL abundance. Furthermore, ATF4 has been implicated in the anticancer effects of ONC201 in hematological malignancies as shown by Ishikawa et al. (23). Nevertheless, the impact of ATF4 abundance on cell viability is complex and context-dependent (10, 12). Increased ATF4 abundance can be cytoprotective, (24, 25), anti-proliferative or cytotoxic, in part due to ATF4’s potential to regulate more than 400 genes (10) and to interact with more than 17 proteins (26). It is not surprising, therefore, that the consequences of ATF4 induction by ONC201 are not identical in all cell types, as we and Ishikawa et al. (23) have shown. In HCT116 colorectal cancer cells and potentially in other solid tumor cell types, ATF4 may suppress the Akt pathway and induce apoptosis, in part through TRB3, TRAIL, and DR5. In hematologic malignancies, ATF4 and CHOP may cause cell death partly by promoting the synthesis of the pro-apoptotic Bcl-2 family members PUMA (27) and Bim (28), consequently activating the intrinsic apoptotic pathway.
Phosphorylation of eIF2α was critical for the ONC201-induced increase in ATF4 abundance, at least in HCT116 colorectal cancer cells, which we determined required the eIF2α kinases HRI and PKR. However, PKR, but not HRI, appeared critical for the ONC201-induced decrease in cyclin D1 abundance. Given the similarities in the kinase domains of the four eIF2α kinases (18), there are various contexts when at least two eIF2α kinases are involved in eIF2α phosphorylation. In fibroblasts, both PERK and GCN2 must be inhibited to counteract eIF2α phosphorylation that occurs during the unfolded protein response (UPR) (19). In another cell type, HT1080 cells, PERK and PKR are critical for the eIF2α-dependent decrease in cyclin D1 abundance in UPR (20). Their unique regulatory domains partially explain why some stresses activate specific eIF2α kinases. HRI is kept inactive by its interaction with heme and is engaged under conditions of heme depletion (29), but can also be activated by bortezomib (30, 31) and arsenite (32) through poorly understood mechanisms. Small molecule (33, 34) activators of HRI have have promising anti-cancer effects (33). PKR has two tandem motifs that bind double-stranded RNA (dsRNA) molecules (35), which are involved in antiviral responses (36). However, it is also critical in other stress conditions (37). The versatility of PKR can be partially explained by its many potential binding partners. For instance, it activates the nuclear factor κ-light-chain enhancer of activated B cells (NFκB) pathway by interacting with inhibitor of kappa B (IκB) kinase α (IKKα) (38). PKR associates with proteins involved in RNA processing, RNA binding, and protein translation and that the identity of the proteins in these complexes can be perturbed (such as in obesity) (39). Future studies are warranted to elucidate how ONC201 engages these two kinases. It is possible that the kinase that primarily phosphorylates eIF2α is HRI, and PKR facilitates this phosphorylation through its kinase-independent capabilities.
The relationship between eIF2α phosphorylation and the increase in ATF4 abundance in response to ONC201 treatment appeared to be cell type-specific. Whereas we found that eIF2α phosphorylation preceded the increase in ATF4 abundance in colorectal cancer cells, Ishizawa et al. (23) found that ATF4 abundance correlated with eIF2α phosphorylation in acute myeloid leukemia cell lines, but not in mantle cell lymphoma cell lines. These findings indicate that ONC201 induced ATF4 through eIF2α-dependent and –independent mechanisms. Other pharmacological inducers of the ISR, fenretidine (a derivative of retinoic acid) and bortezomib (40), also engage more than one mechanism to increase ATF4 abundance. Based on our observations on the role of two eIF2α kinases in the ONC201-mediated increase in ATF4 abundance and Ishikawa et al.’s results, especially from their upstream open reading frame (uORF) reporter assay, we propose that ONC201 may enhance ATF4 abundance primarily at the translational level. Although ATF4 mRNA translation is increased generally as a result of eIF2α phosphorylation, ATF4 abundance can be enhanced in the absence of eIF2α phosphorylation. Reductions in translation termination efficiency, such as in situations when eukaryotic release factor 3a (eRF3a) is depleted, results in increased translation of ATF4 in a manner that is independent of eIF2α phosphorylation (9). Oxidative stress inhibits global protein synthesis (and presumably increases ATF4 abundance) in a manner dependent on phosphorylated eIF2α by inhibiting ribosomal transit (41).
The engagement of the ISR by ONC201 suggests some therapeutic strategies. Cell types that secrete large amounts of protein, such as the immunoglobulin-producing myeloma cells, are susceptible to ER stress and are potentially more sensitive to ISR inducers (42, 43), including ONC201. This may explain at least in part the susceptibility of myeloma cells to ONC201, with EC50s in the nano-molar range. The increase in CHOP abundance that occurs in cancer cells that are sensitive to ONC201 but not in resistant cells suggests that CHOP could be used as a pharmacodynamic marker for ONC201 activity. Although DR5 abundance is also increased, it may be a less suitable marker because DR5 is more ubiquitous in nature (44), whereas CHOP protein is generally present absent under basal conditions. These results suggest that ONC201, which is currently in early phase clinical trials in advanced solid tumors, exerts its therapeutic effects by ISR activation, which may drive the downstream dual inactivation of the AKT and ERK pathways and promote TRAIL-induced apoptosis.
Materials and Methods
Cell culture
The human tumor cell lines were acquired from the American Type Culture Collection. HCT116 p53−/− cells were kind gifts from Dr. Bert Vogelstein of Johns Hopkins University. Wild-type, PERK−/− and GCN2−/− mouse embryonic fibroblasts, and myeloma cells were kindly shared by Dr. Scot Kimball, and Dr. Nathan Dolloff, respectively. The human lung, colorectal, thyroid, liver, and prostate cancer cell lines were cultured in RPMI or McCoy’s 5A medium. Human breast and glioma cell lines and MEFs were maintained in Dulbecco’s minimum essential medium. All basal media were supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution.
Viability and apoptosis assays
Viability was assayed using the CellTiter-Glo (CTG) assay according to the manufacturer’s instructions (Promega). 1 x 104cells were seeded in 96-well black plates and incubated for 24 hours prior to addition of vehicle or ONC201. To assay viability by a different method, trypan blue dye exclusion assays were performed. The cells were cultured in 24-well plates, treated with vehicle or ONC201, harvested and stained with trypan blue. Viable cells were counted using a Cellometer cell counter.
Sub-G1 analyses were performed to quantitate apoptosis. After treatment, floating and adherent cells were fixed in 95% ethanol and stained with propidium iodide in the presence of RNase A. Cell cycle profile and SubG1 analyses were performed using a Coulter-Beckman Elite Epics cytometer. To assess apoptosis by a different method, caspase-3 activity assays were performed using the Caspase-Glo® 3/7 assay reagent (Promega). Cells were cultured in black 96 well plates. Parallel CTG viability assay measurements were collected and used to normalize the luminescence values collected with the Caspase-Glo 3/7 assay.
Cell proliferation was assessed via BrdU proliferation assays. Cells were incubated with 10 μM BrdU for 30 minutes, trypsinized and fixed in 70% ethanol. After fixation, cells were washed and incubated with mouse anti-BrdU primary antibody (BD Biosciences) for 30 minutes and FITC-conjugated anti-rabbit secondary antibody for 30 minutes. Cells were stained with propidium iodide in the presence of RNAse A and analyzed using a Coulter-Beckman Elite Epics cytometer.
Long-term effects of ONC201 were assessed by performing clonogenic assays. A total of 500 or 2500 cells/well were plated in 6-well plates. After 24 h incubation, medium was replaced with that containing DMSO vehicle or ONC201. After 72 h treatment, fresh medium without agents was added. Cells were cultured for 10 days after ONC201 removal, with replenishment of medium after 3–4 days. Cells were washed twice with PBS, fixed and stained with 0.25% crystal violet-methanol solution for 30 minutes. After washing stain off with water, plates were allowed to dry and colonies were counted manually. Plating efficiency was calculated by dividing the number of colonies observed with the expected number of colonies. Expected number of colonies was derived by multiplying the total number of plated cells with observed % viability (in CTG assays) after 72 hours of culture with ONC201.
In vivo study
Animal experiments were conducted in accordance with our Institutional Animal Care and Use Committee. Eight week-old female athymic nu/nu mice (Charles River Laboratories) were inoculated with HCT116 p53−/− cells or sorted Aldefluor (+) DLD1 colorectal cancer stem-like cells (45) in the rear flank as a 200 μl suspension of 1:1 Matrigel (BD Biosciences)/PBS. Subcutaneous tumors were allowed to reach a detectable volume (~125 mm3) prior to ONC201 administration. Mice that were injected with HCT116 p53−/− cells were treated with vehicle or 25 mg/kg ONC201 intravenously via the tail vein and tumor tissues were harvested after 8 hours. Mice that were injected with the colorectal cancer stem-like cells were treated with either vehicle or 50 mg/kg ONC201 intraperitoneally. Tissues were harvested after 72 hours.
Tissue analyses
Mice were humanely sacrificed by cervical dislocation under anesthesia. Tissues for Western blot analyses were minced and homogenized in lysis buffer and processed as described in the immunoblotting section below. For immunohistochemical analyses, excised tumors were fixed in 4% paraformaldehyde in PBS overnight at 4°C. Paraffin-embedding and serial sectioning of slides were performed by the Penn State Hershey Histology Core Facility. Slides were dewaxed in xylene and hydrated in a decreasing gradient of ethanol. Antigen retrieval was carried out by boiling in 10 mM citric acid (pH 6.0) for 6 minutes. Samples were blocked with horse serum (Vector Laboratories) and incubated with primary antibody against CHOP (raised in rabbit; Santa Cruz Biotechnology) overnight at 4°C in a humidity chamber. Incubation with biotinylated secondary antibody and DAB deposition were carried out according to manufacturer’s protocol (Vector Laboratories). Samples were counterstained with hematoxylin, rinsed in dH2O, dehydrated, cleared with xylene, and mounted with Cytoseal XYL. Images were recorded on an Axioskop microscope with QCapture software (QImaging).
Isolation of ONC201-resistant clones
RKO cells were plated in 6-well plates (2 x 105cells/well) and treated with 0.5 μM ONC201 for one week. Cells were cultured in increasing concentrations of ONC201, up to 16 μM. Two of the wells had multiple clones and these were isolated into two separate cultures. Resistance was verified by assaying viability after ONC201 treatment and by assessing ONC201-induced surface TRAIL abundance and FOXO3a, AKT and ERK phosphorylation.
Microarray analyses
HCT116, wild-type RKO and ONC201-resistant RKO cells were plated in 6-well plates and treated with vehicle or 5 μM ONC201. After 18 and 48 hours of treatment, RNA was isolated using the RNeasy kit (Qiagen). Gene expression analyses were performed as previously described (8).
Immunoblotting
Cultures were washed twice with PBS and the cells were lysed into lysis buffer (50 mM HEPES, 100 mM NaCl, 10 mM EDTA, 0.5 % NP40, 10% glycerol, supplemented with 0.0001% Tween 20, 0.1 mM PMSF, 0.1 mM NaVO4, 0.5 mM NaF, 5 μg/ml leupeptin, 0.1 mM DTT). The proteins were quantified with the Bio-Rad protein assay and loaded equally onto 4–12% NuPage SDS-polyacrylamide gels (Invitrogen). Proteins were electrophoretically transferred to PVDF membranes. After blocking with 5% BSA or 5% milk, membranes were incubated with primary antibody overnight. Subsequently, incubation with appropriate secondary antibodies labelled with either horseradish peroxidase or near IR dyes was performed. Signal was visualized using chemiluminescence detection or using an Odyssey Infrared Imaging System (Li-Cor). The primary antibodies used in this study were: antibodies against ATF4 (cat# 11815S), CHOP (cat# 2895S), DR5 (cat# 3696S), total eIF2 alpha (cat# 9722S), BCL2 (cat# 2876S), MCL-1 (cat# 4572S), cIAP-2 (cat# 3130S), BIM (cat# 2819S), BAX (cat# 5023S), phospho-Rb (S795) (cat# 9301S), phospho-p70 S6K (cat# 9205S), cyclin D1 (cat# 2978S), phospho-FOXO3a (S253) (cat# 9466S), phospho-AKT (S473) (cat# 4051S), pan-AKT (cat# 4685), phospho-ERK1/2 (cat# 4377S), ERK (cat# 9102S), PKR (cat# 12297S) and PARP (cat# 9542S) (Cell Signaling); against ATF3 (sc-188), TRB3 (sc-271572), PERK (sc-377400), and HRI (sc-30143) (Santa Cruz Biotechnology); against XIAP (cat#610716) and Ran (cat# 610341) (BD Biosciences); against phospho-eIF2α (ab32157) and FOXO3a (cat# ab12162) (Abcam), and against phospho-PKR (cat# 07-886) and p21 (cat# OP64-100ug) (EMD Millipore). Secondary antibodies were acquired from Pierce (cat# 31430 and 31460) (horseradish peroxide-conjugated), LI-COR Biosciences (cat# 926-32211) (IR dye-conjugated) and Invitrogen (cat# A21057) (AlexaFluor 680-conjugated).
Quantitative RT-PCR (qRT-PCR)
RNA was isolated using RNeasy kit (Qiagen) or Quick-RNA Miniprep kit (Zymo Research) according to manufacturers’ instructions. RNA was quantitated using a Nanodrop spectrophotometer. cDNA was synthesized using a SuperScript II RT kit while real-time PCR was performed using a Quantitect SYBR Green PCR mix. Relative amounts of target mRNA were quantitated using the 2ΔΔCt method using GAPDH as internal control. At least three technical replicates per biological replicate were analyzed.
Knockdown using siRNA
Cells were plated in medium with 10% FBS but without antibiotic and incubated overnight. siRNA (a pool of 3 target-specific siRNA’s) (Santa Cruz Biotechnology) (40 nM final concentration) was transfected into cells using 9 μl Lipofectamine RNAiMax. After 24 hr, vehicle or ONC201 was added. In the case of double knockdowns, the final concentration of each siRNA was 20 nM.
To ensure that observed effects using pooled siRNA’s were not due to off target effects, transfections were done with individual target-specific siRNA’s (Sigma-Aldrich) using conditions as stated above, except that the final siRNA concentration was 13.33 nM.
Plasmid overexpression
Plasmid constructs were acquired from Addgene and amplified according to Addgene recommendations. Plasmid DNA was isolated using QIAprep Spin miniprep kit (Qiagen) according to manufacturer’s instructions. 0.5–1 μg DNA was transfected into cells using Lipofectamine 2000. Vehicle or ONC201 was added 48 hours later.
Statistics
Data are presented as means + SEM. To assess the statistical significance of the differences, unpaired Student’s t-test with Holm-Sidak correction for multiple comparisons (maximum of three comparisons were made), was performed with p< 0.05 deemed as statistically significant. Measurements from three biological replicates per treatment group were compared. Unless otherwise noted in the figure legend, comparisons were made against the vehicle control.
Microarray data were quantile normalized using the Genome Studio software. Differentially expressed genes were identified using volcano plots using a fold-change threshold of 1.4 or greater and p-values <0.05 deemed as statistically significant. Multiple testing correction (Benjamini-Hochberg) was performed when the number of differentially expressed genes was greater than 1000 and false discovery rate was set to p<0.05.
Supplementary Material
Fig. S1. The broad-spectrum anti-cancer activity of ONC201 can be attributed to its anti-proliferative and pro-apoptotic effects
Fig. S2. ONC201 activates ATF4 and CHOP
Fig. S3. The differential response of ONC201-sensitive and resistant lines to ER stress inducers is correlated with ATF4 pathway activation
Fig. S4. ONC201 induces a decrease in cyclin D1 abundance but not in Akt and ERK phosphorylation.
Fig. S5. ONC201 does not activate the eIF2α–ATF4 pathway by inhibiting the eIF2α phosphatase or by activating GCN2 or PERK.
Fig. S6. The abundance of cIAP-2 and Bcl-2 family members is similarly altered regardless of whether cells undergo apoptosis
Fig. S7. Altering the amount of XIAP does not change ONC201’s effects on PARP cleavage.
Table S1. ONC201 concentrations used according to cell type.
Table S2. Top gene expression changes induced by ONC201 in HCT116 cells.
Table S3. Top gene expression changes induced by ONC201 in RKO cells.
Table S4: Gene expression arrays.
Acknowledgments
We gratefully acknowledge the important contribution of the following: for the myeloma cell lines, Dr. Nathan Dolloff (Medical University of South Carolina); for the GCN2−/− and PERK−/− MEF’s, Dr. Scot Kimball (Penn State College of Medicine); for the wild-type and non-phosphorylatable eIF2α plasmid constructs, Dr. David Ron; for microarray analyses, Robert Brucklacher of the Penn State Hershey Genome Sciences Facility; for input on appropriate statistical analyses of results, Dr. Samuel Litwin and Dr. Karthik Devarajan of the Fox Chase Biostatistics and Bioinformatics Facility; for assistance in the preparation of figures for the manuscript, Walter J. Kline II.
Funding: This work was supported by grants from the NIH (CA173453-02) and the American Cancer Society (to W.S.E-D.). W.S.E-D. is an American Cancer Society Research Professor.
Footnotes
Author contributions: C.L.B.K., A.P.J.VdH., J.E.A., V.V.P. and W.S.E-D. conceived the study and participated in the design, analyses and interpretation of experiments. C.L.B.K., A.P.J.VdH., J.E.A., and V.V.P conducted the experiments. C.L.B.K., A.P.J.VdH and J.E.A. wrote the manuscript. D.T.D. conducted flow cytometric analyses. W.S.E-D. supervised the experiments and contributed as senior author including editing of the manuscript and responsibility for oversight and conduct of the research.
Competing interests: J.E.A. and W.S.E-D. hold a patent on ONC201 and are shareholders of Oncoceutics, Inc. W.S.E-D. is a co-founder of Oncoceutics, Inc. Pennsylvania State University has licensed ONC201/TIC10 to Oncoceutics, Inc. for clinical development. W.S.E-D. is fully compliant with institutional disclosure requirements and conflict of interest rules.
Data and Materials Availability: The microarray data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus Database (GSE72865). ONC201 requires a materials transfer agreement from Oncoceutics.
References and Notes
- 1.Ashkenazi A, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. The Journal of clinical investigation. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell death and differentiation. 2014;21:1350–1364. doi: 10.1038/cdd.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merchant MS, et al. Phase I trial and pharmacokinetic study of lexatumumab in pediatric patients with solid tumors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:4141–4147. doi: 10.1200/JCO.2012.44.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner KW, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nature medicine. 2007;13:1070–1077. doi: 10.1038/nm1627. [DOI] [PubMed] [Google Scholar]
- 5.Lu M, et al. Cell death. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. 2014;345:98–101. doi: 10.1126/science.1254312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cazanave SC, et al. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. The Journal of biological chemistry. 2011;286:39336–39348. doi: 10.1074/jbc.M111.280420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang G, et al. Small-molecule activation of the TRAIL receptor DR5 in human cancer cells. Nature chemical biology. 2013;9:84–89. doi: 10.1038/nchembio.1153. [DOI] [PubMed] [Google Scholar]
- 8.Allen JE, et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Science translational medicine. 2013;5:171ra117. doi: 10.1126/scitranslmed.3004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ait Ghezala H, et al. Translation termination efficiency modulates ATF4 response by regulating ATF4 mRNA translation at 5′ short ORFs. Nucleic acids research. 2012;40:9557–9570. doi: 10.1093/nar/gks762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nature cell biology. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zinszner H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes & development. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 13.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. The EMBO journal. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 15.Kiss-Toth E, et al. Human tribbles, a protein family controlling mitogen-activated protein kinase cascades. The Journal of biological chemistry. 2004;279:42703–42708. doi: 10.1074/jbc.M407732200. [DOI] [PubMed] [Google Scholar]
- 16.Brewer JW, Hendershot LM, Sherr CJ, Diehl JA. Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8505–8510. doi: 10.1073/pnas.96.15.8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baird TD, Wek RC. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Advances in nutrition. 2012;3:307–321. doi: 10.3945/an.112.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koromilas AE. Roles of the translation initiation factor eIF2alpha serine 51 phosphorylation in cancer formation and treatment. Biochimica et biophysica acta. 2015;1849:871–880. doi: 10.1016/j.bbagrm.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 19.Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Molecular biology of the cell. 2005;16:5493–5501. doi: 10.1091/mbc.E05-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raven JF, et al. PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2alpha phosphorylation. The Journal of biological chemistry. 2008;283:3097–3108. doi: 10.1074/jbc.M709677200. [DOI] [PubMed] [Google Scholar]
- 21.Ng CP, Zisman A, Bonavida B. Synergy is achieved by complementation with Apo2L/TRAIL and actinomycin D in Apo2L/TRAIL-mediated apoptosis of prostate cancer cells: role of XIAP in resistance. The Prostate. 2002;53:286–299. doi: 10.1002/pros.10155. [DOI] [PubMed] [Google Scholar]
- 22.Kim EH, Kim SU, Shin DY, Choi KS. Roscovitine sensitizes glioma cells to TRAIL-mediated apoptosis by downregulation of survivin and XIAP. Oncogene. 2004;23:446–456. doi: 10.1038/sj.onc.1207025. [DOI] [PubMed] [Google Scholar]
- 23.Ishizawa J, et al. ATF4 induction via an atypical integrated stress response to ONC201 induces p53-independent apoptosis in hematological malignancies. Sci Signal. 2015;8 doi: 10.1126/scisignal.aac4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pike LR, et al. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. The Biochemical journal. 2013;449:389–400. doi: 10.1042/BJ20120972. [DOI] [PubMed] [Google Scholar]
- 25.B’Chir W, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic acids research. 2013;41:7683–7699. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40:14–21. doi: 10.1016/j.biocel.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 27.Galehdar Z, et al. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:16938–16948. doi: 10.1523/JNEUROSCI.1598-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puthalakath H, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 29.Chefalo PJ, Oh J, Rafie-Kolpin M, Kan B, Chen JJ. Heme-regulated eIF-2alpha kinase purifies as a hemoprotein. European journal of biochemistry/FEBS. 1998;258:820–830. doi: 10.1046/j.1432-1327.1998.2580820.x. [DOI] [PubMed] [Google Scholar]
- 30.Fournier MJ, Gareau C, Mazroui R. The chemotherapeutic agent bortezomib induces the formation of stress granules. Cancer cell international. 2010;10:12. doi: 10.1186/1475-2867-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yerlikaya A, Kimball SR, Stanley BA. Phosphorylation of eIF2alpha in response to 26S proteasome inhibition is mediated by the haem-regulated inhibitor (HRI) kinase. The Biochemical journal. 2008;412:579–588. doi: 10.1042/BJ20080324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McEwen E, et al. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. The Journal of biological chemistry. 2005;280:16925–16933. doi: 10.1074/jbc.M412882200. [DOI] [PubMed] [Google Scholar]
- 33.Chen T, et al. Chemical genetics identify eIF2alpha kinase heme-regulated inhibitor as an anticancer target. Nature chemical biology. 2011;7:610–616. doi: 10.1038/nchembio.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen T, et al. Explorations of substituted urea functionality for the discovery of new activators of the heme-regulated inhibitor kinase. Journal of medicinal chemistry. 2013;56:9457–9470. doi: 10.1021/jm400793v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. The EMBO journal. 1998;17:5458–5465. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samuel CE. Antiviral actions of interferon. Interferon-regulated cellular proteins and their surprisingly selective antiviral activities. Virology. 1991;183:1–11. doi: 10.1016/0042-6822(91)90112-o. [DOI] [PubMed] [Google Scholar]
- 37.Marchal JA, et al. The impact of PKR activation: from neurodegeneration to cancer. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2014;28:1965–1974. doi: 10.1096/fj.13-248294. [DOI] [PubMed] [Google Scholar]
- 38.Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Molecular and cellular biology. 2000;20:1278–1290. doi: 10.1128/mcb.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura T, et al. A critical role for PKR complexes with TRBP in Immunometabolic regulation and eIF2alpha phosphorylation in obesity. Cell reports. 2015;11:295–307. doi: 10.1016/j.celrep.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Armstrong JL, Flockhart R, Veal GJ, Lovat PE, Redfern CP. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. The Journal of biological chemistry. 2010;285:6091–6100. doi: 10.1074/jbc.M109.014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shenton D, et al. Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. The Journal of biological chemistry. 2006;281:29011–29021. doi: 10.1074/jbc.M601545200. [DOI] [PubMed] [Google Scholar]
- 42.Obeng EA, et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schonthal AH. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochemical pharmacology. 2013;85:653–666. doi: 10.1016/j.bcp.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 44.Daniels RA, et al. Expression of TRAIL and TRAIL receptors in normal and malignant tissues. Cell research. 2005;15:430–438. doi: 10.1038/sj.cr.7290311. [DOI] [PubMed] [Google Scholar]
- 45.Prabhu VV, Allen JE, Dicker DT, El-Deiry WS. Small molecule ONC201/TIC10 targets chemotherapy-resistant colorectal cancer stem-like cells in an Akt/Foxo3a/TRAIL-dependent manner. Cancer Research OnlineFirst. 2015 doi: 10.1158/0008-5472.CAN-13-3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The broad-spectrum anti-cancer activity of ONC201 can be attributed to its anti-proliferative and pro-apoptotic effects
Fig. S2. ONC201 activates ATF4 and CHOP
Fig. S3. The differential response of ONC201-sensitive and resistant lines to ER stress inducers is correlated with ATF4 pathway activation
Fig. S4. ONC201 induces a decrease in cyclin D1 abundance but not in Akt and ERK phosphorylation.
Fig. S5. ONC201 does not activate the eIF2α–ATF4 pathway by inhibiting the eIF2α phosphatase or by activating GCN2 or PERK.
Fig. S6. The abundance of cIAP-2 and Bcl-2 family members is similarly altered regardless of whether cells undergo apoptosis
Fig. S7. Altering the amount of XIAP does not change ONC201’s effects on PARP cleavage.
Table S1. ONC201 concentrations used according to cell type.
Table S2. Top gene expression changes induced by ONC201 in HCT116 cells.
Table S3. Top gene expression changes induced by ONC201 in RKO cells.
Table S4: Gene expression arrays.