

Abstract
Purpose
Mutations in the coding sequence of the L and M opsin genes are often associated with X-linked cone dysfunction (such as Bornholm Eye Disease, BED), though the exact color vision phenotype associated with these disorders is variable. We examined individuals with L/M opsin gene mutations to clarify the link between color vision deficiency and cone dysfunction.
Methods
We recruited 17 males for imaging. The thickness and integrity of the photoreceptor layers were evaluated using spectral-domain optical coherence tomography. Cone density was measured using high-resolution images of the cone mosaic obtained with adaptive optics scanning light ophthalmoscopy. The L/M opsin gene array was characterized in 16 subjects, including at least one subject from each family.
Results
There were six subjects with the LVAVA haplotype encoded by exon 3, seven with LIAVA, two with the Cys203Arg mutation encoded by exon 4, and two with a novel insertion in exon 2. Foveal cone structure and retinal thickness was disrupted to a variable degree, even among related individuals with the same L/M array.
Conclusions
Our findings provide a direct link between disruption of the cone mosaic and L/M opsin variants. We hypothesize that, in addition to large phenotypic differences between different L/M opsin variants, the ratio of expression of first versus downstream genes in the L/M array contributes to phenotypic diversity. While the L/M opsin mutations underlie the cone dysfunction in all of the subjects tested, the color vision defect can be caused either by the same mutation or a gene rearrangement at the same locus.
Keywords: opsin, myopia, adaptive optics, dichromacy, color vision
The cone dysfunction syndromes are a clinically and genetically heterogeneous group of conditions characterized by early-onset reduction in central vision associated with a variable degree of photophobia and nystagmus, normal fundus examination, and generalized cone dysfunction with normal rod function on electroretinography (ERG).1,2 Bornholm eye disease (BED) is a cone dysfunction syndrome, having been first reported in a single large Danish family with high myopia, astigmatism, optic nerve changes, abnormal cone ERG, and a deutan color vision defect.3,4 Linkage analysis in the Danish family mapped BED to Xq28, and it became the first locus identified for high myopia (MYP1, OMIM number 310460). A second MYP1 family of Danish descent, living in Minnesota, had high myopia like the original Danish family, although they had an accompanying protan (instead of deutan) color vision defect.5 Similarly, five families in the United Kingdom with X-linked cone dysfunction and protanopia were reported in 2005.6 Four additional families with X-linked high myopia, of Chinese7,8 and Asian Indian origin,9 also have been mapped to MYP1; however, affected members all had normal color vision. Thus, the emerging picture for BED is that no single color vision phenotype is a consistent feature of the disorder.
Xq28 (the locus of MYP1) encompasses the long- and middle-wavelength sensitive opsin gene array (OPN1LW and OPN1MW, or L and M, respectively). The initial studies of the Danish, Minnesota, and UK families identified common gene rearrangements within the opsin gene array accounting for the color vision defects, though no mutations were identified to account for the cone dysfunction.5,6,10 However, there is evidence that mutations in the L/M opsin coding sequence can disrupt cone structure/function.11 For example, a specific combination of polymorphisms encoded by exon 3 of the L and M pigment genes (Leu153, Ile171, Ala174, Val178, Ala180 = LIAVA) was found to result in the loss of function of an entire class of cone photoreceptor; leading to dichromacy when occurring in either the L or the M gene,12–14 or blue cone monochromacy (BCM) when occurring in all of the expressed L/M genes.15,16 However, the dichromatic individuals with the LIAVA haplotype were not noted to show other features initially associated with BED (e.g., cone dysfunction and high myopia).12,13 Moreover, in contrast to the variable color vision phenotype of the aforementioned MYP1 BED families, the color vision phenotype in these individuals correlates perfectly with genotype; that is, LIAVA encoded by an L gene in the presence of normal M gene(s) results in protanopia, LIAVA encoded by an M gene in the presence of a normal L gene results in deuteranopia, and individuals with LIAVA encoded by all expressed L/M genes (i.e., at least the first two genes in the array) have BCM.
The Danish and Minnesota BED families had another unique combination of amino acids encoded by exon 3 (Leu153, Val171, Ala174, Val178, Ala180 = LVAVA) in one gene of their L/M opsin arrays.17 However, the Minnesota family had three genes encoding M pigments (conferring an obligate protan phenotype), with LVAVA encoded by exon 3 of the first gene in the array. In contrast, the first two genes in the array of the Danish family encoded L pigments with a spectral separation of 2.5 nm (conferring an obligate deuteranomalous phenotype), with LVAVA encoded by exon 3 of the first gene in the array. The LVAVA haplotype also cosegregated with high myopia in the MYP1 Chinese families.8 It has been shown that LIAVA, LVAVA, and related variants are associated with aberrant splicing,18,19 in which exon 3 is skipped and exon 2 is spliced to exon 4, introducing a frame shift and a premature stop codon. In an in vitro assay, virtually all the mRNA from the LIAVA mutant was missing exon 3, explaining the complete loss of function of that pigment.18,19 However, in the same assay, a small but significant fraction of mRNA from the LVAVA mutant is normally spliced, presumably allowing the production of a reduced amount of LVAVA opsin. Thus, cones expressing this variant should have residual function, which is consistent with the observation of normal color vision in the Chinese LVAVA MYP1 families.7,8
It is becoming appreciated that different OPN1LW/OPN1MW mutations have distinctly different effects on cone function and viability.13,16,20–23 For example, we reported on two single-gene dichromats with LVAVA encoded by exon 3 of their only L/M opsin gene.21 They had a late-onset progressive phenotype in direct contrast to individuals harboring the missense mutation Cys203Arg, which has been shown to lead not only to loss of function,24–26 but also very early degeneration of cones, resulting in reorganization into a regularly packed, albeit sparser, cone mosaic as imaged with adaptive optics ophthalmoscopy.20 There are also differences within genotypes – LIAVA and Cys203Arg often have been associated with dichromatic color vision,12,13,20,27 but only recently have been reported in families diagnosed as having BED.17 Moreover, unlike LIAVA, the LVAVA haplotype has not been reported in individuals with simple dichromacy. In an effort to better characterize the genotype–phenotype relationship in X-linked cone dysfunction, we used quantitative retinal imaging, optical coherence tomography (OCT) and adaptive optics scanning light ophthalmoscopy (AOSLO), to examine 17 males with a diagnosis of BED, including two subjects from the original Danish family and four subjects from the original Minnesota family. We observed variable disruption of the cone mosaic across subjects, with the degree of disruption correlating with axial length. While the opsin variant (e.g., LIAVA, LVAVA, Cys203Arg) appears to be the main determinant of phenotypic differences across subjects, the degree of generalized cone dysfunction and high myopia may also depend on the relative number of cones expressing the aberrant pigment. It has been shown that the relative number of L and M cones (L:M ratio) varies greatly across individuals.28,29 Thus, we propose that genes or sequences that modulate L:M cone ratio in color normal individuals30,31 may be involved in determining the final phenotype in individuals with these opsin variants.1
Methods
Subjects
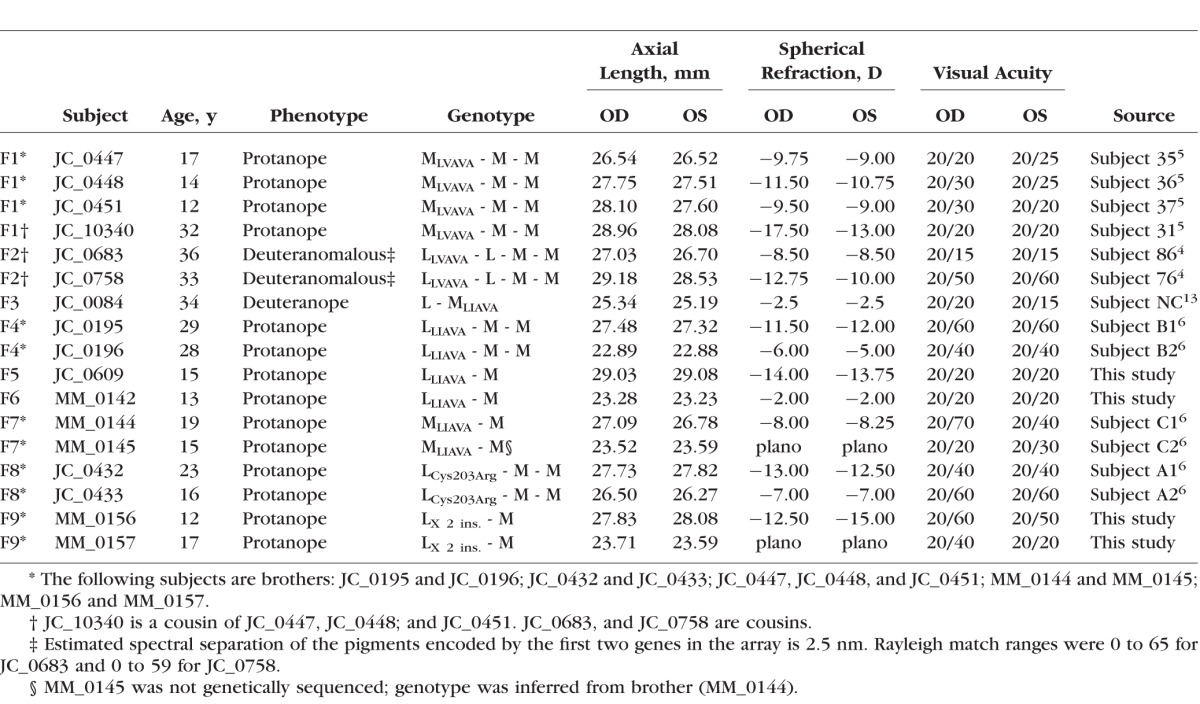
This study followed the tenets of the Declaration of Helsinki and was approved by local Institutional Review Boards. Informed consent was obtained from all subjects, after the nature and possible consequences of the study were explained. We examined 17 patients; the clinical phenotype of 13 of these subjects has been described previously in detail.4–6,13 Although one subject (MM_0145) was not genetically sequenced, the similarity in phenotype between him and his brother (MM_0144) allows his genotype to be reasonably inferred. The four new subjects each provided a blood sample, from which DNA was isolated. The opsin genes were amplified and sequenced using previously described methods to determine the type of mutation, as well as its position within the gene array.12,20 Axial length was measured in each eye imaged using the Zeiss IOL Master (Carl Zeiss Meditec, Dublin, California, USA). Color vision for the 4 newly recruited subjects was assessed using the American Optical Hardy-Rand-Rittler (AO-HRR) and the anomaloscope when available. A brief summary of genetic and clinical findings is provided in Table 1.
Table 1.
A Summary of the Genotype and Clinical Phenotype of Subjects With X-Linked Cone Dysfunction
Adaptive Optics Retinal Imaging
Images of the central photoreceptor mosaic were obtained with one of two previously described confocal AOSLO systems, housed either at the Medical College of Wisconsin or at Moorfields Eye Hospital, London.32–36 Eyes were dilated using one drop each of phenylephrine hydrochloride (2.5%) and tropicamide (1%) before imaging. Images were typically collected at different locations across the central fovea, as well as in strips, up to 10° from fixation in the temporal and superior directions.
Because the system acquires images by scanning the retina, there are distortions inherent to the raw videos that must be removed. Sinusoidal distortions were measured and corrected for using a Ronchi ruling of known spacing. From the resampled image sequences, a set of minimally-distorted frames, judged visually, were selected as references to which all other frames within that sequence would be registered and averaged using previously described strip-based registration software.37 The resultant images were stitched together manually in Photoshop (Adobe Photoshop; Adobe Systems, Inc., San Jose, CA, USA) to produce a larger montage for each of the subjects. Montages were scaled by first determining the degrees per pixel in an image of a ruling with known spacing using the small angle approximation. This value was linearly scaled using the subject's axial length and a reference axial length of 24 mm, and multiplied by the retinal magnification factor, 291 μm/deg, to obtain a final μm/px scale.
Based on differences in image quality, it was necessary to use two methods to estimate the location of the fovea, from which subsequent regions of interest were selected for density analyses. For seven subjects, cones were identified over a large area that reliably encompassed the foveal region using a previously described semiautomated algorithm.38 Next, we used a custom MatLab program to measure cone density at each pixel in the image using each of 10 sampling windows ranging in size from 25 × 25 to 45 × 45 μm. The 10 densities for each pixel were averaged to create an average density map, and the location of peak density in this average density map was used as the location of peak density.39 In nine subjects, peak cone density was not a reliable indicator of foveal location; either due to an extremely sparse foveal cone mosaic or poor image quality resulting in the inability to resolve foveal cones over a sufficiently large area for the topographic mapping method. For nine of these subjects the foveal center was approximated using the preferred retinal locus, identified as follows: Subjects were asked to fixate on the center of a square and each corner in turn; the mean of the coordinates for the center of each frame at each location was taken as the center of fixation. Visual inspection of the montages revealed that the resultant coordinates correlated well with the foveal center. Regions of interest subtending 55 × 55 μm were cropped from the larger montage at the fovea (when possible) and 0.25, 0.5, 1, 2, and 5° along the temporal and superior meridia. Within these regions of interest, cones were identified using a semi-automated algorithm to provide estimates of cone density.20,38 The image quality for JC_10340 was too poor to enable quantitative analysis.
Spectral-Domain OCT (SD-OCT)
For 15 subjects, high-resolution SD-OCT images of the macula were acquired using the Bioptigen SD-OCT system (Morrisville, NC, USA). High-density line scans (either 750 or 1000 A-scans/B-scan, 100-120 repeated B scans) were acquired through the foveal center. Line scans were registered and averaged to reduce speckle noise in the image as previously described.40 In addition, volumetric images of the macula and cross-sectional line scans through the fovea were obtained using a Zeiss Cirrus HD-OCT (Carl Zeiss Meditec, Dublin, California, USA) and used for analysis when Bioptigen images were unavailable (JC_0432 and JC_0433). Volumes were nominally 6 × 6 mm and consisted of 128 B-scans (512 A-scans/B-scan).
The lateral scale of each subjects' OCT data set was determined by dividing the nominal scan length by the assumed axial length (24 mm for the Bioptigen; 24.46 mm for the Cirrus) and multiplying by their measured axial length. Optical coherence tomography images were assessed in terms of total retinal thickness, as well as inner retinal thickness (defined as the distance between the internal limiting membrane [ILM] and the outer plexiform layer/outer nuclear layer [OPL/ONL] junction), and the ONL + Henle Fiber Layer (HFL) thickness (defined as the distance between the OPL/ONL junction and the external limiting membrane [ELM]). As described previously,41 segmentation was done by manually delineating the boundaries of the ILM, OPL, ONL, and RPE – choroid junction (RPE2) using ImageJ42 and custom Matlab software. We then compared our measurements against a subset of a previously published normative database,43 consisting of 70 males, with an average age of 30.7 ± 30 years (range, 7–60 years).
Results
Clinical Phenotype Varies Across Subjects With OPN1LW/OPN1MW Mutations
A summary of the genotype and clinical phenotype for each patient is given in Table 1. Six subjects harbored the LVAVA haplotype, seven had the LIAVA haplotype, and two had the Cys203Arg missense mutation. One family had a novel insertion in exon 2 of their L gene: following nucleotide position 179 in exon 2 (where the first base of exon 2 is position #1) there is a 32-base pair (bp) insertion that corresponds exactly in sequence to nucleotides 149 to 179 of exon 2, thereby creating a direct repeat of 32 bp within the exon. The insertion shifts the protein reading frame and introduces multiple termination codons. Thus, it is likely that the mRNA, if produced, is degraded through one of the many mRNA decay pathways.44 The exact mechanism by which the insertion disrupts cone function is unknown, but possible mechanisms include creating a new RNA secondary structure, altering the protein coding sequence, or altering splicing.
Genes encoding the LIAVA, Cys203Arg, and exon 2 insertion all would be associated with a lack of function for cones expressing those genes; thus, the gene arrays of JC_0084, JC_0195, JC_0196, JC_0609, MM_0142, MM_0144, MM_0145, JC_0432, JC_0433, MM_0156, and MM_0157 all confer an obligate dichromatic phenotype. In contrast, the deutan subjects (JC_0683 and JC_0758) with LLVAVA have the genetic basis for anomalous trichromacy, assuming that their LVAVA-expressing cones retain some function. Subjects from the Minnesota family (JC_0447, JC_0448, JC_0451, and JC_10340) harboring MLVAVA have the genetic basis for either dichromacy or anomalous trichromacy depending on whether their first two genes encode spectrally different pigments. They have three gene-encoding M pigments and the basis for two spectral types, but whether the first two genes specify spectrally different pigments was not determined from the analysis. Across all subjects, axial length (mean ± SD) was on average greater than normal (26.49 ± 4.02 vs. 24.00 ± 1.09 mm).45 Interestingly, the LVAVA subjects had significantly longer axial lengths on average compared to the subjects with the LIAVA haplotype (27.9 vs. 25.5 mm; P = 0.044, unpaired t-test). The LVAVA subjects were more myopic on average than the LIAVA subjects (−11.6 vs. −6.2 diopters [D]); however, this was not statistically significant (P = 0.054, unpaired t-test). Although high myopia often is associated with the mutant opsins studied here, there was a 9 D range in refractive error among the LVAVA subjects and a 14 D range in refractive error among the LIAVA subjects (with one even being emmetropic).
Retinal Lamination
Although retinal thickness was generally reduced across the horizontal meridian, large differences in retinal thickness and lamination integrity were identified between different genotype groups (Fig. 1). Total retinal thickness at the fovea ranged from 142.70 to 212.56 μm with a mean of 181.42 ± 21.25 μm, compared to that of normal male controls who had a mean foveal thickness of 216.08 ± 38.91 μm (Fig. 2A). However, retinal thinning has been associated with myopia,46–48 and our controls had a mean axial length of 24.17 mm; thus, it is difficult to disambiguate retinal thinning due to possible loss of photoreceptors from that owing to axial elongation of the eye. Regardless, there was no apparent correlation between axial length and foveal thickness (R2 = 0.1025, P = 0.21). Subjects with the Cys203Arg mutation and the LIAVA haplotype had the greatest reduction in retinal thickness at the fovea, while thickness for subjects harboring the exon 2 insertion and LVAVA was within the normal range at the fovea. Further examination revealed reduced ONL + HFL thickness (Fig. 2C) but relatively normal inner retinal thickness (Fig. 2B) in all subjects; thus, the overall reduction in retinal thickness can be attributed primarily to ONL + HFL thinning; with the Cys203Arg mutation and LIAVA haplotype being associated with the greatest degree of outer retinal thinning (Fig. 2C). The appearance of the outer hyperreflective bands was highly variable. For example, in some subjects (JC_0758, MM_0145, and JC_0433) there appear to be irregularities in the third hyperreflective band (interdigitation zone [IZ]49,50), similar to those seen in patients with oligocone trichromacy (Fig. 1).43 Two subjects had no clearly resolvable IZ band (JC_0195 and JC_0609), while the rest generally had a normal-appearing IZ (JC_0451, JC_0084, MM_0142, JC_0433, and MM_0157) (Fig. 1). In addition, subjects JC_0609 and JC_0758 showed “mottling” of the second hyperreflective band (ellipsoid zone [EZ]49,50), whereas the remaining subjects had a qualitatively intact EZ band (Fig. 1).
Figure 1.

Variable disruption in retinal lamination in X-linked cone dysfunction. Shown are high-resolution SD-OCT images (horizontal line scans) through the fovea. One image from each family is displayed, labeled with the subject ID and corresponding genotype. There is clear variability in ONL thickness across subjects, with MM_0142 and JC_0433 having the most pronounced ONL thinning at the fovea. Variation in foveal morphology also is apparent, which has been reported previously.64 The integrity of the second (IS/OS or EZ) and third (IZ) hyperreflective bands was variable across subjects, ranging from normal to mottled in appearance. Scale bar: 200 μm.
Figure 2.

Subjects with X-linked cone dysfunction show reduced outer retinal thickness compared to normals. Plotted are the mean SD-OCT retinal thickness measurements along the horizontal meridian for each group of mutations. (A) Total retinal thickness, (B) inner retinal thickness, (C) ONL + HFL thickness. Solid black lines represent the mean values for 70 normal male controls, with the shaded region representing ± 2 SD from the mean. Filled squares represent the six individuals with the LVAVA haplotype (JC_0447, JC_0448, JC_0451, JC_0683, JC_0758, and JC_10340). Filled circles represent the averaged data from the seven individuals with the LIAVA haplotype (JC_0084, JC_0195, JC_0196, JC_0609, MM_0142, MM_0144, and MM_0145). Open triangles represent the two brothers with Cys203Arg mutations (JC_0432 and JC_0433). Open diamonds represent the two brothers with the novel exon 2 insertion (MM_0156 and MM_0157).
Variable Disruption of the Cone Mosaic in X-Linked Cone Dysfunction
All patients had significantly disrupted cone mosaics, with a high degree of intersubject variability (Fig. 3). Cone density was reduced compared to normal,51 at all locations in all subjects, as shown in Table 2. For reference, a normal age-matched retina with a peak cone density of 195,030 cones/mm2 is shown in the top left panel of Figure 3.52
Figure 3.

Photoreceptor mosaic disruption is highly variable in X-linked cone dysfunction, even within a given genotype. Foveal AOSLO images are shown for three subjects with the LIAVA haplotype and, for comparison, a previously published normal control with a peak density of 195,030 cones/mm2 (JC_0138).39 Although many dark spaces are visible in JC_0084's mosaic, there is tight packing at the fovea (peak density = 100,069 cones/mm2). In contrast, MM_0142 has fewer dark spaces but a more loosely packed mosaic (peak density = 53,356 cones/mm2). Subject JC_0609 shows the greatest degree of disruption, with many dark areas, resulting in a much sparser cone mosaic (peak density = 18,927 cones/mm2). Scale bar: 50 μm.
Table 2.
Cone Density for Subjects With X-Linked Cone Dysfunction
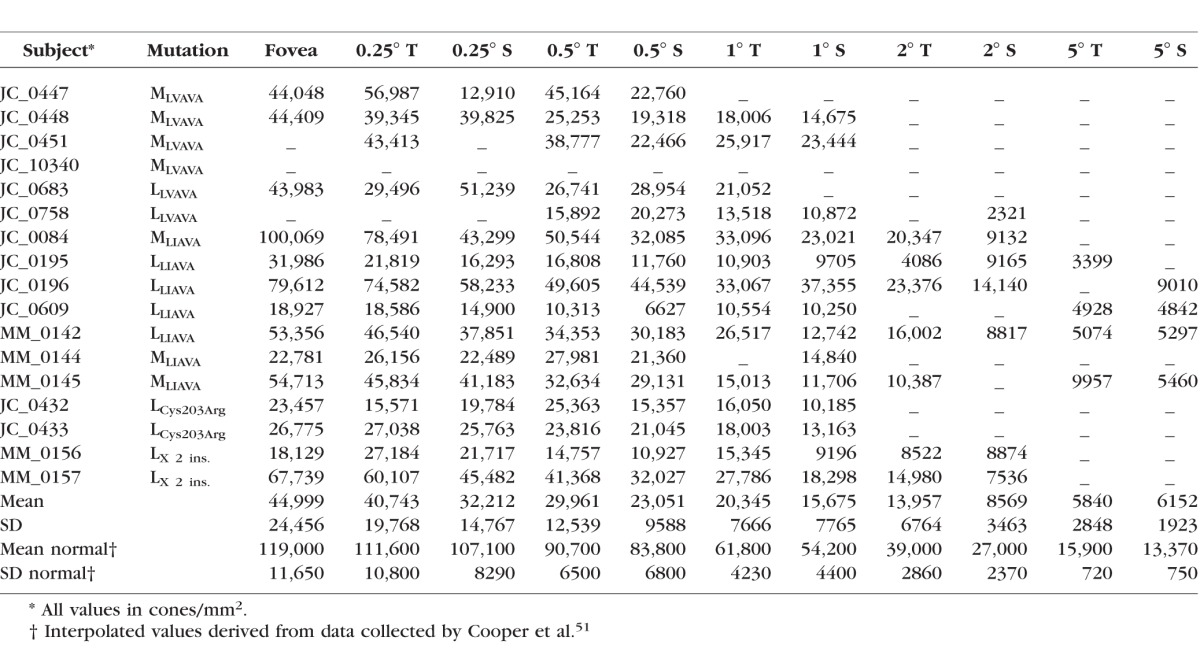
Various studies have reported peak foveal cone density to range from approximately 85,000 to nearly 250,000 cones/mm2 in normal subjects.39,53,54 In our subjects, the cone mosaic was discontiguous and of variably reduced density, ranging from 18,129 to 100,069 cones/mm2 at the fovea. These findings indicate that in subjects with the opsin sequence variants studied here, cone density and topography are significantly disrupted. There are, however, striking differences in the degree of mosaic disruption, not only between subjects who have similar genotypes, but also between related individuals having the same L/M gene array (Table 1, Fig. 4).
Figure 4.

Marked differences in cone mosaic disruption between brothers with X-linked cone dysfunction and OPN1LW/OPN1MW mutations. Foveal AOSLO images are shown from four pairs of brothers. Large variation, not only in the number of cones, but also their apparent size and topography, is evident between JC_0195 and JC_0196, and MM_0156 and MM_0157. For example, MM_0157 shows relatively normal topography, with cone density peaking at the fovea and decreasing with eccentricity (Table 2); MM_0156, on the other hand, has greater disruption at the fovea, with a large area of low reflectivity surrounded by a sparse mosaic of cones. Scale bar: 100 μm.
Regression analysis of the relationship between foveal cone density and the severity of myopia revealed a strong correlation (R2 = 0.46, P < 0.01). As might be expected, given the relatedness between myopia and axial length, there also was a significant inverse relationship between cone density and axial length (R2 = 0.47, P < 0.01), even after correcting for lateral magnification. The relationship between foveal cone density and ONL thickness, on the other hand, was not statistically significant (R2 < 0.01, P = 0.80), in contrast to findings in normal eyes.55 Furthermore, differences in cone density were not apparent functionally, as demonstrated by a null relationship with visual acuity (R2 = 0.15, P = 0.17).
Discussion
In this study, we performed quantitative high-resolution retinal imaging to probe the structural effects of LVAVA, LIAVA, Cys203Arg, and exon insertion variants. In all patients in our series, reduced cone density and reduced ONL thickness was observed, albeit to variable degrees, between and within families. The imaging data here provided a direct link between the retinal mosaic (secondary to the LVAVA/LIAVA/ Cys203Arg/exon insertion variants)12,13,20 and the cone dysfunction, as well as the color vision defect for patients who have otherwise normal L/M array structure (a single L gene followed by one or more M genes).
Cause of Red-Green Color Vision Defects in Patients With X-Linked Cone Dysfunction
An important aspect of the present work is that many patients with X-linked cone dysfunction (previously referred to as BED) can have multiple alterations present within the L/M gene array, although not all of these alterations necessarily cause a color vision defect. The LIAVA opsin gene variant is associated with absence of function of the corresponding cone. Thus, the color vision deficiency is directly attributable to the absence of function of the cone type corresponding to the opsin gene encoding the LIAVA variant in these subjects (JC_0084, JC_0195, JC_0196, and JC_0609, MM_0142, MM_0144, MM_0145). The protan color vision defect in JC_0432 and JC_0433 is attributable to the presence of the inactivating Cys203Arg mutation in the L cone photopigment (leaving only functional M photopigment). Likewise the protan phenotype of subjects MM_0144 (and MM_0145 by inference) is due to the 32 bp insertion in exon 2 of the L gene, meaning only functional M photopigment is predicted in these retinas. For subjects JC_0447, JC_0448, JC_0451, and JC_10340 (from the original MLVAVA Minnesota family) the protan color vision deficiency can be accounted for by the presence of only M opsin genes in the L/M gene array for each subject. For subjects JC_0683 and JC_0758 (from the original LLVAVA Danish family), their reported deutan color vision defect is attributed to L genes being the first two in the array, which encode pigments with a spectral separation of 2.5 nm. In keeping with this, these subjects had Rayleigh match ranges consistent with deuteranomalous trichromacy, not deuteranopia as originally reported in the first descriptions of the family. This, coupled with the normal color vision in the Chinese MYP1 BED families,7,8 is direct evidence that the LVAVA variant is functional, at least to some extent.
The original clinical classification of BED included high myopia, dichromacy, astigmatism, optic nerve changes, and abnormal cone ERG. Presumably, what called attention to the original BED family was the fact that the high myopia was associated with the color vision defect, subsequently inspiring the linkage studies and the identification of the MYP1 locus. However, as previously suggested1,17 and as shown here, the LVAVA mutation causing the cone dysfunction is simply closely linked to L/M gene rearrangements causing the color defect, and so the two cosegregated. Importantly, while all the pathologic features of BED, including cone dysfunction, the high myopia, and abnormal ERG, are directly caused by LVAVA, the color vision defect is not caused by LVAVA. This is consistent with the observation of normal color vision in the Asian Indian families who were mapped to MYP1 as well as the Chinese families who were mapped to MYP1,7 and later shown to harbor LVAVA.8 As approximately 8% of males are color-deficient, we estimate that for every colorblind BED patient with LVAVA, there are likely at least 10 times as many color-normal males with many or all of the same pathologic symptoms seen in BED. However, this is based on the assumption that the LVAVA variant occurs with equal frequency in color-normal and color-deficient populations. Nevertheless, genetic testing is important for identifying LVAVA patients, most of whom have normal color vision and will rarely be associated with a color vision defect. However LVAVA can lead to severe localized degeneration;21 thus, there may be subclinical changes in the retinas of these individuals – it would be interesting to image these color normal individuals with AOSLO and examine the integrity of their photoreceptor mosaic.
Phenotypic Variation and L:M Cone Ratio
There were marked differences in phenotype with regard to the severity of cone mosaic disruption across subjects, even in individuals harboring the same OPN1LW/OPN1MW mutations. We hypothesize that individual differences in the relative expression of first versus downstream genes in the L/M array (which modulates L:M cone ratio in color normal individuals) may underlie this variability.1 Already, large individual differences have been demonstrated in L:M cone ratio in normal subjects, ranging from 28% to 93% L, corresponding to more than a 30-fold range in L:M ratio (0.4–13).28,29 For example, mutations causing loss of function or viability of the cones expressing that pigment (e.g., LIAVA and Cys203Arg), could result in dramatically different phenotypes based on how many cones express the mutant pigment. If only 10% to 30% of the cones are lost or nonfunctional, perhaps none of the other symptoms of BED may be observed. Conversely, the situation may be quite different if 70% to 90% of the cones are lost or nonfunctional as a result of the same mutations; in these cases, many more of the BED-like symptoms, including myopia and abnormal ERG, may be observed. Whereas extreme L:M cone ratios may be relatively rare in normal individuals, the ability of the LIAVA or Cys203Arg mutation to reside in either the first or second gene in the L/M array would result in a higher incidence of skewed ratios of normal:mutant gene expression.
Cone ratio may also modulate the severity of symptoms for subjects with the LVAVA haplotype, although the effect of this haplotype appears generally to be more severe, such that most subjects, even those expressing LVAVA in a minority cone population, may exhibit BED symptoms, particularly later in life if there is a progressive loss of cones due to possible bystander effects.21 In the subjects studied here, greater severity of the LVAVA mutation is evident in the significantly higher axial length and slightly higher myopia among LVAVA patients compared to LIAVA, though interestingly there was not a concomitant difference in retinal thickness. It currently appears that the vast majority of people with the LVAVA haplotype do not have a color vision defect, so they would not be diagnosed as having BED, despite suffering from high myopia and cone dysfunction characteristic of BED.
Interestingly, there was a high degree of variation in the cone mosaic within each set of brothers, despite them having the same opsin gene array in each case; this also was associated with pronounced differences in visual acuity and refractive error/axial length within sibling pairs. One possible explanation for this is that the percentage of L cones has been found to differ by up to 14% among pairs of trichromatic brothers who share the same L/M gene array (see Supplementary Material). Even small differences in L:M cone ratio could lead to significant structural differences. For example, consider two brothers with L cones expressing one of the mutations described herein, with one having an L:M ratio of 90% L and the other of 76% L – this would result in a 2.4-fold difference in the actual number of functional M cones in their retinas.
In addition to the variability in retinal structure, we also found a large range in axial length across our sample, with longer eyes being associated with lower cone density. Although this trend is in agreement with previous AO literature in the case of the diseased retina,56–58 the relationship between axial length and cone density cannot be attributed to retinal stretching alone. Whereas myopia was considered previously to be one of the defining characteristics of BED,1,3–5 many of our subjects were not myopic, having little or no refractive error while still having mosaic disruption; though this was observed in LIAVA, but not LVAVA, subjects.
Progressive/Stationary Nature of X-Linked Cone Dysfunction
Another important factor to consider is whether intrafamilial differences could be due to disease progression. Not only the type of mutation but also its position in the gene array could have important implications for the extent and timing of cone degeneration. For example, the LIAVA haplotype and the Cys203Arg missense mutation have been associated with static mosaic disruption,20,59 whereas the LVAVA haplotype has been associated with later onset disease progression in those who possess a single opsin gene.21 Comparison of cone density values across all subjects revealed no significant correlation with age at the time of imaging; however, any differences in cone mosaic integrity and stability between genotypes could conceal age-related effects in our sample. Nevertheless, longitudinal assessment is needed before we can rule out the possibility of progression in these conditions. Moreover, it is not known whether the dark gaps in the mosaics of our subjects indicate complete degeneration of the photoreceptors or whether there is residual cone structure. In some other retinal conditions, nonconfocal split-detection imaging has revealed remnant inner-segment structure within dark spaces similar to those shown here35,60 (Langlo CS, et al. IOVS 2015;56:ARVO E-Abstract 4017). This indicates that a lack of reflectivity in confocal AOSLO images is not necessarily indicative of complete cone degeneration. This is important, as mosaic disruption similar to that of our subjects has been reported in a number of other retinal conditions.61–63 Longitudinal examination using nonconfocal split-detection imaging is needed to determine the relative stability of the photoreceptor mosaic across the different genotypes reported here, and arguably to better determine the potential for future therapeutic intervention by identifying cones that could be rescued with gene replacement or other therapies.
Conclusions
Our assessment of subjects with mutations in the coding region of OPN1LW/OPN1MW revealed significantly disrupted foveal cone mosaics and reduced retinal thickness. There was a large degree of variation in the number and spacing of photoreceptors, even within families with the same L/M array, which is most likely due to variability in the genetic mechanisms that control L:M cone ratio in color-normal individuals. Our results suggested that mutations that completely disrupt cone function, either by causing early cone cell death (Cys203Arg) or a complete loss of cone function (LIAVA), lead to dichromacy, but not necessarily high myopia and other cone dysfunction symptoms seen in the LVAVA BED patients. Defects caused by such mutations may be more common than previously thought, and their effects on structure and function of cones is yet to be fully appreciated. Investigation of other exon 3 haplotypes combined with systematic comparison with a normative database is needed to fully understand the effects of genotype. Future work using additional measurement techniques, such as more extensive color vision testing and split-detection AOSLO imaging,35 may enable us to establish possible genotype/phenotype correlations in the broad spectrum of disorders associated with OPN1LW/OPN1MW mutations. However, as no single color vision phenotype is a consistent feature of patients with MYP1 BED, we propose that color vision deficiency be removed as a necessary feature of this condition.
Supplementary Material
Acknowledgments
The authors thank Robert Cooper, Ann Holleschau, Brian Higgins, Phyllis Summerfelt, C. Gail Summers, and David R. Williams for their contributions to this work.
Presented in part at the annual meetings of the Association for Research in Vision and Ophthalmology, Ft. Lauderdale, Florida, United States, May 2011 and Denver, Colorado, May 2015.
Supported in part by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health (NIH; Bethesda, MD, USA) Grant UL1TR000055, and by the National Eye Institute and National Institute of General Medical Sciences, NIH Grants R01EY017607, R01EY021242, T32EY014537, T32GM080202, P30EY001931, and P30EY001730. This investigation was conducted in part in a facility constructed with support from the Research Facilities Improvement Program; Grant C06RR016511 from the National Center for Research Resources, NIH. Also supported by an unrestricted grant from Research to Prevent Blindness, Foundation Fighting Blindness (USA), Moorfields Special Trustees, Moorfields Eye Charity, Fight for Sight, the Wellcome Trust [099173/Z/12/Z], and the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, and by a Career Development Award from the Foundation Fighting Blindness (USA; MM). The authors alone are responsible for the content and writing of this paper.
Disclosure: E.J. Patterson, None; M. Wilk, None; C.S. Langlo, None; M. Kasilian, None; M. Ring, None; R.B. Hufnagel, None; A.M. Dubis, None; J.J. Tee, None; A. Kalitzeos, None; J.C. Gardner, None; Z.M. Ahmed, None; R.A. Sisk, None; M. Larsen, None; S. Sjoberg, None; T.B. Connor, None; A. Dubra, Athena Vision (C), P; J. Neitz, P; A.J. Hardcastle, None; M. Neitz, P; M. Michaelides, Athena Vision (C); J. Carroll, AGTC (F), Athena Vision (C), OptoVue (F)
References
- 1. Michaelides M,, Hunt DM,, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol. 2004; 88: 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aboshiha J,, Dubis AM,, Carroll J,, Hardcastle AJ,, Michaelides M. The cone dysfunction syndromes. Br J Ophthalmol. 2016; 100: 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haim M,, Fledelius HC,, Skarsholm D. X-linked myopia in a Danish family. Acta Ophthalmol. 1988; 66: 450–456. [DOI] [PubMed] [Google Scholar]
- 4. Schwartz M,, Haim M,, Skarsholm D. X-Linked myopia: Bornholm Eye Disease - linkage to DNA markers on the distal part of Xq. Clin Genet. 1990; 38: 281–286. [PubMed] [Google Scholar]
- 5. Young TL,, Deeb SS,, Ronan SM,, et al. X-linked high myopia associated with cone dysfunction. Arch Ophthalmol. 2004. ; 122: 897–908. [DOI] [PubMed] [Google Scholar]
- 6. Michaelides M,, Johnson S,, Bradshaw K,, et al. X-linked cone dysfunction syndrome with myopia and protanopia. Ophthalmology. 2005. ; 112: 1448–1454. [DOI] [PubMed] [Google Scholar]
- 7. Guo X,, Xiao X,, Li S,, Wang P,, Jia X,, Zhang Q. Nonsyndromic high myopia in a Chinese family mapped to MYP1: linkage confirmation and phenotypic characterization. Arch Ophthalmol. 2010. ; 128: 1473–1479. [DOI] [PubMed] [Google Scholar]
- 8. Li J,, Gao B,, Guan L,, et al. Unique variants in OPN1LW cause both syndromic and nonsyndromic X-linked high myopia mapped to MYP1. Invest Ophthalmol Vis Sci. 2015. ; 56: 4150–4155. [DOI] [PubMed] [Google Scholar]
- 9. Ratnamala U,, Lyle R,, Rawal R,, et al. Refinement of the X-linked nonsyndromic high-grade myopia locus MYP1 on Xq28 and exclusion of 13 known positional candidate genes by direct sequencing. Invest Ophthalmol Vis Sci. 2011. ; 52: 6814–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Metlapally R,, Michaelides M,, Bulusu A,, et al. Evaluation of the X-linked high-grade myopia locus (MYP1) with cone dysfunction and color vision deficiencies. Invest Ophthalmol Vis Sci. 2009. ; 50: 1552–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neitz J,, Neitz M. The genetics of normal and defective color vision. Vision Res. 2011. ; 51: 633–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neitz M,, Carroll J,, Renner A,, Knau H,, Werner JS,, Neitz J. Variety of genotypes in males diagnosed as dichromatic on a conventional clinical anomaloscope. Vis Neurosci. 2004. ; 21: 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carroll J,, Neitz M,, Hofer H,, Neitz J,, Williams DR. Functional photoreceptor loss revealed with adaptive optics: an alternate cause of color blindness. Proc Natl Acad Sci U S A. 2004; 101: 8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ueyama H,, Kuwayama S,, Imai H,, et al. Analysis of L-cone/M-cone visual pigment gene arrays in Japanese males with protan color-vision deficiency. Vision Res. 2004. ; 44: 2241–2252. [DOI] [PubMed] [Google Scholar]
- 15. Crognale MA,, Fry M,, Highsmith J,, et al. Characterization of a novel form of X-linked incomplete achromatopsia. Vis Neurosci. 2004. ; 21: 197–203. [DOI] [PubMed] [Google Scholar]
- 16. Mizrahi-Meissonnier L,, Merin S,, Banin E,, Sharon D. Variable retinal phenotypes caused by mutations in the X-linked photopigment gene array. Invest Ophthalmol Vis Sci. 2010. ; 51: 3884–3892. [DOI] [PubMed] [Google Scholar]
- 17. McClements M,, Davies WI,, Michaelides M,, et al. Variations in opsin coding sequences cause X-linked cone dysfunction syndrome with myopia and dichromacy. Invest Ophthalmol Vis Sci. 2013. ; 54: 1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gardner JC,, Liew G,, Quan YH,, et al. Three different cone opsin gene array mutational mechanisms with genotype-phenotype correlation and functional investigation of cone opsin variants. Hum Mutat. 2014. ; 35: 1354–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ueyama H,, Muraki-Oda S,, Yamade S,, et al. Unique haplotype in exon 3 of cone opsin mRNA affects splicing of its precursor, leading to congenital color vision defect. Biochem Biophys Res Commun. 2012. ; 424: 152–157. [DOI] [PubMed] [Google Scholar]
- 20. Carroll J,, Baraas RC,, Wagner-Schuman M,, et al. Cone photoreceptor mosaic disruption associated with Cys203Arg mutation in the M-cone opsin. Proc Natl Acad Sci U S A. 2009; 106: 20948–20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carroll J,, Dubra A,, Gardner JC,, et al. The effect of cone opsin mutations on retinal structure and the integrity of the photoreceptor mosaic. Invest Ophthalmol Vis Sci. 2012. ; 53: 8006–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wagner-Schuman M,, Neitz J,, Rha J,, Williams DR,, Neitz M,, Carroll J. Color-deficient cone mosaics associated with Xq28 opsin mutations: A stop codon versus gene deletions. Vision Res. 2010. ; 50: 2396–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McClements M,, Davies WI,, Michaelides M,, et al. X-linked cone dystrophy and colour vision deficiency arising from a missense mutation in a hybrid L/M cone opsin gene. Vision Res. 2013. ; 80: 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kazmi MA,, Sakmar TP,, Ostrer H. Mutation of a conserved cysteine in the X-linked cone opsins causes color vision deficiencies by disrupting protein folding and stablilty. Invest Ophthalmol Vis Sci. 1997. ; 38: 1074–1081. [PubMed] [Google Scholar]
- 25. Karnik SS,, Sakmar TP,, Chen H-B,, Khorana HG. Cysteine residues 110 and 187 are required for the formation of correct structure in bovine rhodopsin. Proc Natl Acad Sci U S A. 1988; 85: 8459–8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hwa J,, Reeves PJ,, Klein-Seetharaman J,, Davidson F,, Khorana HG. Structure and function in rhodopsin: further elucidation of the role of the intradiscal cysteines Cys-110, −185, and −187, in rhodopsin folding and function. Proc Natl Acad Sci U S A. 1999; 96: 1932–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jagla WM,, Jägle H,, Hayashi T,, Sharpe LT,, Deeb SS. The molecular basis of dichromatic color vision in males with multiple red and green visual pigment genes. Hum Mol Genet. 2002. ; 11: 23–32. [DOI] [PubMed] [Google Scholar]
- 28. Carroll J,, Neitz J,, Neitz M. Estimates of L:M cone ratio from ERG flicker photometry and genetics. J Vis. 2002. ; 2: 531–542. [DOI] [PubMed] [Google Scholar]
- 29. Hofer H,, Carroll J,, Neitz J,, Neitz M,, Williams DR. Organization of the human trichromatic cone mosaic. J Neurosci. 2005. ; 25: 9669–9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McMahon C,, Carroll J,, Awua S,, Neitz J,, Neitz M. The L:M cone ratio in males of African descent with normal color vision. J Vis. 2008; 8 (2): 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gunther KL,, Neitz J,, Neitz M. Nucleotide polymorphisms upstream of the X-chromosome opsin gene array tune L:M cone ratio. Vis Neurosci. 2008. ; 25: 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cooper RF,, Dubis AM,, Pavaskar A,, Rha J,, Dubra A,, Spatial Carroll J. and temporal variation of rod photoreceptor reflectance in the human retina. Biomed Opt Express. 2011; 2: 2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dubra A,, Sulai Y. Reflective afocal broadband adaptive optics scanning ophthalmoscope. Biomed Opt Express. 2011. ; 2: 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dubra A,, Sulai Y,, Norris JL,, et al. Noninvasive imaging of the human rod photoreceptor mosaic using a confocal adaptive optics scanning ophthalmoscope. Biomed Opt Express. 2011. ; 2: 1864–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scoles D,, Sulai YN,, Langlo CS,, et al. In vivo imaging of human cone photoreceptor inner segments. Invest Ophthalmol Vis Sci. 2014. ; 55: 4244–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dubis AM,, Cooper RF,, Aboshiha J,, et al. Genotype-dependent variability in residual cone structure in achromatopsia: towards developing metrics for assessing cone health. Invest Ophthalmol Vis Sci. 2014. ; 55: 7303–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dubra A,, Harvey Z. Registration of 2D images from fast scanning ophthalmic instruments. : Fischer B,, Dawant B,, Lorenz C, Biomedical Image Registration. Berlin: Springer-Verlag; 2010: 60–71. [Google Scholar]
- 38. Garrioch R,, Langlo C,, Dubis AM,, Cooper RF,, Dubra A,, Carroll J. Repeatability of in vivo parafoveal cone density and spacing measurements. Optom Vis Sci. 2012. ; 89: 632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilk MA,, McAllister JT,, Cooper RF,, et al. Relationship between foveal cone specialization and pit morphology in albinism. Invest Ophthalmol Vis Sci. 2014. ; 55: 4186–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tanna H,, Dubis AM,, Ayub N,, et al. Retinal imaging using commercial broadband optical coherence tomography. Br J Ophthalmol. 2010. ; 94: 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McAllister JT,, Dubis AM,, Tait DM,, et al. Arrested development: high-resolution imaging of foveal morphology in albinism. Vision Res. 2010. ; 50: 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schneider CA,, Rasband WS,, Eliceiri KW. NIH, Image to ImageJ: 25 years of image analysis. Nat Methods. 2012; 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Michaelides M,, Rha J,, Dees EW,, et al. Integrity of the cone photoreceptor mosaic in oligocone trichromacy. Invest Ophthalmol Vis Sci. 2011. ; 52: 4757–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Akimitsu N. Messenger RNA surveillance systems monitoring proper translation termination. J Biochem. 2008. ; 143: 1–8. [DOI] [PubMed] [Google Scholar]
- 45. Oyster CW. The Human Eye: Structure and Function. Sunderland MA: Sinauer Associates, Inc.; 1999: 765. [Google Scholar]
- 46. Chen RWS,, Greenberg JP,, Lazow MA,, et al. Autofluorescence imaging and spectral-domain optical coherence tomography in incomplete congenital stationary night blindness and comparison with retinitis pigmentosa. Am J Ophthalmol. 2011. ; 153: 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wolsley CJ,, Saunders KJ,, Silvestri G,, Anderson RS. Investigation of changes in the myopic retina using multifocal electroretinograms, optical coherence tomography and peripheral resolution acuity. Vision Res. 2008. ; 48: 1554–1561. [DOI] [PubMed] [Google Scholar]
- 48. Lam DS,, Leung KS,, Mohamed S,, et al. Regional variations in the relationship between macular thickness measurements and myopia. Invest Ophthalmol Vis Sci. 2007. ; 48: 376–382. [DOI] [PubMed] [Google Scholar]
- 49. Spaide RF,, Curcio CA. Anatomical correlates to the bands seen in the outer retina by optical coherence tomography: literature review and model. Retina. 2011; 31: 1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Staurenghi G,, Sadda S,, Chakravarthy U,, Spaide RF. International nomenclature for optical coherence tomography P. Proposed lexicon for anatomic landmarks in normal posterior segment spectral-domain optical coherence tomography: the IN•OCT consensus. Ophthalmology. 2014; 121: 1572–1578. [DOI] [PubMed] [Google Scholar]
- 51. Cooper RF,, Wilk MA,, Tarima S,, Carroll J. Evaluating descriptive metrics of the human cone mosaic. Invest Ophthalmol Vis Sci. 2016; 57: 2992–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilk MA,, Dubis AM,, Cooper RF,, Summerfelt P,, Dubra A,, Carroll J.Assessing the spatial relationships between fixation and foveal specializations. Vision Res. [published online ahead of print June 18, 2016] . doi: http://dx.doi.org/10.1016/j.visres.2016.05.001. [DOI] [PMC free article] [PubMed]
- 53. Curcio CA,, Sloan KR,, Kalina RE,, Hendrickson AE. Human photoreceptor topography. J Comp Neurol. 1990. ; 292: 497–523. [DOI] [PubMed] [Google Scholar]
- 54. Zhang T,, Godara P,, Blancob ER,, et al. Variability in human cone topography assessed by adaptive optics scanning laser ophthalmoscopy. Am J Ophthalmol. 2015. ; 160: 290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Menghini M,, Lujan BJ,, Zayit-Soudry S,, et al. Correlation of outer nuclear layer thickness with cone density values in patients with retinitis pigmentosa and healthy subjects. Invest Ophthalmol Vis Sci. 2015. ; 56: 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li KY,, Tiruveedhula P,, Roorda A. Intersubject variability of foveal cone photoreceptor density in relation to eye length. Invest Ophthalmol Vis Sci. 2010. ; 51: 6858–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dabir S,, Mangalesh S,, Schouten JS,, et al. Axial length and cone density as assessed with adaptive optics in myopia. Indian J Ophthalmol. 2015. ; 63: 423–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Obata R,, Yanagi Y. Quantitative analysis of cone photoreceptor distribution and its relationship with axial length age, and early age-related macular degeneration. PLoS One. 2014. ; 9: e91873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rha J,, Dubis AM,, Wagner-Schuman M,, et al. Spectral domain optical coherence tomography and adaptive optics: imaging photoreceptor layer morphology to interpret preclinical phenotypes. Adv Exp Med Biol. 2010. ; 664: 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Scoles D,, Flatter JA,, Cooper RF,, et al. Assessing photoreceptor structure associated with ellipsoid zone disruptions visualized with optical coherence tomography. Retina. 2016. ; 36: 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hansen SO,, Cooper RF,, Dubra A,, Carroll J,, Weinberg DV. Selective cone photoreceptor injury in acute macular neuroretinopathy. Retina. 2013. ; 33: 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cideciyan AV,, Hufnagel RB,, Carroll J,, et al. Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Hum Gene Ther. 2013. ; 24: 993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sun LW,, Johnson RD,, Langlo C,, et al. Assessing photoreceptor structure in retinitis pigmentosa and Usher syndrome. Invest Ophthalmol Vis Sci. 2016; 57: 2428–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wagner-Schuman M,, Dubis AM,, Nordgren RN,, et al. Race- and sex-related differences in retinal thickness and foveal pit morphology. Invest Ophthalmol Vis Sci. 2011. ; 52: 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.