

ABSTRACT
Mechanistic studies of many processes in Agrobacterium tumefaciens have been hampered by a lack of genetic tools for characterization of essential genes. In this study, we used a Tn7-based method for inducible control of transcription from an engineered site on the chromosome. We demonstrate that this method enables tighter control of inducible promoters than plasmid-based systems and can be used for depletion studies. The method enables the construction of depletion strains to characterize the roles of essential genes in A. tumefaciens. Here, we used the strategy to deplete the alphaproteobacterial master regulator CtrA and found that depletion of this essential gene results in dramatic rounding of cells, which become nonviable.
IMPORTANCE Agrobacterium tumefaciens is a bacterial plant pathogen and natural genetic engineer. Thus, studies of essential processes, including cell cycle progression, DNA replication and segregation, cell growth, and division, may provide insights for limiting disease or improving biotechnology applications.
INTRODUCTION
Members of the genus Agrobacterium are common soil-dwelling bacteria. Most are saprophytic and survive primarily by consuming decaying organic material; but some cause phytopathic diseases, including Agrobacterium rhizogenes (hairy root disease), Agrobacterium rubi (cane gall disease), Agrobacterium vitis (crown gall of grape) and Agrobacterium tumefaciens (crown gall disease). During infection, A. tumefaciens transfers a portion of its DNA (transfer DNA [T-DNA]) to plants, resulting in the formation of crown galls in susceptible flowering plants. Infected plants form tumors that limit transport of water and nutrients, leading to decreased vigor and crop yield (1, 2). Because of the ubiquity of A. tumefaciens and its ability to evade plant defenses, crown gall disease continues to be a problem, with documented economic impact on fruit and nut trees, particularly in nurseries (2). Thus, A. tumefaciens has become a model for the study of host-pathogen interactions (3–5) and processes related to pathogenicity, including type IV secretion (6), quorum sensing (7), and biofilm formation (8). Finally, mechanistic studies of A. tumefaciens-mediated DNA transfer into host cells have been harnessed for the genetic engineering of plants, fungi, and mammals (5, 9–11).
While the processes related to phytopathogenicity have been subject to intense scrutiny, processes essential for A. tumefaciens survival in the environment, both in the rhizosphere and in association with hosts, are poorly understood. Recent studies have shown that A. tumefaciens cells exhibit a striking pattern of polar cell growth (12–14), leading to a vast array of experimental questions (15). Proper spatial and temporal regulation of the cell cycle must be required in order to coordinate key processes, such as chromosome replication and segregation, cell wall biogenesis, and cell division. Understanding the mechanisms underlying these essential processes will provide key insights into the basic biology of A. tumefaciens that can be exploited to limit pathogenesis or promote genetic engineering.
A. tumefaciens is a genetically tractable bacterium, and current methods exist for the construction of transposon mutants and deletion of nonessential genes (16). A plasmid-based complementation system using a reengineered lac promoter is available and has been shown to provide tight regulation of target genes (17). Despite rigorous efforts, depletion of predicted essential genes of A. tumefaciens using these vectors has failed, suggesting that some proteins are expressed at biologically relevant levels even when repressed. Thus, a limitation of the molecular approaches currently available is the inability to construct depletion mutants for the characterization of essential genes. Here, we report the construction of vectors that integrate into a single engineered site on the A. tumefaciens chromosome, enabling single-copy gene complementation and depletion studies. Advantages of the chromosomal insertion system include bypassing the need for continued antibiotic selection, tighter regulation, and single-copy complementation.
Transposon Tn7-based plasmid systems have been described and used for single-copy integration in many bacterial species (18–23). The Tn7 transposon integrates at high frequency and specificity at the attTn7 site downstream of the glmS gene, which encodes an essential glucosamine-fructose-6-phosphate aminotransferase (18, 24). Many bacteria contain a single glmS gene and therefore a single attTn7 site for insertion of the Tn7 element, which is presumed to be a neutral site for insertion. We found that A. tumefaciens has a native attTn7 site; however, the site is not neutral for insertion. Therefore, we have engineered a strain with a second, artificial attTn7 site (a-attTn7), and we show that Tn7 insertion is highly biased toward a-attTn7. In this work, we show that mini-Tn7 vectors can serve as a valuable molecular tool for A. tumefaciens, enabling single-copy gene complementation of complex phenotypes, such as biofilm formation and motility. The degree of complementation and tightness of regulation can be tuned using different promoters on the mini-Tn7 vectors. In this work, we utilize two isopropyl-β-d-1-thiogalactopyranoside (IPTG)-inducible promoters, lac and tac. The lac promoter from Escherichia coli provides tight regulation and a relatively low level of expression (17, 25), whereas the hybrid tac promoter (derived from the lacUV5 and trp promoters) results in approximately 5- to 10-times-stronger expression (18, 26, 27). We found that the single-copy expression of green fluorescent protein (GFP) driven from the lac or tac promoter is dependent on the presence of inducer. Furthermore, we can rapidly deplete GFP from A. tumefaciens cells following the removal of the inducer.
Using the mini-Tn7 system in our engineered A. tumefaciens strain, we depleted an essential gene in A. tumefaciens. Based on evidence from a saturating transposon mutagenesis screen (28) and an inability to construct a deletion strain (29), we hypothesized that the master regulator protein CtrA is essential for A. tumefaciens cell survival. We introduced ctrA under the control of the tac promoter into the a-attTn7 site and subsequently deleted the native ctrA locus. In the presence of inducer, the cells produce CtrA, are viable, and exhibit normal rod-shaped morphology. In the absence of inducer, the cells are depleted of CtrA, are nonviable, and have a swollen, round shape. Our work has validated the utility of the mini-Tn7 system for the construction of A. tumefaciens depletion mutants.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
A list of all the bacterial strains and plasmids used in this study is provided in Table 1. Agrobacterium tumefaciens C58 and derived strains were grown in ATGN medium (30) without exogenous iron at 28°C with shaking. When appropriate, antibiotics were used at the following working concentrations: kanamycin, 300 μg/ml, and gentamicin, 200 μg/ml. When indicated, IPTG was used as an inducer at a concentration of 1 mM.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Reference/source |
|---|---|---|
| Plasmids | ||
| pTNS3 | Apr; helper plasmid encoding the site-specific TnsABCD Tn7 transposition pathway | 32 |
| pUC18-mini-Tn7T-GM-LAC | Apr Gmr; mini-Tn7 vector containing lacIq and tac promoter | 18 |
| pUC18-mini-Tn7T-GM-Plac-HA | Apr Gmr; mini-Tn7 vector containing lacIq and lac promoter | This study |
| pUC18-mini-Tn7T-GM-Ptac-HA | Apr Gmr; mini-Tn7 vector containing lacIq and tac promoter | This study |
| pUC18-mini-Tn7T-GM-Ptac-sfgfp | Apr Gmr; mini-Tn7 vector containing sfGFP under the control of the tac promoter | This study |
| pUC18-mini-Tn7T-GM-Plac-sfgfp | Apr Gmr; mini-Tn7 vector containing sfGFP under the control of the lac promoter | This study |
| pNTPS138/139 | Kmr; Suicide vector containing oriT and sacB | D. Alley |
| pMEH0125 | Suicide plasmid for allelic exchange of tetRA locus | Fuqua laboratory |
| pNTPS139 ΔtetRA::a-attTn7 | Suicide plasmid for allelic exchange of tetRA locus with artificial attTn7 site | This study |
| pSRKKm | Kmr; broad-host-range vector containing lacIq and lac promoter | 17 |
| pSRKKm-Plac-HA | pSRKKm vector containing lacIq and lac promoter with an HA tag | This study |
| pSRKKm-Plac-sfgfp | pSRKKm vector containing lacIq and lac promoter with sfGFP | This study |
| pSRKKm-Ptac-sfgfp | pSRKKm vector containing lacIq and tac promoter with sfGFP | This study |
| pNTPS138Δrem | Kmr Sucs; deletion plasmid for rem | Fuqua laboratory |
| pSRKKm-Plac-rem-HA | Kmr; complementation vector containing rem-HA under the control of the lac promoter | This study |
| pUC18-mini-Tn7T-GM-Plac-rem-HA | Apr Gmr; mini-Tn7 vector containing rem-HA under the control of the lac promoter | This study |
| pNTPS138ΔuppX | Kmr Sucs; deletion plasmid for uppX | Fuqua laboratory |
| pSRKKm-Plac-uppX-HA | Kmr; complementation vector containing uppX-HA under the control of lac promoter | This study |
| pUC18-mini-Tn7T-GM-Plac-uppX-HA | Apr Gmr; mini-Tn7 vector containing uppX-HA under the control of the lac promoter | This study |
| pJEH005 (pNTPS138ΔctA) | Kmr Sucs; deletion plasmid for ctrA | Fuqua laboratory |
| pUC18-mini-Tn7T-GM-Plac-ctrA | Apr Gmr; mini-Tn7 vector containing ctrA under the control of the lac promoter | This study |
| pUC18-mini-Tn7T-GM-Ptac-ctrA | Apr Gmr; mini-Tn7 vector containing ctrA under the control of the tac promoter | This study |
| pKC129 | Source of sfGFP | Huang laboratory |
| pRVGFPC-2 | Source of multiple-cloning-site fragment | 50 |
| E. coli strains | ||
| DH5α | Cloning strain | Life Technologies |
| S17-1 | Smr; RP4-2 Tc::Mu Km-Tn7; for plasmid mobilization | 51 |
| A. tumefaciens strains | ||
| C58 | Nopaline type strain; pTiC58; pAtC58 | 52 |
| C58 pSRKKm-Plac-HA | C58 with empty pSRKKm plasmid | This study |
| C58ΔtetRA::a-attTn7 | Replacement of the tetRA locus with an artificial attTn7 site | This study |
| C58ΔtetRA::a-attTn7 pSRKKm-Plac-HA | C58ΔtetRA::a-attTn7 with empty pSRKm-Plac-HA plasmid | This study |
| C58ΔtetRA::a-attTn7 pSRKKm-Plac-sfgfp | C58ΔtetRA::a-attTn7 with pSRKm expressing sfGFP under the control of the lac promoter | This study |
| C58ΔtetRA::a-attTn7 pSRKKm-Ptac-sfgfp | C58ΔtetRA::a-attTn7 with pSRKm expressing sfGFP under the control of the tac promoter | This study |
| C58ΔtetRA::mini-Tn7T-GM-LAC | Mini-Tn7T-GM-LAC inserted into a-attTn7 site | This study |
| C58ΔtetRA::mini-Tn7T-GM-Plac-sfgfp | Mini-Tn7T-GM-Plac-sfgfp inserted into a-attTn7 site to express sfGFP under the lac promoter | This study |
| C58ΔtetRA::mini-Tn7T-GM-Ptac-sfgfp | Mini-Tn7T-GM-Ptac-sfgfp inserted into a-attTn7 site to express sfGFP under the tac promoter | This study |
| C58ΔtetRA::a-attTn7 Δrem | Δrem | This study |
| C58ΔtetRA::mini-Tn7T-GM-Plac-rem-HA Δrem | Chromosome-based complementation of Δrem with Rem-HA | This study |
| C58ΔtetRA::a-attTn7 Δrem pSRKKm-Plac-HA | Empty pSRKKm-Plac-HA plasmid in Δrem | This study |
| C58ΔtetRA::a-attTn7 Δrem pSRKKm-Plac-rem-HA | Plasmid-based complementation of Δrem with Rem-HA | This study |
| C58ΔtetRA::a-attTn7 ΔuppX | ΔuppX | This study |
| C58ΔtetRA::mini-Tn7T-GM-Plac-uppX-HA ΔuppX | Chromosome-based complementation of ΔuppX with UppX-HA | This study |
| C58ΔtetRA::a-attTn7 ΔuppX pSRKKm-Plac-HA | Empty pSRKKm-Plac-HA plasmid in ΔuppX | This study |
| C58ΔtetRA::a-attTn7 ΔuppX pSRKKm-Plac-uppX-HA | Plasmid-based complementation of ΔuppX with UppX-HA | This study |
| C58ΔtetRA::mini-Tn7T-GM-Plac-ctrA | Mini-Tn7T-GM-Ptac-ctrA inserted into a-attTn7 site to express CtrA under the lac promoter | This study |
| C58ΔtetRA::mini-Tn7T-GM-Ptac-ctrA | Mini-Tn7T-GM-Ptac-ctrA inserted into a-attTn7 site to express CtrA under the tac promoter | This study |
| C58ΔtetRA::mini-Tn7T-GM-Ptac-ctrA ΔctrA | Chromosome-based complementation of ΔctrA with CtrA | This study |
E. coli DH5α and S17-1 λ pir were routinely cultivated in Luria-Bertani (LB) medium at 37°C with shaking. When necessary, antibiotics were used at the following concentrations: kanamycin, 50 μg/ml, and gentamicin, 20 μg/ml.
Construction of an a-attTn7 site in A. tumefaciens.
A list of all the synthesized DNA primers and gene fragments used in this study is provided in Table 2. All the custom-made DNA was made by Integrated DNA Technologies, Inc. The artificial attachment site was introduced into the tetRA locus using allelic replacement as described previously (16). A synthetic double-stranded 373-bp gBlock gene fragment, att miniTn7 tet locus, was ligated into pMEH0125 with the NdeI and NcoI restriction endonuclease sites. The resulting construct, pNTPS139 ΔtetRA::a-attTn7, was used to replace the tetRA locus with the a-attTn7 site. PCR amplification was used to verify the insertion by using primers Tet locus P5 and P6.
TABLE 2.
Synthesized DNA primers and gene fragments used in this study

Insertion of mini-Tn7 vectors in A. tumefaciens.
Standard electroporation protocols (16) were used to introduce mini-Tn7 vectors, along with the helper plasmid, pTNS3, into A. tumefaciens cells. Briefly, 40 μl of electrocompetent cells was mixed with 800 ng mini-Tn7 plasmid and 50 ng pTNS3, electroporated at 25 V/cm; after the addition of fresh medium, the transformed cells were incubated with shaking at 28°C for 4 h and plated on selective medium. Insertion was verified by colony PCR using the upstream tetRA locus primer, tet forward, and the right-end Tn7-specific primer, Tn7R109 (Table 2).
Deletion of target genes in A. tumefaciens.
Nonpolar, markerless deletions of the A. tumefaciens rem (Atu0573), uppX (Atu0480), and ctrA (Atu2434) genes were generated using the plasmids pNPTS138Δrem, pNPTS138ΔuppX, and pNPTS138ΔctrA and following an established allelic-replacement protocol (16). Primary integrants were made by conjugation with E. coli S17-1 cells as the donor strain for pNPTS138/139. The primers used for PCR verifying deletions were Atu0573 P5/Atu0573 P6, Atu0480 P5/Atu0480 P6, and CtrA P5/CtrA P6 (Table 2). For the ctrA depletion, two strains containing a chromosome-based complementation copy of CtrA with either an inducible lac or tac promoter were used with a modified protocol adding 1 mM IPTG to the medium at all steps to maintain expression. Only the strain containing the complementing tac promoter-driven copy of ctrA was successful in deleting the chromosomal copy of ctrA.
Construction of complementation plasmids.
Complementation plasmids of the A. tumefaciens rem (Atu0573), uppX (Atu0480), and ctrA (Atu2434) genes were made by PCR amplification of genomic DNA using the primers Atu0573 NdeI F/Atu0573 BamHI R, Atu0480 NdeI F/Atu0480 BamHI R, and CtrA NdeI F/CtrA BamHI R (Table 2). Amplified gene fragments were digested and ligated into plasmids digested with NdeI/BamHI to make the corresponding plasmids listed for each gene in Table 1. Additional information on plasmid modification can be found in the supplemental material.
Western blot analysis.
For monitoring of cytoplasmic GFP expression, 40 ml of cells was grown to an optical density at 600 nm (OD600) of 0.6 in the presence of IPTG. A 2-ml aliquot of the culture was removed, and the pellet was stored to serve as the time zero (plus-IPTG) point. The remaining 38 ml of culture was washed with ATGN via centrifugation at 4,000 rpm for 10 min three times to remove the IPTG. The washed pellet was resuspended in 20 ml of fresh ATGN normalized to an OD600 of 0.4. Aliquots were collected hourly to monitor the levels of GFP expression during the depletion. The cell pellets were stored at −80°C until further analysis. The cell pellets were incubated with 100 μl of a master mix containing 1 ml of BugBuster protein extraction reagent (Novagen) and supplemented with 1× EDTA-free protease inhibitor cocktail (Sigma), 10 μl of lysonase (Novagen), 2,500 U/ml DNase I (Thermo Scientific), and 1 mM dithiothreitol (DTT) (Thermo Scientific) for 25 min with shaking at room temperature to lyse the cell pellets. The whole-cell lysates were clarified by centrifugation at 10,000 rpm for 15 min. A final concentration of 1× Laemmli buffer was added to the cleared cell lysates. Samples were boiled at 100°C for 5 min prior to loading on an SDS-12% PAGE gel. The separated proteins were electroblotted onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad) and blocked overnight in 5% nonfat dry milk powder solubilized in 1% TBST (1× Tris-buffered saline [TBS], 1% Tween 20). The blocked PVDF membranes were probed with anti-GFP (1:3,000) monoclonal antibody for 1 h in 2.5% milk-TBST, followed by incubation with a donkey anti-mouse horseradish peroxidase-conjugated secondary antibody for 1 h in 2.5% milk-TBST. The secondary antibody was detected using the ECL Plus HRP substrate (Thermo Scientific Pierce).
Swim plate assays.
Swim plate assays were conducted as described previously (31). Briefly, 5 μl of cells normalized to an OD600 of 0.6 was spotted on ATGN plates containing 0.3% agar and incubated in a sealed humid chamber for 5 days. The diameters of the swim rings were measured on days 3 and 5. Each strain was tested in triplicate during three independent experiments.
Biofilm assays.
Biofilm assays were conducted as described previously (31). Bacteria were grown to mid-log phase in ATGN medium and diluted to an OD600 of 0.05. Three milliliters of diluted bacteria was placed in the wells of 12-well polystyrene plates containing polyvinyl chloride coverslips placed vertically in each well. The plates were incubated at room temperature for 48 h. After incubation, the coverslips were removed and rinsed with distilled water (dH2O) to remove planktonic cells. The coverslips were stained with 0.1% crystal violet for 5 to 10 min and destained with 33% acetic acid. The A600 of the solubilized crystal violet and the OD600 of the cultures from each well were measured. The biofilm score was calculated as the A600 divided by the OD600 to allow normalization of strains. Three biological replicates were completed in triplicate for each experiment.
Growth curve analysis.
For the CtrA depletion strain, cells were grown in ATGN medium in the presence of 1 mM IPTG to an OD600 of 0.4. The cells were washed 3 times by centrifugation in ATGN medium to remove the inducer and resuspended to an OD600 of 0.05 in ATGN medium with or without IPTG. For each condition, 200 μl of sample was added in triplicate to a 96-well plate. The OD600 was measured in a BioTek Synergy H1 Hybrid Reader every 15 min for 48 h at 28°C with 1 min of shaking before reads.
Cell viability assays.
For the CtrA depletion strain, cells were grown in ATGN medium in the presence of 1 mM IPTG to an OD600 of 0.4. The cells were washed 3 times by centrifugation in ATGN medium to remove the inducer and resuspended to an OD600 of 0.05 in ATGN medium with or without IPTG. Serial dilutions of the cell culture were plated on ATGN medium containing 1 mM IPTG immediately after washing to determine the number of CFU at the outset of the experiment. Serial dilutions from cultures with and without IPTG were plated after 2, 4, 8, and 24 h of induction or depletion to obtain counts of CFU. Relative cell viability scores were calculated as the number of CFU at the indicated time point (2 h, 4 h, 8 h, or 24 h) divided by the number of CFU at the outset of the experiment (0 h). Each condition was tested in duplicate during three independent experiments.
Fluorescence and time lapse microscopy.
A small volume (∼0.8 μl) of cells in exponential phase was applied to a 1% agarose pad as described previously (12). For time lapse imaging, the cells were imaged every 10 min for the duration of the experiment. Differential interference microscopy and epifluorescence microscopy were performed with an inverted Nikon Eclipse TiE with a QImaging Rolera em-c2 1K electron-multiplying charge-coupled-device (EMCCD) camera and Nikon Elements imaging software.
RESULTS AND DISCUSSION
Construction of A. tumefaciens with an a-attTn7 site enables neutral insertion of mini-Tn7 vectors.
We engineered a Tn7-based method for inducible control of transcription from an engineered site on the chromosome of A. tumefaciens (Fig. 1). Transposon Tn7-based plasmid systems have been described and used for single-copy gene integration in many bacterial species (18–23, 32). In most bacterial species, a single neutral site of Tn7 insertion (attTn7) is present in an intergenic region downstream of the glmS gene (18). We attempted to use the Tn7-based system in A. tumefaciens; however, the native attTn7 is not neutral (Fig. 1A). In A. tumefaciens, the glmS gene overlaps the adjacent gene (Fig. 1A), and insertion of a mini-Tn7 vector into this operon causes reduced transformation efficiency, small colonies, and slow growth. Therefore, we engineered an a-attTn7 site at a cryptic tetracycline locus in the A. tumefaciens genome (Fig. 1B) using a standard allelic-exchange protocol (16). Replacement of the tetRA locus with a-attTn7 and subsequent integration of a Tn7 cassette did not impact growth, motility, or biofilm formation of A. tumefaciens (Fig. 1C; see Fig. S1 in the supplemental material), suggesting that our engineered site is neutral. Indeed, the engineered strain contains two attTn7 sites, but PCR-based assays for transposon insertion at both sites revealed that insertion is strongly biased toward the engineered site, with over 97% (72/74) of integration events detected only at the a-attTn7 site.
FIG 1.
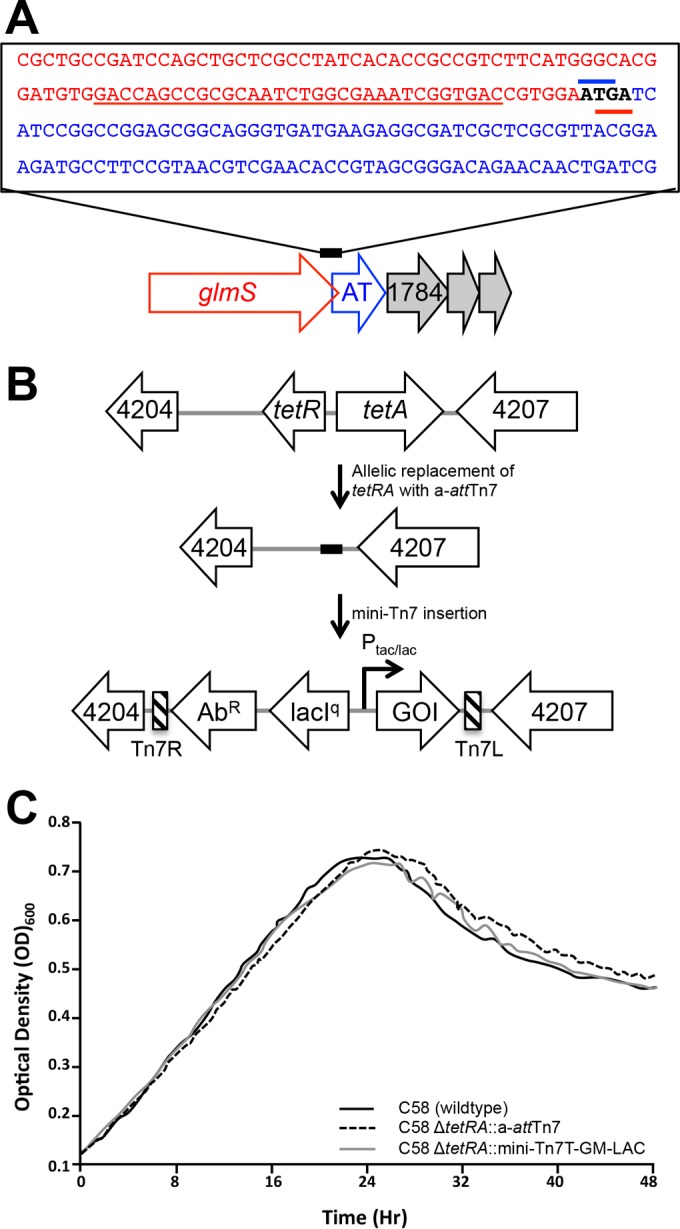
Construction of an a-attTn7 site in A. tumefaciens. (A) The native att site (underlined in red) is located at the 3′ end of the glmS gene (Atu1786) in A. tumefaciens. Insertion of Tn7 constructs into the native att site is not neutral, presumably due to the overlap of the glmS stop codon (red bar) with the start codon (blue bar) of the downstream gene encoding an AT (Atu1785). (B) The entire boxed sequence containing the att site in panel A was introduced into a cryptic tetracycline locus on the A. tumefaciens chromosome. Allelic exchange was used to replace the cryptic tetracycline locus (tetRA; Atu4205 to Atu4206) with the a-attTn7 site. The mini-Tn7 cassette contains an antibiotic resistance gene (AbR), the lacI repressor, and either a strong (Ptac) or weak (Plac) promoter to drive a gene of interest (GOI). The left (Tn7L) and right (Tn7R) ends of Tn7 are indicated by the striped bars. (C) Replacement of the tetRA locus with a-attTn7 (dashed line) and subsequent insertion of a mini-Tn7 cassette (gray line) do not impact cell growth in ATGN compared to the wild-type parental strain (C58; solid black line).
Single-copy chromosomal complementation of complex phenotypes is tighter than plasmid-based complementation.
Single-copy chromosomal complementation provides several advantages in the complementation of complex phenotypes. First, genes inserted in the chromosome are stably maintained in the absence of selection, enabling complementation studies in the absence of antibiotics, which may slow cell growth. Second, the genes are present in single copies, reducing the possible gene dosage effect that can occur when multicopy plasmids are used. Third, the degree of leakiness (expression in the absence of inducer) should be reduced in single copy. Finally, the level of expression from multicopy plasmids using lac promoter derivatives under inducing conditions varies in individual cells (33), and chromosomal insertion reduces the genetic noise. Here, we compared single-copy chromosomal complementation and multicopy plasmid-based complementation with genes under the control of the same promoter in the restoration of two complex phenotypes, motility and biofilm formation.
In the Rhizobiales, a regulatory hierarchy controls the ordered assembly of the basal body, the hook, and the filament to produce functional flagella and drive motility (34–37). Rem (regulator of exponential-growth motility) activates the flagellar genes and is required for assembly of the flagella and swimming. In order to assay for motility, the swim ring diameters of the wild-type, engineered, Δrem, and complementation strains were measured after 5 days on ATGN soft agar (0.3%) (Fig. 2). In A. tumefaciens, the Δrem strain retains only ∼10% of the motility of the wild-type or a-attTn7 strain (Fig. 2B). When a single copy of hemagglutinin-tagged rem (rem-HA) is introduced at the a-attTn7 site under the control of the lac promoter, partial complementation is observed; motility is restored to ∼30% in the presence of IPTG (Fig. 2B). Notably, in the absence of inducer, motility is not restored, indicating that the partial complementation requires the presence of the inducer (Fig. 2B). Plasmid-based complementation of Δrem in the presence of IPTG restores motility to ∼75% of that of the wild-type strain; however, partial complementation to ∼40% occurs in the absence of IPTG (Fig. 2C). Taken together, these results indicate that single-copy complementation of motility on the chromosome is partial but limits leaky expression. A reduction in protein expression from the single-copy complementation likely accounts for the relatively low level of complementation compared to plasmid-based complementation. In addition, since motility is subject to cell cycle regulation in A. tumefaciens (38), it is possible that the constitutive expression of Rem from the inducible promoter (in either the chromosomal or plasmid-based complementation) results in partial complementation.
FIG 2.

Chromosomal and plasmid-based complementation of motility in Δrem. (A) Schematics of strains used, comparing single-copy chromosomal complementation (strains 1 to 4) and multiple-copy plasmid complementation (strains 5 to 8). (B) Strains 1 to 4 were assayed for swimming motility on ATGN soft-agar plates in the presence (black bars) or absence (white bars) of the inducer, IPTG. The data were normalized to the results obtained in the wild-type strain (strain 1). S.E., standard error of the mean. (C) Strains 5 to 8 were assayed for swimming motility on ATGN soft-agar plates containing kanamycin to maintain the plasmid in the presence (black bars) or absence (white bars) of the inducer, IPTG. In the absence of the inducer, we repeatedly observed an increase in the mucoidy at the center of the swim rings. In the Δrem strains, which are nonmotile, this increase in mucoidy leads to a slightly larger swim ring diameter. The data were normalized to the results obtained in the wild-type strain containing an empty vector (strain 5). Swim ring diameters were measured after 5 days of incubation at room temperature. The data shown are the means of the results of three independent experiments completed in triplicate.
In order to determine if the single-copy chromosomal complementation generally provides tighter regulation, we assayed the complementation of a mutant impaired in biofilm formation. In A. tumefaciens, biofilm formation is dependent, at least in part, on the ability to extrude a unipolar polysaccharide (UPP) (38, 39). The UPP plays an essential role in attachment to surfaces and subsequent biofilm formation (37, 40). Here, we compare the abilities of chromosome-based and plasmid-based strategies to complement the biofilm formation of a ΔuppX mutant, which fails to produce a UPP. Adherent biofilm formation on plastic coverslips was measured after 48 h of incubation for the wild-type, a-attTn7, ΔuppX, and complementation strains (Fig. 3). We found that the ΔuppX mutant retained only ∼25% of the biofilm formation of the wild-type or a-attTn7 strain. When uppX-HA was introduced in a single copy at the a-attTn7 site under the control of the lac promoter, IPTG-dependent partial complementation (∼50%) was observed (Fig. 3B). Introduction of uppX-HA under the control of the lac promoter on a plasmid resulted in a similar degree of complementation in the presence of inducer (∼50%); however, partial complementation was also observed in the absence of inducer (∼40%) (Fig. 3C). These observations suggest that single-copy chromosomal insertion enables tight regulation of target genes under the control of the lac promoter, potentially enabling the depletion of target genes.
FIG 3.

Chromosomal and plasmid-based complementation of biofilm formation in ΔuppX. (A) Schematics of the strains used, comparing single-copy chromosomal complementation (strains 1 to 4) and multiple-copy plasmid complementation (strains 5 to 8). (B) Strains 1 to 4 were assayed for biofilm formation on vertical plastic coverslips immersed in ATGN medium in the presence (black bars) or absence (white bars) of the inducer, IPTG. The data were normalized to the results obtained in the wild-type strain (strain 1). (C) Strains 5 to 8 were assayed for biofilm formation in ATGN medium containing kanamycin to maintain the plasmid in the presence (black bars) or absence (white bars) of IPTG. The data were normalized to the results obtained in the wild-type strain containing an empty vector (strain 5). For all the strains, the coverslips were removed after 48 h of incubation at room temperature and rinsed to remove any loosely associated cells. Adherent biomass was determined as the absorbance of solubilized crystal violet (A600), and the optical density of the planktonic culture (OD600) was measured. Biofilm scores were calculated as the A600/OD600 ratio, and the data were normalized. The data shown are the means of three independent experiments completed in triplicate. Representative coverslips prior to crystal violet solubilization are shown for each strain.
Mini-Tn7 vectors expressing sfGFP in a-attTn7 are tightly regulated.
To assess the ability of the mini-Tn7 system to enable depletion studies, we introduced the gene encoding superfolder GFP (sfGFP) (41–43) into the a-attTn7 site under the control of the lac and tac promoters (Fig. 4A). As expected, we observed that the tac promoter drives a much higher level of expression than the lac promoter, as assessed by detection of GFP levels in Western blots and the relative amount of fluorescence emitted by GFP in live cells when IPTG was present (Fig. 4B and C). Similarly, we found that expression of sfgfp under the control of the lac or tac promoter from the chromosomal integration was much lower than that from the same promoter introduced on a plasmid (see Fig. S2 in the supplemental material). These observations suggest that the tac and lac promoters can drive relatively high or low levels of expression as needed for specific target genes.
FIG 4.

GFP driven from Ptac or Plac in the a-att site is rapidly depleted. (A) Schematics of strains used to compare expression of sfgfp from Ptac (top) and Plac (bottom). (B) Western blots illustrating the depletion of GFP following the removal of the inducer (IPTG) as described in Materials and Methods. Expression of GFP driven from Ptac (top) is higher than expression of GFP driven from Plac (bottom). (C) Depletion of GFP signal driven from Ptac (top) and Plac (bottom) following the removal of the inducer (IPTG) was monitored in live cells using fluorescence microscopy.
Next, we wanted to determine if sfGFP could be depleted from cells when the inducer is removed. Cells grown in the presence of IPTG were washed to remove the inducer, and the depletion of GFP from the cell populations was monitored by Western blotting (Fig. 4B; see Fig. S2 in the supplemental material) and fluorescence microscopy (Fig. 4C). Chromosomal insertion of sfgfp under the control of the tac and lac promoters enables rapid depletion of sfGFP in the absence of inducer (Fig. 4B; see Fig. S2 in the supplemental material). Under the control of the tac promoter, the levels of GFP began to be depleted within 2 to 3 h and were barely detectable in Western blots or live-cell imaging after 5 h. Depletion occurred more rapidly (within 2 h) under the lac promoter, presumably due to the reduced level of initial expression. Together, these data suggest that expression of genes of interest from the lac or tac promoter should enable depletion of target proteins following removal of the gene at the native locus.
Depletion of CtrA decreases viability and induces rounding of A. tumefaciens cells.
Coordination of DNA replication and segregation, cell elongation, and cell division must be properly regulated during the cell cycle of A. tumefaciens, yet little is known about the mechanisms of essential processes driving cell cycle progression in A. tumefaciens due to the limited ability to characterize essential genes. In many Alphaproteobacteria, the response regulator protein, CtrA, plays an important role as a transcription factor to mediate proper cell cycle progression (44–49). In A. tumefaciens, CtrA is essential for cell survival (28) and remains uncharacterized. Here, we depleted ctrA as a validation of the mini-Tn7 system for depletion of essential genes.
In order to deplete CtrA from A. tumefaciens cells, ctrA was introduced under the control of the tac promoter at the a-attTn7 site. In the presence of IPTG, ctrA was deleted from the native location by allelic exchange using a standard protocol (16) (Fig. 5A). Growth curve analysis of the ctrA depletion strain in the presence of IPTG indicated that growth was comparable to that of the wild type. In contrast, in the absence of IPTG, the optical density of cells depleted of CtrA increased for several hours before reaching a plateau (Fig. 5B); however, the observed increase in cell density is not a result of net population growth, but rather, is due to the elongation and swelling of individual nonviable cells (Fig. 5C and D). In order to assess the ability of cells depleted of CtrA to recover, we conducted colony-forming assays after 2, 4, 8, and 24 h of incubation in the presence or absence of IPTG. We found that the ctrA depletion rapidly resulted in loss of cell viability with no increase in the number of viable cells (Fig. 5C). In the presence of IPTG, the ctrA depletion strain is capable of cell elongation and division, resulting in the formation of a microcolony within 24 h (Fig. 5D, top row). In contrast, in the absence of IPTG, the depletion of CtrA causes the cells to elongate without dividing (Fig. 5D, bottom two rows). After 10 h of ctrA depletion, most cells begin to swell, resulting in a terminal block in cell division (Fig. 5D, bottom two rows). Between 10 and 24 h of ctrA depletion, although some cells generate constrictions (Fig. 5D, bottom row), only 11% (17/149) of individual cells complete cell division. Notably, the rare cell division events that occur are atypical and often occur at asymmetric sites, resulting in production of minicells. The cells continue to swell from the midcell and ultimately lyse. These observations are consistent with an expected role of CtrA in the regulation of cell division, as described in the closely related bacterium Sinorhizobium meliloti (48).
FIG 5.
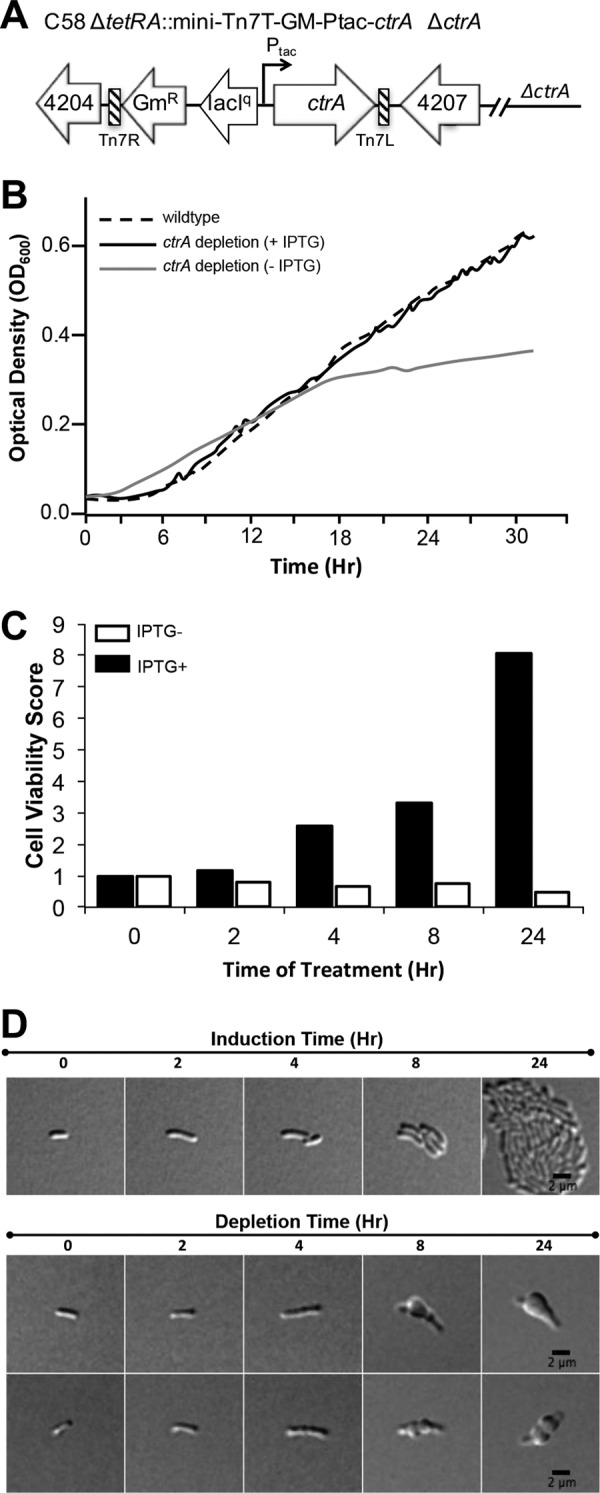
Depletion of the essential cell cycle regulator CtrA results in the production of nonviable, rounded cells. (A) Schematic of the ctrA depletion strain. (B) Growth curves of the wild-type parent strain (dashed line) and the ctrA depletion strain in the presence (solid black line) and absence (gray line) of IPTG. (C) Cell viability was assessed as the ability to produce CFU on ATGN plates containing IPTG after the ctrA depletion strain was cultured in the presence (black bars) or absence (white bars) of IPTG for each indicated time point. The cell viability score was calculated as the number of CFU at the indicated time point divided by the number of CFU at the outset of the experiment. (D) Select images from 24 h of time lapse microscopy of individual ctrA depletion cells grown on ATGN agarose pads in the presence (top row) or absence (bottom two rows) of IPTG.
Conclusions.
We have successfully engineered A. tumefaciens with an artificial attTn7 site at a neutral locus in the chromosome and constructed a set of mini-Tn7 vectors that allow integration and inducible expression of target genes from the a-attTn7 site. This approach allows more precise regulation of target genes than multicopy plasmids using the same promoters, enabling complementation studies that require tight regulation of gene expression. Furthermore, this method enables rapid depletion of target proteins. As proof of concept, we depleted ctrA, an essential gene in A. tumefaciens, and found that cells failed to divide, underwent dramatic changes in morphology, and lost viability. This method provides a necessary advance in our ability to study essential processes, such as cell cycle progression, cell growth, and cell division in A. tumefaciens.
Supplementary Material
ACKNOWLEDGMENTS
We thank Clay Fuqua, Jason Heindl, K. C. Huang, and Herbert Schweizer for providing plasmids used in this study.
This research is supported by start-up funds (P.J.B.B.) from the University of Missouri and by funds from the Initiative for Maximizing Student Diversity in Biomedical Sciences Training Program NIH NIGMS grant R25 GM 056901-16 (W.F.-C.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01392-16.
REFERENCES
- 1.Escobar MA, Dandekar AM. 2003. Agrobacterium tumefaciens as an agent of disease. Trends Plant Sci 8:380–386. doi: 10.1016/S1360-1385(03)00162-6. [DOI] [PubMed] [Google Scholar]
- 2.Pulawska J. 2010. Crown gall of stone fruits and nuts, economic significance and diversity of its causal agents: tumorigenic Agrobacterium spp. J Plant Pathol 92:S87–S98. [Google Scholar]
- 3.Citovsky V, Kozlovsky SV, Lacroix B, Zaltsman A, Dafny-Yelin M, Vyas S, Tovkach A, Tzfira T. 2007. Biological systems of the host cell involved in Agrobacterium infection. Cell Microbiol 9:9–20. doi: 10.1111/j.1462-5822.2006.00830.x. [DOI] [PubMed] [Google Scholar]
- 4.Lacroix B, Citovsky V. 2013. The roles of bacterial and host plant factors in Agrobacterium-mediated genetic transformation. Int J Dev Biol 57:467–481. doi: 10.1387/ijdb.130199bl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nester EW. 2014. Agrobacterium: nature's genetic engineer. Front Plant Sci 5:730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarez-Martinez CE, Christie PJ. 2009. Biological diversity of prokaryotic type IV secretion systems. Microbiol Mol Biol Rev 73:775–808. doi: 10.1128/MMBR.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White CE, Winans SC. 2007. Cell-cell communication in the plant pathogen Agrobacterium tumefaciens. Philos Trans R Soc Lond B Biol Sci 362:1135–1148. doi: 10.1098/rstb.2007.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danhorn T, Fuqua C. 2007. Biofilm formation by plant-associated bacteria. Annu Rev Microbiol 61:401–422. doi: 10.1146/annurev.micro.61.080706.093316. [DOI] [PubMed] [Google Scholar]
- 9.Gelvin SB. 2003. Agrobacterium-mediated plant transformation: the biology behind the “gene-jockeying” tool. Microbiol Mol Biol Rev 67:16–37. doi: 10.1128/MMBR.67.1.16-37.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pacurar DI, Thordal-Christensen H, Pacurar ML, Pamfil D, Botez C, Bellini C. 2011. Agrobacterium tumefaciens: from crown gall tumors to genetic transformation. Physiol Mol Plant Pathol 76:76–81. doi: 10.1016/j.pmpp.2011.06.004. [DOI] [Google Scholar]
- 11.Lacroix B, Tzfira T, Vainstein A, Citovsky V. 2006. A case of promiscuity: Agrobacterium's endless hunt for new partners. Trends Genet 22:29–37. doi: 10.1016/j.tig.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Brown PJ, de Pedro MA, Kysela DT, Van der Henst C, Kim J, De Bolle X, Fuqua C, Brun YV. 2012. Polar growth in the alphaproteobacterial order Rhizobiales. Proc Natl Acad Sci U S A 109:1697–1701. doi: 10.1073/pnas.1114476109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cameron TA, Anderson-Furgeson J, Zupan JR, Zik JJ, Zambryski PC. 2014. Peptidoglycan synthesis machinery in Agrobacterium tumefaciens during unipolar growth and cell division. mBio 5:e01219-14. doi: 10.1128/mBio.01219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zupan JR, Cameron TA, Anderson-Furgeson J, Zambryski PC. 2013. Dynamic FtsA and FtsZ localization and outer membrane alterations during polar growth and cell division in Agrobacterium tumefaciens. Proc Natl Acad Sci U S A 110:9060–9065. doi: 10.1073/pnas.1307241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cameron TA, Zupan JR, Zambryski PC. 2015. The essential features and modes of bacterial polar growth. Trends Microbiol 23:347–353. doi: 10.1016/j.tim.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Morton ER, Fuqua C. 2012. Genetic manipulation of Agrobacterium. Curr Protoc Microbiol Chapter 3:Unit 3D 2. doi: 10.1002/9780471729259.mc03d02s25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan SR, Gaines J, Roop RM II, Farrand SK. 2008. Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl Environ Microbiol 74:5053–5062. doi: 10.1128/AEM.01098-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 19.Choi KH, Schweizer HP. 2006. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc 1:153–161. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 20.Crepin S, Harel J, Dozois CM. 2012. Chromosomal complementation using Tn7 transposon vectors in Enterobacteriaceae. Appl Environ Microbiol 78:6001–6008. doi: 10.1128/AEM.00986-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Damron FH, McKenney ES, Schweizer HP, Goldberg JB. 2013. Construction of a broad-host-range Tn7-based vector for single-copy P(BAD)-controlled gene expression in gram-negative bacteria. Appl Environ Microbiol 79:718–721. doi: 10.1128/AEM.02926-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jittawuttipoka T, Buranajitpakorn S, Fuangthong M, Schweizer HP, Vattanaviboon P, Mongkolsuk S. 2009. Mini-Tn7 vectors as genetic tools for gene cloning at a single copy number in an industrially important and phytopathogenic bacteria, Xanthomonas spp. FEMS Microbiol Lett 298:111–117. doi: 10.1111/j.1574-6968.2009.01707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A, Dalton C, Cortez-Cordova J, Schweizer HP. 2010. Mini-Tn7 vectors as genetic tools for single copy gene cloning in Acinetobacter baumannii. J Microbiol Methods 82:296–300. doi: 10.1016/j.mimet.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Peters JE, Craig NL. 2001. Tn7: smarter than we thought. Nat Rev Mol Cell Biol 2:806–814. [DOI] [PubMed] [Google Scholar]
- 25.Pourcel C, Marchal C, Louise A, Fritsch A, Tiollais P. 1979. Bacteriophage lambda-E. coli K12 vector-host system for gene cloning and expression under lactose promoter control. I. DNA fragment insertion at the lacZ EcoRI restriction site. Mol Gen Genet 170:161–169. [DOI] [PubMed] [Google Scholar]
- 26.de Boer HA, Comstock LJ, Vasser M. 1983. The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Natl Acad Sci U S A 80:21–25. doi: 10.1073/pnas.80.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amann E, Brosius J, Ptashne M. 1983. Vectors bearing a hybrid trp-lac promoter useful for regulated expression of cloned genes in Escherichia coli. Gene 25:167–178. doi: 10.1016/0378-1119(83)90222-6. [DOI] [PubMed] [Google Scholar]
- 28.Curtis PD, Brun YV. 2014. Identification of essential alphaproteobacterial genes reveals operational variability in conserved developmental and cell cycle systems. Mol Microbiol 93:713–735. doi: 10.1111/mmi.12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J, Heindl JE, Fuqua C. 2013. Coordination of division and development influences complex multicellular behavior in Agrobacterium tumefaciens. PLoS One 8:e56682. doi: 10.1371/journal.pone.0056682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morton ER, Fuqua C. 2012. Laboratory maintenance of Agrobacterium. Curr Protoc Microbiol Chapter 1:Unit 3D 1. doi: 10.1002/9780471729259.mc03d01s24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morton ER, Fuqua C. 2012. Phenotypic analyses of Agrobacterium. Curr Protoc Microbiol Chapter 3:Unit 3D 3. doi: 10.1002/9780471729259.mc03d03s25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi KH, Mima T, Casart Y, Rholl D, Kumar A, Beacham IR, Schweizer HP. 2008. Genetic tools for select-agent-compliant manipulation of Burkholderia pseudomallei. Appl Environ Microbiol 74:1064–1075. doi: 10.1128/AEM.02430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuang Y, Biran I, Walt DR. 2004. Simultaneously monitoring gene expression kinetics and genetic noise in single cells by optical well arrays. Anal Chem 76:6282–6286. doi: 10.1021/ac049053f. [DOI] [PubMed] [Google Scholar]
- 34.Rotter C, Muhlbacher S, Salamon D, Schmitt R, Scharf B. 2006. Rem, a new transcriptional activator of motility and chemotaxis in Sinorhizobium meliloti. J Bacteriol 188:6932–6942. doi: 10.1128/JB.01902-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sourjik V, Muschler P, Scharf B, Schmitt R. 2000. VisN and VisR are global regulators of chemotaxis, flagellar, and motility genes in Sinorhizobium (Rhizobium) meliloti. J Bacteriol 182:782–788. doi: 10.1128/JB.182.3.782-788.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tambalo DD, Del Bel KL, Bustard DE, Greenwood PR, Steedman AE, Hynes MF. 2010. Regulation of flagellar, motility and chemotaxis genes in Rhizobium leguminosarum by the VisN/R-Rem cascade. Microbiology 156:1673–1685. doi: 10.1099/mic.0.035386-0. [DOI] [PubMed] [Google Scholar]
- 37.Xu J, Kim J, Koestler BJ, Choi JH, Waters CM, Fuqua C. 2013. Genetic analysis of Agrobacterium tumefaciens unipolar polysaccharide production reveals complex integrated control of the motile-to-sessile switch. Mol Microbiol 89:929–948. doi: 10.1111/mmi.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heindl JE, Wang Y, Heckel BC, Mohari B, Feirer N, Fuqua C. 2014. Mechanisms and regulation of surface interactions and biofilm formation in Agrobacterium. Front Plant Sci 5:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomlinson AD, Fuqua C. 2009. Mechanisms and regulation of polar surface attachment in Agrobacterium tumefaciens. Curr Opin Microbiol 12:708–714. doi: 10.1016/j.mib.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu J, Kim J, Danhorn T, Merritt PM, Fuqua C. 2012. Phosphorus limitation increases attachment in Agrobacterium tumefaciens and reveals a conditional functional redundancy in adhesin biosynthesis. Res Microbiol 163:674–684. doi: 10.1016/j.resmic.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aronson DE, Costantini LM, Snapp EL. 2011. Superfolder GFP is fluorescent in oxidizing environments when targeted via the Sec translocon. Traffic 12:543–548. doi: 10.1111/j.1600-0854.2011.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dinh T, Bernhardt TG. 2011. Using superfolder green fluorescent protein for periplasmic protein localization studies. J Bacteriol 193:4984–4987. doi: 10.1128/JB.00315-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. 2006. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 44.Bellefontaine AF, Pierreux CE, Mertens P, Vandenhaute J, Letesson JJ, De Bolle X. 2002. Plasticity of a transcriptional regulation network among alpha-proteobacteria is supported by the identification of CtrA targets in Brucella abortus. Mol Microbiol 43:945–960. doi: 10.1046/j.1365-2958.2002.02777.x. [DOI] [PubMed] [Google Scholar]
- 45.Bird TH, MacKrell A. 2011. A CtrA homolog affects swarming motility and encystment in Rhodospirillum centenum. Arch Microbiol 193:451–459. doi: 10.1007/s00203-011-0676-y. [DOI] [PubMed] [Google Scholar]
- 46.Brilli M, Fondi M, Fani R, Mengoni A, Ferri L, Bazzicalupo M, Biondi EG. 2010. The diversity and evolution of cell cycle regulation in alpha-proteobacteria: a comparative genomic analysis. BMC Syst Biol 4:52. doi: 10.1186/1752-0509-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greene SE, Brilli M, Biondi EG, Komeili A. 2012. Analysis of the CtrA pathway in Magnetospirillum reveals an ancestral role in motility in alphaproteobacteria. J Bacteriol 194:2973–2986. doi: 10.1128/JB.00170-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pini F, De Nisco NJ, Ferri L, Penterman J, Fioravanti A, Brilli M, Mengoni A, Bazzicalupo M, Viollier PH, Walker GC, Biondi EG. 2015. Cell cycle control by the master regulator CtrA in Sinorhizobium meliloti. PLoS Genet 11:e1005232. doi: 10.1371/journal.pgen.1005232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quon KC, Marczynski GT, Shapiro L. 1996. Cell cycle control by an essential bacterial two-component signal transduction protein. Cell 84:83–93. doi: 10.1016/S0092-8674(00)80995-2. [DOI] [PubMed] [Google Scholar]
- 50.Thanbichler M, Iniesta AA, Shapiro L. 2007. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res 35:e137. doi: 10.1093/nar/gkm818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 52.Watson B, Currier TC, Gordon MP, Chilton MD, Nester EW. 1975. Plasmid required for virulence of Agrobacterium tumefaciens. J Bacteriol 123:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.