

ABSTRACT
Arbuscular mycorrhizal (AM) fungi form mutualisms with plant roots that increase plant growth and shape plant communities. Each AM fungal cell contains a large amount of genetic diversity, but it is unclear if this diversity varies across evolutionary lineages. We found that sequence variation in the nuclear large-subunit (LSU) rRNA gene from 29 isolates representing 21 AM fungal species generally assorted into genus- and species-level clades, with the exception of species of the genera Claroideoglomus and Entrophospora. However, there were significant differences in the levels of sequence variation across the phylogeny and between genera, indicating that it is an evolutionarily constrained trait in AM fungi. These consistent patterns of sequence variation across both phylogenetic and taxonomic groups pose challenges to interpreting operational taxonomic units (OTUs) as approximations of species-level groups of AM fungi. We demonstrate that the OTUs produced by five sequence clustering methods using 97% or equivalent sequence similarity thresholds failed to match the expected species of AM fungi, although OTUs from AbundantOTU, CD-HIT-OTU, and CROP corresponded better to species than did OTUs from mothur or UPARSE. This lack of OTU-to-species correspondence resulted both from sequences of one species being split into multiple OTUs and from sequences of multiple species being lumped into the same OTU. The OTU richness therefore will not reliably correspond to the AM fungal species richness in environmental samples. Conservatively, this error can overestimate species richness by 4-fold or underestimate richness by one-half, and the direction of this error will depend on the genera represented in the sample.
IMPORTANCE Arbuscular mycorrhizal (AM) fungi form important mutualisms with the roots of most plant species. Individual AM fungi are genetically diverse, but it is unclear whether the level of this diversity differs among evolutionary lineages. We found that the amount of sequence variation in an rRNA gene that is commonly used to identify AM fungal species varied significantly between evolutionary groups that correspond to different genera, with the exception of two genera that are genetically indistinguishable from each other. When we clustered groups of similar sequences into operational taxonomic units (OTUs) using five different clustering methods, these patterns of sequence variation caused the number of OTUs to either over- or underestimate the actual number of AM fungal species, depending on the genus. Our results indicate that OTU-based inferences about AM fungal species composition from environmental sequences can be improved if they take these taxonomically structured patterns of sequence variation into account.
INTRODUCTION
Sequences of rRNA genes have revolutionized our understanding of microbial diversity (1, 2) and the factors structuring microbial communities (3). When used in combination with high-throughput sequencing, rRNA gene sequences have provided critical insights into the composition and dynamics of microbial communities in a wide range of environments, from the deep ocean (4) to the microbiomes found within other organisms (5). To provide these insights, it is standard for bacterial sequence-based studies to cluster similar sequences into operational taxonomic units (OTUs) that are meant to approximate species-level groups. These OTUs are typically created by a clustering algorithm that uses a single sequence similarity threshold (usually 97%) to define OTU membership. This use of rRNA gene sequences as species barcodes also represents a powerful, culture-independent way to better understand the diversity of eukaryotic microbes. However, in these organisms, sequence variation cannot always be divided into species-level OTUs, because OTUs can either lump sequences from multiple species together or split sequences from the same species apart (6–9). It is currently unclear how well OTUs correspond to species of arbuscular mycorrhizal (AM) fungi, an ecologically important but poorly described group.
AM fungi (phylum Glomeromycota) are a widely distributed group of fungi that form mutualisms with the roots of most terrestrial plant species (10) and can shape plant communities (11, 12). Because AM fungi are only known to reproduce asexually, species cannot be validated by using the biological species concept and are instead defined by the morphology of their spores formed in the soil. AM fungal species defined in this way can be functionally distinct (13), and the composition of AM fungal communities can change during ecological succession (14). However, in order to identify the AM fungal species participating in active mycorrhizal associations with plants, it is necessary to use DNA sequences from roots. In general, soil-dwelling fungi are genetically diverse (15), and AM fungi have an exceptionally large amount of rRNA gene sequence variation compared to other fungal groups (16, 17). However, in contrast to most other microbes, in AM fungi, this sequence variation can occur within a single multinucleate cell (18, 19). While recent genome sequencing of isolates of the genus Rhizoglomus (20) has expanded our understanding of genetic variation in AM fungi beyond the rRNA genes (21–23), it remains largely unknown how either genome-wide or rRNA gene sequence variation may itself differ across evolutionary lineages. Previous studies that sampled a limited amount of rRNA gene sequence variation found that sequences from morphologically defined species generally formed clades on the gene tree (24–26), which is an essential prerequisite for clustering sequences into species-level OTUs. However, it is unclear whether this correspondence will remain robust to the sampling of additional sequence variation in AM fungi, as other work has called it into question for species of the genus Claroideoglomus (27, 28).
Understanding how the amount of within-species sequence variation may differ across the AM fungal phylogeny is important to more confidently determine species composition from environmental samples. For studies of bacteria, the inclusion of a mixture of DNA from specific strains with known OTU compositions as a “mock community” in a sequencing run has allowed the evaluation of sequence clustering methods (29) and the assessment of OTU clustering accuracy for environmental samples (30). This mock-community approach has not been used for AM fungi, and this may partly be due to uncertainty about how the amount of sequence variation in this group would affect the stability of a mock community's OTU composition across different sequencing runs. Instead, several studies of AM fungi have provided indirect assessments of the abilities of various sequence clustering methods to determine species composition from environmental samples by testing the correlation between specific OTUs and environmental variables (31–33). Direct assessments that use sequences from individual AM fungal species to evaluate the performance of sequence-based methods of species identification are generally lacking for AM fungi. However, one such direct assessment using sequences from isolates of the genus Rhizoglomus found multiple OTUs per species and a mismatch between OTUs and clades of the gene tree (34). Given the large amount of within-species sequence variation that occurs in AM fungi, it is possible that this pattern of multiple OTUs per species of Rhizoglomus is not unique among AM fungal groups, and therefore the key assumption that each OTU corresponds to a species-level group of sequences may not hold for AM fungi in general.
Here we used a phylogenetically broad sampling of AM fungi to first determine the extent to which the rRNA gene tree based on a few sequences per species (25) is representative of a larger range of the genetic diversity that is present within species. We then tested whether the distribution of sequence variation differed both across the phylogeny and across taxonomic groups. Finally, we used the large range of within-species sequence variation that occurs in AM fungi to test how the OTUs generated by five different sequence clustering methods correspond to morphologically defined species.
MATERIALS AND METHODS
Data set of rRNA gene sequences from AM fungal species.
The data set comprised “reference” and “test” rRNA gene sequences. The reference sequences were obtained from two sources: (i) a previously reported multiple-sequence alignment (25), retaining only the sequences spanning a 350-bp portion of the phylogenetically informative D2 region of the nuclear large-subunit (LSU) rRNA gene that corresponded with the test sequences, and (ii) supplemental sequences with confident species attributions obtained from GenBank to expand the phylogenetic breadth of the reference data set. These supplemental sequences were added to the existing alignment by using the −add function in MAFFT v.7.029b (35), which performs well when fungal rRNA gene sequences are added to existing alignments (35).
The test sequences were obtained from spores of 29 isolates from 21 morphologically defined species of AM fungi that were harvested from soil-based cultures. Six species were represented by two to three different geographic isolates, and nine species or geographic isolates had replicate DNA extractions, including both single and multiple spore extractions for some isolates (see Data Set S1 in the supplemental material for culture accession numbers and sampling information). Spore preparation and DNA extraction procedures were performed as described previously (24). Briefly, spores were cleaned by sonication, and either single or multiple spores were crushed in Tris-EDTA (TE) buffer, heated to 100°C, and then frozen until use as a template for PCRs using primer LR1 (36) and barcoded primer FLR2 (37). The purified PCR products were then pyrosequenced (454 Life Sciences, Branford, CT).
The resulting sequences were then quality screened before clustering into OTUs. The clustering method CD-HIT-OTU (38) employs its own sequence quality control and chimera sequence removal pipeline that does not rely on base quality scores, and so the full data set of all raw test sequences (188,713) with the PCR primer sequences still attached and all reference sequences was used directly with CD-HIT-OTU. After quality control screening, 51,135 sequences remained and were automatically used for clustering. For the AbundantOTU (39), CROP (40), mothur (41), and UPARSE (42) clustering methods, a common pipeline for stringent sequence quality control was applied to all raw test sequences. This pipeline removed sequences with any undetermined bases (Ns) or any insertions, any deletions, or more than one substitution in the PCR primer site before trimming of the primer site from the sequences. Sequences were then truncated to 350 bp before being quality filtered by using USEARCH (43), allowing a maximum of 1 expected error per sequence. The remaining sequences were dereplicated to remove identical sequences, which was necessary before chimeric sequences could be removed by using UCHIME de novo (44). We then used the multidimensional scaling (MDS) algorithm described below to visually identify a small number of sequences (253 sequences when dereplicated and 1,238 sequences when rereplicated) that closely grouped with sequences from phylogenetically distant species instead of their expected species affiliation. These sequences were removed from the data set, and the number of affected sequences from each barcode and the corresponding isolate names are listed in Table S1 in the supplemental material. After quality control, 9,094 unique test sequences remained. These sequences were added to the dereplicated reference sequences to form the data set for use with the MDS visualization as well as CROP and UPARSE (10,081 sequences); for clustering with AbundantOTU and mothur, the unique test sequences were rereplicated to reconstruct the abundance of each unique, nonchimeric sequence and were then added to the reference sequences, yielding 51,543 sequences. The number of sequences passing the CD-HIT-OTU quality screening pipeline was within 1% of the number passing the common quality screening pipeline for the other clustering methods, and the distribution of sequences among barcodes was highly linearly correlated between the two quality screening pipelines (r = 0.93), suggesting that the two pipelines are comparable overall and that all clustering results can be compared. The reference sequences extended the phylogenetic coverage of the data set and allowed independent validation of species attributions in phylogenies; however, only the test sequences were used for all analyses involving metrics of sequence variation.
Clustering of sequences into OTUs.
The five different clustering methods that we tested were selected to represent a range of clustering approaches based on three distinct types of underlying clustering algorithms: (i) greedy (or top-down), with AbundantOTU v.0.93b (39), CD-HIT-OTU v.0.0.2 (38), and UPARSE v.8.1.1 (42); (ii) hierarchical (or bottom-up), with mothur v.1.34.0 (41); and (iii) Bayesian, with CROP v.1.33 (40). We used a 97% sequence similarity as the required threshold for the greedy and hierarchical algorithms and ran them with default settings, except that we allowed CD-HIT-OTU to find the consensus PCR primer sequence as the first 21 bp of each sequence. For CROP, similarity levels approximated 97% (see the supplemental material for clustering commands). For each clustering method, we calculated the number of OTUs represented per isolate (OTU richness) and the Shannon diversity index of sequence distribution among OTUs for each isolate (OTU diversity). To correct for uneven numbers of sequences between isolates, we rarefied the OTU richness value for each isolate to the minimum number of sequences per isolate (Diversispora spurca, with 85 sequences for AbundantOTU, CROP, mothur, and UPARSE and 93 sequences for CD-HIT-OTU) by using the vegan package in R (v.3.1.2; R Core Team, Vienna, Austria).
Visualization of sequence variation.
Sequence variation in the data set was visualized by using a novel MDS algorithm (45) that represents each sequence as a point in three-dimensional space, with the location of each point being determined by the pairwise differences between that sequence and all other sequences in the data set. To compare the MDS visualization of sequence variation with the inferred evolutionary relationships between sequences, we interpolated the gene tree into the visualization using a neighbor-joining-based algorithm that we developed previously (46). For clarity, not all sequences were included in this gene tree; instead, the data set was preclustered by using AbundantOTU (39) with a 99% sequence similarity threshold. The resulting sequences still represented all genera in the initial data set. We aligned the sequences with MUSCLE v.3.8.3 (47) and created an unrooted maximum likelihood gene tree using RAxML v.8.0.0 (48) with the generalized time-reversible (GTR) gamma model of nucleotide substitution and 1,000 rapid bootstrap replicates to determine statistical confidence in the tree topology; all subsequent multiple-sequence alignments and gene trees were constructed in the same way, except that the gene trees were rooted by including an outgroup sequence.
To evaluate the sequence similarity between isolates from three clades that contained different amounts of sequence variation, we constructed both a rooted gene tree and a heat map of pairwise sequence divergence for each clade. For the gene trees, all aligned sequences were used from the clades corresponding to the Gigasporaceae and Rhizoglomus, and for the clade corresponding to the genera Claroideoglomus and Entrophospora, the gene tree was made by using representative sequences for each species and each evolutionary history (sequence group) (see Results) that were created by clustering sequences using AbundantOTU (39) with a 97% sequence similarity threshold. Each of the three gene trees was rooted by using a sequence from the most closely related clade in the data set (Fig. 1). Clades on each gene tree that represented a single species were collapsed for clarity and colored by species. For each of the three clades, heat maps of pairwise sequence divergence were made from a random subsample of 240 sequences, split evenly among all species, geographic isolates, and both sequence groups for Claroideoglomus and Entrophospora. Pairwise genetic distances for the aligned sequences were calculated with MEGA 7 (49), using the Kimura 2-parameter model of nucleotide substitution (50) with gamma rate distribution, which was the best-fitting substitution model overall for the three clades, as determined by the substitution model-fitting function in MEGA 7; the use of different substitution models gave qualitatively identical results.
FIG 1.
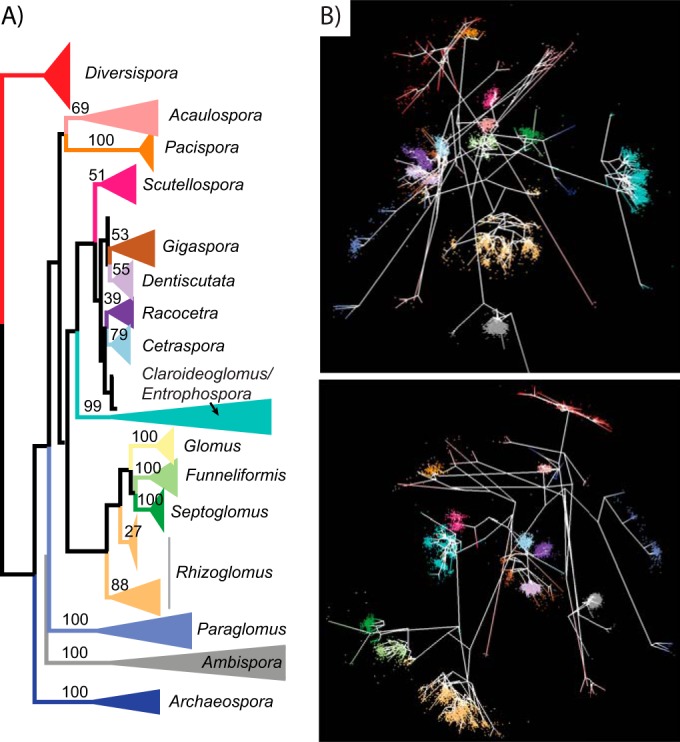
Correspondence between the rRNA gene tree (left) and two different views of the multidimensional scaling (MDS) visualization (right) for sequences from 21 species of AM fungi colored by genus. Branches within genera on the gene tree are collapsed for clarity, with the bootstrap value for each of these genus-level clades being noted on its branch; in contrast, each sequence is represented as a point in the MDS visualization.
Evaluation of the phylogenetic signal in patterns of sequence variation.
To estimate the amount of sequence variation contained in each barcode, which represents sequences from different species, geographic isolates, or replicate DNA extractions, we calculated the mean per-site nucleotide diversity (denoted π). We did this by first aligning all sequences for each barcode and then taking 10 independent, random subsamples from that barcode, with each subsample comprising 85 aligned sequences (the minimum number of sequences per isolate). For each of these subsamples, we calculated the value of π averaged across all sites in the alignment that were represented by two or more sequences by using an extension of the single-site method reported previously (51):
| (1) |
where B is the number of positions (columns) with two or more aligned bases and therefore the possibility of a single nucleotide polymorphism (SNP); x is the number of positions in the alignment where a SNP occurs; ni is the number of different sequences represented in the alignment at each position with a SNP; and FAi, FTi, FCi, and FGi are the frequencies of nucleotides A, T, C, and G, respectively, in the alignment at each position with a SNP. For each barcode, the π values calculated from all 10 subsamples were then averaged and were used for all analyses.
To determine how nucleotide diversity (π), rarefied OTU richness, and OTU diversity may vary across different taxonomic groups, we tested the magnitude and statistical significance of variance components corresponding to AM fungal genera, species, and isolates using mixed models with the MIXED procedure in SAS (v.9.4; SAS Institute, Inc., Cary, NC). We followed the consensus AM fungal genus names proposed previously (52), except for Rhizoglomus instead of Rhizophagus (20) and Claroideoglomus and Entrophospora, which we considered the same genus for statistical tests (see Results and Discussion). Total variance was calculated as the sum of the variances explained by differences among isolates, species, and genera and was considered to be significantly different from zero if at least one of those variance components was significantly different from zero. For rarefied OTU richness and OTU diversity, we repeated this analysis for each of the five clustering methods.
To test for phylogenetic signal in the variation of nucleotide diversity, rarefied OTU richness, and OTU diversity, we adapted an approach developed previously to estimate heritability using genetic markers (53). Phylogenetic heritability is estimated as the slope of the regression line between the pairwise phylogenetic distance (predictor variable) and the pairwise cross product of sequence variation (response variable). The pairwise cross product of sequence variation is calculated as
| (2) |
where yi and yj are the trait (nucleotide diversity, rarefied OTU richness, or OTU diversity) values for each of the samples (sequences from each barcode) in the pairwise comparison, μ is the mean trait value for all the samples, and V is the unbiased variance of the trait value for all of the samples.
Pairwise phylogenetic distances were calculated by using the cophenetic function in the ape package of R on a rooted phylogeny of all species in the test data set using an alignment of extended sequences (∼675 bp) that were either collected from data reported previously (25) (19 species), obtained from GenBank (Cetraspora pellucida and the outgroup Rhodotorula hordea), or sequenced by us (Entrophospora infrequens). These extended sequences were used to obtain better resolution for internal phylogenetic nodes and therefore more accurate phylogenetic distances between genera than would be possible by using our shorter test sequences. Geographic isolates or replicate DNA extractions of the same species were added to the phylogenetic distance matrix as entries with values of zero.
Phylogenetic heritability is estimated by using the following regression:
| (3) |
where ri,j is the pairwise phylogenetic distance for each of the samples in the pairwise comparison, h2 is the phylogenetic heritability, is the pairwise correlation of each of the samples due to environmental factors, and ei,j is measurement error. Because and ei,j are assumed to be independent of ri,j, phylogenetic heritability can be calculated as follows:
| (4) |
We then estimated the amount of variance explained by phylogenetic relationships by multiplying the total variance (calculated above) with the phylogenetic heritability from equation 4.
Assessing the match between OTUs and known AM fungal species.
We visualized the OTUs from each of the four sequence clustering methods by color-coding points (sequences) in the MDS visualization according to their OTU membership for OTUs containing at least 10 sequences, and OTUs were colored according to their size (the number of sequences that they contained). We evaluated how well the OTUs produced by each clustering method matched the known species composition by calculating the adjusted Rand index (54, 55) using the phyclust package in R with the null model assumption that all sequences from each species would be assigned to the same OTU. Rand index values can range from 0 to 1, with a value of 1 indicating a perfect match between OTUs and species.
Accession number(s).
The raw 454 sequences from this study have been deposited in the NCBI Sequence Read Archive (SRA) under accession number SRP067281.
RESULTS
The rRNA gene tree closely corresponds to the MDS visualization.
The gene tree from a representative set of sequences generally assigned sequences from each genus to a single clade, with the exception of Rhizoglomus (Fig. 1A). Within genera, groups of similar sequences in the MDS visualization were connected by branches of the gene tree that had relatively shallow nodes, suggesting that the visualization can help identify phylogenetically meaningful variation in large sequence data sets (Fig. 1B). In both the MDS visualization and the gene tree, sequences from isolates of Entrophospora infrequens and isolates of Claroideoglomus species were indistinguishable, and therefore we considered them to be in the same group.
Variation in rRNA sequences is generally assorted into species-level clades.
For the Gigasporaceae (genera Cetraspora, Dentiscutata, Gigaspora, Racocetra, and Scutellospora) and the genus Rhizoglomus (family Glomeraceae), sequences from isolates of each species generally formed a clade (Fig. 2A and C), consistent with recombination of rRNA gene sequences within species. The two main exceptions were the majority of sequences from Dentiscutata erythropus, which did not form a clade with bootstrap support and were paraphyletic relative to D. heterogama, and sequences from Rhizoglomus irregulare, which formed groups that generally had weak bootstrap support and that were paraphyletic relative to the other Rhizoglomus species. However, the heat maps of pairwise sequence divergence, which were calculated independently of the gene trees, emphasize a pattern of sequence similarity within species. In the Gigasporaceae, sequences from isolates of the same species or from different geographic isolates of Racocetra fulgida were most similar to each other, followed by sequences from isolates of closely related species (Fig. 2B). Sequences from isolates of Rhizoglomus species were also distinct from one another despite the larger amount of within-species sequence variation in this group, as indicated by the less consistent blocks of color in the heat map, including the geographic isolates of Rhizoglomus clarum that were so differentiated that they appear to be separate species (Fig. 2D).
FIG 2.

Comparisons of sequence similarity between the gene tree and a heat map of pairwise sequence divergence for isolates of species of the Gigasporaceae (top row), Rhizoglomus (center row), and Claroideoglomus-Entrophospora (bottom row), with each species or geographic isolate being represented by the same color in both the branches of the gene tree and the colored bars bordering the heat map. For the Gigasporaceae and Rhizoglomus, clades on the gene tree that are formed by sequences from a single species are represented as triangles, where the height of the triangle is proportional to the number of sequences in the clade. On the gene trees for all three groups, the bootstrap value for each clade is noted on its branch for bootstrap values of >15. In each heat map, the sequence divergence for each pairwise comparison is represented by a color ranging from dark red (little divergence) to dark blue (larger divergence), with tiles on the diagonal representing comparisons within each species or geographic isolate and tiles near the diagonal representing comparisons between closely related species or between geographic isolates of the same species. For clarity, for Claroideoglomus-Entrophospora, the color representing each species or geographic isolate is shown in a box to the right of its label in the gene tree; in both the gene tree and the heat map, sequences from group A are marked by pink circles, and those from group B are marked by black triangles. Scu., Scutellospora; Gig., Gigaspora; Den., Dentiscutata; Cet., Cetraspora; Rac., Racocetra; Rhi., Rhizoglomus; Cla., Claroideoglomus; Ent., Entrophospora. Labels IN.1 and IN.2 represent Indiana sites 1 and 2, respectively.
In contrast, sequences from isolates of each Claroideoglomus-Entrophospora species generally did not form a clade but instead assorted into two groups with different evolutionary histories (Fig. 2E). All sequences from Claroideoglomus etunicatum and C. luteum as well as a subset of sequences from C. claroideum and E. infrequens that aggregated in the MDS visualization (see Fig. S1 in the supplemental material) are referred to here as “group A,” but they did not form a clade (Fig. 2E, top). Rather, these sequences were paraphyletic relative to the clade representing other sequences from C. claroideum Indiana isolate 2 and all three geographic isolates of E. infrequens (referred to here as “group B”) (Fig. 2E, bottom) that are peripheral in the MDS visualization (see Fig. S1 in the supplemental material). The checkerboard pattern in the heat map of pairwise sequence divergence for isolates of these two species indicates that sequence similarity is determined more by sequence group than by species (Fig. 2F).
Distribution of rRNA sequence variation across the phylogeny.
Nucleotide diversity (π), rarefied OTU richness, and OTU diversity differed substantially for isolates across the AM fungal phylogeny but remained relatively constant between geographic isolates of the same species (Fig. 3). These phylogenetic patterns of the OTU-based metrics were usually consistent regardless of the clustering method, although all clustering methods had significantly lower values of rarefied OTU richness per isolate than those determined by mothur (F4,140 = 8.52; P < 0.001), and AbundantOTU, CD-HIT-OTU, and CROP had significantly lower values of OTU diversity (F4,140 = 5.50; P < 0.001) per isolate than those determined by mothur or UPARSE, except for the comparison between AbundantOTU and UPARSE (P = 0.13 by Tukey's honestly significant difference [HSD] test). Rarefied OTU richness for all clustering methods consistently overestimated true species richness across the phylogeny, by up to a factor of 4 for CD-HIT-OTU and a factor of 9 for mothur, with the other clustering methods having intermediate levels of overestimation (Fig. 3).
FIG 3.
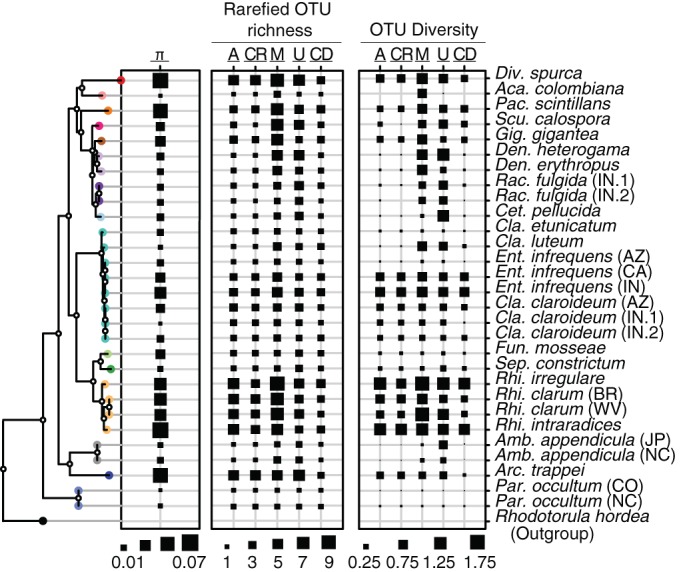
Phylogenetic differences in nucleotide diversity (π) (left) as well as both rarefied OTU richness (middle) and OTU diversity (right) for all isolates and for each of the following five clustering methods: AbundantOTU (A), CROP (CR), mothur (M), UPARSE (U), and CD-HIT-OTU (CD). The rooted phylogeny was made by using representative extended sequences (∼675 bp) from each species. The leaves are colored by genus to match those in Fig. 1. Div, Diversispora; Aca, Acaulospora; Pac, Pacispora; Fun, Funneliformis; Sep., Septoglomus; Amb., Ambispora; Arc., Archaeospora; Par., Paraglomus.
Genera explained a large and statistically significant amount of variance in rarified OTU richness for all methods except CD-HIT-OTU (P = 0.10) and in OTU diversity for all methods except UPARSE (P = 0.12) and CD-HIT-OTU (P = 0.10) (Table 1). Genera also explained a large although not significant amount of the variance in nucleotide diversity (P = 0.07) (Table 1). In contrast, different isolates accounted for a relatively small and nonsignificant amount of variance in both nucleotide diversity and rarefied OTU richness, although they accounted for a significant and much larger amount of the variance in OTU diversity for all methods except UPARSE (P = 0.07) (Table 1). A significant although small amount of the total variance in both OTU richness and diversity was explained by the full phylogeny regardless of the clustering method, but this was not the case for nucleotide diversity (P = 0.13) (Table 1).
TABLE 1.
Total variance in nucleotide diversity, rarefied OTU richness, and OTU diversitya
| Metric | OTU clustering method | Total | Genus |
Species |
Isolate |
Phylogeny |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| s2 (SE) | s2 (SE) | % | s2 (SE) | % | s2 (SE) | % | s2 | h2 | ||
| π | 2.71 × 10−4 (9.4 × 10−3) | 1.8 × 10−4 (1.2 × 10−4) | 66 | 5.6 × 10−5 (7.6 × 10−5) | 21 | 3.5 × 10−5 (4.3 × 10−5) | 13 | 1.58 × 10−5 | 0.06 | |
| OTU richness | AbundantOTU | 2.18 (0.88) | 1.98 (0.86) | 91 | 0.0 | 0 | 0.20 (0.16) | 9 | 0.31 | 0.14 |
| CROP | 1.75 (0.72) | 1.55 (0.70) | 89 | 0.0 | 0 | 0.20 (0.12) | 11 | 0.23 | 0.13 | |
| mothur | 5.70 (2.39) | 4.40 (2.18) | 77 | 0.57 (0.09) | 10 | 0.73 (0.49) | 13 | 0.62 | 0.11 | |
| UPARSE | 2.03 (1.16) | 1.49 (0.89) | 74 | 0.18 (0.65) | 9 | 0.35 (0.37) | 17 | 0.40 | 0.20 | |
| CD-HIT-OTU | 0.51 (0.32) | 0.26 (0.21) | 51 | 0.02 (0.17) | 4 | 0.23 (0.18) | 45 | 0.05 | 0.13 | |
| OTU diversity | AbundantOTU | 0.11 (0.05) | 0.06 (0.03) | 50 | 0.01 (0.03) | 13 | 0.04 (0.02) | 37 | 0.02 | 0.19 |
| CROP | 0.09 (0.04) | 0.04 (0.03) | 48 | 0.01 (0.02) | 7 | 0.04 (0.02) | 45 | 0.02 | 0.22 | |
| mothur | 0.19 (0.07) | 0.11 (0.06) | 61 | 0.00 | 2 | 0.07 (0.03) | 37 | 0.02 | 0.11 | |
| UPARSE | 0.14 (0.08) | 0.05 (0.04) | 36 | 0.03 (0.05) | 21 | 0.06 (0.04) | 43 | 0.01 | 0.11 | |
| CD-HIT-OTU | 0.09 (0.04) | 0.03 (0.02) | 33 | 0.02 (0.03) | 22 | 0.04 (0.02) | 45 | 0.01 | 0.16 | |
Shown are data for the total variance (s2) in nucleotide diversity (π), rarefied OTU richness, and OTU diversity, including the amount of variance and percentage of the total variance explained by genus, species, and isolate as well as the amount of variance explained by phylogeny and phylogenetic heritability (h2). Values in boldface type denote significant differences in variance across the taxonomic group or across the phylogeny at the level of an α value of ≤0.05.
No clustering method produced species-level OTUs for AM fungi.
In the Gigasporaceae and in Rhizoglomus, none of the four sequence clustering methods that used the common sequence processing pipeline consistently gave a single OTU for isolates of each AM fungal species (Fig. 4). We observed both ways in which OTUs can fail to correspond to species-level groups of sequences: (i) sequences from a single species were split into multiple OTUs, particularly for the three Rhizoglomus species (Fig. 4F to J), and (ii) sequences from different species were lumped into a single OTU, especially for sequences from D. erythropus and D. heterogama in the Gigasporaceae (Fig. 4A), which were assigned to the same OTU by AbundantOTU and CROP (Fig. 4B and C) and also by CD-HIT-OTU (results not shown). OTUs that lumped sequences from different species also occurred for Claroideoglomus-Entrophospora (see Fig. S1 in the supplemental material), as expected given that the rRNA sequence variation does not assort into species-level clades in this group (Fig. 2E). Overall, OTU delineations were visually more closely matched to species of the Gigasporaceae (Fig. 4A to E) than Rhizoglomus (Fig. 4F to J). This was corroborated by the substantially higher adjusted Rand index values, which quantify the match between OTUs and species, for all clustering methods for the Gigasporaceae (CD-HIT-OTU value, 0.95) (Fig. 4B to E) than those for Rhizoglomus (CD-HIT-OTU value, 0.87) (Fig. 4G to J). For both AM fungal groups, AbundantOTU, CROP, and CD-HIT-OTU produced OTUs that better matched species than did OTUs from either mothur or UPARSE.
FIG 4.

MDS visualizations of OTU clusters for isolates of species of the family Gigasporaceae (left) and species of the genus Rhizoglomus (right). For both columns, the sequences (points) in the top row are colored by their species or geographic isolate affiliation (the same colors as those used in Fig. 2), and the remaining four rows (top to bottom) show the same sequences colored by OTU for each of the following four clustering methods that used the common sequence processing pipeline: AbundantOTU (second row), CROP (third row), mothur (fourth row), and UPARSE (fifth row). Only OTUs containing >10 sequences are shown, and OTUs are colored according to the number of sequences that they contain. The adjusted Rand index value in the top right corner of each panel for the clustering methods quantifies the fit of the OTU delineations compared to the known species attribution. Higher adjusted Rand index values indicate closer correspondence between each OTU and the sequences originating from each AM fungal species. All four clustering methods had closer OTU-to-species correspondence for the Gigasporaceae, with its smaller amount of within-isolate variation, than for Rhizoglomus, but for both groups, AbundantOTU and CROP consistently generated OTUs that better matched species than OTUs generated from mothur or UPARSE. Interactive three-dimensional versions of these MDS visualizations are available at https://spidal-gw.dsc.soic.indiana.edu/public/groupdashboard/AM%20fungal%20clustering%20AEM.
DISCUSSION
Sequence variation in genera and species is generally assorted into clades.
With the exception of the genus Diversispora, the gene tree made by using the large range of sequence variation contained in isolates of the 21 species surveyed (Fig. 1A) generally agreed with the current consensus phylogeny of AM fungi (52) at the genus level and the most current AM fungal rRNA gene tree (25). Furthermore, the overall concordance between the gene tree and the MDS visualization of sequence variation (Fig. 1B) suggests that the correspondence between morphologically defined taxa and clades on the AM fungal gene tree is robust to differences in the magnitude of sequence variation (Fig. 2A to D). The notable exception to this general agreement between morphologically and phylogenetically defined taxa is the Claroideoglomus-Entrophospora clade.
Claroideoglomus and Entrophospora form one group with gene tree discordance.
Sequences from isolates of Claroideoglomus and Entrophospora species did not correspond to separate clades on the gene tree, and because of this, we identify both genera as a single group. Species in this group are morphologically well defined. For example, spores of E. infrequens develop differently and have a unique wall structure compared to spores of Claroideoglomus species (see Fig. S2 in the supplemental material). However, isolates of both species show little genetic differentiation over the portion of the LSU rRNA gene used here (Fig. 2E and F) and have less variation within species than the Gigasporaceae or Rhizoglomus (Fig. 2B, D, and F). Previous studies also documented a lack of genetic distinction between Claroideoglomus species (27, 28) but did not include E. infrequens in their analyses, nor has the most comprehensive AM fungal rRNA gene tree (25). In Claroideoglomus species, previous work demonstrated the presence of two evolutionary lineages in LSU rRNA gene sequences (“L” and “S” variants) (28). The PCR primer set used here amplified only L sequence variants due to primer site mismatches with S variants. Most L variant sequences reported previously (28) correspond to group A from this study, but 9% (17 of 195) of sequences correspond to group B, including 5 sequences from C. luteum, suggesting that it has the same gene tree discordance that we observed for E. infrequens and C. claroideum (Fig. 2E and F). Because this discordance is shared among species, the sequence variation that underlies it was likely present in a common ancestor of the group and may be due to introgression or to incomplete lineage sorting of standing sequence variation.
Sequence variation differs across clades and the full phylogeny.
We find strong evidence that the amount of genetic variation in AM fungi varies across clades that correspond to genera, as indicated by patterns of nucleotide diversity, rarefied OTU richness, and OTU diversity (Fig. 3 and Table 1). Although these genus-level differences were not statistically significant for CD-HIT-OTU (Table 1), this was likely due to its good overall performance in giving consistently low rarefied OTU richness estimates across nearly all species and geographic isolates (Fig. 3). These patterns were also consistent regardless of how many spores were sampled. This indicates that the same amount of sequence variation that occurs in multiple spores is also contained within a single spore and provides evidence of phylogenetic heritability of intracellular variation. In addition, the small overall amount of variance explained by isolate for nucleotide diversity and rarefied OTU richness is indicative of the presence of relatively little variation between replicate DNA extractions of the same culture, although this variance was larger for OTU diversity (Table 1).
The fact that genus-level clades account for more of the variance in all three metrics of sequence variation than the full phylogeny (Table 1) is perhaps surprising but can be clearly illustrated with the genus Rhizoglomus. Species of Rhizoglomus have markedly higher values for each of the three metrics than do species of the sister genera Septoglomus and Funneliformis (Fig. 3). The lower predictive power of the full phylogeny than of genus-level clades indicates that sequence variation is a trait that evolves quickly relative to the deep history represented by the AM fungal phylogeny (56).
Bidirectional error in linking OTUs to AM fungal species.
None of the sequence clustering methods tested here were effective at consistently creating species-level OTUs, and therefore OTU richness cannot reliably estimate AM fungal species richness. The internal transcribed spacer (ITS) region of the rRNA genes has been proposed as a universal sequence barcode for fungal species, but it has limited use for AM fungi due to the large amount of ITS sequence variation in this group, and either the small-subunit (SSU) or LSU rRNA gene is typically used instead (17, 57). However, even within LSU sequences, we found enough variation across genus-level clades of AM fungi to cause bidirectional errors in the correspondence between OTUs and species.
For groups like Rhizoglomus with a large amount of within-isolate variation (Fig. 2D), sequences from the same species were commonly split into multiple OTUs (Fig. 4G to J), a finding similar to the findings of another recent study (34). In contrast, for genera with little between-isolate variation, sequences from isolates of different species could be lumped into the same OTU. For example, although sequences from isolates of D. erythropus and D. heterogama (Fig. 2 and 4A, red and orange, respectively) showed consistent differences (Fig. 2A and B), they were assigned to the same OTU by AbundantOTU and CROP (Fig. 4B and C) and also by CD-HIT-OTU (results not shown), the three clustering methods that otherwise gave the closest matches between OTUs and species. Sequences from isolates of different Claroideoglomus-Entrophospora species were also lumped together into the same OTU (see Fig. S1 in the supplemental material). An essential assumption of all sequence clustering methods is that sequences from each species form a clade on the gene tree, and therefore, no clustering method can create species-level OTUs for groups like this that have gene tree discordance. A similar bidirectional error in the correspondence between OTUs and species occurs for other groups of fungi (6) and eukaryotic microbes (7, 8), suggesting that it is a shared pattern across a range of genetic systems that is determined by the evolutionary history of the group (9).
No clustering method created OTUs that accommodated the range of sequence variation that occurs across the AM fungal phylogeny (Fig. 3). However, AbundantOTU, CROP, and CD-HIT-OTU performed comparably and gave OTUs better matched to species than either mothur or UPARSE, both across the phylogeny (Fig. 3) and across a range of sequence variation in the Gigasporaceae and Rhizoglomus (see Results for adjusted Rand index values for CD-HIT-OTU) (Fig. 4). Bayesian inference, as used by CROP, potentially has the flexibility to accommodate differences in sequence variation when generating OTUs (40), so it is surprising that AbundantOTU and CD-HIT-OTU, with their fixed similarity threshold, gave clustering results nearly identical to those of CROP.
Implications for environmental sequencing.
Determining AM fungal community composition from environmental sequence data should be guided by knowledge of how sequence variation is distributed across the phylogeny and also how different clustering methods delineate OTUs based on that variation. Several evaluations and guidelines for characterizing AM fungal communities in environmental samples have recently been reported (58–60), but they do not accommodate the systematic differences in within-species sequence variation that occurs in isolates across the phylogeny (Fig. 3). Caution should also be exercised when assigning taxonomic attributions to environmental sequences using phylogenetic differentiation, such as the generalized mixed Yule coalescent (GMYC) (33) and Poisson tree processes (PTP) (61) methods, or using entities like the virtual taxa that are represented by voucher sequences in the MaarjAM database on the basis of both phylogenetic monophyly and high sequence similarity (32, 57, 62). For example, these methods would consistently underrepresent the diversity in groups with a small amount of between-species sequence variation, like some species in the Gigasporaceae (Fig. 2A and B), and in groups that have gene tree discordance, like the Claroideoglomus-Entrophospora clade (Fig. 2E and F). For the isolates considered here, the MDS visualization demonstrates that OTU richness can underestimate species richness by one-half (Fig. 4A to C), although more extensive sequence sampling within these groups is necessary to better understand the magnitude of this effect. Conversely, for AM fungal groups with a large amount of within-species sequence variation, such as species of Rhizoglomus, OTU richness overestimated actual species richness by at least 4-fold across all clustering methods, which was the most of any AM fungal genus (Fig. 3). This phenomenon may partly underlie field observations of phylogenetic aggregation in AM fungal communities over short distances (meters) (31), as this aggregation may represent sequences from a single organism that were assigned to several OTUs instead of the presence of multiple, functionally similar species. Finally, recent evidence documenting variation in the level of heterokaryosis that occurs among isolates of the same AM fungal species (63) may also affect the range of sequence variation observed for AM fungi.
Identification of putative AM fungal species in environmental samples typically is done by using rRNA gene sequences. We find that the level of sequence variation in the LSU rRNA gene consistently differs across clades of AM fungi that correspond to genera and that this sequence variation does not correspond to morphologically defined species of Claroideoglomus-Entrophospora. The different levels of rRNA sequence variation that occur across both genus-level clades and the full phylogeny present genuine problems in the use of OTU richness to estimate species richness for AM fungi. The MDS visualization that we demonstrate here can assist as a diagnostic tool to identify groups that may be especially affected by differences in rRNA sequence variation, but no current method of sequence-based species identification is able to overcome this problem, and we suggest that interpretations of AM fungal OTU composition from environmental sequences should be made in the context of these limitations.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Morton and INVAM for access to AM fungal cultures, E. Kim for laboratory assistance, A. Rosling for initial data exploration, M. Hahn for insightful comments that substantially improved the manuscript, and two anonymous reviewers and S. Hempel for their constructive comments that also improved the manuscript.
Computing resources were provided by the Digital Science Center at Indiana University and in part by Lilly Endowment, Inc., through the Indiana University Pervasive Technology Institute and the Indiana METACyt Initiative.
Wittaya Kaonongbua and James D. Bever designed the research; Wittaya Kaonongbua performed the research; Yang Ruan, Geoffrey Fox, and Yuzhen Ye contributed new analytic methods and tools; Geoffrey L. House, Saliya Ekanayake, Ursel M. E. Schütte, and James D. Bever analyzed the data; Geoffrey L. House and James D. Bever wrote the paper; and all authors contributed to paper revisions.
We have no competing interests to declare.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00816-16.
REFERENCES
- 1.Rappé MS, Giovannoni SJ. 2003. The uncultured microbial majority. Annu Rev Microbiol 57:369–394. doi: 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- 2.Woese CR, Fox GE. 1977. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A 74:5088–5090. doi: 10.1073/pnas.74.11.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere. ”Proc Natl Acad Sci U S A 103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. 2008. Evolution of mammals and their gut microbes. Science 320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaalid R, Kumar S, Nilsson RH, Abarenkov K, Kirk PM, Kauserud H. 2013. ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol Ecol Resour 13:218–224. doi: 10.1111/1755-0998.12065. [DOI] [PubMed] [Google Scholar]
- 7.Caron DA, Countway PD, Savai P, Gast RJ, Schnetzer A, Moorthi SD, Dennett MR, Moran DM, Jones AC. 2009. Defining DNA-based operational taxonomic units for microbial-eukaryote ecology. Appl Environ Microbiol 75:5797–5808. doi: 10.1128/AEM.00298-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nebel M, Pfabel C, Stock A, Dunthorn M, Stoeck T. 2011. Delimiting operational taxonomic units for assessing ciliate environmental diversity using small-subunit rRNA gene sequences. Environ Microbiol Rep 3:154–158. doi: 10.1111/j.1758-2229.2010.00200.x. [DOI] [PubMed] [Google Scholar]
- 9.Ryberg M. 2015. Molecular operational taxonomic units as approximations of species in the light of evolutionary models and empirical data from Fungi. Mol Ecol 24:5770–5777. doi: 10.1111/mec.13444. [DOI] [PubMed] [Google Scholar]
- 10.Smith SE, Read DJ. 2008. Mycorrhizal symbiosis. Academic Press, San Diego, CA. [Google Scholar]
- 11.van der Heijden MGA, Klironomos JN, Ursic M, Moutoglis P, Streitwolf-Engel R, Boller T, Wiemken A, Sanders IR. 1998. Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nature 396:69–72. doi: 10.1038/23932. [DOI] [Google Scholar]
- 12.Vogelsang KM, Reynolds HL, Bever JD. 2006. Mycorrhizal fungal identity and richness determine the diversity and productivity of a tallgrass prairie system. New Phytol 172:554–562. doi: 10.1111/j.1469-8137.2006.01854.x. [DOI] [PubMed] [Google Scholar]
- 13.Bever JD, Morton JB, Antonovics J, Schultz PA. 1996. Host-dependent sporulation and species diversity of arbuscular mycorrhizal fungi in a mown grassland. J Ecol 84:71–82. doi: 10.2307/2261701. [DOI] [Google Scholar]
- 14.Johnson NC, Zak DR, Tilman D, Pfleger FL. 1991. Dynamics of vesicular-arbuscular mycorrhizae during old field succession. Oecologia 86:349–358. doi: 10.1007/BF00317600. [DOI] [PubMed] [Google Scholar]
- 15.Tedersoo L, Bahram M, Põlme S, Kõljalg U, Yorou NS, Wijesundera R, Ruiz LV, Vasco-Palacios AM, Thu PQ, Suija A, Smith ME, Sharp C, Saluveer E, Saitta A, Rosas M, Riit T, Ratkowsky D, Pritsch K, Põldmaa K, Piepenbring M, Phosri C, Peterson M, Parts K, Pärtel K, Otsing E, Nouhra E, Njouonkou AL, Nilsson RH, Morgado LN, Mayor J, May TW, Majuakim L, Lodge DJ, Lee SS, Larsson K-H, Kohout P, Hosaka K, Hiiesalu I, Henkel TW, Harend H, Guo L-D, Greslebin A, Grelet G, Geml J, Gates G, Dunstan W, Dunk C, Drenkhan R, Dearnaley J, De Kesel A, et al. 2014. Global diversity and geography of soil fungi. Science 346:1256688. doi: 10.1126/science.1256688. [DOI] [PubMed] [Google Scholar]
- 16.Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson K-H. 2008. Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol Bioinform Online 4:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Fungal Barcoding Consortium . 2012. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A 109:6241–6246. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clapp JP, Fitter AH, Young JPW. 1999. Ribosomal small subunit sequence variation within spores of an arbuscular mycorrhizal fungus, Scutellospora sp. Mol Ecol 8:915–921. doi: 10.1046/j.1365-294x.1999.00642.x. [DOI] [PubMed] [Google Scholar]
- 19.Sanders IR, Alt M, Groppe K, Boller T, Wiemken A. 1995. Identification of ribosomal DNA polymorphisms among and within spores of the Glomales: application to studies on the genetic diversity of arbuscular mycorrhizal fungal communities. New Phytol 130:419–427. doi: 10.1111/j.1469-8137.1995.tb01836.x. [DOI] [Google Scholar]
- 20.Sieverding E, da Silva GA, Berndt R, Oehl F. 2015. Rhizoglomus, a new genus of the Glomeraceae. Mycotaxon 129:373–386. doi: 10.5248/129.373. [DOI] [Google Scholar]
- 21.Boon E, Halary S, Bapteste E, Hijri M. 2015. Studying genome heterogeneity within the arbuscular mycorrhizal fungal cytoplasm. Genome Biol Evol 7:505–521. doi: 10.1093/gbe/evv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin K, Limpens E, Zhang Z, Ivanov S, Saunders DGO, Mu D, Pang E, Cao H, Cha H, Lin T, Zhou Q, Shang Y, Li Y, Sharma T, van Velzen R, de Ruijter N, Aanen DK, Win J, Kamoun S, Bisseling T, Geurts R, Huang S. 2014. Single nucleus genome sequencing reveals high similarity among nuclei of an endomycorrhizal fungus. PLoS Genet 10:e1004078. doi: 10.1371/journal.pgen.1004078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tisserant E, Malbreil M, Kuo A, Kohler A, Symeonidi A, Balestrini R, Charron P, Duensing N, Frei dit Frey N, Gianinazzi-Pearson V, Gilbert LB, Handa Y, Herr JR, Hijri M, Koul R, Kawaguchi M, Krajinski F, Lammers PJ, Masclaux FG, Murat C, Morin E, Ndikumana S, Pagni M, Petitpierre D, Requena N, Rosikiewicz P, Riley R, Saito K, San Clemente H, Shapiro H, van Tuinen D, Bécard G, Bonfante P, Paszkowski U, Shachar-Hill YY, Tuskan GA, Young JP, Sanders IR, Henrissat B, Rensing SA, Grigoriev IV, Corradi N, Roux C, Martin F. 2013. Genome of an arbuscular mycorrhizal fungus provides insight into the oldest plant symbiosis. Proc Natl Acad Sci U S A 110:20117–20122. doi: 10.1073/pnas.1313452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaonongbua W, Morton JB, Bever JD. 2010. Taxonomic revision transferring species in Kuklospora to Acaulospora (Glomeromycota) and a description of Acaulospora colliculosa sp. nov. from field collected spores. Mycologia 102:1497–1509. doi: 10.3852/10-011. [DOI] [PubMed] [Google Scholar]
- 25.Krüger M, Krüger C, Walker C, Stockinger H, Schüßler A. 2012. Phylogenetic reference data for systematics and phylotaxonomy of arbuscular mycorrhizal fungi from phylum to species level. New Phytol 193:970–984. doi: 10.1111/j.1469-8137.2011.03962.x. [DOI] [PubMed] [Google Scholar]
- 26.Morton J, Msiska Z. 2010. Phylogenies from genetic and morphological characters do not support a revision of Gigasporaceae (Glomeromycota) into four families and five genera. Mycorrhiza 20:483–496. doi: 10.1007/s00572-010-0303-9. [DOI] [PubMed] [Google Scholar]
- 27.den Bakker HC, VanKuren NW, Morton JB, Pawlowska TE. 2010. Clonality and recombination in the life history of an asexual arbuscular mycorrhizal fungus. Mol Biol Evol 27:2474–2486. doi: 10.1093/molbev/msq155. [DOI] [PubMed] [Google Scholar]
- 28.VanKuren NW, den Bakker HC, Morton JB, Pawlowska TE. 2013. Ribosomal RNA gene diversity, effective population size, and evolutionary longevity in asexual Glomeromycota. Evolution 67:207–224. doi: 10.1111/j.1558-5646.2012.01747.x. [DOI] [PubMed] [Google Scholar]
- 29.Huse SM, Welch DM, Morrison HG, Sogin ML. 2010. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. 2013. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horn S, Caruso T, Verbruggen E, Rillig MC, Hempel S. 2014. Arbuscular mycorrhizal fungal communities are phylogenetically clustered at small scales. ISME J 8:2231–2242. doi: 10.1038/ismej.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Öpik M, Vanatoa A, Vanatoa E, Moora M, Davison J, Kalwij JM, Reier Ü, Zobel M. 2010. The online database MaarjAM reveals global and ecosystemic distribution patterns in arbuscular mycorrhizal fungi (Glomeromycota). New Phytol 188:223–241. doi: 10.1111/j.1469-8137.2010.03334.x. [DOI] [PubMed] [Google Scholar]
- 33.Powell JR, Monaghan MT, Öpik M, Rillig MC. 2011. Evolutionary criteria outperform operational approaches in producing ecologically relevant fungal species inventories. Mol Ecol 20:655–666. doi: 10.1111/j.1365-294X.2010.04964.x. [DOI] [PubMed] [Google Scholar]
- 34.Senés-Guerrero C, Schüßler A. 2015. A conserved arbuscular mycorrhizal fungal core-species community colonizes potato roots in the Andes. Fungal Divers 77:317–333. doi: 10.1007/s13225-015-0328-7. [DOI] [Google Scholar]
- 35.Katoh K, Frith MC. 2012. Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics 28:3144–3146. doi: 10.1093/bioinformatics/bts578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Tuinen D, Jacquot E, Zhao B, Gollotte A, Gianinazzi-Pearson V. 1998. Characterization of root colonization profiles by a microcosm community of arbuscular mycorrhizal fungi using 25S rDNA-targeted nested PCR. Mol Ecol 7:879–887. doi: 10.1046/j.1365-294x.1998.00410.x. [DOI] [PubMed] [Google Scholar]
- 37.Trouvelot S, van Tuinen D, Hijri M, Gianinazzi-Pearson V. 1999. Visualization of ribosomal DNA loci in spore interphasic nuclei of glomalean fungi by fluorescence in situ hybridization. Mycorrhiza 8:203–206. doi: 10.1007/s005720050235. [DOI] [Google Scholar]
- 38.Li W, Fu L, Niu B, Wu S, Wooley J. 2012. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief Bioinform 13:656–668. doi: 10.1093/bib/bbs035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye Y. 2010. Identification and quantification of abundant species from pyrosequences of 16S rRNA by consensus alignment. Proc IEEE Int Conf Bioinformatics Biomed 2010:153–157. doi: 10.1109/BIBM.2010.5706555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hao X, Jiang R, Chen T. 2011. Clustering 16S rRNA for OTU prediction: a method of unsupervised Bayesian clustering. Bioinformatics 27:611–618. doi: 10.1093/bioinformatics/btq725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edgar RC. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 43.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 44.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruan Y, Ekanayake S, Rho M, Tang H, Bae S-H, Qiu J, Fox G. 2012. DACIDR: deterministic annealed clustering with interpolative dimension reduction using a large collection of 16S rRNA sequences, p 329–336. In BCB ‘12: proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine ACM, New York, NY. doi: 10.1145/2382936.2382978. [DOI] [Google Scholar]
- 46.Ruan Y, House GL, Ekanayake S, Schütte U, Bever JD, Haixu T, Fox G. 2014. Integration of clustering and multidimensional scaling to determine phylogenetic trees as spherical phylograms visualized in 3 dimensions, p 720–729. In Proceedings of the 14th IEEE/ACM International Symposium on Cluster, Cloud and Grid Computing (CCGrid) IEEE, New York, NY. doi: 10.1109/ccgrid.2014.126. [DOI] [Google Scholar]
- 47.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 51.Nei M, Tajima F. 1981. DNA polymorphism detectable by restriction endonucleases. Genetics 97:145–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Redecker D, Schüßler A, Stockinger H, Stürmer SL, Morton JB, Walker C. 2013. An evidence-based consensus for the classification of arbuscular mycorrhizal fungi (Glomeromycota). Mycorrhiza 23:515–531. doi: 10.1007/s00572-013-0486-y. [DOI] [PubMed] [Google Scholar]
- 53.Ritland K. 2000. Marker-inferred relatedness as a tool for detecting heritability in nature. Mol Ecol 9:1195–1204. doi: 10.1046/j.1365-294x.2000.00971.x. [DOI] [PubMed] [Google Scholar]
- 54.Hubert L, Arabie P. 1985. Comparing partitions. J Classif 2:193–218. doi: 10.1007/BF01908075. [DOI] [Google Scholar]
- 55.Rand WM. 1971. Objective criteria for the evaluation of clustering methods. J Am Stat Assoc 66:846–850. doi: 10.1080/01621459.1971.10482356. [DOI] [Google Scholar]
- 56.Redecker D, Kodner R, Graham LE. 2000. Glomalean fungi from the Ordovician. Science 289:1920–1921. doi: 10.1126/science.289.5486.1920. [DOI] [PubMed] [Google Scholar]
- 57.Öpik M, Davison J, Moora M, Zobel M. 2013. DNA-based detection and identification of Glomeromycota: the virtual taxonomy of environmental sequences. Botany 92:135–147. doi: 10.1139/cjb-2013-0110. [DOI] [Google Scholar]
- 58.Hart MM, Aleklett K, Chagnon P-L, Egan C, Ghignone S, Helgason T, Lekberg Y, Öpik M, Pickles BJ, Waller L. 2015. Navigating the labyrinth: a guide to sequence-based, community ecology of arbuscular mycorrhizal fungi. New Phytol 207:235–247. doi: 10.1111/nph.13340. [DOI] [PubMed] [Google Scholar]
- 59.Lekberg Y, Gibbons SM, Rosendahl S. 2014. Will different OTU delineation methods change interpretation of arbuscular mycorrhizal fungal community patterns? New Phytol 202:1101–1104. doi: 10.1111/nph.12758. [DOI] [PubMed] [Google Scholar]
- 60.Lindahl BD, Nilsson RH, Tedersoo L, Abarenkov K, Carlsen T, Kjøller R, Kõljalg U, Pennanen T, Rosendahl S, Stenlid J, Kauserud H. 2013. Fungal community analysis by high-throughput sequencing of amplified markers—a user's guide. New Phytol 199:288–299. doi: 10.1111/nph.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, Kapli P, Pavlidis P, Stamatakis A. 2013. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29:2869–2876. doi: 10.1093/bioinformatics/btt499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davison J, Moora M, Öpik M, Adholeya A, Ainsaar L, Bâ A, Burla S, Diedhiou AG, Hiiesalu I, Jairus T, Johnson NC, Kane A, Koorem K, Kochar M, Ndiaye C, Pärtel M, Reier Ü, Saks Ü, Singh R, Vasar M, Zobel M. 2015. Global assessment of arbuscular mycorrhizal fungus diversity reveals very low endemism. Science 349:970–973. doi: 10.1126/science.aab1161. [DOI] [PubMed] [Google Scholar]
- 63.Ropars J, Toro KS, Noel J, Pelin A, Charron P, Farinelli L, Marton T, Krüger M, Fuchs J, Brachmann A, Corradi N. 2016. Evidence for the sexual origin of heterokaryosis in arbuscular mycorrhizal fungi. Nat Microbiol 1:16033. doi: 10.1038/nmicrobiol.2016.33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.