

ABSTRACT
The establishment of a clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 system for strain construction in Bacillus subtilis is essential for its progression toward industrial utility. Here we outline the development of a CRISPR-Cas9 tool kit for comprehensive genetic engineering in B. subtilis. In addition to site-specific mutation and gene insertion, our approach enables continuous genome editing and multiplexing and is extended to CRISPR interference (CRISPRi) for transcriptional modulation. Our tool kit employs chromosomal expression of Cas9 and chromosomal transcription of guide RNAs (gRNAs) using a gRNA transcription cassette and counterselectable gRNA delivery vectors. Our design obviates the need for multicopy plasmids, which can be unstable and impede cell viability. Efficiencies of up to 100% and 85% were obtained for single and double gene mutations, respectively. Also, a 2.9-kb hyaluronic acid (HA) biosynthetic operon was chromosomally inserted with an efficiency of 69%. Furthermore, repression of a heterologous reporter gene was achieved, demonstrating the versatility of the tool kit. The performance of our tool kit is comparable with those of systems developed for Escherichia coli and Saccharomyces cerevisiae, which rely on replicating vectors to implement CRISPR-Cas9 machinery.
IMPORTANCE In this paper, as the first approach, we report implementation of the CRISPR-Cas9 system in Bacillus subtilis, which is recognized as a valuable host system for biomanufacturing. The study enables comprehensive engineering of B. subtilis strains with virtually any desired genotypes/phenotypes and biochemical properties for extensive industrial application.
INTRODUCTION
Genetic engineering strategies aimed at converting common microbes to productive cell factories are becoming increasingly common (1). Strain construction entails metabolic design of biosynthetic pathways and genetic manipulations. To enhance productivity, key genes for heightened expression are introduced via plasmid transformation or genomic integration (knock-in [KI]), while divergent pathways are eliminated via gene disruption (knockout [KO]) (2). However, certain genes, particularly those associated with central metabolism or core aspects of physiology (e.g., membrane integrity, ATP generation, etc.), cannot be completely inactivated; otherwise, host viability can be compromised. In such cases, reducing expression levels of the key genes (knockdown [KD]) may ultimately prove effective (3).
While engineering microbial genomes is challenging, recent application of clustered regularly interspaced short palindromic repeats (CRISPRs) and their CRISPR-associated (Cas) proteins has dramatically altered the course of genomic engineering across the spectrum of life. CRISPR arrays are part of an adaptive prokaryotic viral defense system and contain target-specific protospacers that are expressed as CRISPR RNAs (crRNAs) (4). crRNAs direct Cas nucleases to DNA targets based on the presence of a protospacer adjacent motif (PAM) specific to the Cas protein (5, 6) and protospacer homology. In type II systems such as CRISPR-Cas9 from Streptococcus pyogenes, a trans-activating crRNA (tracrRNA) is required for processing a precursor crRNA (pre-crRNA) into a functional crRNA (7). More recently, the synthetic guide RNA (gRNA), comprised of a protospacer, Cas9-binding hairpin (CBH), and transcriptional terminator, has been used for targeting, further simplifying application of the CRISPR-Cas9 system (8). The CRISPR-induced double-stranded breaks (DSBs) enable selection of mutants that evade DNA cleavage via recombination of exogenous editing templates. Accordingly, the CRISPR-Cas9 system has proven to be an indispensable tool for genome editing in bacteria (9–22), yeasts (23, 24) and higher eukaryotes (25–28).
In addition to genome editing, the CRISPR-Cas9 system can be applied to reversibly regulating gene transcription, known as CRISPR interference (CRISPRi), for which a Cas9 variant (dead Cas9 [dCas9]), exhibiting loss of endonucleolytic activity while retaining DNA-binding capability, acts as a transcriptional repressor (29, 30). While various RNA-mediated regulatory mechanisms, such as cis-acting riboswitches (31) and antisense RNAs (asRNAs) with (32, 33) or without (3, 34) recruiting motifs for proteins promoting hybridization, have been developed for tailoring gene expression, inherent complexities exist in the application of these technologies. The use of cis-acting riboswitches requires upstream sequence modifications to the targeted gene (31), limiting their practical utility for metabolic engineering and genetic screening. Similarly, the design of recruiting scaffolds for synthetic asRNAs entails extensive screening of endogenous regulatory RNAs and evaluation of synthetic constructs (32, 33), while off-target effects may also be a concern when applying asRNAs (3). On the other hand, CRISPRi provides excellent transcriptional control and is simple to implement in many organisms. The CRISPR-dCas9 system has been applied to genome-scale transcriptional repression (35) and activation (35, 36) for interrogation of gene function in human cell lines as well as to genetic and metabolic engineering of Escherichia coli (37, 38), Corynebacterium glutamicum (39), and mycobacteria (40).
Bacillus subtilis is a model Gram-positive organism that is sought after for its capacity in the high-level production of biopolymers (41), metabolites (42), and recombinant proteins (43). In contrast to E. coli, B. subtilis has received a “generally regarded as safe” (GRAS) designation, readily secretes products into the extracellular medium, and can metabolize nearly any carbon source, making it an attractive biomanufacturing platform (43). While B. subtilis is an ideal organism for industrial application, available genetic tools are lagging behind those for popular production hosts such as E. coli and Saccharomyces cerevisiae (1, 44). Because markerless genome engineering is essential for the development of commercial B. subtilis strains, a common approach has been the application of counterselectable markers, such as upp (45, 46), blaI (47), and mazF (48), flanked by short direct repeats (DRs) for autoeviction of the selection cassettes by single-crossover recombination. While these methods are simple to use, the editing efficiencies are relatively low, conditional genetic backgrounds are required in some cases, and cloning of integration constructs (i.e., integration vectors or PCR-amplified integration cassettes) can be complicated and/or time-consuming. Moreover, multiplexing is not practical, as the number of available selection and counterselection markers is limited, exposure to multiple antibiotics is not preferable (i.e., may compromise cell physiology), and counterselection will become increasingly difficult (i.e., less efficient or more time-consuming) as the number of simultaneous targets increases. On the other hand, site-specific recombination via the Cre/loxP (49) and FLP/FLP recombination target (FRT) (50) systems has also been applied to markerless recombineering in B. subtilis, with a generally higher efficiency than counterselection methods. Additionally, single-stranded DNA (ssDNA) recombineering mediated by the λ Red phage β-recombinase provides a high editing efficiency (51). However, these systems are not particularly conducive to multiplexing either, given the requirement for multiple selection (and potentially counterselection) markers and subsequent tedious screening.
In this study, we developed a CRISPR-Cas9 tool kit for comprehensive engineering of B. subtilis to overcome major limitations associated with existing genome engineering technologies (e.g., low editing efficiency, tedious cloning, and limited multiplexing capability). While CRISPR-Cas9 offers potential solutions to these technical issues in E. coli and S. cerevisiae (9, 10, 23, 24), the protocols include multicopy plasmids which must be removed from the cell. In that regard, an ideal CRISPR-Cas9 system should facilitate multiple mutations, either in series or simultaneously, while imposing minimal physiological impact on the host. Our approach not only simplifies construction of genetic elements required for CRISPR-Cas9-mediated genome editing and transcriptional interference in B. subtilis but also obviates reliance on potentially unstable multicopy plasmids and subsequent plasmid curing. We demonstrated high editing efficacy of our novel gRNA transcription and delivery system based on a simple counterselection procedure with the capacity for successive genomic manipulations, including site-specific mutations for gene inactivation and gene insertions. The effects of editing template characteristics and PAM site sensitivity were also investigated to increase editing efficiency. Finally, we expanded our tool kit for transcriptional repression of gene expression using dCas9 with our gRNA delivery system. The developed tool kit will advance technologies in engineering of B. subtilis to achieve its full potential as a biomanufacturing platform.
MATERIALS AND METHODS
Bacterial strains, primers, and plasmids.
The B. subtilis strains used in this study are listed in Table 1. E. coli HI-Control 10G chemically competent cells (Lucigen, Middleton, WI, USA) were prepared as electrocompetent cells as described previously (52) and used as the host for plasmid construction. B. subtilis and E. coli strains were maintained as glycerol stocks at −80°C. Primers (Table 1) were synthesized by Integrated DNA Technologies (IDT) (Coralville, IA, USA). Plasmids pIEFBPR (ECE195), pDG1731 (ECE119), and pAX01 (ECE137) were obtained from the Bacillus Genetic Stock Center (BGSC) (Columbus, OH). pCRISPR and pCas9 (Addgene plasmids 42875 and 42876, respectively) were gifts from Luciano Marraffini, and pgRNA-bacteria and pdCas9-bacteria (Addgene plasmids 44251 and 44249, respectively) were gifts from Stanley Qi.
TABLE 1.
Strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Characteristics or sequence (5′→3′)a | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| HI-Control 10G | mcrA Δ(mrr-hsdRMS-mcrBC) endA1 recA1 ϕ80dlacZΔM15 ΔlacX74 araD139 Δ(ara leu)7697 galU galK rpsL (Strr) nupG λ− tonA Mini-F lacIq1 (Gentr) | Lucigen |
| B. subtilis | ||
| 1A751 | his nprR2 nprE18 ΔaprA3 ΔeglS102 ΔbglT bglSRV | 82 |
| AW009 | 1A751 amyE::(Pgrac.UPmod::hasSE:tuaD, Neor) | Our lab |
| AW001-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) | This work |
| AW002-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) thrC::(PxylA.SphI+1::ugtP-gRNA.P395T, ParaE::mazF, Spcr) ΔugtP | This work |
| AW003-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) thrC::(ugtP-CRISPRa.P395T, ParaE::mazF, Spcr) ΔugtP | This work |
| AW004-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) amyE::Pgrac::hasSE:tuaD thrC::(PxylA.SphI+1::amyE-gRNA.P636T, ParaE::mazF, Spcr) | This work |
| AW005-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) amyE::Pgrac::hasSE:tuaD thrC+ | This work |
| AW006-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) amyE::Pgrac::ΔhasSE:tuaD thrC::(PxylA.SphI+1::hasSE-gRNA.P394T, ParaE::mazF, Spcr) | This work |
| AW007-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) amyE::Pgrac::ΔhasSE:tuaD thrC+ | This work |
| AW008-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) amyE::Pgrac::ΔhasSE:tuaD thrC::(PxylA.SphI+1::ugtP-gRNA.P395T, ParaE::mazF, Spcr) ΔugtP | This work |
| AW009-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) thrC::(PxylA.SphI+1::amyE-gRNA.P636T, PxylA.SphI+1::ugtP-gRNA.P395T, ParaE::mazF, Spcr) ΔamyE, ΔugtP | This work |
| AW010-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) thrC::(PxylA.SphI+1::amyE-gRNA.P636T, ParaE::mazF, Spcr) ΔamyE | This work |
| AW011-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) thrC::(PxylA.SphI+1::amyE-gRNA.P25NT, ParaE::mazF, Spcr) ΔamyE | This work |
| AW012-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) thrC::(PxylA.SphI+1::amyE-gRNA.P330T, ParaE::mazF, Spcr) ΔamyE | This work |
| AW013-2 | 1A751 lacA::(cas9, tracrRNA, Ermr) thrC::(PxylA.SphI+1::amyE-gRNA.P1344T, ParaE::mazF, Spcr) ΔamyE | This work |
| AW014-2 | 1A751 amyE::(Pgrac.UPmod::hasSE:tuaD, Neor) lacA::(PxylA, Bm::dcas9, xylR, Ermr) | This work |
| AW015-2 | 1A751 amyE::(Pgrac.UPmod::hasSE:tuaD, Neor) lacA::(PxylA, Bm::dcas9, xylR, Ermr) wprA::(PxylA.SphI+1::lacZ-gRNA.P28NT) | This work |
| AW016-2 | 1A751 amyE::(Pgrac.UPmod::hasSE:tuaD, Neor) lacA::(PxylA, Bm::dcas9, xylR, Ermr), wprA::(PxylA.SphI+1::lacZ-gRNA.P28NT) ugtP::(lacI, Pgrac::lacZ, Spcr) | This work |
| Plasmids | ||
| pIEFBPR | Pspac::mazF lacI Ampr Spcr, B. subtilis autoevicting counterselectable bpr integration vector | 48 |
| pDG1731 | B. subtilis thrC integration vector | 53 |
| pAX01 | PxylA, Bm, xylR Ampr Ermr, B. subtilis lacA integration vector | 54 |
| pCRISPR | E. coli plasmid for CRISPRa transcription | 9 |
| pCas9 | E. coli plasmid for expression of Cas9 | 9 |
| pgRNA-bacteria | E. coli plasmid for gRNA transcription | 29 |
| pdCas9-bacteria | E. coli plasmid for expression of dCas9 | 29 |
| pAW008 | Pgrac::hasSE:tuaD Neor, B. subtilis amyE integration vector | Our lab |
| pAW016 | Pgrac::lacZ lacI Spcr Ermr, B. subtilis ugtP integration vector | Our lab |
| pAW001-2 | Pspac::mazF lacI Spcr Ampr, B. subtilis autoevicting counterselectable thrC integration vector | This work |
| pAW002-2 | Pspac::mazF lacI Spcr Ermr, B. subtilis autoevicting counterselectable thrC integration vector | This work |
| pAW003-2 | PxylA,Bm::mazF xylR Spcr Ermr, B. subtilis autoevicting counterselectable thrC integration vector | This work |
| pAW004-2 | ParaE::mazF araR Spcr Ermr, B. subtilis autoevicting counterselectable thrC integration vector | This work |
| pAW005-2 | PxylA.SphI+1 ParaE::mazF araR Spcr Ermr, B. subtilis counterselectable thrC integration vector | This work |
| pAW006-2 | PxylA.SphI+1::ugtP-gRNA.P395T ParaE::mazF, araR Spcr Ermr, B. subtilis counterselectable thrC integration vector | This work |
| pAW007-2 | PxylA.SphI+1::amyE-gRNA.P25NT ParaE::mazF araR Spcr Ermr, B. subtilis counterselectable thrC integration vector | This work |
| pAW008-2 | PxylA.SphI+1::amyE-gRNA.P330T ParaE::mazF araR Spcr Ermr, B. subtilis counterselectable thrC integration vector | This work |
| pAW009-2 | PxylA.SphI+1::amyE-gRNA.P636T ParaE::mazF araR Spcr Ermr, B. subtilis counterselectable thrC integration vector | This work |
| pAW010-2 | PxylA.SphI+1::amyE-gRNA.P1344T ParaE::mazF araR Spcr Ermr, B. subtilis counterselectable thrC integration vector | This work |
| pAW011-2 | PxylA.SphI+1::hasSE-gRNA.P394T ParaE::mazF araR Spcr Ermr, B. subtilis counterselectable thrC integration vector | This work |
| pAW012-2 | CRISPRa, ParaE::mazF araR Spcr Ermr, B. subtilis counterselectable thrC integration vector | This work |
| pAW013-2 | ugtP-CRISPRa.P395T, ParaE::mazF araR Spcr Ermr, B. subtilis counterselectable thrC integration vector | This work |
| pAW014-2 | (BglII) PxylA.SphI+1::ugtP-gRNA.P395T (NcoI) ParaE::mazF araR Spcr Ermr, B. subtilis multi-gRNA counterselectable thrC integration vector | This work |
| pAW015-2 | (BglII) PxylA.SphI+1::amyE-gRNA.P636T PxylA.SphI+1::ugtP-gRNA.P395T (NcoI) ParaE::mazF araR Spcr Ermr, B. subtilis multi-gRNA counterselectable thrC integration vector | This work |
| pAW016-2 | cas9, tracrRNA, Ampr, Ermr, B. subtilis lacA integration vector | This work |
| pAW017-2 | PxylA.SphI+1 ParaE::mazF araR Spcr Ermr, B. subtilis autoevicting counterselectable wprA integration vector | This work |
| pAW018-2 | PxylA.SphI+1::lacZ--gRNA.P28NT ParaE::mazF araR Spcr Ermr, B. subtilis autoevicting counterselectable wprA integration vector | This work |
| pAW019-2 | PxylA, Bm::dcas9 xylR Ampr Ermr, B. subtilis lacA integration vector | This work |
| pAW020-2 | Pgrac::hasSE:tuaD Ampr, ET for insertion of the HA biosynthetic operon into the amyE locus at amyE.P636T | This work |
| pAW021-2 | hasSE-ET-1330bp-P394T Ampr, editing template for KO of hasSE at hasSE. P394T | This work |
| pAW022-2 | amyE-ET-1330bp-P636T Ampr, editing template for KO of amyE at amyE.P636T | This work |
| Primers | ||
| P1 | TCTACTTTGACCTGCAGGAAGTCATGTAAAAGATGAGGTTGGTTCATTCTC | |
| P2 | CAGATTGAGCTAGCGAAGGCAGCAGTTTTTTGGCC | |
| P3 | GTGACTGACCCGGGGGCGCGCCCTCGAGGCCTTCCGAAAATGC | |
| P4 | GTCACTGAACTAGTACCGGTAATTCCGGCGACTGTTTCTGTTTCAG | |
| P5 | CTCTACTTGAGCTCTAAAGCCTGGGGTGCCTAATGA | |
| P6 | GCTAAAGAGGTCCCTAGAAGCGCTAAGGATCTAGGTGAAGATCCTTTTTGATAA | |
| P7 | AGCGCTTCTAGGGACCTCTTTAGCTCCTTG | |
| P8 | CTACTTTGAGCCGGCTGAAGCATTTATCAGGGTTATTGTCTCATG | |
| P9 | CATACCTGCTTCCTCCTTAAGATCTCATTTCCCCCTTTGATTTTTAGAT | |
| P10 | CTCTATGACAATTGGCTCCTAACTTATAGGGGTAACACTTAA | |
| P11 | CAGATTGAGGATCCCTACCCAATCAGTACGTTAATTTTGGC | |
| P12 | AGATCTTAAGGAGGAAGCAGGTATGGTAAGC | |
| P13 | CATACCTGCTTCCTCCTTAAGATCTATGTTGAGTAAAGCGTTTTCATTTAAACCTTC | |
| P14 | CACTTTGAGAATTCTTATTCATTCAGTTTTCGTGCGGACTG | |
| P15 | GTCATTGAGCTAGCCATAAAAAACTAAAAAAAATATTGA | |
| P16 | GACATTGGATCCTGACATCAGTTACAGCATGCATCTTATATAACCTCGTCAGTATTT | |
| P17 | TATGATGCATGCAAGGAAAAACTGCTGGAGATGTTTTAGAGCTAGAAATAGCAAGTTAAA | |
| P18 | TATGATGCATGCAGCGAATAACGGCAGTAAAGGTTTTAGAGCTAGAAATAGCAAGTTAAA | |
| P19 | TATGATGCATGCCAGCCGACATCGTATCAAATGTTTTAGAGCTAGAAATAGCAAGTTAAA | |
| P20 | TATGATGCATGCGCATTGAATGACGGGGCAGAGTTTTAGAGCTAGAAATAGCAAGTTAAA | |
| P21 | TATGATGCATGCGGTTCATTTCAAGTGAACGAGTTTTAGAGCTAGAAATAGCAAGTTAAA | |
| P22 | TATGATGCATGCCAAGCAATGTCATTGTTCATGTTTTAGAGCTAGAAATAGCAAGTTAAA | |
| P23 | GCTCATGAGGATCCAAAAAAAGCACCGACTCGGT | |
| P24 | GAGACTTGGCTAGCTATTTCTTAATAACTAAAAATATGGTATAATACTC | |
| P25 | CAGATTGAGGATCCATGAGTTCAACTCAAC | |
| P26 | AAACAGATTACGTGAAGGAAAAACTGCTGGAGATG | |
| P27 | AAAACATCTCCAGCAGTTTTTCCTTCACGTAATCT | |
| P28 | GGAACTGCTAGCTGAATCATACAGATCTCATAAAAAACTAAAAAAAATATTGAAAATACT | |
| P29 | TTAACGTACTGATTGGGTAGCCATGGAAAAAAAGCACCGACTCGGT | |
| P30 | CCATGGCTACCCAATCAGTACGTTAATTTTGG | |
| P31 | GCTCATGAGGATCCTAAGGAGGAAGCAGGTATGG | |
| P32 | GCTATTGACCATGGAAAAAAAGCACCGACTCGGT | |
| P33 | GCTCATGAGGATCCAAAAAAAGCACCGA | |
| P34 | GTGTTAGATCTAGATAATGCGGTAGTTTATCACAGTTAAATTGC | |
| P35 | CTAAGATATCTAGATCAGTCACCTCCTAGCTGACTCAAATC | |
| P36 | GCTATTGACCTGCAGGTCGTGATGAGCAGCTGAGCC | |
| P37 | AGTATTTTCAATATTTTTTTTAGTTTTTTATGGCTAGCCAGGCTCATATTTCACATCCGC | |
| P38 | GCTAGCCATAAAAAACTAAAAAAAATATTGAAAATACTG | |
| P39 | GTGTTACCATGGCCTAGGTAGATTGAGCATGCCTTATTTTCATCTTATATAACCTCGTCA | |
| P40 | GACTTAGAGGCGCGCCGTCGCAGGCATTATTGCAGC | |
| P41 | GTGATTGAACCGGTTTCCTCTGACAGCTGTTTGAGTG | |
| P42 | GCATTAGCATGCGTCACGACGTTGTAAAACGAGTTTTAGAGCTAGAAATAGCAAGTTAAA | |
| P43 | GCTATTGAACTAGTAAAGGAGGTATCAAGTATGGATAAGAAATACTCAATAGGCTTAGCT | |
| P44 | GACATTGAAGATCTCCTGCAGGATAAAACGAAAGGCCCAGTCTTTC | |
| P45 | ACAAGAGGTTTGACGGCATGATTATC | |
| P46 | GATTTTTACATTGCTTGGATGTCATGATTATCACAGCAGTTTTTCCTTCACGTAATCTG | |
| P47 | TGATAATCATGACATCCAAGCAATGTAAAAATCACAGG | |
| P48 | CGCAGCTATGGATGATAAAGACTTG | |
| P49 | CCTACCTTCAACGTTATGACTG | |
| P50 | ACTTTTTTGTTTGGTGAAAGATTGTAC | |
| P51 | CTCAATCGGGAAACAGTTTTATCG | |
| P52 | ATCAATGCGCTCCACATAGC | |
| P53 | CATTGAGGTGAATTTACTTGAATACC | |
| P54 | GGAAGTGACTGACTCGAGA | |
| P55 | CGTTTACCAAGAAACTCCTTATGAATG | |
| P56 | TCAAAAGAAGGGCAAGTTCC | |
| P57 | ACATTCTTACCGCATCAAAGGAAGC | |
| P58 | CCATCTCCTTCGATAGCTGTGAAG | |
| P59 | GCTAGCCTGATCTCGACCATCGAATTCTTAGTGG | |
| P60 | CCAATGATTCGGATTTTGATAGCCGATGGT | |
| P61 | GTGATTGACCATGGTTGTTTGGAAAGCGAGGGAAGCGTTC | |
| P62 | GGTCTATTGCTAGCTGTGTGTTTCCATGTGTCCAGTTTGG | |
| P63 | GGCTTTTTGAACGATAGATTGCACCAG | |
| P64 | TCAAGCAATGTCATTGTTTAATAAGAGCTCGTCAATCAAGGAAAGCGTCATGCACAG | |
| P65 | GAGCTCTTATTAAACAATGACATTGCTTGATAGGTCACC | |
| P66 | CAATGATTCGGATTTTGATAGCCGATGG | |
| P67 | CGGGAGGAAGGTCATGAATAATCTGC | |
| P68 | GGCGGCATCAAATCGAAATTAAGTACTTTATCAATTCAATGCCCTGTCTAAGAACCG | |
| P69 | TGATAAAGTACTTAATTTCGATTTGATGCCGCCAAACATATAGA | |
| P70 | GGAAGAGAACCGCTTAAGCCCG | |
| P71 | CCATACATTCTTCGCTTGGCTG | |
| P72 | GTTACACCATCACTGTTCGTTCC | |
| P73 | TTGCCGCCAGCGGTATTCC | |
| P74 | TCCAGCGAATAACGGTTACTCGAGTTATTTGAATCGTTTTGCAAACATTCTTGACACTC | |
| P75 | TAACTCGAGTAACCGTTATTCGCTGGATTTTTATTGC | |
| P76 | AAGAGGCGTACTGCCTGAACG | |
| P77 | AAATCTGGTCGGAGATTGGGATGATAGC | |
| P78 | CTTGTTCAGTACCTAAGTAACGTCATTACTCGAGATACGATGTCGGCTGATACAGCC | |
| P79 | CTCGAGTAATGACGTTACTTAGGTACTGAACAAGAATTTAAAGAAATG | |
| P80 | TTCACTTGAAATGAACCCGCTCCA | |
| P81 | GCAACCGTTACTTAGGTACTGAACAAG | |
| P82 | GATCGTGCCTGTCAGTCATTAGGATCCCACTTGAAATGAACCCGCTCCAG | |
| P83 | GGATCCTAATGACTGACAGGCACGATCAATGCCAG | |
| P84 | GATGTTTTGACCGGTTGTGGCG | |
| P85 | CGAAGCGAAGGAAAATGGATGC | |
| P86 | GGTCAAAGCGTCTACTTCACAATGAG | |
| P87 | TGTATGAACGGTCTGGTCTTTGCC | |
| P88 | CAGGTATTCGCTGGTCACTTCGATG | |
| P89 | GAAACGGCAAAACGTTCTGG | |
| P90 | GTGTTGGGTTCACAATGTCG | |
| P138 | GGTTCACAACGATGGCAGCTC | |
| P139 | ACTGATCTGGATCCATGAGAACATTAAAAAACCTCATAACTGTTGTG | |
| P140 | GATTGATCTAGATTAGTGGTGATGATGGTGGTGTAATAATTTTTTACGTGTTCCCCAGTC | |
| P141 | ACAAGCTTCCTTTAGCACCAAAGAGAT | |
| P142 | AGAGGGATTTTTGACTCCGAAGTAAGTC | |
| P143 | ACGCAAATGCATAACTGCTTCCAACA |
Gene products: amyE, α-amylase; lacA (ganA), β-galactosidase; cas9, CRISPR-associated protein 9 (Cas9; Streptococcus pyogenes); mazF, endoribonuclease (Escherichia coli); thrC, threonine synthase; ugtP, UDP-glucose diacylglyceroltransferase; hasSE, hyaluronan synthase (Streptococcus equisimilis); tuaD, UDP-glucose 6-dehydrogenase; xylR, xylose operon repressor (Bacillus megaterium); wprA, cell wall-associated protease; dcas9, dead Cas9 (derived from S. pyogenes); lacZ, β-galactosidase (E. coli); lacI, lactose operon repressor (E. coli); bpr, bacillopeptidase F; araR, repressor of arabinose operons. Abbreviations: Neor, neomycin resistance cassette; Ermr, erythromycin resistance cassette; Spcr, spectinomycin resistance cassette; Ampr, ampicillin resistance cassette. In the primer sequences, restriction sites used for cloning are underlined, inserted restriction sites are italicized, and protospacer sequences are in bold.
Plasmid and editing template construction.
DNA manipulation was performed using standard cloning techniques (52), and DNA sequencing was conducted by The Centre for Applied Genomics (TCAG) (Toronto, Ontario, Canada). We previously identified the thrC locus as a recombination “hot spot” in the genome of B. subtilis, making it a potential site for integration of gRNA transcription cassettes. To construct the gRNA delivery vector, we began by amplifying the thrC 5′ and 3′ homology lengths (HL-5′ and HL-3′) from plasmid pDG1731 (53) with primers P01/P02 and P03/P04, respectively, followed by insertion in place of the corresponding bpr HL-5′ and HL-3′ of pIEFBPR (48) using the respective SbfI/NheI and XmaI/SpeI restriction sites, yielding pAW001-2. Subsequently, the ampicillin resistance marker (Ampr) in pAW001-2 was replaced with an erythromycin cassette (Ermr). To do this, the ColE1 replicon was amplified from pIEFBPR with primers P05/P06, the Ermr cassette was amplified from pAX01 (54) with primers P07/P08, and the two fragments were spliced via splicing by overlap extension (SOE) PCR (this process is referred to here as splicing), followed by insertion of the spliced fragment into pAW001-2 using the SacI/NgoMIV restriction sites, resulting in pAW002-2.
Potentially due to leaky transcription from the spac promoter (Pspac), the transformation efficiencies of pIEFBPR and pAW002-2 in B. subtilis were unacceptably low. Accordingly, the tightly regulated xylA/R promoter cassette from Bacillus megaterium (PxylA,Bm) was amplified from pAX01 with primers P09/P10 (−/MfeI), mazF was amplified from pIEFBPR with primers P11/P12 (BamHI/−), and the two fragments were spliced. The resulting PxylA,Bm::mazF cassette replaced the Pspac::mazF cassette in BamHI/EcoRI-digested pAW002-2, yielding pAW003-2. Though transformation efficiency was improved in B. subtilis, propagation of pAW003-2 was difficult in E. coli, prompting us to identify another inducible promoter to drive mazF. The araE/R promoter system from B. subtilis (ParaE) was chosen, as it is very tightly repressed in the presence of glucose due to the presence of the catabolite repression element (cre) upstream of the araE open reading frame (ORF) (55). The ParaE cassette was amplified with primers P13/P14 from 1A751 genomic DNA (gDNA) and was spliced with mazF as described above. The resulting ParaE::mazF cassette was used to replace the Pspac::mazF cassette in pAW002-2 using the BamHI/EcoRI restriction sites, resulting in plasmid pAW004-2, a vector found to be stable in E. coli while providing significantly enhanced transformation efficiency in B. subtilis relative to pAW002-2.
The xylA promoter (PxylA) from B. subtilis was used to drive transcription of gRNAs. To do this, PxylA was amplified with primers P15/P16 from 1A751 gDNA, replacing the 6 bp between the −10 and +2 regions of PxylA with a SphI restriction site. The resulting promoter, PxylA.SphI+1 (Fig. 1A), was inserted in place of the direct repeat (DR) adjacent to the thrC HL-5′ of pAW004-2 using the NheI/BamHI restriction sites, yielding pAW005-2 (Fig. 1B). The gRNA cassettes ugtP-gRNA.P395T, amyE-gRNA.P25NT, amyE-gRNA.P330T, amyE-gRNA.P636T, amyE-gRNA.P1344T, and hasSE-gRNA.P394T (where hasSE is the Streptococcus equisimilis hyaluronan synthase gene) were amplified from pgRNA-bacteria (29) with respective forward primers P17 and P22 containing unique protospacers and a common reverse primer, P23. Each gRNA cassette was inserted downstream of PxylA.SphI+1 in pAW005-2 using SphI/BamHI restriction sites to obtain single-gRNA delivery vectors (Fig. 1B). To generate the native CRISPR array (CRISPRa) for ugtP disruption (ugtP-CRISPRa.P395T), the empty CRISPRa was amplified from pCRISPR (9) with primers P24/P25 and inserted in place of the DR adjacent to the thrC HL-5′ of pAW004-2 using the NheI/BamHI restriction sites, yielding pAW012-2. Oligonucleotides P26/P27 were then annealed and ligated into BsaI-digested pAW012-2 as previously described (9), resulting in pAW013-2. The lacA locus was chosen for genomic integration of cas9. To do this, pAW016-2 (Fig. 1C) was constructed by removing the PxylA,Bm cassette from pAX01 by SacI digestion and self-ligation and then inserting the cas9-tracrRNA cassette amplified from pCas9 (9) with primers P34/P35 using the XbaI restriction sites. The orientation of the cas9-tracrRNA cassette was confirmed by DNA sequencing.
FIG 1.

Schematic representation of the PxylA.SphI+1 gRNA transcription cassette and the single-gRNA and Cas9 delivery vectors. (A) Sequences of the native promoter PxylA of B. subtilis and PxylA.SphI+1. The 6 bp between the −10 and +2 regions of PxylA were replaced with a SphI restriction site enabling protospacer (PS) exchange without the need for inverse PCR. A unique protospacer is introduced as an overhang in the forward primer, amplifying the CBH and terminator (ter) as a single fragment, and the resulting gRNA cassette is inserted downstream of PxylA.SphI+1 in the single-gRNA delivery vector. The −35 and −10 regions are in bold, the +1 is in dark bold, and the SphI restriction site is underlined. (B) PxylA.SphI+1 was inserted into pAW004-2, removing the DR adjacent to the thrC HL-5′ and yielding pAW005-2. gRNA cassettes were inserted between the SphI and BamHI restriction sites of pAW005-2, generating respective single-gRNA delivery vectors. Transformation of a linearized single-gRNA delivery vector results in integration of the combined PxylA.SphI+1::gRNA-ParaE::mazF-Spcr (gRNA*) cassette at the thrC locus. The gRNA* cassette is subsequently evicted by transformation of the thrC editing template, followed by arabinose selection to induce mazF expression and screening for spectinomycin sensitivity. (C) The PxylA,Bm cassette was removed from pAX01 and the cas9-tracrRNA cassette inserted, yielding pAW016-2. Transformation of linearized pAW016-2 results in integration of the combined cas9-tracrRNA-Ermr (Cas9*) cassette at the lacA locus.
To construct the multi-gRNA delivery vector, the BamHI restriction site downstream of mazF was replaced with a NcoI restriction site, and the BglII restriction site upstream of mazF was removed and a new BglII restriction site was inserted between the NheI restriction site and PxylA.SphI+1 in pAW006-2 (Fig. 2), to facilitate Biobrick cloning of gRNA transcription cassettes. This was accomplished by amplifying the PxylA.SphI+1::ugtP-gRNA.P395T cassette and mazF from pAW006-2 with primers P28/P29 (NheI/−) and P30/P31 (−/BamHI), respectively, followed by splicing of the two fragments to generate a PxylA.SphI+1::ugtP-gRNA.P395T-mazF cassette. The PxylA.SphI+1::ugtP-gRNA.P395T-mazF cassette was subsequently inserted into NheI/BglII-digested pAW005-2, yielding pAW014-2, and the modifications to pAW005-2 which resulted in pAW014-2 are summarized in Fig. 2. In this arrangement, the first gRNA cassette to be inserted into the multi-gRNA delivery vector is amplified with a forward primer introducing the unique protospacer and reverse primer P32 from pgRNA-bacteria (or any plasmid containing a gRNA) and is cloned into pAW014-2 using the SphI/NcoI restriction sites. Each additional gRNA is cloned into pAW005-2, generating single-gRNA delivery vectors from which the corresponding gRNA transcription cassettes are amplified with primers P28/P33 (NheI/BamHI) and sequentially inserted into the NheI/BglII-digested multi-gRNA delivery vector. All gRNA delivery vectors were linearized via SacI digestion prior to transformation into B. subtilis. To enable continuous genome editing (a procedure which is summarized in Fig. 3), 1A751 was first transformed with BseYI-linearized pAW016-2, resulting in strain AW001-2, which constitutively expresses Cas9 from the lacA locus. Continuous editing is then performed by transforming AW001-2 (or its derivatives) with a single- or multi-gRNA delivery vector, integrating the combined PxylA.SphI+1::gRNA-ParaE::mazF-Spcr cassette (gRNA*) at the thrC locus, and an editing template(s) introducing the desired mutation(s). Cells containing the desired mutation(s) evade the CRISPR-Cas9-mediated chromosomal DSB(s) (as elimination of the PAM site[s] occurs in edited gDNA), and the mutation(s) is verified with genetic screening and sequencing. The gRNA* cassette is subsequently removed via transformation of the thrC editing template, restoring the integration site for the next round of editing.
FIG 2.

Schematic representation of the construction of the multi-gRNA delivery vector. The BamHI restriction site was replaced with a NcoI restriction site and the BglII restriction site moved between the NheI restriction site and PxylA.SphI+1 in pAW006-2 (top) to facilitate Biobrick cloning, yielding pAW014-2 (bottom). The PxylA.SphI+1::amyE-gRNA.P636T cassette was inserted between the NheI/BglII restriction sites of pAW014-2, resulting in pAW015-2. In general, a single-gRNA delivery vector is generated from pAW005-2, and the gRNA transcription cassette is amplified, digested with NheI/BamHI, and inserted into the NheI/BglII-digested multi-gRNA delivery vector. If a single-gRNA delivery vector is not required, PxylA.SphI+1 and the gRNA cassette can be spliced and cloned directly into the multi-gRNA delivery vector. PS, protospacer; ter, terminator.
FIG 3.

Continuous editing with the CRISPR-Cas9 tool kit. The combined cas9-tracrRNA-Ermr (Cas9*) cassette was integrated into the lacA locus of 1A751 via transformation with pAW016-2, generating strain AW001-2, which constitutively expresses Cas9. A linearized single- or multi-gRNA delivery vector and the editing template(s) (ET) are transformed into AW001-2 (or one of its derivatives), resulting in integration of the combined PxylA.SphI+1::gRNA-ParaE::mazF-Spcr (gRNA*) cassette at the thrC locus and introduction of the desired mutation(s) via integration of the editing template(s). Cells containing the desired mutation(s) evade the CRISPR-Cas9-mediated chromosomal DSB(s) due to the elimination of the PAM site(s) in the edited gDNA. The resulting mutant is resistant to spectinomycin and sensitive to arabinose induction of mazF expression. After the desired mutation(s) is verified, the gRNA* cassette is evicted by transformation of the thrC editing template to arabinose resistance and spectinomycin sensitivity, restoring the native thrC locus. The mutant is now ready for the next round of editing using the same procedure. See Fig. 1B and C for schematic representations of the gRNA* and Cas9* cassettes, respectively.
To construct the gRNA delivery vector for dCas9 targeting, the wprA HL-5′ was amplified with primers P36/P37, and PxylA.SphI+1 with P38/P39 from 1A751 gDNA, and the two fragments were spliced, followed by insertion of the spliced fragment in place of the thrC HL-5′ in pAW004-2 using the SbfI/NcoI restriction sites. The vector was completed by amplifying the wprA HL-3′ with primers P40/P41 from 1A751 gDNA, followed by insertion in place of the thrC HL-3′ using the AscI/AgeI restriction sites, resulting in pAW017-2. The wprA locus was chosen as the integration site for dCas9-targeting gRNA transcription cassettes for compatibility purposes based on intended future applications for CRISPRi in other areas of research conducted by our group. The lacZ-gRNA.P28NT cassette was amplified from pgRNA-bacteria with primers P42/P32 and cloned into pAW017-2 using the SphI/NcoI restriction sites, yielding pAW018-2 (Fig. 4A). pAW018-2 was linearized via SacI digestion prior to transformation into B. subtilis. Similar to the case for cas9, the lacA locus was selected for genomic integration of the xylose-inducible dCas9 cassette. To do this, pAW019-2 (Fig. 4B) was constructed by amplifying dcas9 from pdCas9-bacteria (29) with primers P43/P44 (SpeI/BglII), followed by insertion into SpeI/BamHI-digested pAX01. To enable transcriptional interference via dCas9 (a procedure which is summarized in Fig. 5), AW009 was transformed with BseYI-linearized pAW019-2, yielding strain AW014-2, which expresses xylose-inducible dCas9 from the lacA locus. Note that the hyaluronic acid (HA)-producing strain AW009 was selected as the host for CRISPRi demonstration to accommodate our research in strain engineering for enhanced HA production in B. subtilis, and any strain possessing an intact lacA locus (e.g., 1A751) can be used in place of AW009. AW014-2 was then transformed with pAW018-2, resulting in integration of the PxylA.SphI+1::lacZ-gRNA.P28NT cassette and the combined ParaE::mazF-Spcr (CS) cassette at the wprA locus. The CS cassette was subsequently autoevicted via single-crossover recombination between the flanking DRs (48), yielding strain AW015-2, which transcribes lacZ-gRNA.P28NT from the wprA locus. Subsequent genomic integration of an inducible copy of lacZ from E. coli at the ugtP locus was performed to assess CRISPRi. To do this, AW015-2 was transformed with pAW016, a vector previously constructed in our lab for genomic integration of isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible lacZ at the ugtP locus, yielding strain AW016-2 (note that this part is not shown in Fig. 5). The gRNA directs dCas9 to the target (i.e., lacZ) based on the presence of a PAM site and adjacent seed region complementary to the protospacer, and the dCas9-gRNA complex remains bound to the target, blocking transcription by RNA polymerase (RNAP).
FIG 4.

Schematic representation of the gRNA delivery vector for integration of dCas9-targeting gRNA transcription cassettes (A) and the dCas9 delivery vector (B). (A) The wprA HL-5′ and PxylA.SphI+1 were spliced and inserted in place of the thrC HL-5′ of pAW004-2 preserving the adjacent DR, and the thrC HL-3′ was replaced with the wprA HL-3′, generating pAW017-2. lacZ-gRNA.P28NT was inserted between the SphI and NcoI restriction sites of pAW017-2, yielding pAW018-2. Transformation of linearized pAW018-2 results in integration of the PxylA.SphI+1::lacZ-gRNA.P28NT (gRNA‡) cassette and the combined ParaE::mazF-Spcr (CS) cassette at the wprA locus. The CS cassette is autoevicted via single-crossover recombination between the flanking DRs. (B) dcas9 was inserted downstream of PxylA,Bm in pAX01, yielding pAW019-2. Transformation of linearized pAW019-2 results in integration of the combined PxylA,Bm::dcas9-Ermr (dCas9*) cassette at the lacA locus. PS, protospacer; ter, terminator.
FIG 5.

Implementing CRISPRi with the CRISPR-Cas9 tool kit. The combined PxylA,Bm::dcas9-Ermr (dCas9*) cassette was integrated into the lacA locus of strain AW009 via transformation with pAW019-2, yielding strain AW014-2, which expresses xylose-inducible dCas9 (note that any strain possessing an intact lacA locus [e.g., 1A751] can be used in place of AW009). AW014-2 was then transformed with pAW018-2, resulting in integration of the PxylA.SphI+1::lacZ-gRNA.P28NT (gRNA‡) cassette and the combined ParaE::mazF-Spcr (CS) cassette at the wprA locus (the resulting mutant was spectinomycin resistant and arabinose sensitive). The CS cassette was subsequently autoevicted via single-crossover recombination between the flanking DRs (black rectangles), yielding strain AW015-2 (spectinomycin sensitive and arabinose resistant), which transcribes lacZ-gRNA.P28NT from the wprA locus (subsequent integration of IPTG-inducible lacZ [E. coli] at the ugtP locus was performed to assess CRISPRi and is not shown in the figure). The gRNA directs dCas9 to the target based on the presence of a PAM site and adjacent seed region complementary to the protospacer, and the dCas9-gRNA complex remains bound to the target, blocking transcription by RNA polymerase (RNAP). See Fig. 4A and B for schematic representations of the gRNA‡ and dCas9* cassettes, respectively.
For the ugtP KO, PAM site ugtP.P395T was selected, where the number in the nomenclature corresponds to the position of the first base pair of the PAM site (P) relative to the beginning of the ugtP ORF (i.e., CGG, where the cytosine is the 395th bp in the ORF) and base-pairing occurs with the template (T) strand (or the nontemplate [NT] strand for other PAM sites). The full-length ugtP editing template was generated by splicing the two 1,337-bp and 1,332-bp HLs (flanking a 12-bp mutation region) (Fig. 6A) amplified with primers P45/P46 and P47/P48, respectively, from 1A751 gDNA. The ugtP editing templates of HLs 100, 300, 500, 750, and 1,000 bp (Fig. 6A) were amplified with primers P49/P50, P51/P52, P53/P54, P55/P56, and P57/P58, respectively, from the full-length ugtP editing template and therefore preserved the original 12-bp mutation region. The full-length amyE editing template (amyE.P636T) was constructed by splicing the two 1,368-bp and 1,335-bp HLs (flanking a 15-bp mutation region) (Fig. 6B) amplified with primers P67/P68 and P69/P70, respectively, from 1A751 gDNA. The full-length amyE editing template was inserted into pJET1.2/blunt using the CloneJET PCR cloning kit (ThermoFisher Scientific, Waltham, MA, USA) as per the manufacturer's instructions, yielding pAW022-2, which was linearized via BsaI digestion prior to transformation into B. subtilis as an editing template. The 1,000-bp HL amyE editing template (amyE.P636T) was amplified from the full-length amyE editing template with primers P71/P72, preserving the original 15-bp mutation region. The amyE editing templates for PAM sites amyE.P25NT (primers P73/P74 and P75/P76), amyE.P330T (primers P77/P78 and P79/P80), and amyE.P1344T (primers P81/P82 and P83/P84) were constructed in the same way as the full-length amyE editing template (amyE.P636T), except that only 1,000-bp HLs flanked 11-bp (amyE.P330T) or 12-bp (amyE.P25NT and amyE.P1344T) mutation regions (Fig. 6B). Editing templates introduced premature stop codons and restriction sites in place of PAM sites and adjacent nucleotides. Alternatively, the HA operon (i.e., Pgrac::hasSE:tuaD) flanked by the amyE HL-5′ and HL-3′ was also used as the editing template for evading the DSB associated with PAM site amyE.P636T, resulting in KI of the HA operon at the amyE locus. To construct this editing template, the partial HA operon KI cassette, containing the 759-bp amyE HL-3′ and 2,909-bp Pgrac::hasSE:tuaD HA operon, was amplified from pAW008, a vector previously constructed in our lab, with primers P59/P60 and inserted into pJET1.2/blunt, yielding an intermediate vector. To complete the HA operon KI cassette, the 738-bp amyE HL-5′ was amplified from 1A751 gDNA with primers P61/P62 and inserted into the intermediate vector using the NcoI/NheI restriction sites, yielding pAW020-2. pAW020-2 was linearized via ScaI digestion prior to transformation into B. subtilis as an editing template. The hasSE editing template (hasSE. P394T) was constructed by splicing the two 1,303-bp and 1,338-bp HLs (flanking a 15-bp mutation region) (Fig. 6C) amplified with primers P63/P64 and P65/P66, respectively, from pAW020-2. The hasSE editing template was inserted into pJET1.2/blunt to enhance the transformation efficiency of the poorly transformable HA-producing strain AW005-2, and the resulting plasmid was pAW021-2. pAW021-2 was linearized via ScaI digestion prior to transformation into B. subtilis as an editing template. The thrC editing template used to evict gRNA transcription cassettes was generated as a 2,876-bp PCR product (i.e., 1,452-bp and 1,306-bp fragments flanking the deleted 118-bp region of thrC) amplified with primers P85/P86 from 1A751 gDNA.
FIG 6.

Unaltered sequences and mutation regions of editing templates for ugtP, hasSE, and amyE KOs and schematic representation of the KI of the HA biosynthetic operon at the amyE locus. (A) The native (ugtP) and modified (editing template) sequences of the mutation region for ugtP KO at ugtP.P395T are in uppercase, and the adjacent 20 bp of flanking homology is in lowercase. In the native sequence, the PAM site is underlined and the two base pairs between which the DSB occurs are in bold. The BspHI restriction site is italicized in the modified sequence, and a summary of HLs analyzed during editing template (ET) HL optimization is shown. (B) The native (amyE) and modified (editing template) sequences of the mutation regions for amyE KO at amyE.P25NT, amyE.P330T, amyE.P636T, and amyE.P1344T are in uppercase, and the adjacent 18 to 21 bp of flanking homology is in lowercase. In the native sequences, PAM sites are underlined and the two base pairs between which the DSBs occur are in bold. The XhoI (amyE.P25NT and amyE.P330T), ScaI (amyE.P636T), and BamHI (amyE.P1344T) restriction sites are italicized in the modified sequences. (C) The unaltered (hasSE) and modified (editing template) sequences of the mutation region for hasSE KO at hasSE. P394T are in uppercase, and the adjacent 18 bp of flanking homology is in lowercase. In the unaltered sequence, the PAM site is underlined, and the two base pairs between which the chromosomal DSB occurs are in bold.
Competent-cell preparation and transformation.
Transformation of B. subtilis was performed using a standard protocol for natural competence (56). SpC medium contained the following: (NH4)2SO4, 1.67 g/liter; K2HPO4, 11.64 g/liter; KH2PO4, 5.0 g/liter; trisodium citrate dihydrate, 833 mg/liter; glucose, 4.17 g/liter; MgSO4 · 7H2O, 151 mg/liter; yeast extract, 1.67 g/liter; Casamino Acids, 208 mg/liter; Arg, 7.5 g/liter; His, 383 mg/liter; and Trp, 48 mg/liter. SpII medium contained the following: (NH4)2SO4, 1.67 g/liter; K2HPO4, 11.64 g/liter; KH2PO4, 5.0 g/liter; trisodium citrate dihydrate, 833 mg/liter; glucose, 4.17 g/liter; MgSO4 · 7H2O, 725 mg/liter; yeast extract, 858 mg/liter; Casamino Acids, 86 mg/liter; Arg, 3.78 g/liter; His, 189 mg/liter; Trp, 24 mg/liter; and CaCl2, 48 mg/liter. To improve transformation efficiency, the following modifications were made to the cited protocol: (i) yeast extract was increased by 30% in SpC (2.17 g/liter) and SpII (1.12 g/liter) media, (ii) glycerol was removed from the resuspension media, and (iii) cells were resuspended in 1/40 of the initial volume of SpII medium (the cited protocol specifies 1/10 of the initial volume). B. subtilis strains were plated on nonselect lysogeny broth (LB) containing 5 g/liter NaCl, 5 g/liter yeast extract, and 10 g/liter tryptone and incubated overnight (O/N). Prewarmed SpC medium was inoculated by cell patches from the O/N plate to an optical density at 600 nm (OD600) of 0.5 to 0.7. Seventy-five minutes after the logarithmic growth phase ended, cultured cells were diluted 100-fold in prewarmed SpII medium and incubated for 110 min before harvesting. A 2-μg quantity of gRNA delivery vector and 2 μg of each editing template were used per transformation (400 μl total volume), and transformed cells were incubated for 80 min (260 revolutions per min [rpm]) and then plated on LB agar containing 12 g/liter glucose (LBG) and 85 μg/ml spectinomycin to select recombinants. All cultivation steps were conducted at 37°C and 300 rpm unless otherwise indicated, and all experiments were performed in triplicate. To remove the gRNA transcription cassettes by transformation of the thrC editing template, cells were transformed with 1.5 μg of the thrC editing template, plated on LB agar containing 20 g/liter arabinose (LBA) to select recombinants, and screened for spectinomycin sensitivity. To facilitate autoeviction of the combined ParaE::mazF-Spcr cassette after transformation of pAW018-2, cells were grown for ∼20 h in nonselect LB at 37°C and 260 rpm, plated on LBA, and screened for spectinomycin sensitivity.
HA production, purification, and analysis.
To assess HA production, AW005-2 was plated on nonselect LB and grown O/N at 37°C. A single colony was used to inoculate 25 ml nonselect LB, and the culture was grown for ∼14 h at 37°C and 280 rpm. The culture was then used to inoculate 20 ml prewarmed nonselect cultivation medium (4%, vol/vol) with the following composition: (NH4)2SO4, 1 g; K2HPO4 · 3H2O, 9.15 g; KH2PO4, 3 g; trisodium citrate · 2H2O, 1 g; yeast extract, 10 g; Casamino Acids, 2.5 g; CaCl2, 5.5 mg; FeCl2 · 6H2O, 13.5 mg; MnCl2· 4H2O, 1 mg; ZnCl2, 1.7 mg; CuCl2 · 2H2O, 0.43 mg; CoCl2 · 6H2O, 0.6 mg; and Na2MoO4 · 2H2O, 0.6 mg. Glucose or sucrose was used as the primary carbon source (20 g/liter), and the cultures were grown at 37°C and 280 rpm in triplicate. Samples were diluted 2-fold in phosphate-buffered saline, and HA was purified with cetylpyridinium chloride as previously described (57). The HA titer was determined using the modified carbazole assay (58), and the molecular mass was analyzed via agarose gel electrophoresis as described previously (59) with slight modifications. Two micrograms of purified HA was loaded per well, and gels stained O/N in 0.005% Stains-All (50% [vol/vol] ethanol) were destained for ∼8 h in 20% (vol/vol) ethanol, followed by destaining for ∼16 h in 10% (vol/vol) ethanol. Gels were then photobleached for 20 min on an LED light box and scanned with an Epson Perfection V600 photo scanner (Epson, Nagano, Japan). Scanned images were analyzed using ImageJ (60), and data analysis was performed as previously described (59). All samples were analyzed in duplicate.
Sample preparation and evaluation of β-galactosidase activity.
To assess transcriptional interference of lacZ, a single colony was used to inoculate 25 ml LB (85 μg/ml spectinomycin), and the seed culture was incubated for ∼14 h at 37°C and 280 rpm. A 0.5-ml volume of the seed culture was transferred into 50 ml LB containing 85 μg/ml spectinomycin, 1 mM IPTG for induction of lacZ, and 1.2% (wt/vol) xylose for induction of dCas9 and was grown to OD600 of ∼1.6. To obtain cell extract, cells in the amount of 30 OD600 units (defined as the product of cell density in OD600 and sample volume in ml) were centrifuged at 10,000 × g for 10 min at room temperature. The cell pellet was resuspended in 1.5 ml Z buffer and sonicated intermittently (0.5/0.5 s on/off) for 4 min in an ice water environment with a Sonicator 3000 ultrasonic liquid processor and microtip (Misonix, Farmingdale, NY, USA). The raw cell extract was then used to determine β-galactosidase activity as previously described (61). All experiments were performed in triplicate.
qRT-PCR.
For RNA isolation, cells were grown as described in the preceding section. Total RNA was prepared using the High Pure RNA isolation kit (Roche Diagnostics, Basel, Switzerland) as per the manufacturer's instructions. cDNAs were synthesized using the high-capacity cDNA reverse transcription kit (ThermoFisher Scientific, Waltham, MA, USA). Sequence specific primers were used for reverse transcription of the lacZ (P88) mRNA and internal control rpsJ (P90) mRNA, encoding the 30S ribosomal protein S10, at a final concentration of 1 μM. One hundred nanograms of total RNA and 20 units of murine RNase inhibitor (New England BioLabs [NEB], Ipswich, MA, USA) were used per 20-μl reaction mixture. Real-time quantitative reverse transcription-PCR (qRT-PCR) was carried out using the Power SYBR green PCR master mix (ThermoFisher Scientific, Waltham, MA, USA) in an Applied Biosystems StepOnePlus system as per the manufacturers' instructions. Sequence-specific primers were used for amplification of lacZ (P87/P88) and rpsJ (P89/P90). Data analysis to quantify relative expression between cultures with or without induction of dCas9 was performed as previously described (62). All experiments were performed in triplicate.
RESULTS
Design and evaluation of the PxylA.SphI+1 gRNA transcription cassette.
Compared to the native pre-crRNA/tracrRNA duplex, the chimeric gRNA has been preferred for CRISPR-Cas9-mediated genome editing and transcriptional interference (12, 14, 28, 29, 35–37, 63) since its introduction. We first developed a gRNA transcription cassette facilitating simple replacement of the protospacer without the requirement for inverse PCR, which is a procedure often employed to replace the existing protospacer (29, 64). Given the requirement for a precise 5′ end to the protospacer (64), we chose the native promoter PxylA for its considerable strength and annotated transcriptional start site (65). To facilitate insertion of the gRNA transcription cassette, we introduced a SphI restriction site between the −10 and +2 regions of PxylA, yielding PxylA.SphI+1 (Fig. 1A). This arrangement allowed the addition of a unique protospacer as an overhang in the forward primer amplifying the combined CBH terminator fragment, generating a gRNA cassette that can be inserted downstream of PxylA.SphI+1 using restriction/ligation cloning. To construct the base B. subtilis strain for evaluation of CRISPR-Cas9 tool kit components, we transformed pAW016-2 into 1A751, resulting in AW001-2, which constitutively expressed cas9 and transcribed the tracrRNA from the lacA locus. On the other hand, the gRNA transcription cassette(s) was integrated into the thrC locus of the B. subtilis genome to ensure gRNA stability and to allow simple eviction of the cassette with a subsequent integration event using the thrC editing template once the CRISPR-Cas9-mediated mutation was complete. Also, mazF was included in the gRNA delivery vectors for genomic cointegration with the gRNA transcription cassette, as it is an effective counterselectable marker in B. subtilis (48). To assess the vector design, we chose to knock out ugtP (encoding a UDP-glucose diacylglyceroltransferase), since the mutation causes a distinct morphological change. For comparison purposes, AW001-2 was transformed with either pAW006-2 (transcribing a gRNA targeting ugtP.P395T) or pAW013-2 (transcribing a CRISPR array targeting ugtP.P395T) and the full-length ugtP editing template, generating AW002-2 and AW003-2, respectively. The editing efficiency was evaluated via phenotypical screening, followed by colony PCR, subsequent BspHI digestion (Fig. 7A), and sequencing of selected colonies. Similar editing efficiencies were observed when transforming pAW006-2 (79%) and pAW013-2 (82%) (Fig. 7B), suggesting functional promoter activity of PxylA.SphI+1. Nevertheless, the transformation efficiency remained low at less than 20 CFU μg−1 editing template. We then modified the competence protocol to increase transformation efficiency as described in Materials and Methods.
FIG 7.

Assessment of the PxylA.SphI+1 gRNA transcription cassette and continuous editing with the CRISPR-Cas9 tool kit. (A) Colony PCR screening of ugtP, hasSE, and amyE KOs and KI of the HA operon. To screen for the ugtP KO (ugtP.P395T), primers P45/P138 amplified a 1,817-bp product, and successful recombination of the editing template generated products of 1,344 bp and 473 bp upon BspHI digestion. To screen for the hasSE KO (hasSE. P394T), primers P139/P140 amplified a 1,284-bp product, and successful recombination of the editing template generated products of 407 bp and 877 bp upon SacI digestion. To screen for the KI of the HA operon (amyE.P636T), primers P141/P143 amplified a 1,559-bp product upon successful recombination of the editing template (no product is observed in the absence of recombination). To screen for the amyE KO (amyE.P636T), primers P142/P143 amplified a 2,286-bp product, and successful recombination of the editing template generated products of 690 bp and 1,596 bp upon ScaI digestion. Lane 1, marker; lanes 2 and 3, modified and unmodified colonies screened for the ugtP KO, respectively; lanes 4 and 5, modified and unmodified colonies screened for the KI of the HA operon, respectively; lanes 6 and 7, modified and unmodified colonies screened for the hasSE KO, respectively; lanes 8 and 9, modified and unmodified colonies screened for the amyE KO, respectively. Images of multiple agarose gels have been spliced together for the purpose of condensing the data presented. (B) The PxylA.SphI+1 gRNA transcription cassette was assessed in a parallel comparison with the native CRISPRa by transforming AW001-2 with either pAW006-2 (transcribing a gRNA targeting ugtP.P395T) or pAW013-2 (transcribing a CRISPRa targeting ugtP.P395T) and the full-length ugtP editing template to knock out ugtP. Editing efficiency was evaluated via phenotypical screening and colony PCR (and subsequent BspHI digestion). Transformation efficiency is defined as the total number of CFU containing the desired mutation generated per microgram of editing template (ET) DNA. Standard deviations (SD) from experiments performed in triplicate are shown. (C) The capacity of the tool kit for continuous editing was evaluated by introducing three successive mutations into the same background. First, the HA operon (Pgrac::hasSE:tuaD) was inserted into the amyE locus (amyE.P636T) of AW001-2 via transformation of pAW009-2 and pAW020-2, resulting in mucoid strain AW004-2. KI efficiency was evaluated via phenotypical screening followed by colony PCR. The combined PxylA.SphI+1::amyE-gRNA.P636T-ParaE::mazF-Spcr cassette was evicted from AW004-2 by transformation of the thrC editing template to arabinose resistance (and spectinomycin sensitivity), resulting in AW005-2. Next, hasSE was mutated (at hasSE.P394T) in AW005-2 via transformation of pAW011-2 and pAW021-2, resulting in AW006-2, a strain exhibiting the wild-type (WT) morphology. Editing efficiency was evaluated by phenotypical screening followed by colony PCR (and subsequent SacI digestion). The combined PxylA.SphI+1::hasSE-gRNA.P394T-ParaE::mazF-Spcr cassette was evicted from AW006-2 as previously described, yielding AW007-2. Finally, ugtP was mutated (at ugtP.P395T) in AW007-2 via transformation of pAW006-2 and the full-length ugtP editing template. Editing efficiency was evaluated by phenotypical screening followed by colony PCR (and subsequent BspHI digestion). SD from experiments performed in triplicate are shown.
Continuous editing for gene KI and KOs.
With the modified transformation protocol, we exploited the capacity of our tool kit for continuous editing. We first conducted genomic insertion of the HA operon (Pgrac::hasSE:tuaD) into the amyE locus (amyE.P636T). HA is a linear, unbranched polysaccharide composed of alternating N-acetyl-d-glucosamine (GlcNAc) and d-glucuronic acid (GlcUA), reaching up to 8 MDa in size (41). The hyaluronan synthase (HasA) autonomously synthesizes HA from UDP-GlcNAc and UDP-GlcUA (66), which are precursors for cell wall synthesis in B. subtilis. As a result, HasA is the only heterologous enzyme required to produce HA in this organism (41). UDP-GlcUA availability has been shown to limit HA production in B. subtilis, such that constitutive expression of the UDP-glucose 6-dehydrogenase is required to achieve high-level production (41). Pgrac is a strong hybrid promoter developed for B. subtilis (67), whereas hasSE and tuaD encode the HasA from Streptococcus equisimilis (68) and native UDP-glucose 6-dehydrogenase (TuaD) (41), respectively. HA was chosen for demonstration for the following reasons. First, it is a high-value therapeutic biopolymer, and only two genes (i.e., hasSE and tuaD, or equivalent homologues) need to be expressed in B. subtilis to achieve significant production. Additionally, HA-producing strains have a prominent and observable mucoid phenotype (41), facilitating evaluation of editing efficiency. AW001-2 was transformed with pAW009-2 (transcribing a gRNA targeting amyE.P636T) and pAW020-2 (as an editing template), resulting in a mucoid strain (AW004-2) upon successful KI. The high KI efficiency for the HA operon (69%) (Fig. 7C) was attributed to the enhanced transformation, which led to a 5-fold increase in the number of successful mutants relative to those from the initial ugtP KO demonstration. To prepare for the next round of editing, AW004-2 was transformed to be arabinose resistant with the thrC editing template (see Fig. 3 for the continuous-editing procedure), producing AW005-2. Eviction of the combined PxylA.SphI+1::amyE-gRNA.P636T-ParaE::mazF-Spcr cassette was confirmed by screening for spectinomycin sensitivity. The efficiency of mazF counterselection was, however, low (6%) compared to that in the initial demonstration (48), due to the significant reduction (∼30-fold) in transformation efficiency observed for HA-encapsulated strains (data not shown). For the next round of editing, hasSE was mutated by transformation of AW005-2 with pAW011-2 (transcribing a gRNA targeting hasSE.P394T) and pAW021-2 (as an editing template), abolishing HA production and the mucoid phenotype in the resulting strain (AW006-2). The hasSE editing efficiency was also low (Fig. 7C), owing to transformation interference from the HA capsule. To further challenge the tool kit, we removed the combined PxylA.SphI+1::hasSE-gRNA.P394T-ParaE::mazF-Spcr cassette using the same counterselection procedure, yielding AW007-2, and subsequently mutated ugtP by transformation of AW007-2 with the full-length ugtP editing template and pAW006-2 (transcribing a gRNA targeting ugtP.P395T), generating AW008-2. The mazF counterselection efficiency was significantly higher when generating AW007-2 (31%) than when generating AW005-2 (6%), supporting the conclusion that poor transformability led to low editing efficiency. The high editing efficiency for the ugtP KO (99%) (Fig. 7C) represented a substantial improvement over the initial ugtP KO demonstration (79%) (Fig. 7B), and this observation coincides with the 276-fold increase in transformation efficiency obtained with the enhanced competence protocol (4.0 × 103 CFU μg−1 editing template versus 14.5 CFU μg−1 editing template). Note that the KI efficiency of the HA operon was comparable to that of single gene insertions reported in E. coli (12). Also, the ugtP editing efficiency was in line with systems developed for E. coli (12, 14) and S. cerevisiae (23, 24), all of which rely on multicopy vectors to deliver CRISPR-Cas9 machinery.
We assessed the capacity of AW005-2 to produce high-molecular-mass HA using glucose or sucrose as the primary carbon source. Similar growth patterns were observed during cultivation on either carbon source (OD600 of ∼8 after 8 h) (Fig. 8A), and these were similar to those of our HA-producing strains constructed using traditional cloning techniques (data not shown). The HA titer was slightly higher for the 8-h cultivation sample with sucrose (717 ± 99 mg/liter) as the carbon source than for that with glucose (530 ± 139 mg/liter) (Fig. 8B), although sucrose metabolism generally led to a significantly higher molecular mass (1.67 ± 0.05 MDa and 1.15 ± 0.09 MDa for sucrose and glucose, respectively) (Fig. 8C). Moreover, a higher maximum molecular mass was obtained (2.1 ± 0.22 MDa and 1.60 ± 0.07 MDa for sucrose and glucose, respectively) and the molecular mass peaked later (and declined to a lesser extent) for the sucrose cultivation. The molecular mass observed during cultivation on either carbon source compares favorably to that from a previous report of HA production in B. subtilis (41), and both the HA titer and molecular mass were similar to those obtained with our HA-producing strains developed through conventional cloning. The titers obtained with AW005-2 also compared favorably to the titer reported for a similar strain of B. subtilis over the same cultivation period (57). A significantly longer cultivation was required to achieve a similar titer, and molecular mass was not assessed in the aforementioned study. In addition, the titer and molecular mass of HA produced by AW005-2 were similar to those obtained with strains of B. subtilis overexpressing additional enzymes of the HA biosynthetic pathway (i.e., GtaB, GlmM, GlmS, and GlmU) in combination with HasA and TuaD (69). Accordingly, it appears that chromosomal expression of Cas9 does not hinder HA production, and this feature is critical for the tool kit to be applied to industrial strain development. Finally, the HA titer increased by extending the cultivation, although a concomitant decrease in molecular mass was observed. Declining molecular mass during extended cultures of HA-producing strains of B. subtilis has been reported (41, 70).
FIG 8.
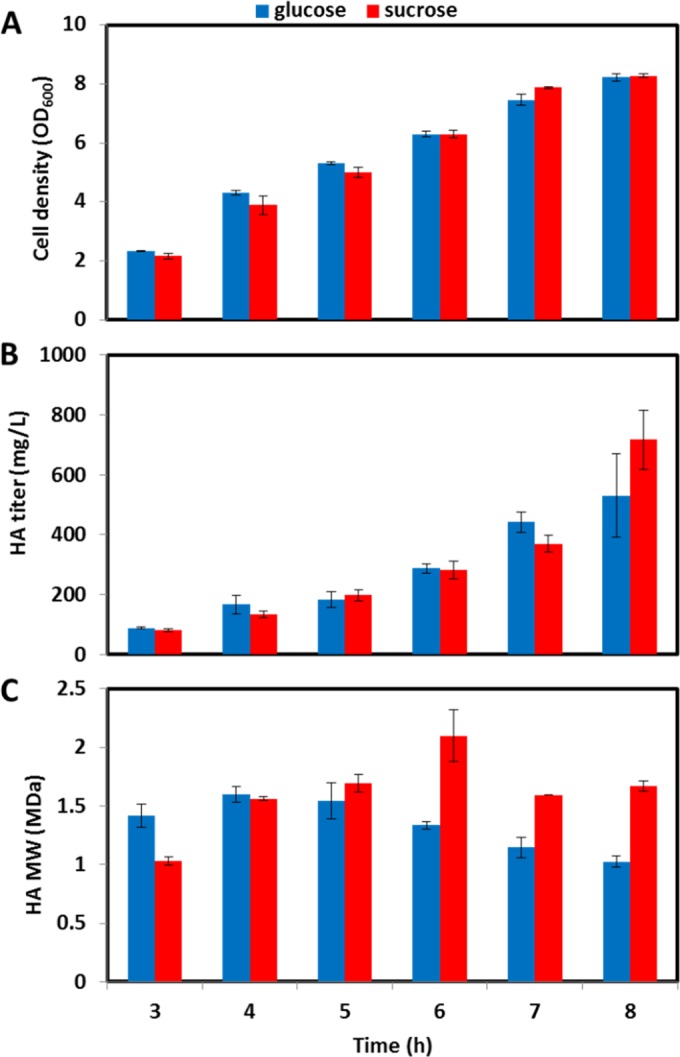
Cultivation of AW005-2 for HA production. (A) Cell density; (B) HA titer; (C) HA molecular mass. SD from experiments performed in triplicate are shown in panels A and B, and SD from duplicate samples are shown in panel C.
Application of the CRISPR-Cas9 tool kit to multiplexing.
For simultaneous editing of B. subtilis genomic targets, we constructed a multi-gRNA delivery vector to accommodate multiple gRNAs using the Biobrick assembly approach (71). Each gRNA transcription cassette can be transferred from its respective single-gRNA delivery vector to the multi-gRNA delivery vector or by direct cloning of the spliced PxylA.SphI+1 and gRNA cassette as described in Materials and Methods. To assess multiplexing capability, the PxylA.SphI+1::amyE-gRNA.P636T cassette was inserted into pAW014-2 using NheI and BglII restriction sites, yielding pAW015-2, to enable simultaneous KO of ugtP and amyE. AW001-2 was transformed with pAW015-2 (transcribing two gRNAs targeting ugtP.P395T and amyE.P636T) and the full-length ugtP and amyE editing templates, which each contain ∼1,330-bp HLs. Colonies were first assessed for the ugtP-null phenotype, after which ugtP mutant and nonmutant colonies were screened for α-amylase (encoded by amyE) deficiency via iodine staining. Colonies from each of the phenotype subsets (i.e., ugtP+ amyE+, ΔugtP amyE+, ugtP+ ΔamyE, and ΔugtP ΔamyE) were screened via colony PCR and subsequent BspHI (ugtP) or ScaI (amyE) digestion, and selected colonies were sequenced. While simultaneous KO of ugtP and amyE was successful, the multiplexing efficiency was only 36% (Fig. 9A), owing to a much lower editing efficiency for amyE than for ugtP (38 and 86%, respectively). Hence, several genome editing factors potentially limiting multiplexing efficiency, specifically the editing template type (i.e., PCR product versus linearized plasmid), HL size, and PAM site sensitivity, were investigated.
FIG 9.

Application of the CRISPR-Cas9 tool kit to multiplexing. (A) The preliminary evaluation of multiplexing efficiency was performed by simultaneously mutating ugtP and amyE via transformation of AW001-2 with pAW015-2 and the full-length ugtP (ugtP.P395T) and amyE (amyE.P636T) editing templates. Mutants were first screened for the ugtP-null phenotype, followed by iodine staining of mutant and WT colonies to evaluate amyE editing efficiency. Colonies from each of the phenotype subsets (i.e., ugtP+ amyE+, ΔugtP amyE+, ugtP+ ΔamyE, and ΔugtP ΔamyE) were screened via colony PCR and subsequent BspHI (ugtP) or ScaI (amyE) digestion. Transformation efficiency is defined as the total number of CFU containing the desired mutation generated per microgram of editing template (ET) DNA. SD from experiments performed in triplicate are shown. (B) amyE was evaluated as a single KO (at amyE.P636T) by transforming AW001-2 with either the full-length amyE editing template or linearized pAW022-2 and pAW009-2. Editing efficiency was evaluated via iodine staining followed by colony PCR and subsequent ScaI digestion. SD from experiments performed in triplicate are shown. (C) Editing template HL was optimized using ugtP as a KO target (ugtP.P395T). Editing templates containing HLs of 100, 300, 500, 750, and 1,000 bp (in addition to the full-length ugtP editing template) were assessed by transforming AW007-2 with pAW006-2 and the corresponding editing templates. Editing efficiency was evaluated by phenotypical screening followed by colony PCR (and subsequent BspHI digestion). SD from experiments performed in triplicate are shown. (D) PAM site sensitivity analysis for amyE. Three PAM sites in the amyE ORF were evaluated (amyE.P25NT, amyE.P330T, and amyE.P1344T), in addition to amyE.P636T, using the optimized editing template HL of 1,000 bp. AW001-2 was transformed with pAW007-2 (amyE.P25NT), pAW008-2 (amyE.P330T), pAW009-2 (amyE.P636T), or pAW010-2 (amyE.P1344T) and the corresponding editing templates. Editing efficiency was evaluated via iodine staining followed by colony PCR and subsequent XhoI (amyE.P25NT and amyE.P330T), ScaI (amyE.P636T), or BamHI (amyE.P1344T) digestion. SD from experiments performed in triplicate are shown. (E) Colony PCR screening of amyE KO at amyE.P25NT, amyE.P330T, amyE.P636T, or amyE.P1344T. To screen for the amyE KO at amyE.P25NT, primers P77/P80 amplified a 2,001-bp product, and successful recombination of the editing template generated products of 685 bp and 1,316 bp upon XhoI digestion. To screen for the amyE KO at amyE.P330T, primers P67/P76 amplified a 1,772-bp product, and successful recombination of the editing template generated products of 703 bp and 1,069 bp upon XhoI digestion. To screen for the amyE KO at amyE.P636T, primers P142/P143 amplified a 2,286-bp product, and successful recombination of the editing template generated products of 690 bp and 1,596 bp upon ScaI digestion. To screen for the amyE KO at amyE.P1344T, primers P142/P143 amplified a 2,286-bp product, and successful recombination of the editing template generated products of 1,396 bp and 890 bp upon BamHI digestion. Lane 1, marker; lanes 2 and 3, modified and unmodified colonies screened for the amyE KO at amyE.P25NT, respectively; lanes 4 and 5, modified and unmodified colonies screened for the amyE KO at amyE.P330T, respectively; lanes 6 and 7, modified and unmodified colonies screened for the amyE KO at amyE.P636T, respectively; lanes 8 and 9, modified and unmodified colonies screened for the amyE KO at amyE.P1344T, respectively. Images of multiple agarose gels have been spliced together for the purpose of condensing the data presented. (F) Enhanced multiplexing using editing template combination 1. ugtP and amyE were simultaneously mutated by transforming AW001-2 with pAW015-2 and the 1,000-bp HL ugtP (ugtP.P395T) and amyE (amyE.P636T) editing templates. Editing efficiency was evaluated as described for panel A, and SD from experiments performed in triplicate are shown. (G) Enhanced multiplexing using editing template combination 2. ugtP and amyE were simultaneously mutated by transforming AW001-2 with pAW015-2 and the 1,000-bp HL ugtP (ugtP.P395T) editing template and pAW022-2 (amyE.P636T). Editing efficiency was evaluated in the same way as for editing template combination 1. SD from experiments performed in triplicate are shown.
Effect of editing template type.
Due to the distinctively low amyE editing efficiency and given that transformation efficiencies for PCR products are expectedly lower than those for linearized plasmids (46), the amyE single KO was evaluated by transforming AW001-2 with pAW009-2 (transcribing a gRNA targeting amyE.P636T) and the full-length amyE editing template (as a PCR product editing template) or pAW022-2 (as a plasmid editing template). While the amyE editing efficiency was significantly higher as a single KO (93%) (Fig. 9B), the transformation efficiency (2.69 × 102 CFU μg−1 editing template) was low. On the other hand, the use of pAW022-2 as an editing template increased the transformation efficiency by nearly 6-fold (1.55 ×103 CFU μg−1 editing template) and, in turn, the editing efficiency (100%) (Fig. 9B) compared to those for the full-length amyE editing template.
Effect of HL size.
The optimal HL was determined by targeting ugtP, as it was perceived to be a recombination “hot spot” based on generally high editing and transformation efficiencies. Editing templates containing 100-, 300-, 500, 750-, and 1,000-bp HLs were constructed from the full-length ugtP editing template such that the same mutation region was flanked by the specified HL (Fig. 6A). Various editing templates were transformed with pAW006-2 (transcribing a gRNA targeting ugtP.P395T) into AW007-2. The editing efficiency remained high for HLs between 500 and 1,000 bp (>97%) (Fig. 9C) but decreased dramatically when the HL was reduced to 300 bp. No transformants were obtained for the 100-bp HL. Our results are consistent with earlier reports suggesting that 400- to 500-bp HLs are sufficient for acceptable transformation efficiency of linear DNA in B. subtilis (49). The optimal HL was determined to be 1,000 bp, for which editing efficiency reached ∼100%.
PAM site sensitivity.
To further improve amyE editing efficiency, we assessed three PAM sites in the amyE ORF, in addition to the original PAM site (amyE.P636T). The PAM sites were selected based on the purine content of the last four bp of the 3′ end of the protospacer (minimum of 75%) (63) and the location relative to the initial PAM site. P25NT was the first available site in the ORF; P330T was approximately half the distance from P25NT to P636T, and P1344T was approximately half the distance from P636T to the stop codon. Due to the moderate GC content of the B. subtilis genome (43.5%), all protospacers were 40 to 55% GC, and the targeting strand was not considered a priority due to a modest effect on gRNA efficacy (63). AW001-2 was transformed with pAW007-2 (transcribing a gRNA targeting amyE.P25NT), pAW008-2 (transcribing a gRNA targeting amyE.P330T), pAW009-2 (transcribing a gRNA targeting amyE.P636T), or pAW010-2 (transcribing a gRNA targeting amyE.P1344T), using the optimized editing template HL of 1,000 bp. Editing efficiency was evaluated via iodine staining followed by colony PCR and subsequent digestion with XhoI (amyE.P25NT and amyE.P330T), ScaI (amyE.P636T), or BamHI (amyE.P1344T) (Fig. 9E). The editing efficiencies for the first three PAM sites from the start codon were similar (87, 85, and 91%, respectively) (Fig. 9D), with amyE.P636T being targeted most effectively, suggesting minimal bias for the targeted strand. The observation of the low editing efficiency when targeting P1344T (23%) is consistent with the previous report that editing efficiency can vary dramatically between PAM sites in a single gene (23).
Enhanced multiplexing efficiency under optimized conditions.
To enhance the multiplexing capacity of the tool kit, we reexplored the double KO of amyE and ugtP (by targeting ugtP.P395T and amyE.P636T) under the optimized conditions for editing template and amyE PAM site. Two editing template combinations were evaluated: PCR products containing 1,000-bp HLs (amyE and ugtP) (combination 1) and a PCR product containing 1,000-bp HLs (ugtP) and pAW022-2 (amyE) (combination 2). AW001-2 was transformed with pAW015-2 (transcribing two gRNAs targeting ugtP.P395T and amyE.P636T) and either editing template combination 1 or 2. Relative to the initial multiplexing experiment (Fig. 9A), using editing template combination 1 resulted in an improved editing efficiency for both the amyE KO (45%) and the double KO (45%), although a substantial reduction was observed for the ugtP KO (46%) (Fig. 9F). On the other hand, using editing template combination 2 led to drastic improvements in editing efficiency for both the amyE KO (86%) and the double KO (85%), with a similarly high efficiency for the ugtP KO (85%) (Fig. 9G). The high multiplexing efficiency observed for editing template combination 2 was similar to reports of the double editing efficiency with ssDNA as an editing template (83%) (14) but was somewhat lower than the editing with a double-stranded DNA (dsDNA) editing template (97%) (12) in E. coli. These results suggest that our CRISPR-Cas9 tool kit can achieve high multiplexing efficiency in B. subtilis, even when challenging targets (such as amyE) are chosen.
Extension of the CRISPR-Cas9 tool kit to transcriptional interference.
To exploit the full utility of the tool kit, we extended our existing CRISPR-Cas9 platform from genome editing to CRISPRi for transcriptional interference. The lacZ gene was used as a reporter to assess repression at the level of transcription and protein expression in AW016-2. To construct AW016-2, we began by transforming AW009 with pAW019-2, yielding strain AW014-2, which expresses xylose-inducible dCas9 from the lacA locus. AW014-2 was then transformed with pAW018-2, followed by autoeviction of the CS cassette (Fig. 4A), yielding strain AW015-2, in which a gRNA targeting lacZ.P28NT was transcribed from the wprA locus. Finally, IPTG-inducible lacZ was integrated into the ugtP locus of AW015-2 via transformation of pAW016, generating strain AW016-2. Cultures of AW016-2 in which dCas9 expression was induced with xylose were compared with uninduced cultures to assess CRISPRi efficiency. Nearly an 8-fold reduction in both lacZ mRNA and β-galactosidase activity was observed in AW016-2 upon induction of dCas9 (Fig. 10), demonstrating the efficacy of our tool kit for reducing gene expression.
FIG 10.
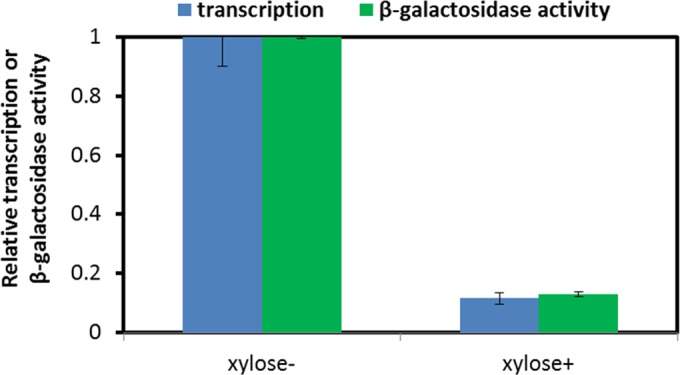
Evaluation of CRISPRi-mediated repression of lacZ expression at the level of transcription and protein expression. AW016-2 was grown in LB supplemented with 85 μg/ml spectinomycin, 1 mM IPTG to induce lacZ expression, and 1.2% (wt/vol) xylose to induce dCas9 expression (xylose+) or without xylose (xylose−). rpsJ, encoding the 30S ribosomal protein S10, served as an internal control for analysis of transcriptional repression via real-time qRT-PCR. β-Galactosidase activity was evaluated using the Miller assay to assess repression at the level of protein expression. Relative transcription (i.e., transcription relative to that of rpsJ) and protein expression were normalized to the values obtained from cultures in which dCas9 expression was not induced (xylose−). SD from experiments performed in triplicate are shown.
DISCUSSION
The recent advent of the CRISPR-Cas9 system has significantly increased the capacity for genome editing and transcriptional modulation in a selection of organisms (13, 14, 24, 26, 28, 72). B. subtilis shows considerable promise as an established industrial workhorse (73, 74), such that a CRISPR-Cas9 tool kit is essential to its progression toward full industrial utility. Traditional methods employed in B. subtilis, such as autoevicting counterselectable markers and site-specific recombination, suffer from low editing efficiency (47, 48) and/or limited capacity for multiplexing. Furthermore, existing technologies for transcriptional metering require extensive characterization or sequence modification prior to deployment, making their adoption cumbersome and time-consuming (3, 31–34). Here we propose an effective and scalable CRISPR-Cas9 tool kit for comprehensive engineering of B. subtilis, including targeted single-gene KO and multiple-gene KI, continuous genome editing, multiplexing, and targeted transcriptional repression. We employed chromosomal maintenance of CRISPR-Cas9 machinery for several reasons: (i) multicopy plasmids are potentially unstable, an issue that is of particular concern in B. subtilis (75–77); (ii) multicopy plasmids can impose a fitness burden on the host, particularly when selection is required to maintain them; (iii) CRISPR-Cas systems naturally exist in many bacteria and presumably do not impede cell viability in this context; (iv) the transformation efficiency of monomeric plasmids obtained from traditional cloning procedures is typically low in B. subtilis (56, 78); and (v) plasmids must be cured from the engineered cell.
For the development of the counterselectable gRNA delivery vectors, we tested two promoters for inducible mazF expression, in addition to Pspac, whose leaky nature presumably resulted in low transformation efficiency in B. subtilis. The resulting vector (pAW003-2) based on the use of PxylA,Bm, a stronger and more tightly controlled promoter (54), was difficult to maintain in E. coli. The reduced viability of the E. coli strain carrying pAW003-2 could not be resolved, even by replacing mazF with mazE, encoding the antitoxin of MazF (MazE). On the other hand, the use of ParaE resulted in a vector (pAW004-2) that was stable in E. coli and effectively transformed into B. subtilis. The presence of the native cre between ParaE and the ribosome-binding site (RBS) of mazF provides additional regulation of transcription in the presence of glucose, and this feature is desirable given the strong dependence of tool kit performance on transformation efficiency (as discussed below). To exploit the simplicity of the CRISPR-Cas9 system, we developed a gRNA transcription cassette using a modified version of the native promoter PxylA, i.e., PxylA.SphI+1, facilitating the construction of gRNA delivery vectors. The transcriptional start site (+1) of PxylA was determined to be 4 to 6 bp downstream of the −10 region (65), while that of the similar PxylA,Bm was found to be located 6 bp downstream of the −10 region (79). Accordingly, a single mismatch at the 5′ end of the gRNA (i.e., bp 6 of the SphI restriction site [Fig. 1A]) appears to have a negligible effect on Cas9 targeting, given the high editing efficiencies and transcriptional repression observed in general.
As the CRISPR-Cas9 mechanism involves at least two simultaneous recombination events (i.e., integration of gRNA[s] and editing template[s]), effective transformation becomes critical. Our enhanced competence protocol significantly improved DNA transformation into B. subtilis, leading to high editing efficiencies with our CRISPR-Cas9 tool kit. Using the improved protocol, the transformation efficiency for the ugtP KO with the 1,000-bp HL editing template (8.87 × 103 CFU μg−1 editing template) (Fig. 9C) exceeds that from a recent report of enhanced transformation efficiency of individual dsDNA PCR products (∼4.0 × 103 CFU μg−1 dsDNA) and is similar to the efficiency reported for linearized plasmid (∼1.0 × 104 CFU μg−1 plasmid) via induction of the master competence regulator ComK (46). Our tool kit also provided high editing efficiencies for both a multiple-gene KI and a single-gene KO over sequential rounds of editing using the counterselectable marker mazF. While our tool kit is not necessarily a convenient option to replace existing technologies for single KIs and KOs, it is more efficient for continuous genome engineering in B. subtilis. Using successive knockouts as an example, this procedure would entail two rounds of restriction/ligation cloning to replace the HLs of an existing vector or using an advanced cloning technique (e.g., Gibson assembly) to fuse various DNA fragments, (i.e., the plasmid backbone, the 5′-HL, the selection and counterselection markers, and the 3′-HL). These methods either are time-consuming or may result in unwanted mutations. The situation becomes more complicated for conventional cloning when mutations are introduced into the targeted ORF. This will require that either (i) the DRs of the integration vector be redesigned such that the single-crossover event between them needed to excise the selection and counterselection markers results in the introduction of the desired mutation or (ii) an editing template be designed to replace the selection and counterselection markers with the desired mutation. Another option would be to construct an integration cassette using multiple rounds of SOE PCR, which can be unreliable due to large DNA sizes (80). On the other hand, our tool kit requires only a single restriction/ligation step to insert the new gRNA (102 bp) into the gRNA delivery vector and a single round of SOE PCR to generate the editing template (which can be as small as 1 kb). Furthermore, our tool kit requires no additional effort to introduce specific mutations, beyond designing the mutation region of the editing template to introduce the desired mutation and, if necessary, an additional silent mutation to remove the PAM site (72).
The improved editing efficiency observed for the ugtP KO compared to the KI of the HA operon (Fig. 7C) was expected given that increasing insertion size has been shown to correlate with decreasing editing efficiency in CRISPR-Cas9 systems (10, 14). Moreover, the reduced recombination frequency of the HA operon KI cassette could have been exacerbated by an excessive metabolic burden associated with HA synthesis, which is an energy- and carbon-intensive process directly competing with central metabolism and cell wall synthesis (41). This conclusion was supported by our observation of a few transformants with the KI of the HA operon but showing no mucoid phenotype, implying that expression of functional hasSE and/or tuaD can be inactivated. In addition to the expected reduction in recombination frequency associated with large insertions, recombination may occur less effectively at certain genomic loci. Considering that the number of escapers were similar for the KI of the HA operon (at amyE.P636T), the ugtP KO, and the single amyE KOs (except at amyE.P1344T), discrepancies in transformation and editing efficiencies at the amyE and ugtP loci (Fig. 7C and 8B and D) could have been influenced by different recombination frequencies at these sites. Interestingly, our KI efficiency for the 2,909-bp HA operon (69%) exceeds that reported for a 3,000-bp CRISPR-Cas9-mediated insertion in E. coli (59%) (14). This comparison corroborates the efficacy of our tool kit for large chromosomal insertions, given that the insertion location of amyE appears to be a difficult recombination site and the inserted operon imparts a significant metabolic burden on the host. Finally, chromosomal expression of Cas9 appeared to have a minimal effect on cell viability, given that the specific growth rates and final cell densities were similar for 1A751 and AW001-2 (data not shown), and Cas9 was stably maintained in the chromosome across three sequential rounds of editing.
The low efficiency of the initial multiplexing trial (36%) suggests that amyE is a difficult recombination target. As discussed above, the similar number of escapers observed when targeting amyE and ugtP suggests that CRISPR-Cas9-mediated cleavage was not the limiting factor for the low amyE editing efficiency. Various factors limiting the editing efficiency were systematically evaluated to enhance the performance of our tool kit when targeting difficult sites. Our results for the ugtP KO (Fig. 9C) support the previous conclusion that 400- to 500-bp HLs are sufficient to achieve an acceptable transformation efficiency of linear DNA in B. subtilis (49). Decline in transformation efficiency for increasing homology beyond an optimal length has been reported elsewhere (81) and was attributed to the reduced number of plasmids transformed at larger HLs (based on an equivalent quantity of DNA per transformation). A stark decline in CRISPR-Cas9 editing efficiency in S. cerevisiae was also observed upon increasing the editing template HL from 50 to 60 bp, and sequence-specific features of the longer editing template causing premature termination of the hybrid editing template-crRNA transcript were the proposed cause (23).
Targeting amyE.P25NT, amyE.P330T, and amyE.P636T resulted in comparable transformation and editing efficiencies, with amyE.P636T being targeted most effectively (Fig. 9D). Small discrepancies in the number of escapers were observed, suggesting that Cas9 accessibility to the amyE locus was not limiting the editing efficiency. On the other hand, it has been perceived that certain PAM sites are less susceptible to CRISPR-Cas9-mediated DSBs since the editing efficiency can vary substantially between PAM sites in a single gene (23). The poor editing efficiency when targeting amyE.P1344T supports this theory, although gRNA sequence characteristics can also affect targeting efficacy (63). With the critical design parameters (except the targeting strand, for which minimal bias appears to exist) in mind (63), the underlying problem in gRNA design may be associated with potential secondary structures formed in vivo. Several secondary structures are generally possible for each gRNA sequence such that unwanted secondary structures may form, reducing the binding capacity (or frequency of binding) of Cas9 to the gRNA.
The multiplexing efficiency of our tool kit reached 85% through optimization of various editing template parameters and PAM sites (Fig. 9G). A high multiplexing efficiency (up to 97%) was reported for the double KO of maeA and maeB in E. coli, although the multiplexing capacity was limited by the inclusion of gRNA transcription cassettes and editing templates in a single plasmid (12). A CRISPR-Cas9 system recently developed for S. cerevisiae provided a high multiplexing efficiency for three targets (up to 87%); however, a long period of cultivation after transformation was required, significantly increasing the duration of a single round of editing (23). In contrast to these systems and other improved methods for use in E. coli (14) and S. cerevisiae (24), our tool kit provides comparably high multiplexing efficiencies via CRISPR-Cas9 elements maintained in the chromosome. Our approach is more similar to the chromosomal arrangement of CRISPR-Cas systems in native hosts and takes advantage of the simplicity of the gRNA for Cas9 targeting.
The extension of our tool kit to CRISPRi provides an effective strategy for transcriptional modulation in B. subtilis. We demonstrate that expressing dCas9 and transcribing gRNAs chromosomally are sufficient to achieve efficient transcriptional repression in B. subtilis, which is likely the case in E. coli and many other bacterial species (64). This is an attractive aspect of our approach due to potential plasmid instability (75–77), which is a greater concern when applying CRISPRi, as dCas9 and gRNA transcription cassettes must be stably maintained in the cell. The extent of repression achieved with our tool kit is comparable to that obtained with existing asRNA protocols. Repression levels of as high as 90% were obtained using an asRNA targeting buk mRNA (encoding the butyrate kinase) in Clostridium acetobutylicum (3). However, significant repression of the acetate kinase (encoded by ack) was observed in the same strain, owing to the large degree of homology between buk and ack, demonstrating the potential for off-target effects when applying asRNA strategies. Similar repression of DsRed2 expression was observed in E. coli using an asRNA containing an Hfq-recruiting scaffold to enhance hybridization, although the assessment of multiple scaffold sequences from endogenous asRNAs was necessary to achieve maximum repression (32). Higher repression levels (>98%) were obtained in E. coli with 5′ cis sequences inserted upstream of the RBS (to which the sequences were complementary) of a green fluorescent protein, blocking recognition of the RBS by the 30S ribosomal subunit via a stem-loop structure (31). Repression could be deactivated with a trans-activating RNA, although application of this strategy requires upstream sequence modification of the target gene, making it tedious to apply, particularly for multiplexing. A significant level of repression of β-galactosidase expression via CRISPRi was reported in E. coli in which dCas9 and a lacZ-targeting gRNA were maintained in plasmids (29). In the same study, up to 300-fold repression of expression of a monomeric red fluorescent protein (mRFP) was observed, which is similar to the CRISPR-dCas9-mediated RFP repression levels achieved in Corynebacterium glutamicum (39). The differences in repression efficiency observed between β-galactosidase and RFP suggests that certain targets may be more susceptible to CRISPRi. Various degrees of repression between different targets have also been observed in mycobacteria when applying CRISPRi, although the differences were less dramatic (40). To allow targeting of multiple genes for multiplexing or targeting multiple sites in the same gene for enhanced repression, a multi-gRNA delivery vector was also constructed to enable autoeviction of the ParaE::mazF-Spcr cassette, while the multi-gRNA array is retained in the chromosome, using the same approach as outlined for pAW014-2 (Fig. 2). Finally, the repression level can be adjusted by tuning gRNA design parameters established previously (29) rather than adjusting xylose concentration for inducing dCas9 expression (64). For example, mismatches introduced in bp 8 to 12 of the protospacer (relative to the 3′ end) cause a dramatic reduction in repression, while mismatches in bp 13 to 20 have a less significant effect on repression efficiency (the first 7 bp of the protospacer should not be altered) (29). Moreover, the repression level is inversely proportional to the distance of the targeted PAM site from the start codon of the target ORF (29).
ACKNOWLEDGMENTS
This work was supported in part by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canada Research Chairs (CRC) program.
We thank Steve George and Marc Aucoin for their assistance in performing real-time qRT-PCR. We thank Duane Chung for drawing our attention to CRISPR as a tool for genetic manipulation.
REFERENCES
- 1.Keasling JD. 2010. Manufacturing molecules through metabolic engineering. Science 330:1355–1358. doi: 10.1126/science.1193990. [DOI] [PubMed] [Google Scholar]
- 2.Lee JW, Na D, Park JM, Lee J, Choi S, Lee SY. 2012. Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol 8:536–546. doi: 10.1038/nchembio.970. [DOI] [PubMed] [Google Scholar]
- 3.Desai RP, Papoutsakis ET. 1999. Antisense RNA strategies for metabolic engineering of Clostridium acetobutylicum. Appl Environ Microbiol 65:936–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. 2008. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C. 2009. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 7.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pyne ME, Moo-Young M, Chung DA, Chou CP. 22 May 2015. Coupling the CRISPR/Cas9 system to lambda Red recombineering enables simplified chromosomal gene replacement in Escherichia coli. Appl Environ Microbiol doi: 10.1128/aem.01248-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang H, Zheng G, Jiang W, Hu H, Lu Y. 2015. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim Biophys Sin 47:231–243. doi: 10.1093/abbs/gmv007. [DOI] [PubMed] [Google Scholar]
- 12.Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S. 2015. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol 81:2506–2514. doi: 10.1128/AEM.04023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh J-H, van Pijkeren J-P. 2014. CRISPR-Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res 42:e131. doi: 10.1093/nar/gku623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Lin Z, Huang C, Zhang Y, Wang Z, Tang Y-J, Chen T, Zhao X. 2015. Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab Eng 31:13–21. doi: 10.1016/j.ymben.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Xu T, Li Y, Shi Z, Hemme CL, Li Y, Zhu Y, Van Nostrand JD, He Z, Zhou J. 2015. Efficient genome editing in Clostridium cellulolyticum via CRISPR-Cas9 nickase. Appl Environ Microbiol 81:4423–4431. doi: 10.1128/AEM.00873-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cobb RE, Wang Y, Zhao H. 2015. High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol 4:723–728. doi: 10.1021/sb500351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Zhang Z-T, Seo S-O, Choi K, Lu T, Jin Y-S, Blaschek HP. 2015. Markerless chromosomal gene deletion in Clostridium beijerinckii using CRISPR/Cas9 system. J Biotechnol 200:1–5. doi: 10.1016/j.jbiotec.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Vercoe RB, Chang JT, Dy RL, Taylor C, Gristwood T, Clulow JS, Richter C, Przybilski R, Pitman AR, Fineran PC. 2013. Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet 9:e1003454. doi: 10.1371/journal.pgen.1003454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reisch CR, Prather KLJ. 2015. The no-SCAR (Scarless Cas9 Assisted Recombineering) system for genome editing in Escherichia coli. Sci Rep 5:15096. doi: 10.1038/srep15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia J, Wang L, Zhu J-B, Sun C-J, Zheng M-G, Zheng L, Lou Y-H, Shi L. 2016. Expression of Shewanella frigidimarina fatty acid metabolic genes in E. coli by CRISPR/cas9-coupled lambda Red recombineering. Biotechnol Lett 38:117–122. [DOI] [PubMed] [Google Scholar]
- 21.Zeng H, Wen S, Xu W, He Z, Zhai G, Liu Y, Deng Z, Sun Y. 2015. Highly efficient editing of the actinorhodin polyketide chain length factor gene in Streptomyces coelicolor M145 using CRISPR/Cas9-CodA(sm) combined system. Appl Microbiol Biotechnol 99:10575–10585. doi: 10.1007/s00253-015-6931-4. [DOI] [PubMed] [Google Scholar]
- 22.Tong Y, Charusanti P, Zhang L, Weber T, Lee SY. 2015. CRISPR-Cas9 based engineering of actinomycetal genomes. ACS Synth Biol 4:1020–1029. doi: 10.1021/acssynbio.5b00038. [DOI] [PubMed] [Google Scholar]
- 23.Bao Z, Xiao H, Liang J, Zhang L, Xiong X, Sun N, Si T, Zhao H. 2014. Homology-integrated CRISPR–Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth Biol doi: 10.1021/sb500255k. [DOI] [PubMed] [Google Scholar]
- 24.Horwitz AA, Walter JM, Schubert MG, Kung SH, Hawkins K, Platt DM, Hernday AD, Mahatdejkul-Meadows T, Szeto W, Chandran SS, Newman JD. 2015. Efficient multiplexed integration of synergistic alleles and metabolic pathways in yeasts via CRISPR-Cas. Cell Syst 1:88–96. doi: 10.1016/j.cels.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Bassett AR, Tibbit C, Ponting CP, Liu J-L. 2013. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep 4:220–228. doi: 10.1016/j.celrep.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JRJ, Joung JK. 2013. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J-F, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM, Sheen J. 2013. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31:688–691. doi: 10.1038/nbt.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. 2013. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41:7429–7437. doi: 10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isaacs FJ, Dwyer DJ, Ding C, Pervouchine DD, Cantor CR, Collins JJ. 2004. Engineered riboregulators enable post-transcriptional control of gene expression. Nat Biotechnol 22:841–847. doi: 10.1038/nbt986. [DOI] [PubMed] [Google Scholar]
- 32.Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY. 2013. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol 31:170–174. doi: 10.1038/nbt.2461. [DOI] [PubMed] [Google Scholar]
- 33.Man S, Cheng R, Miao C, Gong Q, Gu Y, Lu X, Han F, Yu W. 2011. Artificial trans-encoded small non-coding RNAs specifically silence the selected gene expression in bacteria. Nucleic Acids Res 39:e50. doi: 10.1093/nar/gkr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim JYH, Cha HJ. 2003. Down-regulation of acetate pathway through antisense strategy in Escherichia coli: improved foreign protein production. Biotechnol Bioeng 83:841–853. doi: 10.1002/bit.10735. [DOI] [PubMed] [Google Scholar]
- 35.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. 2014. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lv L, Ren Y-L, Chen J-C, Wu Q, Chen G-Q. 2015. Application of CRISPRi for prokaryotic metabolic engineering involving multiple genes, a case study: controllable P(3HB-co-4HB) biosynthesis. Metab Eng 29:160–168. doi: 10.1016/j.ymben.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Wu J, Du G, Chen J, Zhou J. 2015. Enhancing flavonoid production by systematically tuning the central metabolic pathways based on a CRISPR interference system in Escherichia coli. Sci Rep 5:13477. doi: 10.1038/srep13477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cleto S, Jensen JVK, Wendisch VF, Lu TK. 2016. Corynebacterium glutamicum metabolic engineering with CRISPR interference (CRISPRi). ACS Synth Biol doi: 10.1021/acssynbio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choudhary E, Thakur P, Pareek M, Agarwal N. 2015. Gene silencing by CRISPR interference in mycobacteria. Nat Commun 6:6267. doi: 10.1038/ncomms7267. [DOI] [PubMed] [Google Scholar]
- 41.Widner B, Behr R, Von Dollen S, Tang M, Heu T, Sloma A, Sternberg D, DeAngelis PL, Weigel PH, Brown S. 2005. Hyaluronic acid production in Bacillus subtilis. Appl Environ Microbiol 71:3747–3752. doi: 10.1128/AEM.71.7.3747-3752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li S, Wen J, Jia X. 2011. Engineering Bacillus subtilis for isobutanol production by heterologous Ehrlich pathway construction and the biosynthetic 2-ketoisovalerate precursor pathway overexpression. Appl Microbiol Biotechnol 91:577–589. doi: 10.1007/s00253-011-3280-9. [DOI] [PubMed] [Google Scholar]
- 43.Zweers JC, Barák I, Becher D, Driessen AJ, Hecker M, Kontinen VP, Saller MJ, Vavrová L, van Dijl JM. 2008. Towards the development of Bacillus subtilis as a cell factory for membrane proteins and protein complexes. Microb Cell Fact 7:10. doi: 10.1186/1475-2859-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong H, Zhang D. 2014. Current development in genetic engineering strategies of Bacillus species. Microb Cell Fact 13:63. doi: 10.1186/1475-2859-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fabret C, Ehrlich SD, Noirot P. 2002. A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol Microbiol 46:25–36. doi: 10.1046/j.1365-2958.2002.03140.x. [DOI] [PubMed] [Google Scholar]
- 46.Shi T, Wang G, Wang Z, Fu J, Chen T, Zhao X. 2013. Establishment of a markerless mutation delivery system in Bacillus subtilis stimulated by a double-strand break in the chromosome. PLoS One 8:e81370. doi: 10.1371/journal.pone.0081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brans A, Filée P, Chevigné A, Claessens A, Joris B. 2004. New integrative method to generate Bacillus subtilis recombinant strains free of selection markers. Appl Environ Microbiol 70:7241–7250. doi: 10.1128/AEM.70.12.7241-7250.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X-Z, Yan X, Cui Z-L, Hong Q, Li S-P. 2006. mazF, a novel counter-selectable marker for unmarked chromosomal manipulation in Bacillus subtilis. Nucleic Acids Res 34:e71–e71. doi: 10.1093/nar/gkl358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan X, Yu H-J, Hong Q, Li S-P. 2008. Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl Environ Microbiol 74:5556–5562. doi: 10.1128/AEM.01156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen PT, Shaw J-F, Chao Y-P, David Ho T-H, Yu S-M. 2010. Construction of chromosomally located T7 expression system for production of heterologous secreted proteins in Bacillus subtilis. J Agric Food Chem 58:5392–5399. doi: 10.1021/jf100445a. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Weng J, Waseem R, Yin X, Zhang R, Shen Q. 2012. Bacillus subtilis genome editing using ssDNA with short homology regions. Nucleic Acids Res 40:e91. doi: 10.1093/nar/gks248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sambrook J, Russell D. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 53.Guérout-Fleury A-M, Frandsen N, Stragier P. 1996. Plasmids for ectopic integration in Bacillus subtilis. Gene 180:57–61. doi: 10.1016/S0378-1119(96)00404-0. [DOI] [PubMed] [Google Scholar]
- 54.Härtl B, Wehrl W, Wiegert T, Homuth G, Schumann W. 2001. Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol 183:2696–2699. doi: 10.1128/JB.183.8.2696-2699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inácio JM, Costa C, de Sá-Nogueira I. 2003. Distinct molecular mechanisms involved in carbon catabolite repression of the arabinose regulon in Bacillus subtilis. Microbiology 149:2345–2355. doi: 10.1099/mic.0.26326-0. [DOI] [PubMed] [Google Scholar]
- 56.Harwood CR, Cutting SM. 1990. Molecular biological methods for Bacillus. Wiley, Chichester, United Kingdom. [Google Scholar]
- 57.Chien L-J, Lee C-K. 2007. Enhanced hyaluronic acid production in Bacillus subtilis by coexpressing bacterial hemoglobin. Biotechnol Prog 23:1017–1022. [DOI] [PubMed] [Google Scholar]
- 58.Bitter T, Muir HM. 1962. A modified uronic acid carbazole reaction. Anal Biochem 4:330–334. doi: 10.1016/0003-2697(62)90095-7. [DOI] [PubMed] [Google Scholar]
- 59.Cowman MK, Chen CC, Pandya M, Yuan H, Ramkishun D, LoBello J, Bhilocha S, Russell-Puleri S, Skendaj E, Mijovic J, Jing W. 2011. Improved agarose gel electrophoresis method and molecular mass calculation for high molecular mass hyaluronan. Anal Biochem 417:50–56. doi: 10.1016/j.ab.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 60.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 62.Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, van den Hoff MJB, Moorman AFM. 2009. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37:e45. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang T, Wei JJ, Sabatini DM, Lander ES. 2014. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS. 2013. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc 8:2180–2196. doi: 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gärtner D, Geissendörfer M, Hillen W. 1988. Expression of the Bacillus subtilis xyl operon is repressed at the level of transcription and is induced by xylose. J Bacteriol 170:3102–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weigel PH. 2002. Functional characteristics and catalytic mechanisms of the bacterial hyaluronan synthases. IUBMB Life 54:201–211. doi: 10.1080/15216540214931. [DOI] [PubMed] [Google Scholar]
- 67.Phan TTP, Nguyen HD, Schumann W. 2006. Novel plasmid-based expression vectors for intra- and extracellular production of recombinant proteins in Bacillus subtilis. Protein Expr Purif 46:189–195. doi: 10.1016/j.pep.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 68.Kumari K, Weigel PH. 1997. Molecular cloning, expression, and characterization of the authentic hyaluronan synthase from group C Streptococcus equisimilis. J Biol Chem 272:32539–32546. doi: 10.1074/jbc.272.51.32539. [DOI] [PubMed] [Google Scholar]
- 69.Jin P, Kang Z, Yuan P, Du G, Chen J. 2016. Production of specific-molecular-weight hyaluronan by metabolically engineered Bacillus subtilis 168. Metab Eng 35:21–30. doi: 10.1016/j.ymben.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 70.Jia Y, Zhu J, Chen X, Tang D, Su D, Yao W, Gao X. 2013. Metabolic engineering of Bacillus subtilis for the efficient biosynthesis of uniform hyaluronic acid with controlled molecular weights. Bioresour Technol 132:427–431. doi: 10.1016/j.biortech.2012.12.150. [DOI] [PubMed] [Google Scholar]
- 71.Shetty R, Endy D, Knight T. 2008. Engineering BioBrick vectors from BioBrick parts. J Biol Eng 2:5. doi: 10.1186/1754-1611-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim H, Ishidate T, Ghanta KS, Seth M, Conte D, Shirayama M, Mello CC. 2014. A Co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics 197:1069–1080. doi: 10.1534/genetics.114.166389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harwood CR. 1992. Bacillus subtilis and its relatives: molecular biological and industrial workhorses. Trends Biotechnol 10:247–256. doi: 10.1016/0167-7799(92)90233-L. [DOI] [PubMed] [Google Scholar]
- 74.van Dijl JM, Hecker M. 2013. Bacillus subtilis: from soil bacterium to super-secreting cell factory. Microb Cell Fact 12:3. doi: 10.1186/1475-2859-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leonhardt H, Alonso JC. 1991. Parameters affecting plasmid stability in Bacillus subtilis. Gene 103:107–111. doi: 10.1016/0378-1119(91)90400-6. [DOI] [PubMed] [Google Scholar]
- 76.Bron S, Holsappel S, Venema G, Peeters BH. 1991. Plasmid deletion formation between short direct repeats in Bacillus subtilis is stimulated by single-stranded rolling-circle replication intermediates. Mol Gen Genet 226:88–96. [DOI] [PubMed] [Google Scholar]
- 77.Bron S, Meijer W, Holsappel S, Haima P. 1991. Plasmid instability and molecular cloning in Bacillus subtilis. Res Microbiol 142:875–883. doi: 10.1016/0923-2508(91)90068-L. [DOI] [PubMed] [Google Scholar]
- 78.Zhang XZ, Zhang YHP. 2011. Simple, fast and high-efficiency transformation system for directed evolution of cellulase in Bacillus subtilis. Microb Biotechnol 4:98–105. doi: 10.1111/j.1751-7915.2010.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rygus T, Scheler A, Allmansberger R, Hillen W. 1991. Molecular cloning, structure, promoters and regulatory elements for transcription of the Bacillus megaterium encoded regulon for xylose utilization. Arch Microbiol 155:535–542. doi: 10.1007/BF00245346. [DOI] [PubMed] [Google Scholar]
- 80.Shevchuk NA, Bryksin AV, Nusinovich YA, Cabello FC, Sutherland M, Ladisch S. 2004. Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res 32:e19. doi: 10.1093/nar/gnh014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kung SH, Retchless AC, Kwan JY, Almeida RPP. 2013. Effects of DNA size on transformation and recombination efficiencies in Xylella fastidiosa. Appl Environ Microbiol 79:1712–1717. doi: 10.1128/AEM.03525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wolf M, Geczi A, Simon O, Borriss R. 1995. Genes encoding xylan and β-glucan hydrolysing enzymes in Bacillus subtilis: characterization, mapping and construction of strains deficient in lichenase, cellulase and xylanase. Microbiology 141:281–290. doi: 10.1099/13500872-141-2-281. [DOI] [PubMed] [Google Scholar]