

Abstract
Recently, it was suggested that neurons can release and transfer damaged mitochondria to astrocytes for disposal and recycling 1. This ability to exchange mitochondria may represent a potential mode of cell-cell signaling in the central nervous system (CNS). Here, we show that astrocytes can also release functional mitochondria that enter into neurons. Astrocytic release of extracellular mitochondria particles was mediated by a calcium-dependent mechanism involving CD38/cyclic ADP ribose signaling. Transient focal cerebral ischemia in mice induced astrocytic mitochondria entry to adjacent neurons that amplified cell survival signals. Suppression of CD38 signaling with siRNA reduced extracellular mitochondria transfer and worsened neurological outcomes. These findings suggest a new mitochondrial mechanism of neuroglial crosstalk that may contribute to endogenous neuroprotective and neurorecovery mechanisms after stroke.
Astrocytes play broad roles in the CNS, and are involved in the regulation of neurodevelopment, neurotransmission, cerebral metabolism and blood flow 2–4. Normal astrocytes protect neurons against oxidative stress and excitotoxicity 5–7. In contrast, unhealthy astrocytes may release deleterious factors that damage neurons 8,9. Healthy mitochondria may be essential for these neuroglial protective mechanisms because inhibition of astrocytic mitochondria makes neurons vulnerable to cell death 10. Mitochondria comprise the intracellular cores for energetics and viability 11, but under some conditions mitochondria might also be released into extracellular space 12. For instance, retinal neurons may transfer mitochondria to astrocytes for disposal and recycling 1, and bone-marrow derived stromal cells may transfer mitochondria into pulmonary alveoli to suppress acute lung injury 13.
In this study, we asked whether astrocytes can produce functional extracellular mitochondria to support neuronal viability after ischemic stroke. Electron microscopy confirmed the presence of extracellular particles containing mitochondria in conditioned media from rat cortical astrocytes (Fig. 1a, Extended Data Fig. 1a). qNano analysis revealed that astrocyte-derived mitochondria particles following FACS isolation spanned a range of sizes from 300 to 1100 nm (Extended Data Fig. 1b–d), and included populations that were positive for β1-integrin (79%) and CD63 (43%) (Extended Data Fig. 2). Mitotracker-labeling suggested that these extracellular mitochondria may still be functional (Fig. 1b), and filtration of astrocyte conditioned media through 0.2 μm filters depleted the amounts of functional mitochondria and reduced measurements of mitochondrial ATP, membrane potential and oxygen consumption (Fig. 1b–e).
Fig. 1. Astrocytic CD38 and extracellular mitochondria.

a, Transmission electron microscopy (TEM) of extracellular mitochondria in astrocyte-conditioned medium (ACM). Scale: 500 nm. b, Rat cortical astrocytes were labeled by Mitotracker Red CMXRos. FACS showed that 0.2 μm filter depleted extracellular mitochondria in ACM (mdACM). c–e, 0.2 μm filters reduced markers of extracellular mitochondrial function in ACM - c, extracellular ATP (n=4), d, membrane potential (n=4), e, oxygen consumption (n= 9 or 6). f, Western blot confirmed higher CD38 in rat cortical astrocytes compared to neurons. g, High and low levels of CD38 cyclase activity in astrocytes and neurons respectively (n= 8 or 5). h,. Experimental schematic for testing CRISPR/Cas9-mediated CD38 activation. i, Twenty four hours after transfection, CD38 cyclase activity was upregulated by CD38 activation plasmid (n=4). j, k, Extracellular ATP production (j) and oxygen consumption (k) were significantly increased by CD38 activation (n=5). l, FACS showed that extracellular mitochondria were increased by cADPR (1 μM) stimulation in astrocytes (n=3). m, cADPR (1 μM) increased extracellular mitochondria membrane potential at 24 hours (n=7). n, Oxygen consumption in extracellular mitochondria was increased by cADPR (n=4). o, cADPR did not cause astrocyte toxicity (n=4). All values are mean +/− SEM.
An important question at this point is whether extracellular mitochondria represent active signals or merely cellular debris. To address this question, we asked whether stimulated astrocytes could actively produce extracellular mitochondria. CD38 catalyzes the synthesis of a calcium messenger, cyclic ADP-ribose (cADPR) in mitochondrial membranes 14,15. In brain, CD38 is mainly expressed in glial cells, and may have a role in neuroglial crosstalk since astrocytes increase CD38 expression in response to glutamate release from neurons 16. Based on this background literature and the fact that most actively secreted cellular events involve calcium regulation, we decided to assess CD38-cADPR-calcium signaling as a candidate mechanism for the astrocytic production of extracellular mitochondria. First, we confirmed that rat cortical astrocytes expressed CD38 protein and CD38/cADPR cyclase activity (Fig. 1f, g). Then, we tried two methods to modify this pathway. When astrocytic CD38 was upregulated using CRISPR/Cas9 activation plasmids, functional endpoints of extracellular mitochondria were significantly increased in conditioned media (Fig. 1h–k). When astrocytes were stimulated by cADPR to activate CD38 signaling, extracellular mitochondria were increased in conditioned media along with enhancement of functional endpoints in a calcium-dependent manner (Fig. 1l–n, Extended Data Fig. 3). Stimulation with cADPR did not appear to damage astrocyte viability (Fig. 1o), suggesting that this release of extracellular mitochondria was not due to nonspecific cytotoxicity.
If astrocytes can produce functional extracellular mitochondria, then is it possible that these signals may affect adjacent neurons? When rat cortical neurons were subjected to oxygen-glucose deprivation, intracellular ATP levels fell and neuronal viability decreased, as expected (Fig. 2a–c, Extended Data Fig. 4). When astrocyte-conditioned media containing extracellular mitochondrial particles was added to neurons, ATP levels were increased and neuronal viability was recovered (Fig. 2a–c, Extended Data Fig. 4). But when extracellular mitochondria were removed from the astrocyte-conditioned media, neuroprotection was no longer observed (Fig. 2a–c, Extended Data Fig. 4). Similar results were obtained with immunostaining-based cell counts (Fig. 2d). As a control, ATP-liposomes were not significantly protective (Fig. 2e), suggesting that the astrocytic mitochondria entry into neurons may generate additional benefits beyond ATP energetics per se. Fluorescent microscopy confirmed that astrocyte-derived mitochondria appeared to be present within treated neurons (Fig. 2f).
Fig. 2. Astrocytic extracellular mitochondria and neuroprotection.
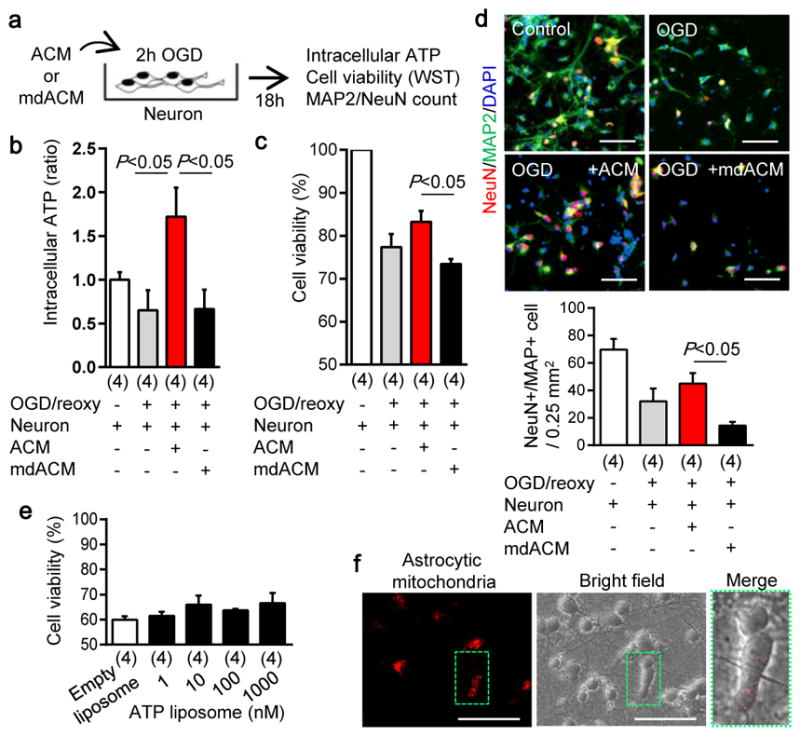
a, Experimental schematic to test neuroprotective effects of astrocyte conditioned media (ACM) or mitochondria-depleted astrocyte conditioned media (mdACM) against oxygen-glucose deprivation (OGD) in rat cortical neurons. b, ACM but not mdACM rescued ATP levels in damaged neurons (n=4). c, ACM but not mdACM recovered neuronal viability after OGD (n=4). d, Immunostaining confirmed that neuroprotective effect of ACM but not mdACM (n=4). Scale: 100 μm. e, No statistically significant neuroprotection with liposomal ATP (1–1000 nM) after OGD. f, Fluorescent microscopy suggests the presence of astrocyte mitochondria (labeled with Mitotracker Red CMXRos, 200 nM) within neurons. Scale: 100 μm. All values are mean +/− SEM.
Beyond the prevention of acute neuronal death, delayed neuroplasticity is also important for stroke outcomes. CD38 may be important for brain plasticity because CD38-deficient mice show worsened recovery after brain injury 17 and CD38 mutations may comprise risk factors for behavioral dysfunction 18. Hence, we asked whether CD38-mediated astrocyte-into-neuron mitochondrial transfer may also influence neuroplasticity. Neurons were labeled with CellLight Mitochondria-GFP and astrocytes were separately labeled with Mitotracker Red CMXRos, and then the two cell types were co-cultured together for 24 hours. Confocal microscopy indicated that astrocyte-derived mitochondria were detected within soma and axon (Fig. 3a), and in these co-culture conditions, astrocytes supported neuronal survival after serum/glucose starvation in a CD38-dependent manner (Extended Data Fig. 5). When astrocytic mitochondria were made dysfunctional via inhibition of mitochondrial aconitase, cADPR-stimulated astrocytes no longer supported neuronal survival and axonal extension (Extended Data Fig. 6). To further assess our hypothesis, we asked whether this ability of astrocytes to transfer mitochondria could in fact enhance neuroplasticity under pathological conditions. Control or CD38-silenced astrocytes were co-cultured with surviving neurons after oxygen-glucose deprivation (Fig. 3b). siRNA suppression of CD38 in astrocytes reduced mitochondria transfer (Fig. 3c) and dendrite regrowth after injury (Fig. 3d).
Fig. 3. Astrocytic mitochondria and neuroplasticity after ischemic stress.

a, Confocal microscopy revealed that astrocytic mitochondria (red, Mitotracker Red CMXRos) may be transferred into neural soma (a) and axon (b), and some may fuse with neuronal mitochondria (c, green, Cell-light Mito-GFP). b, Experimental schematic for co-culture studies. c, Soma size was unchanged but astrocytic mitochondrial density in neuronal soma was significantly decreased when CD38 was suppressed in astrocytes (n=54 or 41 soma were counted). Scale: 20 μm. d, Quantification of dendrite elongation (MAP2 staining) (n=5 or 6). e, Male C57Bl6 mice were subjected to 60 min transient focal ischemia. Three days later, astrocyte mitochondria particles (1,000 particles/2μl, MitoTracker Red CMXRos) were infused into cerebral cortex. Confocal images showed transplanted astrocytic mitochondria (red) within peri-infarct neurons at 24 hrs. f, FVB/N-Tg (GFAPGFP)14Mes/J transgenic mice with fluorescently labeled astrocytes were subjected to 30 min transient focal ischemia. Immunohistochemistry at 24 hrs suggested that GFP (GFAP)-positive particles co-stained with mitochondrial TOM40 were present in MAP2-positive neurons in peri-infarct cortex. g, Western blot indicated that GFP-positive neurons upregulated cell survival-related proteins (phospho-Akt, Bcl-xl) but not apoptosis-related proteins (caspase 3, AIF) along with an increase of mitochondrial TOM40 (n=3). Isolated neurons expressed mature (neurofilament) but not neural stem cell markers (nestin) (Extended Data Fig. 7f). All values are mean +/− SEM.
Taken together, these cellular findings appear consistent with the overall hypothesis that CD38 signaling may help astrocytes transfer mitochondria into neurons and promote survival and plasticity after injury. But does this mechanism work in vivo? To answer this question, we turned to a mouse model of focal cerebral ischemia. First, primary mouse cortical astrocyte cultures were labeled with MitoTracker Red CMXRos and extracellular mitochondria particles were collected. Then mice were subjected to focal cerebral ischemia, and 3 days later, extracellular mitochondria particles were directly injected into peri-infarct cortex. After 24 hrs, immunostaining suggested that transplanted astrocytic mitochondria were indeed present in neurons (Fig. 3e). Next, we turned to FVB/N-Tg (GFAPGFP)14Mes/J transgenic mice where astrocytes are fluorescently labeled. When these mice were subjected to focal cerebral ischemia, fluorescent mitochondrial particle signals appeared within adjacent neurons at 24 hours post-stroke (Fig. 3f). Neurons that were collected from ischemic peri-infarct cortex via flow cytometry showed a general upregulation of cell survival-related signals such as phosphorylated Akt and Bcl-xl along with an increase of the mitochondria marker TOM4 (Fig. 3g, Extended Data Fig. 7).
Finally, we attempted loss-of-function experiments to ask whether blocking CD38 signaling results in worsened outcomes after stroke. In our mouse models of focal cerebral ischemia, CD38 was upregulated in the peri-infarct cortex (Extended Data Fig. 8a). At 5 days post-stroke, CD38 siRNA or control siRNA were injected into cerebral ventricles (Fig. 4a). By 2 days after siRNA injections, total CD38 expression in the peri-infarct cortex was successfully downregulated (Extended Data Fig. 8b). There were no clear differences in infarct area nor the total levels of GFAP-positive reactive astrocytes (Fig. 4b, c), but astrocyte subsets that expressed CD38 were significantly decreased without affecting the number of other CD38 expressing cells such as CD8 T cells and microglia/macrophages 19 (Fig. 4c, Extended Data Fig. 8c–g). To assess the levels of extracellular mitochondrial particles in this in vivo model, flow cytometry was used to analyze cerebrospinal fluid (CSF). GFAP-positive mitochondria were detected in CSF, and CD38 siRNA injections appeared to reduce this extracellular population of astrocyte-derived mitochondria (Fig. 4d). At the same time, flow cytometry was used to quantify levels of MAP2 neuronal mitochondria (Extended Data Fig. 9). Brains treated with CD38 siRNA showed a significant reduction in neuronal mitochondria (Fig. 4e), suggesting that interfering with CD38 signaling may have suppressed endogenous astrocyte-to-neuron mitochondrial transfer. These effects were accompanied by a reduction in peri-infarct GAP43 (a surrogate marker of neuroplasticity, Fig. 4f, g) as well as worsened neurologic outcomes (Fig. 4h, i). Furthermore, CD38 suppression significantly decreased oxygen consumption measurements in CSF-derived extracellular mitochondrial particles (Fig. 4j), and neurologic outcomes seemed to be negatively correlated with these functional endpoints (Fig. 4k, l), suggesting that CSF mitochondrial function may be a potential biomarker of neuroglial signaling after stroke.
Fig. 4. Effects of CD38 siRNA in focal cerebral ischemia.

a, Male C57Bl6 mice were subjected to transient 60 min focal ischemia and control siRNA or CD38 siRNA was injected into lateral ventricles at 5 days post-stroke. Immunostaining showed that CD38 siRNA decreased HMGB1 astrocytes in peri-infarct cortex. b, Nissl staining showed no difference in infarct size (n=8 or 10). c, Immunostaining demonstrated that astrocytic CD38 was diminished by CD38 siRNA. d, Astrocytic CD38 suppression with siRNA reduced GFAP-positive mitochondria in CSF at 7 days (n=6). e, Neuronal mitochondria were decreased by CD38 siRNA (n= 8 or 5). f, CD38 siRNA attenuated peri-infarct GAP43 immunostaining. g, Western blot confirmed a reduction of peri-infarct GAP43 protein within CD38 siRNA-treated brains (n=5). h, i, Suppression of CD38 signaling worsened neurological outcomes in neuroscore (h) and grid walking test (i) (n=7 or 9).*P<0.05 vs day 3 control siRNA, #P<0.05 vs day 7 CD38 siRNA. j, CD38 suppression decreased oxygen consumption in CSF mitochondria (n=7 or 9). k, l, Mitochondrial function in CSF was negatively correlated with neurological outcomes. All values are mean +/− SEM. m, Schematic of CD38 regulation of mitochondria release/transfer hypothesis between astrocytes and neurons.
Taken together, these findings suggest that astrocytes may release extracellular mitochondrial particles via CD38-mediated mechanisms that enter into neurons after stroke (Fig. 4m). But there a few caveats and the detailed mechanisms and generalizability of these proof-of-concept findings should warrant further investigation. First, the dynamics of extracellular mitochondria release and entry into neurons as well as quantitative thresholds for functional benefit remain to be fully defined (Extended Data Fig. 10a–i). A second caveat relates to mitochondrial entry mechanisms. In neurons, endocytosis may be regulated by dynamin/clathrin 20 or integrin pathways 21. In our models, integrin-mediated src/syk signaling may be involved (Extended Data Fig. 10j–m). How integrin-mediated mitochondrial transfer is modulated under different disease conditions requires further study. Third, CD38 is also expressed in immune cells. In this study, CD38 suppression with siRNA in vivo did not appear to affect T cells or microglia/macrophages, but the balance between potentially beneficial CD38 signals in astrocytes versus deleterious CD38 signals in immune cells should be carefully considered. A fourth caveat is whether other glial cells may participate. Microglia, oligodendrocytes and pericytes are activated after stroke 22,23, so their potential roles in mitochondrial exchange warrants further investigation. Finally, astrocytes can produce many factors for protecting and restoring neurons, including tPA, high-mobility group box 1 (HMGB1), extracellular microvesicles containing VEGF and FGF-2, and various microRNAs 24–27. How mitochondrial particles may interact with these other extracellular signals should be explored.
Non-cell autonomous signaling is vital for CNS recovery after injury or disease 28,29. In the context of cerebral ischemia, the present study suggests that astrocytes may release extracellular mitochondrial particles that enter into neurons to support cell viability and recovery after stroke.
Methods
Reagents
BAPTA-AM (A1076), cyclic ADP ribose (C7344) and dynasore hydrate (D7693) were purchased from Sigma, and RGDS peptide (3498) and MNS (2877/50) were purchased from R&D systems.
Mouse Focal Cerebral Ischemia Models
All experiments were performed following an institutionally approved protocol in accordance with National Institutes of Health guidelines and with the United States Public Health Service’s Policy on Human Care and Use of Laboratory Animals. Our methods also included randomization, blinding and statistical criteria consistent with ARRIVE guidelines (Animals in Research: Reporting In vivo Experiments). Basically, male C57Bl6 mice (12–14 weeks) or FVB/N-Tg (GFAPGFP)14Mes/J mice are anesthetized with 5% to 1% isoflurane, and rectal temperatures and cerebral blood flow are monitored. After midline skin incision, 7-0 nylon monofilament coated with silicon resin was introduced through a small incision into the common carotid artery. Adequate cerebral ischemia was assessed by Laser Doppler flowmetry and by examining forelimb flexion after the mice recovered from anesthesia. The mice were re-anesthetized, and reperfusion was established by withdrawal of the filament. Functional outcome after stroke was assessed by neurological severity scores and foot-fault test 25.
Primary neuron cultures
Primary neuron cultures were prepared from cerebral cortices of E17-day-old Sprague-Dawley rat embryos or E17-day-old FVB/N-Tg (GFAPGFP)14Mes/J mouse embryos. Briefly, cortices were dissected and dissociated using papain dissociation system (Worthington Biochemical Corporation, LK003150). Cells were spread on plates coated with poly-D-lysine (Sigma, P7886) and cultured in Dulbecco’s modified Eagle medium (NBM, Life Technology, 11965-084) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate, and 5% fetal bovine serum at a density of 2 × 105 cells/mL (1mL for 12 well format, 0.5 mL for 24 well format). At 24 hours after seeding, the medium was changed to Neurobasal medium (Invitrogen, 21103-049) supplemented with B-27 (Invitrogen, 17504044) and 0.5 mM glutamine. Cells were cultured at 37°C in a humidified chamber of 95% air and 5% CO2. Cultures were used for experiments from 7 to 10 days after seeding.
Primary astrocyte cultures
Primary astrocyte cultures were prepared from cerebral cortices of 2-day-old neonatal Sprague-Dawley rats or E17 C57Bl6 mice. Briefly, dissociated cortical cells were suspended in Dulbecco’s modified Eagle medium (DMEM, Life Technology, 11965-084) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate, and 10% fetal bovine serum and plated on uncoated 25 cm2 flasks at a density of 6×105 cells/cm2. Monolayers of type 1 astrocytes were obtained 12–14 days after plating. Non-astrocytic cells such as microglia and neurons were detached from the flasks by shaking and removed by changing the medium. Astrocytes were dissociated by trypsinization and then reseeded on uncoated T75 flasks. After the cells reached 70–80% confluence, cultures were switched to Neurobasal medium containing 1% penicillin/streptomycin or DMEM containing 1% penicillin/streptomycin, and astrocyte-conditioned media were collected 24h later. Collected astrocyte-conditioned medium (ACM) was treated by spin cell debris down with centrifuging at 2,000g for 10 minutes or by filtrating through a 1.2-μm syringe filter for further experiments.
Oxygen-glucose deprivation (OGD) and reoxygenation
OGD experiments were performed using a specialized, humidified chamber (Heidolph, incubator 1000, Brinkmann Instruments, Westbury, NY) kept at 37 °C, which contained an anaerobic gas mixture (90% N2, 5% H2, and 5% CO2). To initiate OGD, culture medium was replaced with deoxygenated, glucose-free Dulbecco’s modified Eagle medium (Life Technology, 11966-025). After 2 h challenge, cultures were removed from the anaerobic chamber, and the OGD solution in the cultures was replaced with maintenance medium. Cells were then allowed to recover for 18 h (for neurotoxicity assay) and 72 h (for siRNA/astrocyte coculture) in a regular incubator.
Cell Viability Assays
Neuronal injury was measured by standard cell cytotoxicity assays such as lactate dehydrogenase (LDH) using the Cytotoxicity Detection Kit (Roche Applied Science, 11644793001) and/or Cell Counting Kit 8 cytotoxicity assay (DOJINDO, CK04-13). For LDH assay, 100% cell death was induced with 0.5% triton X in sister culture. The relative assessments of neuronal injury were normalized by comparison with 100% cell death (LDH assay) or with control cell as 100% cell survival (CCK8).
Determination of CD38/ADPR-cyclase activity
ADPR cyclase activity was determined by fluorometrically using nicotinamide guanine dinucleotide (NGD+) (Sigma, N5131) as a substrate as described before 30,31. Astrocytes or neurons were incubated with 200 μM NGD+, and the production of cGDPR was determined at excitation/emission wavelengths of Ex 300 nm/Em 410 nm with a microplate reader.
ATP measurement
Intracellular or extracellular ATP was determined by CellTiter-Glo luminescence (Promega, G7570) which can perform cell lysis and generate a luminescent signal proportional to the amount of ATP present. Briefly, opaque-walled 96-well plates with culture media (50μl) or cell lysate (50μl) were prepared. CellTiter-Glo luminescence test solution (50μl) was added and incubated for 30 min at room temperature. Luminescent signal was determined by luminescence microplate reader.
Liposomal ATP treatment
Liposomal ATP was obtained from Encapsula NanoScience. Briefly, we used lyophilized proliposomes compose of 7:3 molar ratio of L-alpha-phosphatidylcholine: L-±-phosphatidylserine containing ATP which forms 100 nm liposomal ATP upon hydration. ATP-loaded liposome (1–1000 nM) was co-incubated with neurons following oxygen-glucose deprivation, and cell viability was analyzed after 18 h reoxygenation.
Mitochondria membrane potential measurement
To monitor mitochondrial health, JC-1 dye (invitrogen, T-3168) was used to assess mitochondrial membrane potential. Rat cortical astrocytes or media were incubated with JC1 (5 μM or 1 μM) for 30 min at 37 °C. JC1 dye exhibits potential-dependent accumulation in mitochondria, indicated by fluorescence emission shift from green (Ex 485 nm/Em 516 nm) to red (Ex 579 nm/Em 599 nm). Mitochondria membrane potential was determined by the fluorescent ratio with a fluorescent microplate reader.
Oxygen consumption analysis
Real time oxygen consumption in astrocytic particles or in CSF samples were measured by Mito-ID Extracellular O2 sensor kit (Enzo Life Science, ENZ-51045) according to the instruction provided Enzo Life Science. Briefly, astrocytes (70–80% confluent cells/well/100 μL ) or particle fraction (100μL; 25-fold concentrated astrocytic conditioned media) were prepared in non-coated regular 96 wells, and O2 sensor probe (10 μL) was added into each well. Each CSF sample (8 – 20 μL) was collected from cisterna magna at day 7 after focal cerebral ischemia. Following centrifugation at 2,000g for 10 minutes, 6 μL CSF was diluted in 54 μL PBS, and 6 μL O2 sensor probe was added into each well. After covering with 100 μL (50 μL for CSF sample) of Mito-ID HS Oil, the plate were read with filter combination of 340 nm for excitation and 642 nm of emission at 30 °C.
Electron microscopy analysis
Rat cortical astrocytes or pellets from astrocyte-conditioned media were fixed in 2.0% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 (Electron Microscopy Sciences, Hatfield, PA) for one hour at room temperature on a rocker. They were rinsed in cacodylate buffer, gently scraped and pelleted and post-fixed in 1.0% osmium tetroxide in cacodylate buffer for one hour on ice. They were rinsed in buffer and stabilized with a small amount of 2% agarose in PBS to hold them together. They were then dehydrated through a graded series of ethanol to 100%, followed by propylene oxide, 100%. They were infiltrated with Epon resin (Ted Pella, Redding, CA) in a 1:1 solution of Epon:propylene oxide overnight on a rocker at room temperature. The following day they were placed in fresh Epon for several hours and then embedded in Epon overnight at 60 C. Thin sections were cut on a Leica EM UC7 ultramicrotome, collected on formvar-coated grids, stained with uranyl acetate and lead citrate and examined in a JEOL JEM 1011 transmission electron microscope at 80 kV. Images were collected using an AMT digital imaging system (Advanced Microscopy Techniques, Danvers, MA). These methods are similar to previous descriptions of extracellular particle and mitochondria detection in astrocyte cultures 12.
FACS analysis
Standard FACS analysis was performed by BD LSR II or BD Fortessa as described before 25,32,33. Astrocyte-conditioned medium (ACM) was collected from rat cortical astrocytes labeled with Mitotracker Red CMXRos followed by filtrating through a 1.2-μm syringe filter. The supernatant was used to sort labeled mitochondria fraction by FACSAriaII cell sorter configured with 561 nm air cooled laser. Brain cells were collected from peri-infarct cortex after stroke. Briefly, tissues are gently minced and then digested at 37°C for 30 min with an enzyme cocktail (Collagenase type I, DNase I, Sigma Aldrich). CSF samples were prepared for further staining after centrifugation at 2,000g for 10 minutes. FACS analysis was performed using an unstained or phenotype control for determining appropriate gates, voltages, and compensations required in multivariate flow cytometry.
Measurement of particle size
Particle size following extracellular mitochondria isolation by FACS was determined by qNano (iZON). Nanopore-based detection allows particle-by-particle assessment of complex mixtures. Optimization of pore size to particle size, by adjusting the stretch of the pore, allows highly accurate measurement 34. Particles containing mitochondria were sorted using FACS analysis, then particle sizes were quantified by NP400 and using CPC400 calibration particle.
Western blot analysis
Western blot was performed as previously reported 25. Each sample was loaded onto 4–20% Tris-glycine gels. After electorophoresis and transferring to nitrocellulose membranes, the membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 and 0.2% I-block (Tropix, T2015) for 90 min at room temperature. Membranes were then incubated overnight at 4°C with following primary antibodies, anti-β-actin (1:1,000, Sigma-aldrich A5441), anti-GFAP antibody (1:1,000, BD biosciences, 556328), anti-MAP2 antibody (1:500, Abcam, ab11267 or ab32454), anti-CD38 antibody (1:500, Santacruz, sc-7049), anti-TOM40 (1:200, Santacruz, sc-11414), anti-phosphorylated Akt (1:500, Cell signaling, 9271), anti-Bcl-xl (1:500, Cell signaling, 2764), anti-active caspase 3 (1:200, Abcam, ab32042), anti-AIF (1:500, Abcam, ab32516), anti-GAP43 (1:500, Santacruz, sc-17790). After incubation with peroxidase-conjugated secondary antibodies, visualization was enhanced by chemiluminescence (GE Healthcare, NA931- anti-mouse, or NA934- anti-rabbit, or NA935- anti-rat). Optical density was assessed using the NIH Image analysis software.
Immunocytochemistry and immunohistochemistry
Immunocytochemistry and immunohistochemistry performed as described before 35,36. After staining with primary antibody, fluorescent-tagged secondary antibody, nuclei were counterstained with or without 4,6-diamidino-2-phenylindole (DAPI), and coverslips were placed. Immunostaining images or time lapse images were obtained with a fluorescence microscope (Nikon ECLIPSE Ti-S) interfaced with a digital charge-coupled device camera and an image analysis system or confocal microscope analysis using Carl Zeiss Laser Scanning Confocal Microscope Pascal 5 LSM and Pascal 5 LSM software Version 3.2. Dendrite elongation was assessed following MAP2 staining followed by NeuriteQuant analysis 37.
CRISPR activation plasmid transfection
Control CRISPR activation plasmid (sc-437275), rat CD38 CRISPR activation plasmid (sc-437321-ACT) were obtained from Santa Cruz Biotechnology. Transfection was performed according to the transfection protocol for cell cultures from Santa Cruz Biotechnology. Briefly, Plasmid transfection reagent mixture of 1ml (Transfection reagent, sc-395739, Transfection medium, sc-108062) was co-incubated with astrocytes for 24 hours in a 5% CO2 incubator at 37°C, and then CD38 cyclase activity was assessed in order to confirm efficiency of transfection.
siRNA experiment
Control siRNA, CD38 siRNA were obtained from Santa Cruz Biotechnology. Control siRNA (sc-37007) consists of a scrambled sequence that will not lead to the specific degradation of any known cellular mRNA. Mouse CD38 siRNA (sc-37246) or Rat CD38 siRNA (sc-270394) is each pool of 3 target-specific 19–25 nt siRNAs designed to knock down gene expression. The sequences for mouse CD38 siRNAs are designed as followed; 5′-GUGUACUACCAACAUUCAA-3′, 5′-GUGUGUCUUUAGUAGGUAU-3′, 5′-CCAGUUUGUGAUUGUUGA-3′. Rat CD38 siRNAs are designed as followed; Sequence 1: 5′-CUCAAACCAUACCAUGUAA-3′, Sequence 2: 5′-GGAAGAGCUUCCCAAUACA-3′, Sequence 3: 5′-GUGUUAUCGUCUAGCAAUA-3′.
siRNA were prepared according to the transfection protocol for cell cultures from Santa Cruz Biotechnology. Briefly, siRNA transfection reagent mixture of 1ml (Transfection reagent, sc-29528, Transfection medium, sc-36868) was co-incubated with astrocytes for 6 hours in a 5% CO2 incubator at 37°C, and then same amount of DMEM 20% FBS was added. An additional incubation was performed for 18 hours.
Statistical analysis
Results were expressed as mean±SEM. When only two groups were compared, unpaired t-test was used. Multiple comparisons were evaluated by one-way ANOVA followed by Tukey’s Kramer or two-way ANOVA. P<0.05 was considered to be statistically significant.
Extended Data
Extended Data Figure 1. Astrocytic mitochondria particle detection.

a, Electron microscopic analysis demonstrated that mitochondria were detected within extracellular astrocyte-derived particles. Free mitochondria were also found in astrocyte-conditioned medium. b, In FACS analysis, control beads were used to gate population ranging in size from 500 nm to 900 nm. c, In astrocyte-derived conditioned media, approximately 53% of particles in the range of size were positive for functional mitochondria (n=5). d, After FACS analysis to isolate extracellular mitochondria fraction from astrocyte-conditioned media, particle size was measured with qNano analysis. Consistent with electron microscope analysis, a range of size distributions were observed (~25%: 300 – 400 nm, ~75%: 400 – 1100 nm). All values are mean +/− SEM.
Extended Data Figure 2. Characteristics of astrocytic mitochondria particle in FACS analysis.

a, Mitochondrial particles were identified by FACS. b, Of these mitochondrial particles, FACS analysis identified that approximately 79% and 43% of particles express β1-integrin and CD63, respectively (n=4). cADPR (1 μM) did not appear to affect these distributions (n=4). All values are mean +/− SEM.
Extended Data Figure 3. Production of astrocytic mitochondria particle in a Ca2+-dependent mechanism.

a, The known CD38 downstream signal, cADPR increased intracellular calcium shown in Fluo-4 intensity in a concentration-dependent manner (n=3). b, Intracellular ATP in astrocytes was upregulated by cADPR stimulation (n=4). **P<0.01 vs cADPR 0 μM. c, To measure ATP levels in extracellular particles, astrocyte-conditioned media were collected and large debris were excluded by centrifugation and filtration using 1.2 μm filter. Following another centrifugation at 20,000g for 30 min, each 100 μl from top or bottom fractions were used for ATP assay. d, The bottom fraction had higher ATP content, and cADPR (1 μM) increased ATP content in this bottom fraction (n= 6 or 8). e, cADPR-induced extracellular ATP levels within extracellular particles was diminished by intracellular calcium blocker, BAPTA-AM (n= 4 or 6). All values are mean +/− SEM.
Extended Data Figure 4. Summary of experiment on Figure 2c.

a, We repeated the experiment in Fig. 2c with n=4 independent primary cultures per group. Similar results were obtained. The extracellular mitochondria-depleted astrocyte media (mdACM) group was significantly different compared to the ACM group. Furthermore, in this repeated experiment, there was also statistical significance between controls (OGD-damaged neurons alone) versus those treated with mitochondria-containing astrocyte media (ACM), and there was no statistically significant worsening when comparing control versus mitochondria-depleted groups (mdACM). Taken together, these two separate experiments suggest a modest but statistically significant neuroprotection induced by astrocyte-derived mitochondria. All values are mean +/− SEM. b, Mitotracker Red CMXRos (200 nM) was incubated without astrocytes to obtain no-cell-derived media (negative control). Media was collected and further incubated with neurons following oxygen-glucose deprivation. After 24 hours, there was no mitochondrial signal observed. Scale: 100 μm.
Extended Data Figure 5. Role of astrocytic CD38 in mitochondria transfer during starvation in vitro.

a, Immunocytochemistry in neuron-astrocyte co-cultures demonstrated that CD38 was primarily expressed within astrocytes. b, Extracellular ATP levels were higher in media collected from neurons co-cultured with astrocytes compared to neuron-alone cultures alone (n=9 or 11). c, After serum/glucose starvation, neurons were significantly damaged, as expected. But neurons co-cultured with astrocytes were protected (n=6 or 4). d, CD38 suppression with siRNA significantly decreased extracellular ATP levels in neuron-astrocyte co-culture, but CD38 suppression did not affect extracellular ATP level in neuron-alone cultures (n=9 or 6). e, Blockade of astrocytic CD38 with siRNA significantly increased LDH release (indicative of cell damage) in the co-culture, suggesting that CD38 may be important to maintain neuroglial homeostasis (n=6). f, Rat primary neurons were co-cultured with rat astrocytes. Immunocytochemistry showed that CD38 suppression with siRNA reduced astrocytic mitochondria (red) transfer into neurons compared to control. g, h, Western blot analysis indicated that CD38 suppression with siRNA can be successfully performed in astrocyte culture without affecting cell viability (n=4 or 3). All values are mean +/− SEM.
Extended Data Figure 6. Metabolic inhibition in astrocyte causes neuronal cell death and retards neurite outgrowth in vitro.

a, Astrocytic aconitase was inhibited by fluorocitrate (FC) which disrupted astrocyte metabolism that was accompanied by SA-β-gal signal. b, Intracellular ATP was decreased in these metabolically-disrupted astrocytes (n=6). *P<0.05, **P<0.01 vs FC 0 mM. c, PI staining showed that fluorocitrate (0.5 mM) did not induce cell death in astrocytes. d, Metabolically-disrupted astrocytes significantly decreased mitochondrial membrane potential. Red: aggregated JC1, Green: monomer JC1. Scale: 20 μm. e, Rat cortical neurons were co-cultured with JC1-labeled astrocytes. After 24 hours co-culture, control astrocytes transferred mitochondria which had a high-membrane potential (aggregated JC1), but metabolically-disrupted astrocytes released and transferred dysfunctional mitochondria into neurons (n=3). f, Metabolically-disrupted astrocytes could not support neural viability under starvation in the co-culture (n=4). g, Co-culture between astrocytes and neurons was conducted for 48 hours to test neurite outgrowth. Immunocytochemistry showed that metabolically-disrupted astrocytes retarded neurite outgrowth and increased neuronal cell death (n=3). h, LDH assay indicated that fluorocitrate (0.5 mM) did not affect cell viability in either rat cortical astrocytes (n=4) or rat cortical neurons (n=4). All values are mean +/− SEM.
Extended Data Figure 7. FACS analysis using E17 FVB/N-Tg (GFAPGFP)14Mes/J transgenic mice.

a, Cortical neurons were isolated from E17 FVB/N-Tg (GFAPGFP)14Mes/J transgenic mice. Immunocytochemistry showed that cultured neurons did not express either GFP or GFAP protein following oxygen-glucose deprivation, suggesting that stroke-like stress may not likely lead to “leakiness” in this astrocyte-specific GFP mouse. b, Brain cell suspension was prepared from FVB/N-Tg (GFAPGFP)14Mes/J mice subjected to transient ischemia, then FACS analysis was performed. c, Representative image before cell sorting. d, Purity after cell sorting. e, Either MAP2+/GFP- or MAP2+/GFP+ population was positive for DAPI as 92.5% or 85.9%, respectively. f, Western blot analysis demonstrated that both GFP-positive and negative neurons expressed mature neuron marker (neurofilament) but not neuronal stem cell marker (nestin). These data exclude the possibility that GFAP-positive cells included subsets of neuronal precursor cells that are known to also express GFAP.
Extended Data Figure 8. Effects of CD38 suppression with siRNA in vivo and in vitro.

a, Western blot showed that CD38 expression was increased in peri-infarct cortex at days 1 to 7 after stroke. b, CD38 siRNA or a scrambled control was injected into lateral ventricles at 5 days after stroke. Western blot analysis confirmed that CD38 expression was successfully decreased in peri-infarct cortex at 7 days. c, In peri-infarct cortex, CD8 T cell and Iba1 positive microglia/macrophage were detected by immunohistochemistry. d, Quantification of the number of CD8 positive cells or Iba1 positive cells indicated that there was no difference between control siRNA and CD38 siRNA (n=6). All values are mean +/− SEM. e, Cultured rat cortical astrocytes were subjected to oxygen-glucose deprivation for 2 hours followed by treating with control siRNA or CD38 siRNA. Astrocyte cell morphology or GFAP expression was assessed by immunocytochemistry or western blot after 22-h reoxygenation. f, Morphology change was not clearly observed in cultured astrocytes suppressed CD38 with siRNA compared to control siRNA. g, Western blot analysis showed that CD38 was successfully decreased by siRNA transfection but GFAP expression was not clearly changed.
Extended Data Figure 9. Neuronal purity confirmed by FACS analysis in vivo.

To be sure about our FACS findings, we used two different standard approaches that have been published in the literature (Bi et al, J Neurosci 2011; Cruz et al, Nat Neurosci Rev 2013) a, By FACS, MAP2 positive population were gated and further assessed by other markers such as Iba1 (microglia/macrophage) and GFAP (astrocyte) in brain cell samples isolated from C57Bl6 mice. These comparisons confirmed that the MAP2+ population did not contain any appreciable amounts of microglia or astrocyte, whereas another neuron marker (NeuN) was highly enriched. b, Similar findings were obtained using an alternative gating method to isolate neurons.
Extended Data Figure 10. Involvement of integrin-mediate src/syk mechanisms in astrocytic mitochondrial entry into neurons in vitro.

a and b, Cultured rat cortical astrocytes were stimulated by cADPR (1 μM) for 24 hours. Intracellular mitochondria labeled by mitotracker dye was significantly increased in astrocytes stimulated with cADPR (1 μM) (n=7). **P<0.01 vs 0h. c, Some of mitochondria were found outside of cells. d, FACS analysis revealed that approximately 5×105 mitochondria were contained in 1mL of astrocyte-derived conditioned media (n=6). cADPR (1 μM) significantly increased the number of mitochondria in the media (n=6). e, Experimental schedule to quantify the mitochondrial entry into neurons following oxygen-glucose deprivation. Rat cortical neurons (1×105 cells/well) were prepared in 24-well culture plate. ACM or cADPR-ACM (each 1 mL) was co-incubated with neurons for 18 hours. Mitochondrial entry into neurons were calculated by mitochondrial intensity measured before and after washing cells with PBS. Phenol red free culture media were used to decrease back ground signal. Back ground signal was subtracted from fluorescent intensity obtained from each sample. f, Oxygen-glucose deprivation for 2 hours decreased approximately 50% of mitochondria in neurons after 18 h reoxygenation (n=4). g, All data are expressed as relative values, with total neuronal mitochondria after 2 h OGD/18 h reoxygenation being 100%. Mitochondrial entry into neurons was slightly higher in cADPR-ACM treatment (18%) compared to ACM treatment (11%), although there was no statistically significance (n=4). h, There was no difference in the percentage of mitochondrial entry between ACM treatment and cADPR-ACM treatment (n=4). i, cADPR-ACM treatment supported neuronal viability better than ACM treatment (n=4). j, Co-culture between rat cortical astrocytes in the upper chamber and rat cortical neurons in the lower chamber was performed for 18 hours following oxygen-glucose deprivation for 2 hours in neurons. Then, mitochondrial entry into neurons was measured. k, Immediately after oxygen-glucose deprivation, dynasore (5 μM), RGDS peptide (50 μg/ml), or MNS (1 μM) was initially added in neurons for 30 min, then astrocyte co-culture was performed for 18 hours. The data are expressed as relative values, with astrocytic extracellular mitochondria plus entered mitochondria into neurons being 100%. RGDS peptide and MNS significantly decreased mitochondrial entry into neurons, but dynasore did not inhibit the entry. l, MNS treatment significantly decreased astrocyte-mediated neuroprotection (n=4). m, Dynasore (5 μM), RGDS peptide (50 μg/ml), or MNS (1 μM) did not affect neuronal viability after 2 h oxygen-glucose deprivation (n=4). All values are mean +/− SEM. These data suggest that astrocyte into neuron mitochondrial particle entry may involve integrin-mediate src/syk mechanisms. However, we acknowledge that these pathways may be multifactorial and deeper analyses are warranted to dissect entry mechanisms under various physiologic and pathologic conditions.
Acknowledgments
This work was supported in part by grants from NIH, the Rappaport Foundation, and the China National Natural Science Foundation Award For Distinguished Young Scholars. Electron microscopy was performed in the Center for Systems Biology. Cytometric assessments were supported by the Department of Pathology Flow and Image Cytometry Core. The authors thank Jim Felton and Jeffrey Zwicker for assistance with qNano analysis.
Footnotes
Competing Interests: The authors declare they have no competing financial interest.
Contributions
K.H. contributed to manuscript preparation, hypothesis generation, experimental design/analysis and conducted experiments. E.E., X.W., Y.T., Y.L., and C.X. conducted experiments and helped with data analysis. X.J. and E.H.L. contributed to manuscript preparation, hypothesis generation and experimental design.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Davis CH, et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014;111:9633–9638. doi: 10.1073/pnas.1404651111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 3.Attwell D, et al. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khakh BS, Sofroniew MV. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci. 2015;18:942–952. doi: 10.1038/nn.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- 6.Wang XF, Cynader MS. Pyruvate released by astrocytes protects neurons from copper-catalyzed cysteine neurotoxicity. J Neurosci. 2001;21:3322–3331. doi: 10.1523/JNEUROSCI.21-10-03322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouyang YB, et al. Astrocyte-enriched miR-29a targets PUMA and reduces neuronal vulnerability to forebrain ischemia. Glia. 2013;61:1784–1794. doi: 10.1002/glia.22556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagai M, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haidet-Phillips AM, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voloboueva LA, Suh SW, Swanson RA, Giffard RG. Inhibition of mitochondrial function in astrocytes: implications for neuroprotection. J Neurochem. 2007;102:1383–1394. doi: 10.1111/j.1471-4159.2007.4634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anne Stetler R, Leak RK, Gao Y, Chen J. The dynamics of the mitochondrial organelle as a potential therapeutic target. J Cereb Blood Flow Metab. 2013;33:22–32. doi: 10.1038/jcbfm.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falchi AM, et al. Astrocytes shed large membrane vesicles that contain mitochondria, lipid droplets and ATP. Histochem Cell Biol. 2013;139:221–231. doi: 10.1007/s00418-012-1045-x. [DOI] [PubMed] [Google Scholar]
- 13.Islam MN, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012;18:759–765. doi: 10.1038/nm.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun. 2006;345:1386–1392. doi: 10.1016/j.bbrc.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 15.Guse AH, Lee HC. NAADP: a universal Ca2+ trigger. Sci Signal. 2008;1:re10. doi: 10.1126/scisignal.144re10. [DOI] [PubMed] [Google Scholar]
- 16.Bruzzone S, et al. Glutamate-mediated overexpression of CD38 in astrocytes cultured with neurones. J Neurochem. 2004;89:264–272. doi: 10.1111/j.1471-4159.2003.02326.x. [DOI] [PubMed] [Google Scholar]
- 17.Levy A, et al. CD38 facilitates recovery from traumatic brain injury. J Neurotrauma. 2009;26:1521–1533. doi: 10.1089/neu.2008.0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higashida H, et al. Social memory, amnesia, and autism: brain oxytocin secretion is regulated by NAD+ metabolites and single nucleotide polymorphisms of CD38. Neurochem Int. 2012;61:828–838. doi: 10.1016/j.neuint.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 19.Choe CU, et al. CD38 exacerbates focal cytokine production, postischemic inflammation and brain injury after focal cerebral ischemia. PLoS One. 2011;6:e19046. doi: 10.1371/journal.pone.0019046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fruhbeis C, et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013;11:e1001604. doi: 10.1371/journal.pbio.1001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowen S, et al. The phagocytic capacity of neurones. Eur J Neurosci. 2007;25:2947–2955. doi: 10.1111/j.1460-9568.2007.05554.x. [DOI] [PubMed] [Google Scholar]
- 22.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu X, et al. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11:56–64. doi: 10.1038/nrneurol.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xin H, et al. Increasing tPA activity in astrocytes induced by multipotent mesenchymal stromal cells facilitate neurite outgrowth after stroke in the mouse. PLoS One. 2010;5:e9027. doi: 10.1371/journal.pone.0009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayakawa K, Pham LD, Katusic ZS, Arai K, Lo EH. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A. 2012;109:7505–7510. doi: 10.1073/pnas.1121146109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proia P, et al. Astrocytes shed extracellular vesicles that contain fibroblast growth factor-2 and vascular endothelial growth factor. Int J Mol Med. 2008;21:63–67. [PubMed] [Google Scholar]
- 27.Li Y, Liu Z, Xin H, Chopp M. The role of astrocytes in mediating exogenous cell-based restorative therapy for stroke. Glia. 2014;62:1–16. doi: 10.1002/glia.22585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lo EH. Degeneration and repair in central nervous system disease. Nat Med. 2010;16:1205–1209. doi: 10.1038/nm.2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing C, Lo EH. Help-me signaling: Non-cell autonomous mechanisms of neuroprotection and neurorecovery. Prog Neurobiol. 2016 doi: 10.1016/j.pneurobio.2016.04.004. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rah SY, Park KH, Han MK, Im MJ, Kim UH. Activation of CD38 by interleukin-8 signaling regulates intracellular Ca2+ level and motility of lymphokine-activated killer cells. J Biol Chem. 2005;280:2888–2895. doi: 10.1074/jbc.M409592200. [DOI] [PubMed] [Google Scholar]
- 31.Graeff RM, Walseth TF, Fryxell K, Branton WD, Lee HC. Enzymatic synthesis and characterizations of cyclic GDP-ribose. A procedure for distinguishing enzymes with ADP-ribosyl cyclase activity. J Biol Chem. 1994;269:30260–30267. [PubMed] [Google Scholar]
- 32.Bi B, et al. Cortical glial fibrillary acidic protein-positive cells generate neurons after perinatal hypoxic injury. J Neurosci. 2011;31:9205–9221. doi: 10.1523/JNEUROSCI.0518-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cruz FC, et al. New technologies for examining the role of neuronal ensembles in drug addiction and fear. Nat Rev Neurosci. 2013;14:743–754. doi: 10.1038/nrn3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vogel R, et al. Quantitative sizing of nano/microparticles with a tunable elastomeric pore sensor. Anal Chem. 2011;83:3499–3506. doi: 10.1021/ac200195n. [DOI] [PubMed] [Google Scholar]
- 35.Hayakawa K, et al. Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2010;30:871–882. doi: 10.1038/jcbfm.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayakawa K, Arai K, Lo EH. Role of ERK map kinase and CRM1 in IL-1beta-stimulated release of HMGB1 from cortical astrocytes. Glia. 2010;58:1007–1015. doi: 10.1002/glia.20982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dehmelt L, Poplawski G, Hwang E, Halpain S. NeuriteQuant: an open source toolkit for high content screens of neuronal morphogenesis. BMC Neurosci. 2011;12:100. doi: 10.1186/1471-2202-12-100. [DOI] [PMC free article] [PubMed] [Google Scholar]