

Abstract
The tumor suppressor p53 functions by inducing the transcription of a collection of target genes. We previously attempted to identify p53 target genes by microarray expression and ChIP‐sequencing analyses. In this study, we describe a novel p53 target gene, FUCA1, which encodes a fucosidase. Although fucosidase, α‐l‐1 (FUCA1) has been reported to be a lysosomal protein, we detected it outside of lysosomes and observed that its activity is highest at physiological pH. As there is a reported association between fucosylation and tumorigenesis, we investigated the potential role of FUCA1 in cancer. We found that overexpression of FUCA1, but not a mutant defective in enzyme activity, suppressed the growth of cancer cells and induced cell death. Furthermore, we showed that FUCA1 reduced fucosylation and activation of epidermal growth factor receptor, and concomitantly suppressed epidermal growth factor signaling pathways. FUCA1 loss‐of‐function mutations are found in several cancers, its expression is reduced in cancers of the large intestine, and low FUCA1 expression is associated with poorer prognosis in several cancers. These results show that protein defucosylation mediated by FUCA1 is involved in tumor suppression.
Keywords: EGFR, FUCA1, fucosidase, fucosylation, p53
The tumor suppressor gene p53 encodes a transcription factor and is the most frequently mutated tumor suppressor gene in human cancer.1, 2, 3, 4 It has been called “the guardian of the genome” and it exerts its tumor suppression function by inducing a collection of target genes. The high incidence of p53 mutations in human cancers illustrates its importance in maintaining normal cell proliferation. In order to discover potentially novel, cancer‐associated genes, we previously undertook a comprehensive search for p53 target genes, and analyzed several target genes whose functions had been unknown.5, 6, 7, 8, 9, 10 This study focuses on one of these newly identified genes, FUCA1.
FUCA1 encodes an α‐l‐fucosidase that removes terminal l‐fucose residues present in glycoproteins.11 The function of FUCA1 in human metabolism is well known, due to its involvement in a malignant, genetic disease called fucosidosis, which is caused by mutation of the FUCA1 gene.12, 13 Fucosidosis patients have symptoms of neurodegeneration with progressive mental and motor deterioration. These symptoms are caused by a lack of fucosidase activity in cells, which leads to the accumulation of fucosyl‐glycopeptides in various tissues. However, the function of FUCA1 in tumorigenesis is not well understood, although there are several studies that indicate a link between fucosylation and tumorigenesis. For example, abnormal fucosylation is known to occur during tumor development, and several well‐known tumor markers such as CA19‐9, α‐fetoprotein‐L3 fraction, and haptoglobin are fucosylated glycoproteins that are over‐represented in tumors.14, 15 In addition, a number of signaling proteins, such as EGFR, and the transforming growth factor‐β1 receptors, E‐cadherin and integrin, are fucosylated, and this modification plays a key role in the regulation of their functions.16, 17, 18, 19, 20 Furthermore, there are reports that enhanced protein fucosylation is associated with breast and colorectal cancers.21, 22
Our study shows that FUCA1 functions downstream of p53, and is the first report showing that the p53 pathway can modulate protein glycosylation. We also show that FUCA1 removes fucose from EGFR and contributes to the repression of EGFR signaling. Furthermore, we show that various cancers carry FUCA1 loss‐of‐function mutations, that FUCA1 expression is decreased in breast and colorectal cancers, and that low expression of FUCA1 is associated with poorer prognosis in these cancer patients.
Materials and Methods
Cell culture and transfection
Cell culture was carried out as previously described.6 COS7, 293T, Saos2, HCT116, H1299, T98G, HeLa, HepG2, Huh7, and MRC5 cells were cultured in DMEM supplemented with 10% FBS. H1648 and HCC2935 cells were cultured in RPMI‐1640 medium supplemented with 10% FBS. Epidermal growth factor was added at 100 ng/mL. Transient transfection assays were carried out using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA).
Northern blot analysis and microarray expression analysis
RNA was prepared using an RNeasy Midi kit (Qiagen, Hilden, Germany). Northern blotting was carried out as previously described.6 Probes were prepared using a BcaBEST labeling kit (Takara Bio, Shiga, Japan), and purified using a Probe Quant G‐50 MicroColumn (Amersham, Little Chalfont, UK) followed by a NICK Column (Amersham). An expressed sequence tag clone containing the full ORF of FUCA1 (IMAGE ID 4871788, purchased from Open Biosystems; Dharmacon, Lafayette, CO, USA) was used for probe preparation. Microarray expression analysis was carried out as previously described.6
Reverse transcription and real‐time PCR
Reverse transcription was carried out using the SuperScript First‐Strand Synthesis System for RT‐PCR (Life Technologies; Thermo Fisher Scientific, Waltham, MA, USA) or ReverTra Ace (Toyobo, Osaka, Japan) following the manufacturer's instructions. Total RNA (0.2–1.0 μg) was used for RT. Reverse‐transcribed cDNAs were subjected to real‐time PCR, which was carried out with a CFX96 Touch Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA, USA). For the detection of FUCA1, PHLDA3, and GAPDH, custom‐designed TaqMan Dual‐labeled Probes from Applied Biosystems (Foster City, CA, USA) (FUCA1: Hs00609173_m1) or from Sigma‐Aldrich (St. Louis, MO, USA) (PHLDA3 and GAPDH) were used. Data are shown as the mean fold expression ±SD.
Chromatin immunoprecipitation–chip assay and ChIP‐seq assay
The ChIP–chip assays were carried out as previously reported.23 For p53 induction, cells were treated with 5‐FU (0.375 mM for 9 h) or UV (10 J or 45 J, harvested 9 h after irradiation). Antibodies against p53 (FL393; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and trimethyl H3 Lys4 (Abcam, Cambridge, UK) were used to precipitate immune complexes. The p53‐binding consensus regions were computationally determined using the TRANSFAC database.
Chromatin immunoprecipitation assay
The ChIP assay was carried out as previously described.23 Control or ts‐FL‐p53‐expressing Saos2 cell lines were tested for p53 binding to FUCA1 enhancer after a shift to the permissive temperature. Cells were collected 6 h after temperature shift. Prepared cell lysates were immunoprecipitated using EZview Red ANTI‐FLAG M2 Affinity Gel (Sigma‐Aldrich), and used for subsequent analyses. Both input and bound (p53‐IP) fractions were analyzed for FUCA1 DNA content; forward, 5′‐GTGACTGCAGCAGCTTCCTGGATA‐3′, and reverse, 5′‐GTGGACAGCAAAACCACATGA‐3′.
Luciferase reporter assay
For the luciferase reporter assay, Saos2 cells were seeded in 96‐well dishes and cotransfected with 60 ng firefly luciferase reporter DNA and 1 ng of each p53 gene cloned into the pcDNA3 vector, together with 15 ng Renilla luciferase expression vector (pGL474 [hRluc/TK] vector; Promega, Tokyo, Japan) as an internal control for transfection efficiency. Cells were harvested 24 h post‐transfection and analyzed using the Dual‐Luciferase Reporter Assay System (Promega). All of the luciferase reporter assay data are the mean‐fold activation ±SD of three independent experiments.
Plasmids
p53 Constructs. Each p53 gene (wild‐type or V143A) was cloned into the pcDNA3 vector as described.6
Promoter–reporter constructs
A 131‐bp fragment of intron 1 of the FUCA1 gene was amplified by PCR using the primers shown below and cloned between the KpnI and SmaI sites of the pGL3‐promotor vector; forward, 5′‐CTAGCAAAATAGGCTGTCCC‐3′ and reverse, 5′‐GTGGGCAGTGAACTTGCATGAACTTGCCCAGCAT‐3′.
Polymerase chain reaction primers
Plasmids carrying one copy of FUCA1‐p53RE or FUCA1‐p53RE mut were obtained by cloning double‐stranded oligonucleotides into the PicaGene basic vector (Wako, Osaka, Japan) containing a minimal promoter. Oligonucleotide sequences: FUCA1‐p53RE, 5′‐AGGCATGCTGGGCAAGTTC‐3′; and FUCA1‐p53RE mut, 5′‐AGGCATGCTGGGAAATTTC‐3′.
Human wild‐type FUCA1, mutant FUCA1 (Q422X; 422–466 a.a. of human FUCA1 deleted, N329Y; a.a. 329 of human FUCA1 mutated to tyrosine), and EGFR were tagged with BamHI and XhoI sites at the 5′‐ and 3′‐ends, respectively, and cloned into the BamHI/XhoI site of the pcDNA3 vector.
Western blot analysis
Cells were lysed in lysis buffer containing 0.1 M Tris–HCl (pH 6.8), 2% SDS, 10% (v/v) glycerol, 0.01% bromophenol blue (BPB), and 0.1 M DTT. Whole cell lysates were analyzed by Western blotting. Antibodies used in this study were as follows: anti‐FUCA1 mouse mAb (sc‐365496), anti‐EGFR rabbit polyclonal antibody (sc‐03) (Santa Cruz Biotechnology), anti‐phospho‐EGFR rabbit polyclonal antibody (2220), anti‐cleaved caspase‐3 rabbit mAb (9664), anti‐p21 rabbit mAb (2947) (Cell Signaling Technology, Danvers, MA, USA), anti‐FLAG mouse mAb conjugated to HRP (PM020‐7) (MBL, Aichi, Japan), and anti‐β‐actin mouse mAb (A2228; Sigma‐Aldrich). After washing with TBS‐Tween (0.05%), membranes were incubated with mouse (NA931V) or rabbit (NA934V) secondary antibodies (GE Healthcare Life Sciences, Piscataway, NJ, USA) for 6 h at room temperature and were visualized with an LAS4000 imaging system (GE Healthcare Life Sciences).
Immunoprecipitation
Cells were lysed in lysis buffer containing 50 mM HEPES (pH 7.0), 150 mM NaCl, 1.5 mM MgCl2, 10% glycerol, 1% Triton‐X, 1 mM EGTA (pH 8.0), 100 mM NaF, 1 mM NaVO4, and a protease inhibitor mix. For immunoprecipitation of FLAG‐tagged proteins, cell lysates were immunoprecipitated with M2 agarose beads (Sigma‐Aldrich) for 15 h at 4°C. For elution of FLAG‐tagged proteins, FLAG‐tagged protein and M2 agarose beads mix solution were incubated with 3× FLAG peptide for 3 h at 4°C.
Fucosidase enzyme activity assay
Activity of α‐l‐fucosidase was assayed as described previously24 using 1 mmol/L 4‐nitrophenyl α‐l‐fucopyranoside (Sigma‐Aldrich) as substrate in PBS (Fig 3b,g,i) or in 0.1 M citrate/0.2 M sodium phosphate buffer at the indicated pH (Figs 3c,d). Absorbance was read on a Tecan plate reader (Wako, Tokyo, Japan) using wavelengths of 405 nm. Absorbance from control wells containing no substrate were taken as background and subtracted from the test wells. Deoxyfuconojirimycin, a potent, specific, and competitive inhibitor of α‐l‐fucosidase, was used to inhibit fucosidase activity.25
Figure 3.
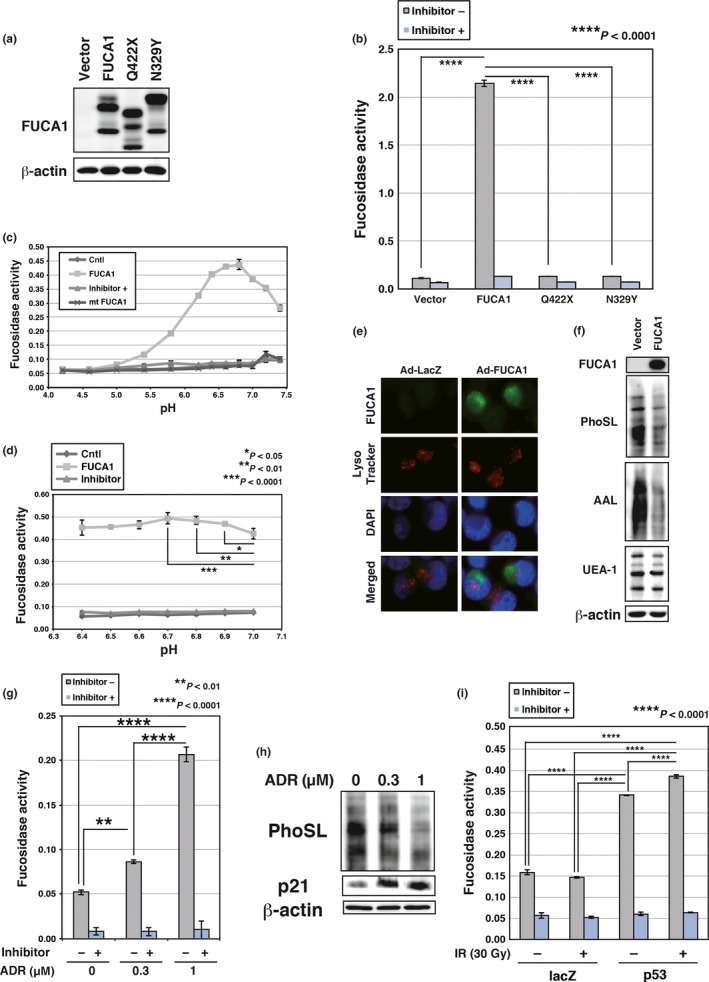
Fucosidase, α‐l‐1 (FUCA1) is active at physiological pH. (a) COS7 cells were transduced with FUCA1, N329Y, Q422X, or control vector. Western blotting was carried out using whole cell lysates. FUCA1 protein has several glycosylation sites and FUCA1 undergoes glycosylation (predicted N‐glycosylation sites N241, N251, N268, and N382; N268 is an identified N‐glycosylation site). The difference in the protein size between wild‐type and N329Y proteins may be the result of abnormal glycosylation of the N329Y mutant protein. (b) Enzyme assay was carried out with (+) or without (−) an inhibitor of FUCA1, using lysates of COS7 cells transduced with FUCA1, N329Y, Q422X, or control vector. The reaction was carried out for 3 h at 37°C in PBS. (c,d) FUCA1‐FLAG and N329Y‐FLAG were purified by immunoprecipitation from whole cell lysates of 293T cells transduced with empty vector (Cntl), FUCA1‐FLAG, or N329Y‐FLAG. Enzyme assays were carried out with or without an inhibitor of FUCA1, using purified wild‐type or mutant (mt) FUCA1. The reaction was carried out for 9 h (c) or 11 h (d) at 37°C in 0.1 M citrate/0.2 M sodium phosphate (McIlvaine) buffer at the indicated pH. Significant enzymatic activities (P < 0.05) were detected between pH 5.4 and 7.4 (c). (e) Subcellular localization of FUCA1 was analyzed. H1299 cells were infected with control (Ad‐LacZ) or FUCA1 (Ad‐FUCA1) expressing adenoviruses. Cells were harvested 37 h post‐infection. Lysosomes were stained with LysoTracker Red and nuclei were stained by DAPI. (f) H1299 cells were infected with control or FUCA1‐expressing adenoviruses. Cells were harvested 62 h post‐infection. Lectin blotting and Western blotting was undertaken using whole cell lysates of H1299. AAL, Argiope aurantia lectin; PhoSL, Pholiota squarrosa lectin; UEA‐1, Ulex europaeus lectin. (g) Enzyme assays were undertaken with or without an inhibitor of FUCA1, using lysates of MRC5 cells subjected to adriamycin (ADR; 0.3 or 1 μM). Cells were harvested 120 h post‐ADR treatment. The enzyme assay reaction was carried out for 52 h at 37°C in PBS. (h) MRC5 cells were subjected to ADR (0.3 or 1 μM). Cells were harvested 120 h post‐ADR treatment. Lectin blotting and Western blotting were carried out using whole cell lysates of MRC5. (i) Enzyme assays were carried out with or without an inhibitor of FUCA1, using lysates of H1648 cells infected with control or p53‐expressing adenoviruses. Cells were subjected to γ‐ray irradiation (IR; 30 Gy) 24 h after virus infection. Cells were harvested 60 h post‐irradiation. The enzyme assay reaction was carried out for 24 h at 37°C in PBS.
Lectin blot analysis
Cells were lysed in lysis buffer containing 0.1 M Tris–HCl (pH 6.8), 2% SDS, 10% (v/v) glycerol, 0.01% bromophenol blue, and 0.1 M DTT. Whole cell lysates were analyzed by lectin blotting with the following biotinylated lectins (J‐Oil Mills, Tokyo, Japan): PhoSL, recognizing α‐1,6‐fucosylation; AAL, recognizing α‐fucosylation; and UEA‐1, recognizing α‐1,2‐fucosylation. Membranes were incubated with each lectin overnight at 4°C. Membranes were washed with TBS‐Tween (0.05%), incubated with avidin–HRP (Vectastain; Vector Laboratories, Burlingame, CA, USA) for 6 h at room temperature and then lectins were visualized with an LAS4000 imaging system (GE Healthcare Life Sciences).
Construction of recombinant adenovirus expressing p53 and FUCA1
Recombinant adenovirus was constructed as reported.7 The Adenovirus Dual Expression Vector Kit (Takara Bio, Shiga, Japan) and Adenovirus Genome DNA‐TPC (Takara Bio) were used to obtain adenovirus. The full ORF of p53 and FUCA1 were inserted into the SmiI site of the pAxCAwtit2 vector. Infectious titer and optimal m.o.i. were determined by the 50% tissue culture infectious dose method using 293 cells, the E1‐complementing helper cell line, according to the manufacturer's instructions.
Immunostaining
H1299 cells were infected with control LacZ or FUCA1‐expressing adenoviruses (m.o.i. 0.2). Cells were incubated with LysoTracker Red (Thermo Fisher Scientific) for 40 min at 36 h post‐infection. At 37 h post‐infection, cells were harvested and fixed with 4% paraformaldehyde for 10 min, washed twice with PBS, permeabilized in 0.2% Triton‐X in PBS for 2 min, and washed with PBS‐Tween (0.05%). The fixed cells were blocked in PBS containing 0.05% Tween, 5 mg/mL BSA, and 50 mM glycine for 1 h at room temperature. The cells were sequentially incubated with anti‐FUCA1 mouse mAb (sc‐365496) for 1 h at room temperature, then Alexa Fluor 488‐labeled secondary antibody (Molecular Probes; Thermo Fisher Scientific). Images were obtained with a BZ‐9000 fluorescence microscope (Keyence, Osaka, Japan).
Colony formation assay
T98G, HCT116 p53−/−, HepG2, and H1299 cells were transduced with control lentivirus or lentivirus expressing FUCA1 or mutant FUCA1 (Q422X). Colony formation assay was carried out using cells selected in blasticidin‐containing medium for 6–10 days in 6‐well plates (2 × 104 cells per well). Subsequently, cells were fixed with 100% methanol for 10 min and stained with Giemsa for 40 min. Images were obtained with a GT‐X980 scanner (Epson, Nagano, Japan). Colonies were analyzed using ImageJ software. Briefly, the images were converted into binary format using ImageJ's binary convert function, and analyzed by ImageJ's batch measure function.
Quantitation of cell death by flow cytometry
Cells were collected and fixed with 70% ethanol overnight, then washed with PBS and incubated with 25 μg/mL propidium iodide and 20 μg/mL RNase A. Flow cytometry analysis was carried out using a FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA), and the proportion of cells in sub‐G1 (chromosome fragmentation) was calculated.
Analysis of FUCA1 mutations
FUCA1 mutations found in human cancers were analyzed using the COSMIC database (http://cancer.sanger.ac.uk/cosmic). Expression of FUCA1 (NM_000147) was analyzed using the ONCOMINE database (https://www.oncomine.org/).
Expression of FUCA1 and correlation to cancer patient prognosis were analyzed using the PrognoScan database (http://www.abren.net/PrognoScan/), a large collection of publicly available cancer microarray datasets with clinical annotations and a tool for assessing the biological relationships between gene expression and prognosis.
Statistical analysis
Data were calculated and shown as mean ± SD. Significance of differences was determined by Student's t‐test (Figs 1c,g,S4c), one‐way anova (Fig. 4a), two‐way anova (Figs 1f,3b–d,g,i) or Mann–Whitney U‐test (Fig. 1h). Statistical significance was defined as P < 0.05.
Figure 1.
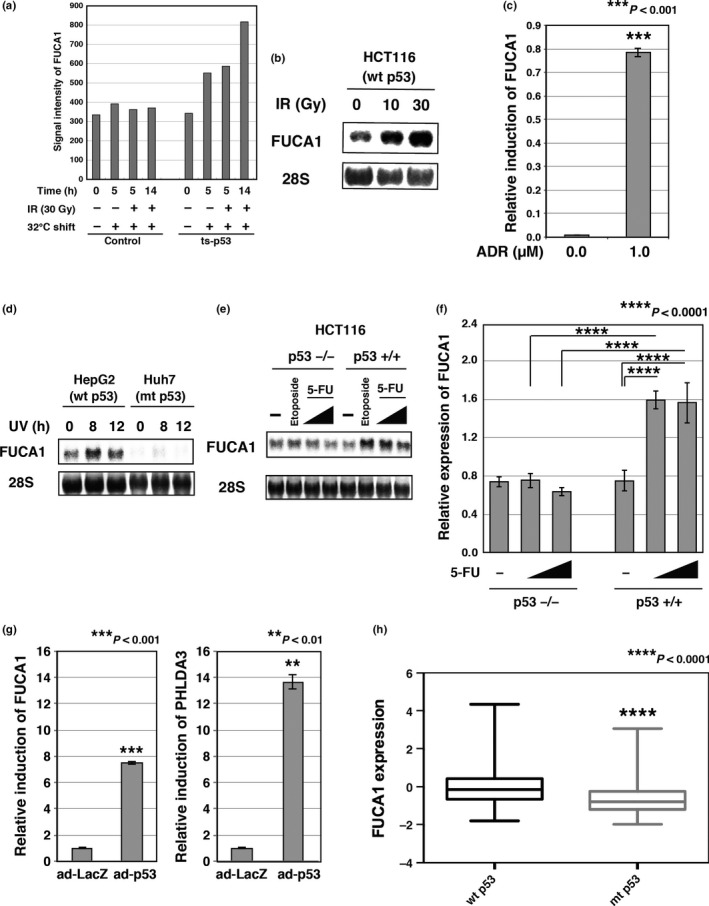
FUCA1 is a p53‐inducible gene. (a) FUCA1 expression was analyzed by microarray analysis. Temperature‐sensitive, p53‐expressing Saos2 cells (ts‐p53) were tested for FUCA1 induction after temperature shift to the permissive temperature with or without γ‐ray irradiation (IR). Cells were subjected to γ‐ray irradiation (30 Gy) 2 h after temperature shift to 32°C. Cells were collected 5 or 14 h post‐temperature shift. (b) HCT116 p53+/+ cells were subjected to γ‐ray irradiation (10 Gy or 30 Gy). Cells were collected 18 h post‐temperature shift. FUCA1 expression was analyzed by Northern blotting. (c) MRC5 cells were subjected to adriamycin (ADR; 1 μM) treatment. Cells were harvested 24 h post‐treatment. FUCA1 expression was analyzed by quantitative RT‐PCR. The mRNA levels of FUCA1 were normalized to GAPDH mRNA levels. (d) HepG2 or Huh7 cells were subjected to UV treatment (45 J/m2). Cells were collected at the indicated time points post‐treatment. FUCA1 expression was analyzed by Northern blotting. (e) HCT116 p53+/+ or p53−/− cells were subjected to etoposide (20 mM) or 5‐fluorouracil (5‐FU) (0.13 or 0.38 mM) treatment. Cells were collected 21 h post‐treatment. FUCA1 expression was analyzed by Northern blotting. (f) HCT116 p53+/+ or p53−/− cells were subjected to 5‐FU (0.38 or 0.76 mM) treatment for 18 h. FUCA1 expression was analyzed by quantitative RT‐PCR. mRNA levels of FUCA1 were normalized to GAPDH mRNA levels. (g) HeLa cells were infected with an adenovirus expressing p53 (ad‐p53) or control LacZ (ad‐LacZ). Cells were harvested 48 h post‐infection. FUCA1 and PHLDA3 expression was analyzed by quantitative RT‐PCR. mRNA levels of FUCA1 and PHLDA3 were normalized to GAPDH mRNA levels. (h) Using the cBioPortal database, FUCA1 expression and the p53 status of breast invasive carcinoma were obtained.39 In total, 971 samples were analyzed. P‐values was calculated using the Mann–Whitney U‐test (P < 0.0001). mt, mutant.
Results
FUCA1 is a p53 target gene
We first identified the FUCA1 gene as a p53‐inducible gene by comprehensive microarray expression analysis of a cell line that expresses ts‐p53.6 As shown in Figure 1(a), FUCA1 mRNA is induced by ts‐p53 at the permissive temperature. We further confirmed that FUCA1 mRNA is induced by DNA damage caused by γ‐ray irradiation, adriamycin, UV, etoposide, or 5‐FU in cell lines that have wild‐type p53 (Fig. 1b–f). Induction of FUCA1 mRNA was not observed in cell lines that do not have functional p53 (Fig. 1d–f). We further analyzed whether FUCA1 mRNA is upregulated by exogenous p53 expression. As shown in Figure 1(g), expression of p53 strongly induced mRNA expression of both FUCA1 and a representative p53 target gene, PHLDA3, in p53‐null Saos2 cells. In addition, we analyzed the correlation between p53 status and FUCA1 mRNA expression in breast cancer tissues. As shown in Figure 1(h), FUCA1 expression was significantly lower in cancers with mutant p53 compared to p53 wild‐type cancer tissues. Collectively, these results indicate that FUCA1 expression is regulated by p53.
The FUCA1 genomic region was analyzed by ChIP‐seq analysis of HCT116 p53 +/+ and HCT116 p53−/− cells that were treated with 5‐FU or untreated (Fig. 2a). This analysis identified a p53‐binding site in intron 1 of the FUCA1 gene. After 5‐FU treatment, H3K27 acetylation was detected around this p53 binding site in HCT116 p53+/+ cells but not in HCT116 p53−/− cells, indicating that this p53‐binding region functions as an active enhancer in cells expressing p53. The DNA surrounding the p53‐binding region is marked by H3K4 mono‐methylation in the absence of 5‐FU treatment, and this increases following 5‐FU treatment. Phospho‐RNA pol II binding at the transcription initiation site is also detected in the absence of 5‐FU treatment, and is increased after 5‐FU treatment in HCT116 p53+/+ but not in HCT116 p53−/− cells. H3K4 trimethylation at the transcription initiation site is also detected and does not change with 5‐FU treatment. These data show that FUCA1 is transcribed under unstressed conditions but is more actively transcribed under stressed conditions in a p53‐dependent manner.
Figure 2.
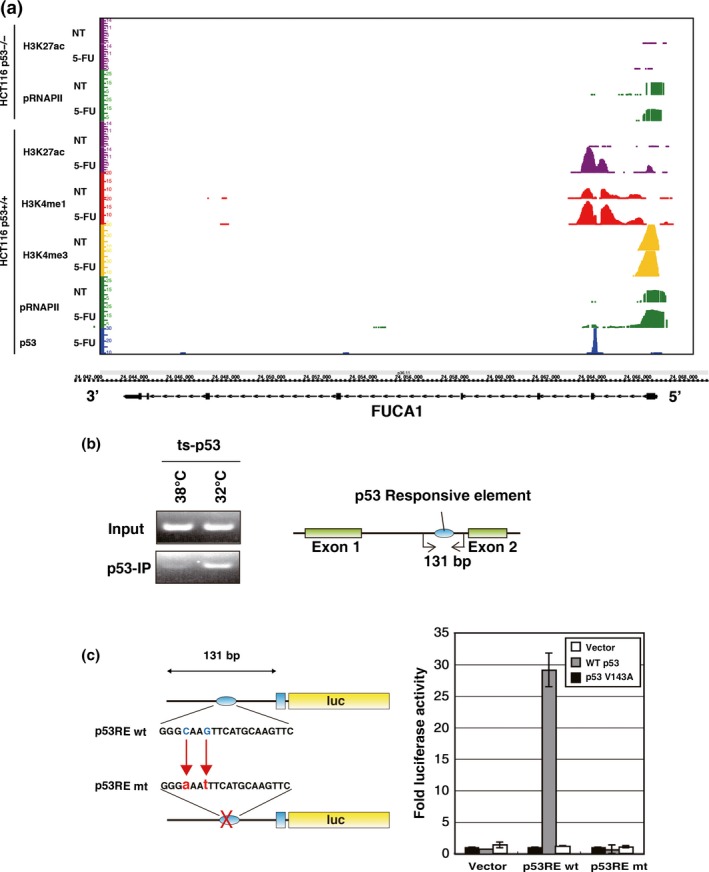
FUCA1 is a direct target gene of p53. (a) HCT116 p53+/+ or p53−/− cells were treated with or without 5‐fluorouracil (5‐FU), and p53 ChIP sequencing analysis was carried out. Genomic locus of FUCA1 is shown together with the results obtained. ChIP sequencing analyses were carried out using antibodies against p53, H3K27ac, H3K4me1, H3K4me3, and phospho‐RNAP II. A p53 binding site (p53RE) was identified within intron 1 of the FUCA1 gene. (b) ChIP assay was carried out for FUCA1 intron 1, which contains the p53RE. A Saos2 cell line that stably expresses a temperature‐sensitive p53 (ts‐p53) was used to analyze p53 binding to the FUCA1 promoter. p53 binding to p53RE was analyzed at the non‐permissive (38°C) and permissive (32°C) temperatures. The positions amplified by PCR (131‐bp fragment was amplified) are shown. (c) The 131‐bp fragments within intron 1 containing wild‐type or mutant p53RE were cloned upstream of a firefly luciferase reporter gene with a minimal promoter, and a luciferase reporter assay was carried out. Constructs were tested for transactivation by WT p53 and p53‐V143A. The assay was undertaken 24 h post‐transfection. mt, mutant.
We further confirmed p53 binding to the FUCA1 intron 1 site by ChIP analysis using primers surrounding the p53‐binding region. As shown in Figure 2(b), p53 bound to the p53‐binding region in Saos2 cells (carrying ts‐p53) at the permissive temperature, but did so much less efficiently at the non‐permissive temperature. The positive and negative controls for this ChIP analysis are shown in Figure S2. As a positive control, we analyzed the binding of p53 to the p53‐responsive element of a previously reported p53 target gene, IER5 (RE2). As a negative control, we also analyzed the binding of p53 to a region near the IER5 gene (RE1) that does not function as a p53‐responsive element.26 We also carried out p53 ChIP–chip analysis using HCT116 p53+/+ or p53−/− cells treated with 5‐FU or UV (15 J and 45 J, respectively), and observed that p53 binds to FUCA1 intron 1 under these conditions (Fig. S1). These data suggest that the p53‐binding region contains a p53‐responsive element that may regulate FUCA1. Examination of the sequences within this region of intron 1 revealed the presence of sequences highly similar to the p53 consensus binding sequence (p53RE; TRANSFAC match score, 0.61) at the center of the region to which p53 was shown to bind. We generated oligonucleotides containing this sequence (p53RE), as well as a version mutated in the p53 consensus sequence (p53RE mut), and cloned each upstream of a luciferase reporter gene containing a minimal promoter (the sequences of wild‐type and mutant p53RE are shown in Fig. 2c). As shown in Figure 2(c), p53 strongly activated the reporter containing the wild‐type p53RE but not p53RE mut. These results collectively demonstrate that p53RE is a p53‐responsive element in the FUCA1 gene, and confirms that FUCA1 is a p53 target gene.
We also compared the p53RE sequence with the consensus response sequences recognized by different p53 family proteins. As shown in Figure S3, p53RE showed high identity with the consensus sequences for p53, p63, and p73.27, 28, 29 In addition, p53RE has been identified as a p63 binding site in a genome‐wide comprehensive analysis of p63 binding sites using cervical carcinoma cells.28 Thus, the FUCA1 gene may be a common target of various p53 family proteins, and it would be interesting to ask in a future study if FUCA1 is regulated by p63 or p73.
FUCA1 encodes a fucosidase that removes α‐l‐fucose
FUCA1 has been reported to be a fucosidase that localizes in the lysosome.30 In addition, fucosidase activity is reported to be lost in fucosidosis patients carrying mutations in the FUCA1 gene.13 We therefore analyzed the fucosidase activity of the wild‐type FUCA1 protein, as well as two mutants (N329Y and Q422X) that are found in fucosidosis patients and which are believed to be defective in fucosidase activity. We expressed each of these in COS7 cells and analyzed their enzymatic activities. As shown in Figure 3(a,b), similar amounts of each protein were expressed, but only wild‐type FUCA1 showed fucosidase activity. This activity was efficiently inhibited by deoxyfuconojirimycin, a potent, specific, and competitive inhibitor of α‐l‐fucosidase.25 We next analyzed the optimum pH for FUCA1 enzymatic activity and found it to be high, between pH 6.4 and 7.0, and the highest at pH 6.7 (Fig. 3c,d). FUCA1 has been reported to reside in the lysosome,30 which has a pH in the 4.5–5.0 range; however, we observed that at pH 5.0 FUCA1 enzymatic activity was very low (Fig. 3c). This suggests that FUCA1 may be optimized for activity in the cytoplasm or another organelle, where the pH is closer to 6.7.
This observation prompted us to examine the subcellular localization of FUCA1 protein by immunofluorescence analysis. We expressed FUCA1 ectopically in H1299 cells and observed that FUCA1 was mainly detected in areas that did not overlap with the lysosomotropic dye LysoTracker Red (Fig. 3e). Rather, FUCA1 appeared as spots in the perinuclear region, which suggests that it may be localized in some organelle surrounding the nucleus.
Fucosylation may be divided into several types according to the linkages it produces, including α1,2‐, α1,3‐, α1,4‐, and α1,6‐fucosylation.31 We analyzed the specificity of FUCA1‐mediated fucosylation by probing blots of fractionated cellular proteins with lectins that can detect and distinguish some of these different forms of fucosylation. Lectins PhoSL and UEA‐1 are specific for α1,6‐ and α1,2‐fucosylation, respectively,32, 33 whereas AAL is a lectin that detects all types of fucosyl linkages.33 As shown in Figure 3(f), PhoSL and AAL staining revealed significant differences between cells expressing vector versus FUCA1, whereas no obvious differences were seen by UEA‐1 staining. These data indicate that FUCA1 efficiently removes α1,6‐fucosylation but not α1,2‐fucosylation. We note that AAL staining was significantly decreased in FUCA1‐expressing sample, suggesting that FUCA1 may also remove other types of common fucosyl linkages such as α1,3‐ and α1,4‐fucosylation, which can be detected by AAL.
As FUCA1 is a p53 target gene, we also analyzed whether fucosidase activity is induced by p53. As shown in Figure 3(g,h), DNA damage by adriamycin treatment resulted in increased fucosidase activity and decreased fucosylation levels in normal human fibroblasts that have wild‐type p53. In addition, fucosidase activity was induced by exogenous p53 expression and this activity was further enhanced by DNA damage (Fig. 3i). Therefore, p53‐regulated FUCA1 expression leads to increase in fucosidase activity and results in a decrease in fucosylated proteins.
Cell death induced by FUCA1 in a manner dependent on its glycosidase activity
As our data indicates that FUCA1 is a p53 target gene, we next asked if FUCA1 has a tumor suppressive function. As shown in Figure 4(a), stable expression of wild‐type FUCA1 significantly inhibited cell proliferation in both p53‐proficient (HepG2) and p53‐deficient (H1299, T98G, and HCT116 p53−/−) cell lines. In contrast, expression of the fucosidase‐deficient mutant FUCA1 had a smaller effect on proliferation in all of these cell lines. We next used flow cytometry to analyze cell cycle and cell death in cells transiently expressing FUCA1. As shown in Figure 4(b), there was no significant difference in the cell cycle distribution between samples. However, the fraction of cells in sub‐G1, indicative of chromosomal fragmentation and cell death, increased significantly in cells expressing wild‐type FUCA1 but not in cells expressing vector or mutant FUCA1. These data show that, while fucosidase activity is required for the induction of cell death and suppression of cell proliferation, the p53 status of the cells does not affect these functions of FUCA1. This is consistent with the notion that FUCA1 is a downstream mediator of p53 action. We also analyzed whether the cell death induced by FUCA1 may be classified as apoptosis. As shown in Figure S4(a), we could detect cleaved caspase‐3 in cells dying as a result of FUCA1 overexpression, indicating that FUCA1 expression results in the induction of apoptosis. Furthermore, we analyzed the effect of FUCA1 knockdown in H1648 cells that express relatively high levels of FUCA1 (Fig. S4b). As shown in Fig. S4(c), FUCA1 knockdown enhanced the proliferation of H1648 cells. We are very much interested in the effect of loss of FUCA1 function, and plan to analyze this further using FUCA1 knockout mice in future studies.
Figure 4.
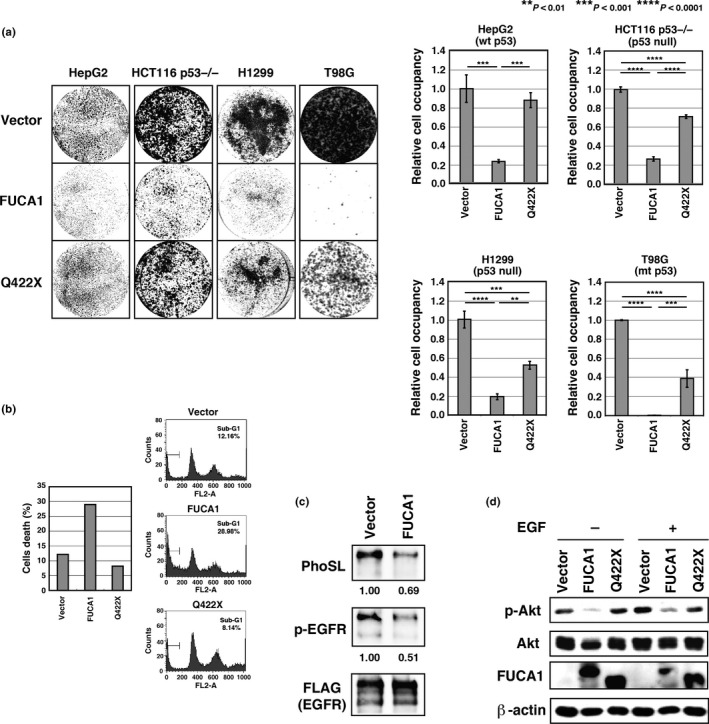
Fucosidase, α‐l‐1 (FUCA1) induces cell death in a glycosidase activity‐dependent manner and inhibits epidermal growth factor (EGF) signaling. (a) T98G cells transduced with control lentivirus or lentivirus expressing FUCA1 or Q422X were selected in blasticidin‐containing medium for 10 days, and subsequently stained with Giemsa. HCT116 p53−/−, HepG2, and H1299 cells transduced with control lentivirus or lentivirus expressing FUCA1 or Q422X were selected in blasticidin‐containing medium for 6 days, and subsequently stained with Giemsa. Colonies were analyzed by ImageJ software. (b) FUCA1 induces cell death. COS7 cells were transfected with FUCA1, Q422X, or control vector. Cells were harvested 48 h post‐transfection and analyzed by FACS. Percentages of cells with sub‐G1 DNA content are shown. Experiments were repeated more than 3 times and representative data is shown. mt, mutant. (c) COS7 cells were co‐transduced with EGF receptor (EGFR)‐FLAG and FUCA1 or control vector. Lectin blotting with Pholiota squarrosa lectin (PhoSL) and Western blotting were carried out using equal amounts of immunoprecipitated EGFR sample. (d) COS7 cells were transfected with the indicated plasmids for 24 h and subsequently stimulated with EGF for 5 min. Protein kinase B (Akt) activity was assessed by Akt phosphorylation and analyzed by Western blotting.
α1,6‐Fucosyl linkages on EGFR cleaved by FUCA1 and EGF signaling inhibited
Several reports have described enhanced fucosylation of proteins in cancer (i.e., CA19‐9, α‐fetoprotein‐L3 fraction, haptoglobin, EGFR, the TGF‐β1 receptors, and E‐cadherin).14, 15, 16, 17, 18, 19, 20 As p53 is frequently mutated in various cancers, which should lead to a decrease in FUCA1 expression, we next turned our attention to proteins that are highly fucosylated in cancers as potential targets of FUCA1. We considered the possibility that FUCA1 may function outside of the lysosome, as its glycosidase activity has an optimal pH of 6.7. We also noted the effect of FUCA1 expression in suppressing proliferation and inducing cell death. One candidate target that would be consistent with these criteria is EGFR, which has been reported to be a α1,6‐fucosylated protein that plays an important role in cell growth and survival.17 We immunoprecipitated EGFR from control or FUCA1‐expressing cells and analyzed EGFR fucosylation by lectin blotting. As shown in Figure 4(c), FUCA1 expression resulted in reduced α1,6‐fucosylation of EGFR, as revealed by PhoSL blotting. In addition, FUCA1 expression resulted in reduced phosphorylation of EGFR, an indicator of EGFR activity. We next asked whether FUCA1 inhibits EGFR downstream signaling by examining phosphorylation of Akt, which is essential for Akt activation. As shown in Figure 4(d), wild‐type but not mutant, FUCA1 efficiently repressed phosphorylation of Akt. These results show that FUCA1 inhibits EGFR signaling by removing α1,6‐fucosyl linkages on EGFR.
Tumor suppressive function of FUCA1
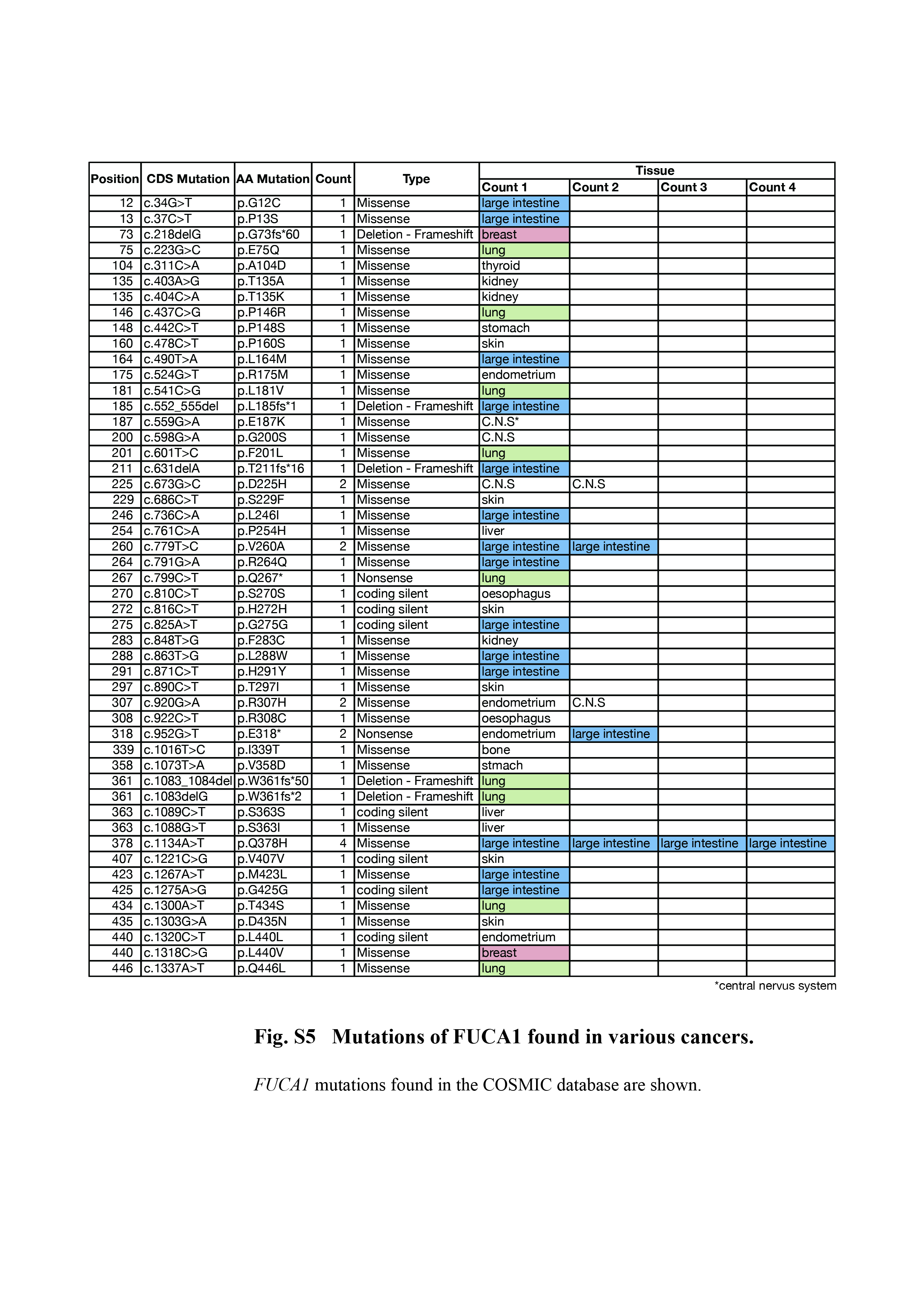
As shown above, FUCA1 can repress EGFR signaling. In addition, the FUCA1 gene is located at 1p36, a chromosome locus frequently deleted in various cancers.34 We therefore asked if FUCA1 has any function as a tumor suppressor. We first searched the publicly available COSMIC database, which listed a total of 57 FUCA1 mutations found in various cancers (Figs 5a,S5). It has been reported that several frame shift and nonsense mutations found in fucosidosis patients generating a stop codon before a.a. Q422 result in the loss of FUCA1 function.12, 13 We also showed above that the Q422X mutant does not have fucosidase activity. Although we have not examined the function of other FUCA1 mutants, we expect that the frame shift mutants (G73fs*60, L185fs*1, and T211fs*16) and nonsense mutants (Q267* and E318*), which are found in cancers, result in the loss of FUCA1 function. To understand how these other mutations may affect FUCA1 function, we examined the reported crystal structure for α‐l‐fucosidase from Thermotoga maritima.35 This fucosidase is the closest bacterial relative to mammalian α‐l‐fucosidase, sharing 38% identity with its human counterpart.35 As shown in Figure S6, 9 out of 10 amino acids in the catalytic pocket are conserved between human FUCA1 and the T. maritima α‐l‐fucosidase. Among the FUCA1 genes mutated in human cancers, several are mutated in the catalytic pocket (n = 3), and one of them, S229F, is located within the catalytic nucleophile residues of FUCA1. Furthermore, 10 amino acids that are conserved between human FUCA1 and T. maritima α‐l‐fucosidase were mutated in human cancers, and one of them, L288W, corresponds to a.a. residues that are highly conserved among vertebrates, invertebrates, and T. maritima (Fig. 5a). Interestingly, 20 out of 57 mutations were found in cancers of the large intestine, suggesting the possibility that FUCA1 is particularly important for suppressing cancers of the large intestine (Fig. S5). We further queried the ONCOMINE database to obtain information about FUCA1 expression levels in normal and cancer tissues. As shown in Figure 5(b,c), FUCA1 expression is significantly lower in cancers of the large intestine compared to normal large intestine tissue. We next searched the PrognoScan database, a large collection of publicly available cancer microarray datasets that includes clinical annotations and a tool for assessing the biological relationships between gene expression and prognosis.36 As shown in Figure 5(d–j), lower FUCA1 expression is associated with poorer prognosis in cancer patients. This association was especially strong in colorectal and breast cancers (Fig. 5e–j). These results suggest that loss of FUCA1 expression may be involved in cancer progression, especially in cancers of the large intestine and breast.
Figure 5.
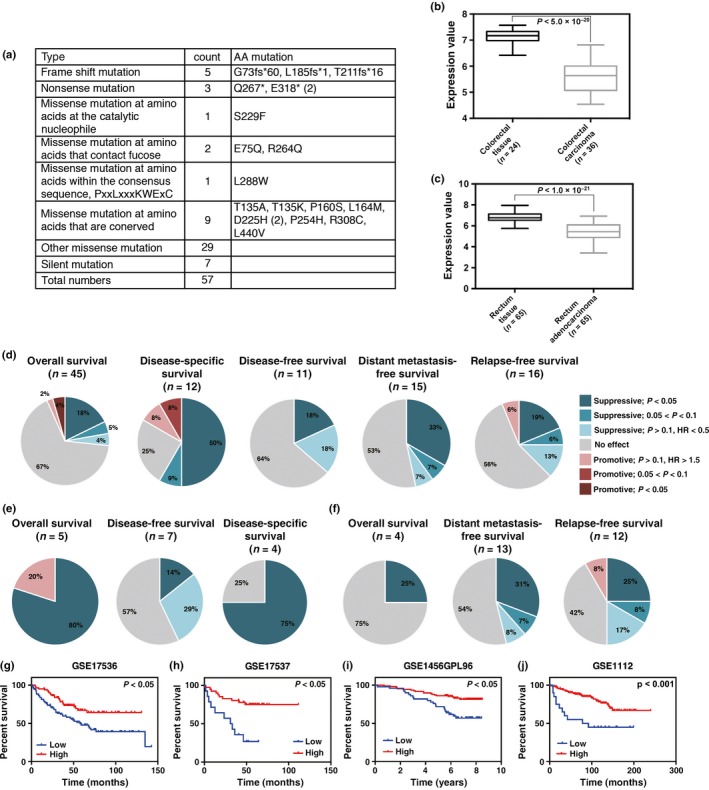
Fucosidase, α‐l‐1 (FUCA1) function is lost/decreased in human cancers and low FUCA1 expression is related to poorer prognosis of cancer patients. (a) FUCA1 mutations found in human cancers are shown. Data were obtained from the COSMIC database. (b,c) FUCA1 (NM_000147) expression in colorectal and rectal cancer was analyzed using the ONCOMINE database. Left and right boxes show the results obtained with normal and cancer tissues, respectively. (b) FUCA1 (probe A_23_P11543) expression in Gaedcke colorectal adenocarcinoma versus normal colorectal tissue was analyzed. FUCA1 was positioned 896th in a ranking of underexpressed genes (in top 5%). P‐value, 7.29E−22; fold‐change, –2.676. (c) FUCA1 (probe 202838_at) expression in Skrzypczak colorectal carcinoma versus normal rectal tissue was analyzed. FUCA1 was positioned 6th in a ranking of underexpressed genes (in top 1%). P‐value, 4.98E−20; fold‐change, –2.976. (d–f) FUCA1 prognostic analysis was carried out using the PrognoScan database. (d) Using the PrognoScan database, FUCA1 expression was analyzed using two probes, 202838_at and 229137_at. In total, 101 datasets were available for FUCA1 expression. Pie charts are shown for each end point. The datasets were categorized into seven categories according to Cox P‐values and hazard ratios (HR) (d, right). Lower FUCA1 expression showed significant associations with poorer prognosis in patients with cancers of the large intestine, breast, or lung. (e,f) Datasets for colorectal (e) and breast (f) cancers are shown. (g–j) Representative survival curves showing the prognosis of colorectal (g,h) and breast (i,j) cancer patients with high or low FUCA1 expression. (g,h) Overall survival of colorectal cancer patients of datasets GSE17536 (n = 177; HR (95% confidence interval [CI]) = 0.15 [0.03–0.68], P = 0.01) and GSE17537 (n = 55; HR [95% CI] = 0.04 [0.00–0.61], P = 0.02). (i) Overall survival of breast cancer patients of dataset GSE1456GPL96 (n = 159; HR [95% CI] = 0.42 [0.20–0.89], P = 0.02). (j) Distant metastasis‐free survival of breast cancer patients of dataset GSE11121 (n = 200; HR [95% CI] = 0.29 [0.14–0.60], P = 0.0007).
Discussion
FUCA1 is a p53 target gene
In this study, we have identified FUCA1 as a novel p53 target gene. FUCA1 mRNA is induced under various DNA‐damaging conditions, and in a p53‐dependent manner. It is also induced by ectopic expression of p53. In addition, we have identified a p53‐responsive element within intron 1 of the FUCA1 gene. This p53‐binding site acts as an active enhancer and several chromatin modifications that are associated with active enhancers, such as H3K27 acetylation and H3K4 mono‐methylation, are found at this site and are enhanced following DNA damage. This is the first report showing that p53 is involved in protein glycosylation and suggests a novel pathway by which p53 might exert its effects on cell growth and death.
FUCA1 encodes a fucosidase that shows highest activity at physiological pH
Previously, FUCA1 was reported to be a lysosomal protein.30 However, the subcellular localization and the optimal pH of FUCA1 suggest that FUCA1 functions not only in the lysosome but in other components within the cell. In this report, we have identified EGFR as one FUCA1 substrate, and shown that FUCA1‐mediated removal of fucose from EGFR leads to decreased activation of the EGFR signaling pathway. However, it can be assumed that FUCA1 may have other target proteins both inside and outside of the lysosome. As FUCA1 expression induces cell death and inhibits growth, identification of other FUCA1 target proteins may reveal novel pathways of tumor suppression that involve the removal of protein fucosyl linkages.
Tumor‐suppressive activity of FUCA1
Although the link between enhanced fucosylation and tumorigenesis has been previously reported, a direct link between the removal of fucosyl linkages and tumor suppression has not.14, 15, 16, 17, 18, 19, 20, 21, 22 Our data suggest that FUCA1, a fucosidase, has tumor‐suppressing activity. We further observed that loss‐of‐function mutations in the FUCA1 gene occur in various human cancers. In addition, FUCA1 expression is significantly reduced in cancers of the large intestine, and low FUCA1 expression is related to poorer prognosis in patients with cancer of the large intestine or breast. It has previously been reported that FUCA1 mRNA expression is decreased in colorectal cancers.37 In addition, it was reported that fucosylation promotes cancer development and malignant progression in breast cancers.38 These reports are in agreement with our results, and suggest that FUCA1 may have tumor‐suppressive activity, and could be a therapeutic target in the treatment of cancers in the clinic. To understand the importance of protein fucosylation and defucosylation in cancers, it will be important to study the function of FUCA1 in vivo using genetically modified mice.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- a.a.
amino acid
- AAL
Aleuria aurantia lectin
- Akt
protein kinase B
- CA19‐9
carbohydrate antigen 19‐9
- ChIP–chip
ChIP with DNA microarray chip
- ChIP‐seq
ChIP sequencing
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- 5‐FU
5‐fluorouracil
- FUCA1
fucosidase, α‐l‐1
- PhoSL
Pholiota squarrosa lectin
- ts‐p53
temperature‐sensitive p53 mutant
- UEA‐1
Ulex europaeus lectin
Supporting information
Fig. S1. p53 Binds to the first intron of the FUCA1 gene.
{kind=link}
Fig. S2. Chromatin immunoprecipitation assay carried out using the p53 responsive element of the p53 target gene IER5.
{kind=link}
Fig. S3. Alignment of the FUCA1 p53RE with p53, p63, and p73 consensus response elements.
{kind=link}
Fig. S4. Analysis of the effect of FUCA1 ectopic expression or FUCA1 knockdown in cancer cells.
{kind=link}
Fig. S5. Mutations of FUCA1 found in various cancers.
{kind=link}
Fig. S6. Alignment of Thermotoga maritima α‐l‐fucosidase and human FUCA1 protein sequences.
{kind=link}
Acknowledgments
We thank Marc Lamphier for critical reading of the manuscript. This study was partly supported by a Grant‐in‐Aid for Scientific Research (C) (#26430133, to R.O.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Applied Research for Innovative Treatment of Cancer from the Ministry of Health, Labor and Welfare (to R.O.), the Project for Development of Innovative Research on Cancer Therapeutics (P‐DIRECT)/Ministry of Education, Culture, Sports, Science and Technology of Japan (to R.O.), the Takeda Science Foundation (to R.O.), Astellas Foundation for Research on Metabolic Disorders (to R.O.), Daiichi‐Sankyo Foundation of Life Science (to R.O.), Extramural Collaborative Research Grant of Cancer Research Institute, Kanazawa University (to R.O.), and Cooperative Research Program of Institute for Frontier Medical Sciences, Kyoto University (to R.O.).
Cancer Sci 107 (2016) 734–745
Funding Information
Ministry of Education, Culture, Sports, Science and Technology of Japan; Ministry of Health, Labor and Welfare of Japan; Project for Development of Innovative Research on Cancer Therapeutics; Takeda Science Foundation; Astellas Foundation for Research on Metabolic Disorders; Daiichi‐Sankyo Foundation of Life Science; Cancer Research Institute, Kanazawa University; Institute for Frontier Medical Sciences, Kyoto University.
References
- 1. Aylon Y, Oren M. Living with p53, dying of p53. Cell 2007; 130: 597–600. [DOI] [PubMed] [Google Scholar]
- 2. Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene 2005; 24: 2899–908. [DOI] [PubMed] [Google Scholar]
- 3. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–31. [DOI] [PubMed] [Google Scholar]
- 4. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell 2009; 137: 413–31. [DOI] [PubMed] [Google Scholar]
- 5. Oda E, Ohki R, Murasawa H et al Noxa, a BH3‐only member of the Bcl‐2 family and candidate mediator of p53‐induced apoptosis. Science 2000; 288: 1053–8. [DOI] [PubMed] [Google Scholar]
- 6. Ohki R, Kawase T, Ohta T, Ichikawa H, Taya Y. Dissecting functional roles of p53 N‐terminal transactivation domains by microarray expression analysis. Cancer Sci 2007; 98: 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7. Ohki R, Nemoto J, Murasawa H et al Reprimo, a new candidate mediator of the p53‐mediated cell cycle arrest at the G2 phase. J Biol Chem 2000; 275: 22627–30. [DOI] [PubMed] [Google Scholar]
- 8. Ohki R, Saito K, Chen Y et al PHLDA3 is a novel tumor suppressor of pancreatic neuroendocrine tumors. Proc Natl Acad Sci USA 2014; 111: E2404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawase T, Ichikawa H, Ohta T et al p53 target gene AEN is a nuclear exonuclease required for p53‐dependent apoptosis. Oncogene 2008; 27: 3797–810. [DOI] [PubMed] [Google Scholar]
- 10. Kawase T, Ohki R, Shibata T et al PH domain‐only protein PHLDA3 is a p53‐regulated repressor of Akt. Cell 2009; 136: 535–50. [DOI] [PubMed] [Google Scholar]
- 11. Darby JK, Johnsen J, Nakashima P et al Pvu II RFLP at the human chromosome 1 alpha‐l‐fucosidase gene locus (FUCA1). Nucleic Acids Res 1986; 14: 9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Willems PJ, Darby JK, DiCioccio RA et al Identification of a mutation in the structural alpha‐l‐fucosidase gene in fucosidosis. Am J Hum Genet 1988; 43: 756–63. [PMC free article] [PubMed] [Google Scholar]
- 13. Willems PJ, Seo HC, Coucke P, Tonlorenzi R, O'Brien JS. Spectrum of mutations in fucosidosis. Eur J Hum Genet 1999; 7: 60–7. [DOI] [PubMed] [Google Scholar]
- 14. Miyoshi E, Moriwaki K, Nakagawa T. Biological function of fucosylation in cancer biology. J Biochem 2008; 143: 725–9. [DOI] [PubMed] [Google Scholar]
- 15. Takeda Y, Shinzaki S, Okudo K, Moriwaki K, Murata K, Miyoshi E. Fucosylated haptoglobin is a novel type of cancer biomarker linked to the prognosis after an operation in colorectal cancer. Cancer 2012; 118: 3036–43. [DOI] [PubMed] [Google Scholar]
- 16. Lin H, Wang D, Wu T et al Blocking core fucosylation of TGF‐beta1 receptors downregulates their functions and attenuates the epithelial‐mesenchymal transition of renal tubular cells. Am J Physiol Renal Physiol 2011; 300: F1017–25. [DOI] [PubMed] [Google Scholar]
- 17. Wang X, Gu J, Ihara H, Miyoshi E, Honke K, Taniguchi N. Core fucosylation regulates epidermal growth factor receptor‐mediated intracellular signaling. J Biol Chem 2006; 281: 2572–7. [DOI] [PubMed] [Google Scholar]
- 18. Zhao Y, Itoh S, Wang X et al Deletion of core fucosylation on alpha3beta1 integrin down‐regulates its functions. J Biol Chem 2006; 281: 38343–50. [DOI] [PubMed] [Google Scholar]
- 19. Hu P, Shi B, Geng F, Zhang C, Wu W, Wu XZ. E‐cadherin core fucosylation regulates nuclear beta‐catenin accumulation in lung cancer cells. Glycoconj J 2008; 25: 843–50. [DOI] [PubMed] [Google Scholar]
- 20. Osumi D, Takahashi M, Miyoshi E et al Core fucosylation of E‐cadherin enhances cell‐cell adhesion in human colon carcinoma WiDr cells. Cancer Sci 2009; 100: 888–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kyselova Z, Mechref Y, Kang P et al Breast cancer diagnosis and prognosis through quantitative measurements of serum glycan profiles. Clin Chem 2008; 54: 1166–75. [DOI] [PubMed] [Google Scholar]
- 22. Muinelo‐Romay L, Vazquez‐Martin C, Villar‐Portela S, Cuevas E, Gil‐Martin E, Fernandez‐Briera A. Expression and enzyme activity of alpha(1,6)fucosyltransferase in human colorectal cancer. Int J Cancer 2008; 123: 641–6. [DOI] [PubMed] [Google Scholar]
- 23. Kaneshiro K, Tsutsumi S, Tsuji S, Shirahige K, Aburatani H. An integrated map of p53‐binding sites and histone modification in the human ENCODE regions. Genomics 2007; 89: 178–88. [DOI] [PubMed] [Google Scholar]
- 24. Abascal I, Skalaban SR, Grimm KM et al Alteration of the isoform composition of plasma‐membrane‐associated rat sperm alpha‐l‐fucosidase during late epididymal maturation: comparative characterization of the acidic and neutral isoforms. Biochem J 1998; 333(Pt 1): 201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winchester B, Barker C, Baines S et al Inhibition of alpha‐l‐fucosidase by derivatives of deoxyfuconojirimycin and deoxymannojirimycin. Biochem J 1990; 265: 277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Asano Y, Kawase T, Okabe A et al IER5 generates a novel hypo‐phosphorylated active form of HSF1 and contributes to tumorigenesis. Sci Rep 2016; 6: 19174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang B, Xiao Z, Ren EC. Redefining the p53 response element. Proc Natl Acad Sci USA 2009; 106: 14373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang A, Zhu Z, Kapranov P et al Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell 2006; 24: 593–602. [DOI] [PubMed] [Google Scholar]
- 29. Yang A, Zhu Z, Kettenbach A et al Genome‐wide mapping indicates that p73 and p63 co‐occupy target sites and have similar dna‐binding profiles in vivo. PLoS One 2010; 5: e11572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sleat DE, Jadot M, Lobel P. Lysosomal proteomics and disease. Proteomics Clin Appl 2007; 1: 1134–46. [DOI] [PubMed] [Google Scholar]
- 31. Becker DJ, Lowe JB. Fucose: biosynthesis and biological function in mammals. Glycobiology 2003; 13: 41R–53R. [DOI] [PubMed] [Google Scholar]
- 32. Kobayashi Y, Tateno H, Dohra H et al A novel core fucose‐specific lectin from the mushroom Pholiota squarrosa. J Biol Chem 2012; 287: 33973–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kobayashi Y, Kawagishi H. Fungal lectins: a growing family. Methods Mol Biol 2014; 1200: 15–38. [DOI] [PubMed] [Google Scholar]
- 34. Ragnarsson G, Eiriksdottir G, Johannsdottir JT et al Loss of heterozygosity at chromosome 1p in different solid human tumours: association with survival. Br J Cancer 1999; 79: 1468–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sulzenbacher G, Bignon C, Nishimura T et al Crystal structure of Thermotoga maritima alpha‐l‐fucosidase. Insights into the catalytic mechanism and the molecular basis for fucosidosis. J Biol Chem 2004; 279: 13119–28. [DOI] [PubMed] [Google Scholar]
- 36. Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: a new database for meta‐analysis of the prognostic value of genes. BMC Med Genomics 2009; 2: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Otero‐Estevez O, Martinez‐Fernandez M, Vazquez‐Iglesias L, Paez de la Cadena M, Rodriguez‐Berrocal FJ, Martinez‐Zorzano VS. Decreased expression of alpha‐l‐fucosidase gene FUCA1 in human colorectal tumors. Int J Mol Sci 2013; 14: 16986–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Listinsky JJ, Siegal GP, Listinsky CM. The emerging importance of alpha‐l‐fucose in human breast cancer: a review. Am J Transl Res 2011; 3: 292–322. [PMC free article] [PubMed] [Google Scholar]
- 39. Ciriello G, Gatza ML, Beck AH et al Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015; 163: 506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. p53 Binds to the first intron of the FUCA1 gene.
Fig. S2. Chromatin immunoprecipitation assay carried out using the p53 responsive element of the p53 target gene IER5.
Fig. S3. Alignment of the FUCA1 p53RE with p53, p63, and p73 consensus response elements.
Fig. S4. Analysis of the effect of FUCA1 ectopic expression or FUCA1 knockdown in cancer cells.
Fig. S5. Mutations of FUCA1 found in various cancers.
Fig. S6. Alignment of Thermotoga maritima α‐l‐fucosidase and human FUCA1 protein sequences.