

Abstract
Acute lymphoblastic leukemia (ALL) occurs with high frequency in childhood and is associated with high mortality in adults. Recent technical advances in next‐generation sequencing have shed light on genetic abnormalities in hematopoietic stem/progenitor cells as the precursor to ALL pathogenesis. Based on these genetic abnormalities, ALL is now being reclassified into newly identified subtypes. Philadelphia chromosome‐like B‐lineage ALL is one of the new high‐risk subtypes characterized by genetic alterations that activate various signaling pathways, including those involving cytokine receptors, tyrosine kinases, and epigenetic modifiers. Philadelphia chromosome‐like ALL is essentially heterogeneous; however, deletion mutations in the IKZF1 gene encoding the transcription factor IKAROS underlie many cases as a key factor inducing aggressive phenotypes and poor treatment responses. Whole‐genome sequencing studies of ALL patients and ethnically matched controls also identified inherited genetic variations in lymphoid neoplasm‐related genes, which are likely to increase ALL susceptibility. These findings are directly relevant to clinical hematology, and further studies on this aspect could contribute to accurate diagnosis, effective monitoring of residual disease, and patient‐oriented therapies.
Keywords: Acute lymphoblastic leukemia, genetic abnormalities, hematopoietic stem cells, IKAROS, Ph‐like ALL
Lymphopoietic potential is an essential feature of authentic HSCs. Hematopoietic stem cells supply multiple lymphocytes, including B, T, and NK cells, which form an exquisite network in the immune system for protection from external pathogens and internal neoplastic cells. B and T lymphocytes, which play pivotal roles in acquired immunity, are supplied by HSCs throughout life and mature by a step‐wise process in the BM or thymus (Fig. 1).1, 2 The lymphopoietic potential of HSCs, however, is not durable. Although HSCs produce large quantities of lymphocytes during fetal and neonatal periods, lymphocyte production dramatically attenuates after adolescence.3, 4 Murine HSCs obtained from aged BM do not produce lymphocytes effectively in vitro or in vivo.5 In humans, adult BM HSCs are far less capable of producing lymphocytes compared to those of cord blood.6 The attenuation of lymphopoietic potential after reproductive age might be intrinsically inherent to the HSC characteristics shared between most existing species.
Figure 1.
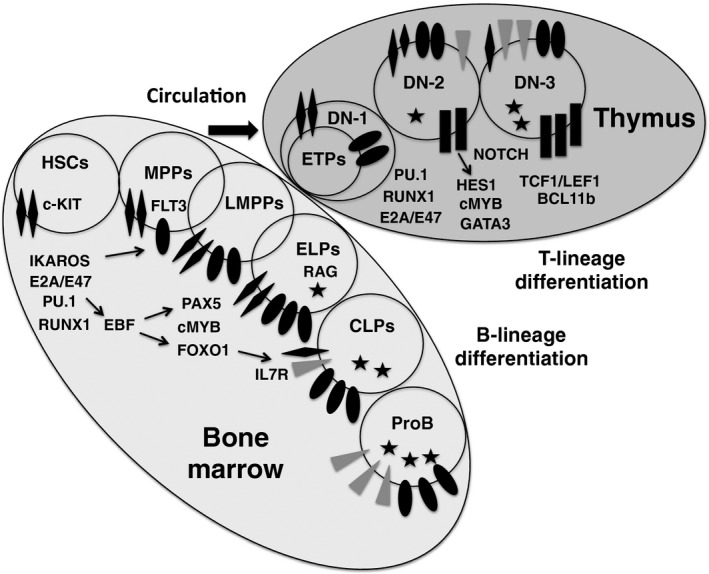
Early lymphoid development in bone marrow and thymus. This schematic shows early differentiation routes of B‐ and T‐lineage cells from hematopoietic stem cells (HSCs). In the bone marrow, the most primitive progenitors with lymphoid specification are contained within the c‐KITHigh FMS‐like tyrosine kinase‐3 (FLT3)+ fraction, termed lymphoid‐primed multipotent progenitors (LMPPs). The LMPP population overlaps multipotent progenitors (MPPs) and early lymphoid progenitors (ELPs), and is thought to be a major source for thymus‐immigrating progenitors, including early T‐lineage progenitors (ETPs). In the thymus, ETPs differentiate to double negative (DN)‐2 and DN‐3 cells with the activation of NOTCH signaling. ELPs differentiate to common lymphoid progenitor (CLP) and ProB cells in the bone marrow with the activation of interleukin‐7 receptor (IL7R) signaling. Transcription factors are indicated near the stages where they are most active. Note that these data were taken from mouse studies. Human counterparts to each progenitor category remain to be identified. EBF, early B cell factor; RAG, recombination‐activating gene.
Fetal and neonatal HSCs are equipped to establish the immune system within a short period by producing a tremendous number of lymphocytes, whereas adult and elderly HSCs maintain homeostatic states of lymphopoiesis and become myeloid‐lineage biased. The incidence of ALL reflects, in part, such age‐related changes in HSCs. Acute lymphoblastic leukemia and AML account for approximately 80% and 20% of acute leukemia cases, respectively, in childhood, whereas the ratio of ALL/AML begins to decrease after adolescence. Additionally, the biological features of ALL cells seem to differ between children and adults, and the age at diagnosis of ALL patients is negatively correlated with survival rates and response to chemotherapy.7 Although the application of pediatric chemotherapy regimens to young adults and elderly ALL patients has improved treatment outcomes, the significant difference in survival rates between childhood and adult ALL cases persists.8, 9
The earliest evidence indicating that ALL pathogenesis involved genetic abnormality in HSCs or their more primitive ancestors was likely the observation by Wiemels et al.10 that chromosomal translocations and rearrangements of the TEL‐AML1 fusion gene were already detectable in neonatal blood cells of identical twin children with ALL, even several years before clinical manifestation of the disease. In recent years, the development of next‐generation sequencing methods has significantly advanced the knowledge regarding ALL pathogenesis.11 This review describes the current understanding of ALL pathogenesis and its new classifications, particularly from the viewpoint of genetic abnormalities in HSCs and somatic stem cells. Furthermore, perspectives on how these findings can be applied to improve ALL treatment are also discussed.
Philadelphia Chromosome‐like ALL Cases, a New ALL Category, and Their Genetic Abnormalities
Gene‐expression profiling approaches divided ALL into several subcategories, in which prognoses and frequencies according to age differ significantly.12 Herein, we introduce one of the new ALL categories, Ph‐like ALL, which is related to high‐risk ALL.
Philadelphia chromosome positivity signifies the result of a translocation that gives rise to the BCR‐ABL1 oncogene and is one of the most detrimental hallmarks observed in ALL patients. Two groups independently proposed that multiple patients with Ph‐negative B‐lineage ALL had gene‐expression profiles similar to those of patients with Ph‐positive ALL.13, 14 Such Ph‐negative ALL cases were categorized as Philadelphia chromosome‐like ALL (Ph‐like ALL). Philadelphia chromosome‐like ALL comprises 10% and 13% of standard and high‐risk childhood B‐lineage ALL, respectively.15 The frequency of Ph‐like ALL increases with age, accounting for >25% of young adult cases, whose event‐free and overall survival expectation is extremely poor, similar to that of Ph‐positive cases.15
Given that the definition of Ph‐like ALL was based only on the similarity of gene‐expression profiles to Ph‐positive ALL, the genetic abnormalities associated with Ph‐like ALL cases were unlikely to be homogeneous. Therefore, transcriptome and whole‐genome sequencing was carried out to understand genetic alterations underlying Ph‐like ALL.15, 16 Among 1725 B‐lineage ALL cases, 154 patients were determined as Ph‐like ALL and underwent genomic analyses. These approaches subcategorized Ph‐like ALL into seven groups: type I, ABL‐class fusions (ABL1, ABL2, CSF1R, PDGRB); type II, erythropoietin‐receptor (EPOR) or JAK2 rearrangements; type III, cytokine receptor‐like factor 2 (CRLF2) rearrangements (often accompanied by JAK2 mutations and JAK‐STAT signal activation); type IV, other mutations activating JAK‐STAT signaling (IL7R, FLT3, SH2B3, TYK2, IL2RB); type V, uncommon miscellaneous kinase mutations (NTRK3, DGKH); type VI, RAS‐pathway mutations (KRAS, NRAS, PTPN11, NF1); and type VII, no mutations in kinase genes (Fig. 2).
Figure 2.
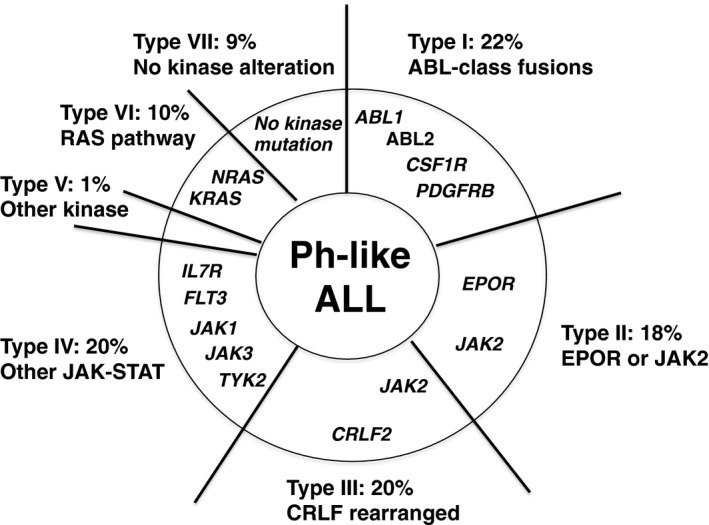
Categorization of Philadelphia chromosome‐like acute lymphoblastic leukemia (Ph‐like ALL) according to genetic abnormalities. Genomic analyses have shown that Ph‐like ALL is heterogeneous, but involves a high frequency of kinase gene alterations. The data summarized in this figure are from Roberts et al.15
The type I category with ABL‐class fusions accounted for 22% of Ph‐like ALL cases and was independent from the abnormalities associated with the JAK‐STAT signaling pathway. The type II category accounted for 18% of Ph‐like ALL, while type III cases showed the most frequently observed genetic abnormality (approximately 20%) in Ph‐like ALL cases. Interestingly, more than half of the CRLF2 rearrangements associated with type III cases also harbored missense or multiple mutations in JAK2, which activate JAK‐STAT signaling. The type IV category was related with other JAK‐STAT‐activating abnormalities, in which IL7R and/or FLT3 mutations were common. Cases involving the type V and type VII categories were infrequent and difficult to characterize, and type VI occurred in a minority of Ph‐like ALL cases, exhibiting genomic abnormalities activating RAS signaling.
Importantly, the high frequency of such kinase‐activating mutations in Ph‐like ALL suggested that the treatment outcomes of Ph‐like ALL might improve with specific inhibitors (Fig. 3). Indeed, the Mullighan group reported that the addition of imatinib induced remission in a refractory Ph‐like ALL patient who had an activating rearrangement of PDGFB.17
Figure 3.
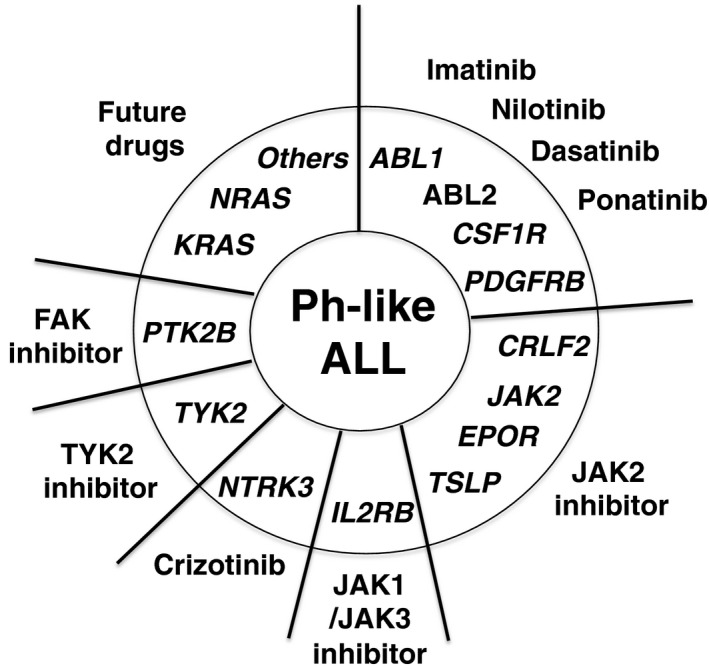
Kinase gene alterations and their inhibitors in Philadelphia chromosome‐like acute lymphoblastic leukemia (Ph‐like ALL). The tyrosine kinase inhibitors for each kinase mutation in Ph‐like ALL are indicated as a possible therapy.
IKZF1 Mutations as a Key Factor Influencing the Development of High‐Risk ALL
Deletions, amplifications, mutations, and structural rearrangements in key transcription factors promoting early lymphoid differentiation (e.g., TCF3/E2A, EBF, LEF1, IKZF1, IKZF3, PAX5, and BLNK) were detected in 40% of B‐lineage ALL cases.18 Among these, mutations in the IKZF1 gene encoding the IKAROS transcription factor were frequently observed and more highly correlated with poor prognosis associated with ALL than were mutations in genes encoding other transcription factors. Notably, many Ph‐like ALL cases, regardless of the subcategories mentioned above, revealed mutations in the IKZF1 gene, which is also a common finding in Ph‐positive ALL. IKZF1 encodes the transcription factor IKAROS, which is indispensable for the induction of B‐lineage differentiation in HSCs.19, 20 Its mutations are also strongly associated with lymphoid blast crisis of CML.21 Therefore, here, we introduce accumulating data pertaining to IKZF1 mutations associated with high‐risk ALL (Table 1).
Table 1.
Features of IKZF1‐mutated acute lymphoblastic leukemia (ALL)
| High‐risk ALL (except for ERG‐deleted cases38, 39) |
| 84% of Philadelphia chromosome‐positive ALL21 |
| 68% of Philadelphia chromosome‐like ALL15 |
| High adhesion potential to hematopoietic stem cell niche through integrins30, 32 |
| Possible response to focal adhesion kinase inhibitors and retinoid receptor agonists30, 32 |
Aberrant IKZF1 mutations are likely some of the most detrimental driver mutations, accounting for >80% of Ph‐positive ALL. IKZF1 deletions were not detectable in chronic‐phase CML, but emerged simultaneously when CML transformed to lymphoid blast crisis.21 IKZF1 alterations were also common in Ph‐like ALL cases, regardless of the type of kinase gene mutation described above,15 and suggested significantly lower 5‐year event‐free survival rates of Ph‐like ALL patients compared to those without an IKZF1 alteration. With respect to T‐lineage ALL, IKZF1 mutations were also observed more frequently in ETP‐ALL, the phenotype of which is characterized as T‐lineage marker‐negative and HSC/myeloid marker‐positive, than in other T‐lineage ALL cases.22 Homozygous germline IKZF1‐null mice lacked T, B, and NK lymphocytes and their early progenitors, and heterozygous dominant‐negative IKZF1‐mutated mice rapidly acquired T‐lineage ALL.19, 23, 24 Dominant‐negative IKZF1 mutations are more deleterious and oncogenic, likely due to cross‐interference with other IKAROS family members.
Most IKZF1 deletions identified in Ph‐positive ALL were monoallelic and lacked exons 3–6 of the IKZF1 gene, which encode the N‐terminal zinc finger DNA‐binding domain.21 This deletion results in dominant‐negative isoforms that inhibit both wild‐type IKAROS and other family members.25 Interestingly, genomic breakpoints in the IKZF1 gene are located in the vicinity of cryptic heptamer‐recombination‐signal sequences, which are recognized by the RAG enzyme complex.21 These observations suggested that IKZF1 deletions likely occurred with RAG expression, which marked the specification of hematopoietic stem/progenitor cells toward the lymphoid lineage.26, 27, 28 Recent studies have indicated that histone H3, trimethylated on lysine at position 4, in proximity to cryptic recombination‐signal sequences, might contribute to misleading the RAG complex into producing aberrant recombination and promoting oncogenes.29 Thus, epigenetic instability in Ph‐positive HSCs or their proximate progenitors could underlie the incidence of IKZF1 mutations.
Although IKZF1 alterations strongly correlate with refractory ALL, the underlying mechanism of which remains unknown, recent efforts to investigate the biological features of IKZF1‐mutated cells offered clues to overcome this intractable disease. The Georgopoulos group showed that induction of dominant‐negative IKAROS isoforms in early pre‐B cells arrested their differentiation at the proliferative large pre‐B cell stage and culminated in oligoclonal expansion with the occurrence of B‐lineage ALL in transplanted recipients.30, 31 These IKAROS‐deleted pre‐leukemic and leukemic cells expressed higher levels of integrins than those in normal counterpart cells, and were more dependent on BM stromal cells through integrin‐mediated adhesion for their growth and survival. Notably, inhibitors for focal adhesion kinase, which transduces integrin signaling into cells, significantly abrogated adhesion and induced apoptosis in IKAROS‐deleted leukemic cells.30
The Mullighan group also reported that IKZF1 alterations induced adhesive potential and HSC‐related characteristics in Ph‐positive ALL cells, while reducing their responsiveness to tyrosine kinase inhibitors.32 IKZF1‐altered ALL cells infiltrated BM and interacted with perivascular mesenchymal cells and arterial endothelial cells, which are thought to comprise the HSC niche.33 It is noteworthy that treatment with retinoid‐receptor agonists enhanced IKZF1 expression, reversed HSC‐like features of IKZF1‐altered Ph‐positive ALL cells, and recovered their sensitivity to tyrosine kinase inhibitors.32 While retinoids have long been known to affect the integrity of HSCs and the differentiation of lympho‐hematopoietic progenitors by directly or indirectly regulating a number of transcription factors,34, 35, 36 numerous nuclear receptors also potentially regulate transcription factors in HSCs.37 Thus, it is worth examining the types of nuclear‐receptor signals that upregulate IKZF1 expression.
Intragenic deletions of ERG, which encodes an ETS family member transcription factor, were recently identified in a subset of childhood B‐lineage ALL.38 Intriguingly, although dominant‐negative IKZF1 deletions were frequently associated with ERG‐deleted ALL cases, the response to treatment and survival of these patients were unexpectedly positive. In fact, within B‐lineage ALL cases involving IKZF1 deletion, 8‐year event‐free survival improved to >85% in cases involving ERG deletions, whereas the rate was only 51% without the deletions.38 An independent group also reported similar results, suggesting that ERG deletions might mitigate the negative impact of IKZF1 deletion in ALL prognoses.39 Future studies on the molecular mechanisms underlying such exceptional cases might discover more sophisticated strategies to overcome IKZF1 alterations in ALL cells.
Inherited Germline Variations as a Risk Factor for ALL Development
Historically, germline variations inherited from ancestors were rarely considered to influence ALL incidence or features. Until a decade ago, ALL pathogenesis was thought to be mostly attributed to acquired mutations in HSCs or lymphoid progenitors. However, recent studies have identified multiple genomic variants that increase ALL susceptibility and affect prognosis.
Genome‐wide association studies for childhood ALL cases identified multiple germline polymorphisms showing high association with ALL incidence and characteristics.11 Some of the inheritable variants were found in lymphoid neoplasm‐related genes, including IKZF1, ARID5B, and CDKN2.40, 41, 42, 43, 44 IKZF1 encodes the lymphoid‐lineage transcription factor IKAROS mutation, which is intimately associated with ALL as previously discussed. Notably, this protein is also an integral component combining transcription factors with the chromatin‐remodeling network.45 ARID5B is a member of the AT‐rich DNA‐interaction domain family,46 and although the ARID5B function in lympho‐hematopoiesis has not been well studied, it may be involved in epigenetic regulation of gene expression in HSCs and early lymphoid progenitors, similar to other AT‐rich DNA‐binding proteins.47, 48, 49, 50 Interestingly, ARID5B polymorphisms are associated with B‐lineage hyperdiploid ALL and racial differences associated with ALL incidence.40, 51 CDKN2 encodes INK4a/ARF, which regulates HSC self‐renewal and differentiation under the control of the transcriptional repressor and polycomb group protein Bmi1.52, 53 Therefore, hereditary predisposition to ALL may be related to the epigenetic instability of HSCs and lymphoid progenitors.
Heteroploid chromosomal abnormality is often observed in ALL cases. A hyperdiploid karyotype is more common in childhood ALL compared to adult ALL; however, the associated mechanisms remain unknown. Moriyama and colleagues studied familial ALL and identified a nonsense variant of ETV6, a member of the erythroblast transformation‐specific family, which is involved in hematopoiesis and oncogenesis, with high prevalence among familial ALL cases.54, 55, 56 They undertook a broad survey for ETV6 mutations in >4405 ALL children and identified 31 somatic ETV6 variants potentially related to ALL susceptibility. Interestingly, ALL children with ETV6 variants were significantly older than those without such variants when diagnosed, and more often had a hyperdiploid karyotype. Additionally, a hypodiploid karyotype is a hallmark of poor ALL outcomes. Holmfeldt and colleagues used genomic profiling on 124 childhood ALL cases with a hypodiploid karyotype57 and identified inherited alterations in TP53, as well as RAS‐, DNA‐repair‐, and receptor tyrosine kinase signaling‐related genes.
The higher frequency of ALL in children relative to adults suggests that the influence of genetic predisposition to the disease might be more profound at younger ages. However, a recent study showed that inherited GATA3 variants strongly enhanced susceptibility to ALL in adolescents and young adults.58 The detected GATA3 polymorphisms were also detectable in childhood Ph‐like ALL,59 and the frequency of ALL patients with those GATA3 variations was positively correlated with patient age at diagnosis. Furthermore, GATA3 variations were also associated with poor treatment response and high risk of relapse. These findings suggested that age‐related differences in ALL biology might reflect, at least in part, the genetic variations resident in HSCs.
Conclusions and Perspectives
In the last decade, advances in gene‐expression profiling and genome‐wide sequencing have revolutionized our understanding of ALL pathogenesis. As described in this short review, accumulating information has revamped ALL classifications according to genetic variations. Many pathogenic ALL mutations have been identified in HSC‐related genes, which are often associated with ALL treatment failure and early relapse. As some of the mutations are also associated with activation of certain kinase pathways, the invention of simple, convenient, and cost‐effective sequencing technologies would enable earlier and more sophisticated therapeutic intervention with specific inhibitors. On the other hand, we must stress that leukemia‐initiating mutations are still undetectable in >10% of childhood ALL patients, and that the genetic information of older adult ALL patients has not been catalogued at this stage. Nonetheless, we believe that studies in the coming decade will completely describe the genomic landscape of ALL across all generations and refine the therapeutic algorithm to be more targeted and individualized.
Disclosure Statement
The authors have no conflicts of interest relating to the topic of this article.
Abbreviations
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- BM
bone marrow
- CML
chronic myeloid leukemia
- ETP
early T‐lineage progenitor
- FLT3
FMS‐like tyrosine kinase‐3
- HSC
hematopoietic stem cell
- IL7R
interleukin‐7 receptor
- NK
natural killer
- Ph‐like
Philadelphia chromosome‐like
- RAG
recombination‐activating gene
- STAT
signal transducer and activator of transcription
Acknowledgments
Research work in the Department of Hematology and Oncology, Osaka University Graduate School of Medicine, is supported by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Cancer Sci 107 (2016) 721–725
Funding Information
Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Pelayo R, Welner R, Perry SS et al Lymphoid progenitors and primary routes to becoming cells of the immune system. Curr Opin Immunol 2005; 17: 100–7. [DOI] [PubMed] [Google Scholar]
- 2. Zhang Q, Iida R, Yokota T, Kincade PW. Early events in lymphopoiesis: an update. Curr Opin Hematol 2013; 20: 265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Montecino‐Rodriguez E, Dorshkind K. Evolving patterns of lymphopoiesis from embryogenesis through senescence. Immunity 2006; 24: 659–62. [DOI] [PubMed] [Google Scholar]
- 4. Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol 2013; 13: 376–89. [DOI] [PubMed] [Google Scholar]
- 5. Sudo K, Ema H, Morita Y, Nakauchi H. Age‐associated characteristics of murine hematopoietic stem cells. J Exp Med 2000; 192: 1273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rossi MI, Yokota T, Medina KL et al B lymphopoiesis is active throughout human life, but there are developmental age‐related changes. Blood 2003; 101: 576–84. [DOI] [PubMed] [Google Scholar]
- 7. Hunger SP, Lu X, Devidas M et al Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol 2012; 30: 1663–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schafer ES, Hunger SP. Optimal therapy for acute lymphoblastic leukemia in adolescents and young adults. Nat Rev Clin Oncol 2011; 8: 417–24. [DOI] [PubMed] [Google Scholar]
- 9. Ram R, Wolach O, Vidal L, Gafter‐Gvili A, Shpilberg O, Raanani P. Adolescents and young adults with acute lymphoblastic leukemia have a better outcome when treated with pediatric‐inspired regimens: systematic review and meta‐analysis. Am J Hematol 2012; 87: 472–8. [DOI] [PubMed] [Google Scholar]
- 10. Wiemels JL, Cazzaniga G, Daniotti M et al Prenatal origin of acute lymphoblastic leukaemia in children. Lancet 1999; 354: 1499–503. [DOI] [PubMed] [Google Scholar]
- 11. Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol 2015; 12: 344–57. [DOI] [PubMed] [Google Scholar]
- 12. Mullighan CG. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematology Am Soc Hematol Educ Program 2014; 2014: 174–80. [DOI] [PubMed] [Google Scholar]
- 13. Mullighan CG, Su X, Zhang J et al Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009; 360: 470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Den Boer ML, van Slegtenhorst M, De Menezes RX et al A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome‐wide classifi cation study. Lancet Oncol 2009; 10: 125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roberts KG, Li Y, Payne‐Turner D et al Targetable kinase‐activating lesions in Ph‐like acute lymphoblastic leukemia. N Engl J Med 2014; 371: 1005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roberts KG, Morin RD, Zhang J et al Genetic alterations activating kinase and cytokine receptor signaling in high‐risk acute lymphoblastic leukemia. Cancer Cell 2012; 22: 153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weston BW, Hayden MA, Roberts KG et al Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1‐PDGFRB‐positive acute lymphoblastic leukemia. J Clin Oncol 2013; 31: e413–6. [DOI] [PubMed] [Google Scholar]
- 18. Mullighan CG, Goorha S, Radtke I et al Genome‐wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007; 446: 758–64. [DOI] [PubMed] [Google Scholar]
- 19. Georgopoulos K, Bigby M, Wang JH et al The Ikaros gene is required for the development of all lymphoid lineages. Cell 1994; 79: 143–56. [DOI] [PubMed] [Google Scholar]
- 20. Ng SY, Yoshida T, Zhang J, Georgopoulos K. Genome‐wide lineage‐specific transcriptional networks underscore Ikaros‐dependent lymphoid priming in hematopoietic stem cells. Immunity 2009; 30: 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mullighan CG, Miller CB, Radtke I et al BCR‐ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008; 453: 110–4. [DOI] [PubMed] [Google Scholar]
- 22. Zhang J, Ding L, Holmfeldt L et al The genetic basis of early T‐cell precursor acute lymphoblastic leukaemia. Nature 2012; 481: 157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winandy S, Wu P, Georgopoulos K. A dominant mutation in the Ikaros gene leads to rapid development of leukemia and lymphoma. Cell 1995; 83: 289–99. [DOI] [PubMed] [Google Scholar]
- 24. Wang JH, Nichogiannopoulou A, Wu L et al Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity 1996; 5: 537–49. [DOI] [PubMed] [Google Scholar]
- 25. Sun L, Liu A, Georgopoulos K. Zinc finger‐mediated protein interactions modulate Ikaros activity, a molecular control of lymphocyte development. EMBO J 1996; 15: 5358–69. [PMC free article] [PubMed] [Google Scholar]
- 26. Igarashi H, Gregory SC, Yokota T, Sakaguchi N, Kincade PW. Transcription from the RAG1 locus marks the earliest lymphocyte progenitors in bone marrow. Immunity 2002; 17: 117–30. [DOI] [PubMed] [Google Scholar]
- 27. Yokota T, Kouro T, Hirose J et al Unique properties of fetal lymphoid progenitors identified according to RAG1 gene expression. Immunity 2003; 19: 365–75. [DOI] [PubMed] [Google Scholar]
- 28. Yokota T, Sudo T, Ishibashi T et al Complementary regulation of early B‐lymphoid differentiation by genetic and epigenetic mechanisms. Int J Hematol 2013; 98: 382–9. [DOI] [PubMed] [Google Scholar]
- 29. Shimazaki N, Lieber MR. Histone methylation and V(D)J recombination. Int J Hematol 2014; 100: 230–7. [DOI] [PubMed] [Google Scholar]
- 30. Joshi I, Yoshida T, Jena N et al Loss of Ikaros DNA‐binding function confers integrin‐dependent survival on pre‐B cells and progression to acute lymphoblastic leukemia. Nat Immunol 2014; 15: 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoshida T, Georgopoulos K. Ikaros fingers on lymphocyte differentiation. Int J Hematol 2014; 100: 220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Churchman ML, Low J, Qu C et al Efficacy of retinoids in IKZF1‐Mutated BCR‐ABL1 acute lymphoblastic leukemia. Cancer Cell 2015; 28: 343–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature 2014; 505: 327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Collins SJ. The role of retinoids and retinoic acid receptors in normal hematopoiesis. Leukemia 2002; 16: 1896–905. [DOI] [PubMed] [Google Scholar]
- 35. Purton LE, Dworkin S, Olsen GH et al RARgamma is critical for maintaining a balance between hematopoietic stem cell self‐renewal and differentiation. J Exp Med 2006; 203: 1283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen X, Esplin BL, Garrett KP, Welner RS, Webb CF, Kincade PW. Retinoids accelerate B lineage lymphoid differentiation. J Immunol 2008; 180: 138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chute JP, Ross JR, McDonnell DP. Minireview: nuclear receptors, hematopoiesis, and stem cells. Mol Endocrinol 2010; 24: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clappier E, Auclerc MF, Rapion J et al An intragenic ERG deletion is a marker of an oncogenic subtype of B‐cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 2014; 28: 70–7. [DOI] [PubMed] [Google Scholar]
- 39. Zaliova M, Zimmermannova O, Dorge P et al ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 2014; 28: 182–5. [DOI] [PubMed] [Google Scholar]
- 40. Trevino LR, Yang W, French D et al Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet 2009; 41: 1001–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sherborne AL, Hosking FJ, Prasad RB et al Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet 2010; 42: 492–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang JJ, Cheng C, Devidas M et al Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet 2011; 43: 237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang JJ, Cheng C, Devidas M et al Genome‐wide association study identifies germline polymorphisms associated with relapse of childhood acute lymphoblastic leukemia. Blood 2012; 120: 4197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu H, Zhang H, Yang W et al Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nat Commun 2015; 6: 7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Georgopoulos K. Haematopoietic cell‐fate decisions, chromatin regulation and ikaros. Nat Rev Immunol 2002; 2: 162–74. [DOI] [PubMed] [Google Scholar]
- 46. Wilsker D, Probst L, Wain HM, Maltais L, Tucker PW, Moran E. Nomenclature of the ARID family of DNA‐binding proteins. Genomics 2005; 86: 242–51. [DOI] [PubMed] [Google Scholar]
- 47. Webb CF, Bryant J, Popowski M et al The ARID family transcription factor bright is required for both hematopoietic stem cell and B lineage development. Mol Cell Biol 2011; 31: 1041–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Will B, Vogler TO, Bartholdy B et al Satb1 regulates the self‐renewal of hematopoietic stem cells by promoting quiescence and repressing differentiation commitment. Nat Immunol 2013; 14: 437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Satoh Y, Yokota T, Sudo T et al The Satb1 protein directs hematopoietic stem cell differentiation toward lymphoid lineages. Immunity 2013; 38: 1105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yokota T, Kanakura Y. Role of tissue‐specific AT‐rich DNA sequence‐binding proteins in lymphocyte differentiation. Int J Hematol 2014; 100: 238–45. [DOI] [PubMed] [Google Scholar]
- 51. Yang W, Trevino LR, Yang JJ et al ARID5B SNP rs10821936 is associated with risk of childhood acute lymphoblastic leukemia in blacks and contributes to racial differences in leukemia incidence. Leukemia 2010; 24: 894–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb‐group gene bmi‐1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999; 397: 164–8. [DOI] [PubMed] [Google Scholar]
- 53. Oguro H, Iwama A, Morita Y, Kamijo T, van Lohuizen M, Nakauchi H. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1‐deficient mice. J Exp Med 2006; 203: 2247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moriyama T, Metzger ML, Wu G et al Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol 2015; 16: 1659–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang LC, Swat W, Fujiwara Y et al The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes Dev 1998; 12: 2392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kar A, Gutierrez‐Hartmann A. Molecular mechanisms of ETS transcription factor‐mediated tumorigenesis. Crit Rev Biochem Mol Biol 2013; 48: 522–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Holmfeldt L, Wei L, Diaz‐Flores E et al The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 2013; 45: 242–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Perez‐Andreu V, Roberts KG, Xu H et al A genome‐wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood 2015; 125: 680–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Perez‐Andreu V, Roberts KG, Harvey RC et al Inherited GATA3 variants are associated with Ph‐like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet 2013; 45: 1494–8. [DOI] [PMC free article] [PubMed] [Google Scholar]