

Abstract
Cisplatin‐resistant A549 and H157 (A549CisR and H157CisR) non‐small cell lung cancer cells show increased stemness of cancer stem cells (CSCs) compared to their parental cells. We investigated whether interleukin‐6 (IL‐6) signaling contributes to this increased stemness in cisplatin‐resistant cells. When A549CisR and H157CisR cells were treated with neutralizing IL‐6 antibody, decreased cisplatin resistance was observed, whereas IL‐6 treatment of parental cells resulted in increased cisplatin resistance. Expression of the CSC markers was significantly upregulated in IL‐6‐expressing scramble cells (in vitro) and scramble cell‐derived tumor tissues (in vivo) after cisplatin treatment, but not in IL‐6 knocked down (IL‐6si) (in vitro) cells and in IL‐6si cell‐derived tumor tissues (in vivo), suggesting the importance of IL‐6 signaling in triggering increased stemness during cisplatin resistance development. Hypoxia inducible factors (HIFs) were upregulated by IL‐6 and responsible for the increased CSC stemness on cisplatin treatment. Mechanism dissection studies found that upregulation of HIFs by IL‐6 was through transcriptional control and inhibition of HIF degradation. Treatment of HIF inhibitor (FM19G11) abolished the upregulation of CSC markers and increased sphere formations in IL‐6 expressing cells on cisplatin treatment. In all, IL‐6‐mediated HIF upregulation is important in increasing stemness during cisplatin resistance development, and we suggest that the strategies of inhibiting IL‐6 signaling or its downstream HIF molecules can be used as future therapeutic approaches to target CSCs after cisplatin treatment for lung cancer.
Keywords: Cancer stem cell, cisplatin‐resistance, hypoxia‐inducible factor, interleukin‐6, non‐small cell lung cancer
Lung cancer is a predominant cause of cancer death in both men and women.1 Lung cancer is heterogeneous and histologically divided into two types, small‐cell lung carcinomas and NSCLCs. The latter comprises 85% of lung cancer cases2 and constitutes a heterogeneous population of squamous, adenocarcinoma, and large‐cell carcinomas.1 Despite recent progress, lung cancer therapeutic outcomes remain unsatisfactory.
Platinum‐based drugs, particularly cis‐diamminedichloroplatinum (II) (cisplatin), are used in the treatment of many cancers, including lung cancer. However, cisplatin treatment initially seems successful, but often chemoresistance develops and the therapy fails.3, 4 Cisplatin treatment induces DNA lesions, which can lead to cell cycle arrest and apoptotic death.4, 5 Lai et al.6 found that DNA repair activity was enhanced in cisplatin‐resistant cell lines. Cisplatin resistance can also be triggered by altered drug delivery system and metabolism, and tumor microenvironment changes such as hypoxia.7
Interleukin‐6 is detectable in a high percentage of lung cancer patients,8 and the circulating IL‐6 level has been suggested as a prognostic marker for survival in advanced NSCLC patients treated with chemotherapy.9 In previous studies, we have shown that the intracellular IL‐6 level is important in developing cisplatin resistance, and this effect was partially through upregulation of the molecules associated with anti‐apoptotic and DNA repair pathways.10 In this study, we investigated the IL‐6 effect on expanding the CSC population, or stemness, during development of cisplatin resistance, as it has been suggested that cisplatin‐resistant cells enable a higher CSC population11 and our laboratory and others have found that IL‐6 promotes growth of CSCs.12
Hypoxia‐inducible factor is a transcription factor that is responsible for induction of genes associated with cell survival under hypoxia.13 Overexpression of HIF has been shown in several types of cancers,14, 15 and therefore targeting HIF has been suggested to be a novel approach to treat cancer.16 In addition, the hypoxia‐induced resistance against drugs through upregulation of HIF1α in lung cancer17 and HIF1α‐induced chemoresistance in the non‐Hodgkin's lymphoma cell line18 has been reported. In this study, we investigated whether IL‐6‐mediated upregulation of HIFs is important in the increase of CSC stemness in NSCLC cells on cisplatin treatment. We also studied whether upregulation of HIFs by IL‐6 following cisplatin treatment is through direct transcriptional control or by suppressing HIF degradation, as HIF expression was reported to be regulated by the proteasome pathway in hypoxic conditions.19
Materials and Methods
Cell culture
A549 and H157 cell lines were purchased from ATCC (Manassas, VA, USA) and cultured in RPMI‐1640 containing 10% FBS. All cells were maintained in a humidified 5% CO2 environment at 37°C. For inhibition studies, FM19G11 (0.5 μM) (Sigma, St. Louis, MO, USA) that inhibits the HIF1α/HIF2α pathways was added into the culture with cisplatin treatment. The IL‐6 antibody (1:400; Thermo Fisher Scientific, Waltham, MA, USA) and the isotype matched rabbit IgG (control) (Sigma) were used in neutralization experiments and MG132 (10 μM) (Sigma) was used in ubiquitination experiments.
Development of IL‐6 knockdown and sc control cells by lentiviral transduction
For incorporation of IL‐6 siRNA or sc control plasmids into A549 and H157 cells, lentivirus construct carrying either sc or IL‐6 siRNA (pLenti‐II vector; Applied Biological Materials, Richmond, Canada) was transfected into 293T cells with a mixture of pLent‐II‐IL‐6 siRNA, psPAX2 (virus‐packaging plasmid), and pMD2G (envelope plasmid) (4:3:2 ratio) using PolyFect Transfection reagent (Qiagen, Valencia, CA, USA). After A549 and H157 cells were virally infected overnight, the culture media containing the virus was replaced with normal culture media, and maintained under normal cell culture conditions. After subculturing, the IL‐6 knockdown cells were selected by culturing cells in the presence of puromycin (2 μg/mL) (Sigma) and then maintained in media containing 0.1 μg/mL puromycin.
Development of cisplatin‐resistant cell lines
Parental A549, H157, and IL‐6 knockdown A549IL‐6si/sc, and H157IL‐6si/sc cells were continuously treated with gradually increased doses of cisplatin for 6 months according to the method described by Barr et al.11 Briefly, cells were treated with 1 μM cisplatin for 72 h and allowed to recover for the following 72 h. After repeating one more cycle at 1 μM cisplatin concentration, the cells were then treated with 2 μM cisplatin in the following two cycles. This procedure was continued with increasing cisplatin concentration up to 30 μM. During the cisplatin‐resistance induction procedure, the IC50 values of every five passage cells were accessed in comparison with those of the parental cells until the IC50 value remained constant. The IC50 values were calculated using GraphPad Prism 5.0 software (San Diego, CA, USA). The cisplatin‐resistant cell lines obtained by this method were maintained in growth media containing 10 μM cisplatin.
Cisplatin cytotoxicity test
Cisplatin cytotoxicity was analyzed by MTT (5 mg/mL, Sigma) assay. Cells (A549IL‐6si/sc and H157IL‐6si/sc) were seeded on 96‐well plates (7 × 103 cells/well) and treated with various concentrations of cisplatin for 48 h. The MTT test was then carried out and absorbance at 490 nm was measured. Cell viability was calculated using the formula: OD sample/OD blank control × 100. Triplicate experiments were carried out and average values with mean ± SEM were represented.
Sphere formation assay
Single‐cell suspensions (1 × 103 cells) were mixed with cold Matrigel (BD, Franklin Lakes, NJ, USA) (1:1 ratio, v/v, total volume of 100 μL) and the mixture was placed along the rim of the 24‐well plates. The culture plates were placed in a 37°C incubator for 10 min to let the mixture solidify and 500 μL medium was then added to the wells. In testing inhibitor effects, appropriate concentrations of individual inhibitor were added to the medium. The number of spheres with diameter greater than 50 μm was counted after 7–14 days using an Olympus light microscope (Center Valley, PA, USA). A minimum of three triplicate experiments was carried out.
In vivo xenograft studies
The A549sc and A549IL‐6si cells (1 × 106/tumor site) were s.c. injected into flanks of 8‐week‐old female nude mice (NCI) (10 mice per group, total 20 mice). Tumor development and volumes were measured twice a week. When tumor volumes reached 400 mm3, cisplatin (3 mg/kg) were i.p. injected two times per week and tumor growth was monitored. At the end of 2 weeks of treatment, mice were sacrificed and tumor tissues were processed for staining. All animal studies were performed under the supervision and guidelines of the University of Rochester Medical Center's Animal Care and Use Committee.
RNA extraction and qPCR analysis
Total RNA (1 μg) was subjected to reverse transcription using Superscript III transcriptase (Invitrogen, Carlsbad, CA, USA). The qPCR was carried out using appropriate primers and a Bio‐Rad CFX96 system (Hercules, CA, USA) with SYBR green to determine the mRNA expression levels of genes of interest. Expression levels were normalized to GAPDH level.
Western blot analysis
Cells were lysed in RIPA buffer (50 mM Tris‐Cl at pH 7.5, 150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 1 mM EDTA, 1 μg/mL leupeptin, 1 μg/mL aprotinin, 0.2 mM PMSF) and proteins (20–40 μg) were separated on 8–10% SDS/PAGE gel and then transferred onto PVDF membranes (Millipore, Billerica, MA, USA). After the blocking procedure, membranes were incubated with primary antibodies (1:1000), HRP‐conjugated secondary antibodies (1:5000), and visualized in Imager (Bio‐Rad) using the ECL system (Thermo Fisher Scientific, Rochester, NY, USA). Antibodies of HIF1α and HIF2α were from Gene Tex (Irvine, CA, USA) and the VHL antibody was purchased from Abgent (San Diego, CA, USA). Antibodies of CD44, Oct4, Notch, and Sox2 were from Cell Signaling Technology (Danvers, MA, USA) and the ALDH antibody was obtained from BD Biosciences (San Jose, CA, USA). The GAPDH antibody was purchased from Abcam (Cambridge, UK).
Plasmid HRE–luciferase assay
Cells in 24‐well plates were transfected with 2 μg/mL HRE reporter plasmid (Addgene, Cambridge, MA, USA) and 0.02 μg/mL phRL‐CMV Renilla luciferase plasmid (used as control for normalizing transfection efficiencies) using PolyFect (Qiagen). After transfection, cells were incubated with or without IL‐6. Twenty‐four hours later, luciferase activities were measured using the Dual‐Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer's instructions. Luciferase activity was measured using the GloMax 20/20 luminometer (Promega). For data analysis, the experimental reporter was normalized to the level of constitutive reporter to adjust for the differences in transfection efficiency.
Statistical analysis
The data values were presented as the mean ± SEM. Differences in mean values between two groups were analyzed by two‐tailed Student's t‐test. P ≤ 0.05 was considered statistically significant.
Results
Cisplatin‐resistant cells showed increased CSC stemness versus parental cells
We developed two cisplatin‐resistant NSCLC cell lines, A549CisR and H157CisR, by treating parental A549 and H157 cells with an increasing dose of cisplatin over 6 months.10 These cells showed four to five times higher IC50 values than parental cells (Fig. 1a). We compared self‐renewal capacity of CSCs and expression of the CSC markers in parental and cisplatin‐resistant cells. In sphere formation assays monitoring the self‐renewal of CSCs,20, 21 we detected significantly larger numbers of CSC‐derived spheres in A549CisR and H157CisR cells than in parental cells (Fig. 1b) and detected significantly higher mRNA expression of the CSC markers CD133,22, 23 ALDH,24 Nanog,22, 24 Oct4,25 Sox2,22 in A549CisR and H157CisR cells than in parental cells (Fig. 1c). These data suggest that cisplatin‐resistant cells showed increased CSC stemness versus parental cells.
Figure 1.
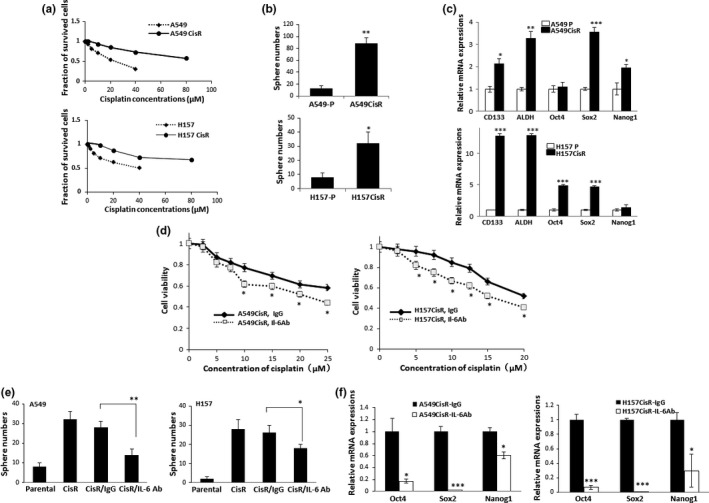
Cancer stem cell (CSC) stemness was enriched in cisplatin‐resistant non‐small‐cell lung carcinoma cells compared to parental cells, and interleukin‐6 (IL‐6) Ab treatment reduced CSC numbers and CSC marker expression in cisplatin‐resistant (CisR) lung cancer cells. (a) Cytotoxicity test of A549 and H157 cells against cisplatin treatment showing development of CisR cells. CisR cells were obtained by continuous treatment of cells with increasing dose of cisplatin for 6 months. Cisplatin cytotoxicity tests (MTT assay) were carried out using parental and CisR cells. (b) Sphere formation assay. Parental (‐P) and CisR cells (5 × 103) were seeded in a mixture of medium and Matrigel (1:1, v/v). Ten days later, spheres larger than 50 μm in diameter were counted. (c) Quantitative real‐time PCR analysis of CSC markers. Total RNAs were extracted from A549/A549CisR and H157/H157CisR cells, cDNA converted, and mRNA expressions of indicated CSC markers were analyzed. (d) Cisplatin cytotoxicity tests in the presence of either IL‐6 Ab or IgG. A549CisR and H157CisR cells were treated with cisplatin in the presence of either the IL‐6 neutralizing antibody or the isotype matched control IgG (for 48 h) and cell survival was analyzed in MTT assays. (e) Sphere formation assays of A549CisR and H157CisR cells in the presence of either IL‐6 Ab or IgG. CisR cells were treated with either IL‐6 neutralizing Ab or control IgG for 48 h. At the end of incubation, sphere formation assays were carried out. (f) Quantitative real‐time PCR analyses of CSC markers in A549CisR and H157CisR cells following IL‐6 Ab/IgG treatment. Cells were treated with either IL‐6 Ab or control IgG and mRNA expressions of CSC markers were analyzed. *P < 0.05; **P < 0.01; ***P < 0.001.
Interleukin‐6 signaling is important in increasing CSC stemness in cisplatin‐resistant cells
To investigate whether IL‐6 signaling is responsible for the increased stemness in cisplatin‐resistant cells, we carried out cisplatin cytotoxicity tests using A549CisR and H157CisR cells in the presence of either IL‐6 Ab or the isotype matched IgG control. As shown in Figure 1c, we observed decreased cell survival against cisplatin treatment when IL‐6 Ab was added to the culture. We also observed significant reduction in CSC‐derived sphere numbers (Fig. 1e) and decreased CSC marker expression (Fig. 1f) after IL‐6 Ab addition.
When we treated parental cells with human recombinant IL‐6, we observed increased cell survival following cisplatin treatment (Fig. 2a). We also found IL‐6 level increases in parental cells when treated with cisplatin (Fig. 2b). These results indicate that IL‐6 signaling is important in increasing stemness of CSCs and in developing cisplatin resistance. To further investigate whether IL‐6 contributes to the increase of CSC stemness during the development of cisplatin resistance, we developed IL‐6‐suppressing cells from A549 and H157 cell lines. Figure 2c (left panels) shows the successful IL‐6 knockdown in A549IL‐6si and H157IL‐6si cell lines. When we investigated the profiles of CSC markers following cisplatin treatment in IL‐6si/sc A549 and H157 cells, there was a significant increase in CSC marker expression in A549sc and H157sc cells, but not in A549IL‐6si and H157IL‐6si cells (Fig. 2c, right panels), confirming that IL‐6 signaling is important in the development of cisplatin resistance in CSCs.
Figure 2.
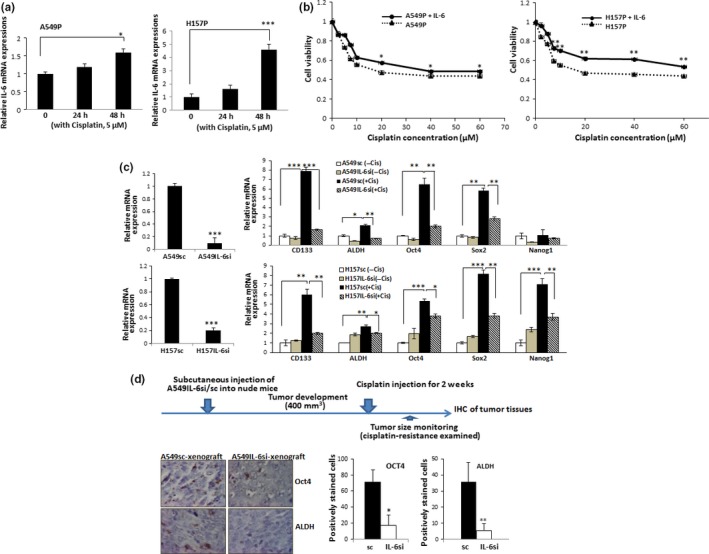
Interleukin‐6 (IL‐6) is important in triggering cancer stem cell (CSC) enrichment during development of cisplatin resistance. (a) A549 and H157 parental cells were treated with indicated amounts of cisplatin and IL‐6 mRNA levels were analyzed by quantitative real‐time PCR. (b) IL‐6 addition rendered parental cells more resistant to cisplatin. Cisplatin cytotoxicity test was carried out with parental A549 and H157 cells in the absence or presence of IL‐6 (10 ng/mL). (c) Quantitative real‐time PCR analysis of CSC markers in IL‐6 knockdown and control cells following cisplatin (Cis) treatment. Left panels, IL‐6 mRNA levels in A549IL‐6si/sc and H157IL‐6si/sc cells. mRNA expression of CSC markers in A549IL‐6si/sc and H157IL‐6si/sc cells, with or without cisplatin treatment (5 μM, 72 h) were analyzed. (d) Immunohistochemical (IHC) staining of tumor tissues obtained from cisplatin‐treated A549IL‐6si/sc cell‐derived xenografts. Tumor tissues were obtained and staining was carried out using Oct4 and A549 aldehyde dehydrogenase (ALDH) Ab. Quantitation is shown on right. *P < 0.05; **P < 0.01; ***P < 0.001.
The in vitro IL‐6 effect was confirmed in mice studies. An s.c. xenograft mouse model was established by injecting the A549IL‐6si/sc cell set and tumor development was monitored twice a week. When tumors developed to 400 mm3, mice were treated with cisplatin for 2 weeks (i.p. injection) and then sacrificed. We observed higher tumor regression (in tumor volume) in mice injected with A549IL‐6si cells compared to the mice injected with A549sc cells (Table 1). The IHC staining results of tumor tissues showed that expression of CSC markers was much lower in tumor tissues of IL‐6si cell‐derived xenografts than in sc cell‐derived xenografts (Fig. 2d). These results confirm our in vitro results (Fig. 2c) showing the IL‐6 is important in enriching CSCs after cisplatin treatment.
Table 1.
Volume of non‐small cell lung carcinoma tumor regression in mice treated with cisplatin for 2 weeks
| Time following cisplatin treatment, weeks | |||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| A549IL‐6si | 9.2% | 20.2% | 28.0% | 32.2% | 48.0% |
| A549sc | 7.0% | 15.8% | 13.9% | 20.3% | 19.0% |
A549IL‐6si, interleukin‐6 siRNA cell‐derived tumor tissue; A549sc, scramble (control) cell‐derived tumor tissue.
Interleukin‐6‐mediated upregulation of HIFs is important in the increase of CSC stemness after cisplatin treatment
While we were searching for the IL‐6 downstream molecules that are responsible for triggering the increase of CSC stemness following cisplatin treatment, we found significant upregulation of HIF molecules after cisplatin treatment in IL‐6‐expressing cells. We found higher expression of HIF1α and HIF2α in A549CisR and H157CisR cells than counterpart parental cells (Fig. 3a), and upregulation of HIFs was suppressed when we added IL‐6 Ab to cisplatin‐resistant cell culture (Fig. 3b), confirming the IL‐6 regulation of HIFs in cisplatin‐resistant cells. We also detected upregulation of these molecules in IL‐6‐expressing A549sc/H157sc cells following cisplatin treatment, but not in IL‐6 knocked down A549IL‐6si/H157IL‐6si cells (protein level, Fig. 3c; mRNA level, Fig. 3d). This result was confirmed in IHC staining data of tumor tissues obtained in mice studies. We detected higher numbers of positive‐stained cells with HIFs antibodies in tumor tissues of A549sc cell‐derived xenografts than tissues of A549IL‐6si cell‐derived xenografts (Fig. 3e).
Figure 3.
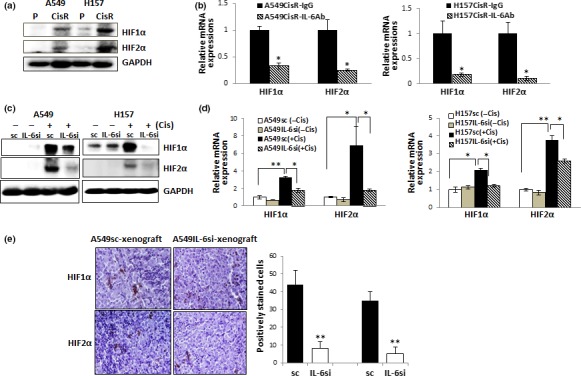
Interleukin‐6 (IL‐6) regulation of hypoxia‐inducible factor (HIF) expression in non‐small‐cell lung carcinoma cells following cisplatin treatment. (a) Western blot analysis of HIF1α and HIF2α in parental (P) and cisplatin‐resistant (CisR) cells. (b) Quantitative real‐time PCR analysis of HIF1α and HIF2α in A549CisR and H157CisR cells after IL‐6 Ab/IgG treatment. Cells were treated with either IL‐6 Ab or control IgG (for 48 h) and HIF mRNA expressions were analyzed. (c) HIF1α and HIF2α protein levels in A549IL‐6si/sc and H157IL‐6si/sc cell sets, with or without cisplatin treatment. Cells were either non‐treated or treated with cisplatin (5 μM, 72 h), cell extracts were obtained, and HIF expressions were analyzed in Western blot analyses. (d) HIF1α and HIF2α mRNA levels in A549IL‐6si/sc and H157IL‐6si/sc cell sets, with or without cisplatin treatment. Cells were treated similarly as in (c), total RNAs extracted, cDNA converted, and mRNA expressions of HIF1α and HIF2α in A549IL‐6si/sc and H157IL‐6si/sc cells, with or without cisplatin treatment were analyzed. (e) Immunohistochemical staining of tumor tissues obtained from cisplatin‐treated A549IL‐6si/sc cell‐derived xenografts. Tumor tissues were obtained and staining was carried out using HIF1α and HIF2α Ab. Quantitation is shown on right. *P < 0.05; **P < 0.01.
Molecular mechanisms of IL‐6‐mediated HIF upregulation following cisplatin treatment
To determine the molecular mechanism of IL‐6 regulation of HIFs, we first investigated whether IL‐6 can regulate HIF expression at the transcriptional level. We measured HRE‐containing luciferase activities in HEK293 cells and H1299 NSCLC cells that do not express IL‐6. We observed an increase in HRE‐luc activities with IL‐6 addition in both cell lines (Fig. 4a), suggesting the IL‐6 regulation of HIFs at the transcriptional level. We then investigated HRE‐luc activities in A549IL‐6si/sc and H157IL‐6si/sc cell sets after cisplatin treatment. Interleukin‐6‐expressing sc cells showed higher HRE‐luc activities than IL‐6si cells (Fig. 4b), implying IL‐6‐mediated upregulation of HIFs at the transcriptional level may contribute to cisplatin resistance.
Figure 4.
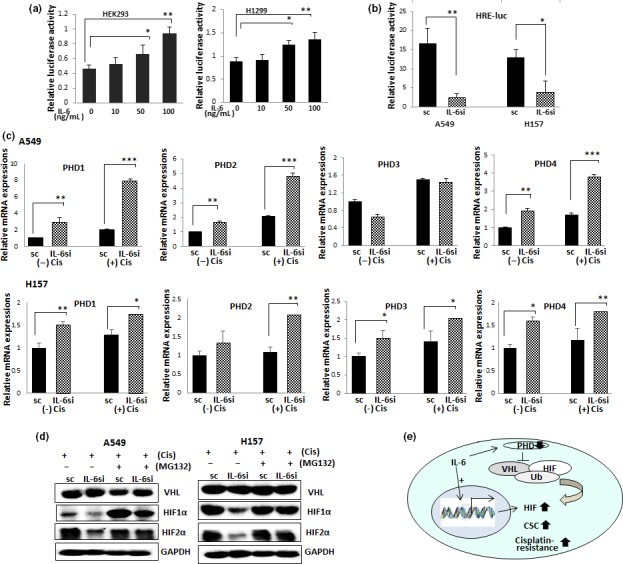
Mechanism dissection studies on interleukin‐6 (IL‐6) regulation of hypoxia‐inducible factor (HIF) following cisplatin treatment. (a) Hypoxia response element–luciferase (HRE‐luc) assay. HEK293 cells were transfected with HRE‐luc‐containing plasmids and incubated with various amounts of IL‐6. After 24 h of incubation, luciferase activities were measured. (b) HRE‐luc assay. A549IL‐6si/sc and H157IL‐6si/sc pairs were transfected with equal amounts of HRE‐luc‐containing plasmids. After 24 h of transfection, luciferase activities were measured. (c) Prolyl hydroxylase (PHD) mRNA levels in A549IL‐6si/sc and H157IL‐6si/sc cell sets, with or without cisplatin treatment. Cells were either non‐treated or treated with cisplatin (5 μM, 72 h), total RNAs extracted, cDNA converted, and mRNA expressions were analyzed. (d) Western blot analysis of von Hippel‐Lindau disease tumor suppressor (VHL), HIF1α, and HIF2α levels in A549IL‐6si/sc and H157IL‐6si/sc cell sets, with or without cisplatin treatment, in the presence or absence of MG132. Cells were either non‐treated or treated with cisplatin (5 μM, 72 h) in the absence or presence of MG132 (10 μM), cell extracts were obtained, and HIF1α and HIF2α levels were analyzed in Western blot. (e) Summary of the results of mechanism studies. *P < 0.05; **P < 0.01; ***P < 0.001. CSC, cancer stem cell.
We next tested whether the IL‐6‐mediated upregulation of HIFs on cisplatin treatment is also due to inhibition of HIF degradation. We tested whether the levels of PHDs that are known to be associated with VHL‐mediated HIF ubiquitination are different in IL‐6si/sc cells after cisplatin treatment. When we investigated mRNA levels of PHDs in A549IL‐6si/sc and H157IL‐6si/sc cell sets, we found the PHD levels were significantly higher in IL‐6si cells than in sc cells, and that the difference was more significant in the presence of cisplatin (Fig. 4c). The upregulation of HIFs after cisplatin treatment observed in sc cells was abolished when cells were treated with the proteasomal degradation inhibitor MG132 (Fig. 4d). However, we found that the VHL level was not affected by IL‐6. These results suggest that IL‐6 triggers the suppression of ubiquitination by mediating PHD level decrease after cisplatin treatment. Figure 4e summarizes these results.
Targeting HIFs abolished the IL‐6 effect of increasing CSCs after cisplatin treatment
To investigate whether targeting HIFs can block the CSC increase in cisplatin‐resistant cells, A549CisR and H157CisR cells were incubated with HIF inhibitor FM19G11 (vehicle as control), and CSC marker expression and CSC‐derived sphere forming ability were analyzed. The inhibitory effects of HIFs were shown in qPCR analyses (Fig. 5a, left panels). When the HIF inhibitor was added to the A549sc cells, we found the increase of CSC marker following cisplatin treatment was abolished (Fig. 5a, right panel) along with a significant reduction in sphere numbers (Fig. 5b). These results provide a potential strategy for targeting HIFs instead of blocking IL‐6 to reduce cisplatin‐induced increase in CSCs.
Figure 5.
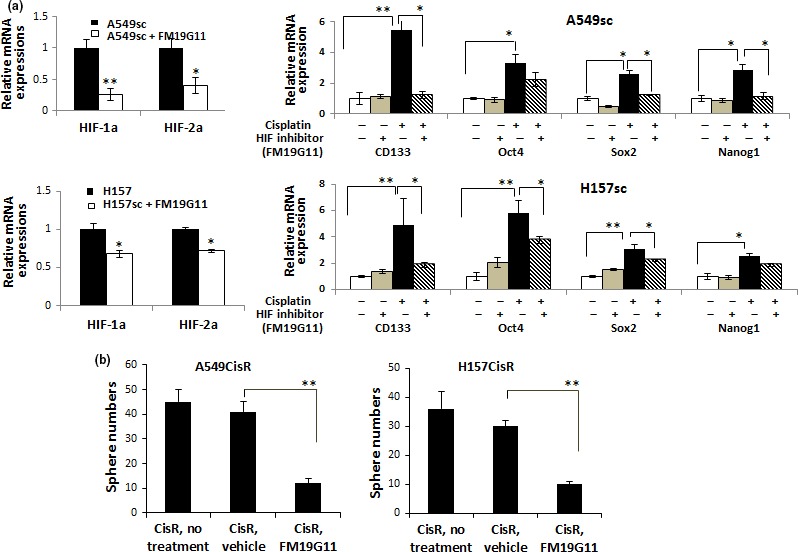
Hypoxia‐inducible factor (HIF) inhibitor treatment inhibited the upregulation of cancer stem cell (CSC) markers following cisplatin treatment, reduced sphere formation in cisplatin‐resistant (CisR) cells, and lowered cisplatin cytotoxicity of CD133+ cells. (a) Effect of HIF inhibitor FM19G11 on expression of CSC markers in A549sc and H157sc cells following cisplatin treatment. A549sc and H157sc cells were treated with cisplatin (5 μM, 72 h) in the presence of either vehicle or FM19G11 (0.5 μM) and mRNA expressions of indicated CSC markers were analyzed. Left panels show the inhibitory effect of FM19G11 on inhibition of HIF1α and HIF2α. (b) Effect of HIF inhibitor FM19G11 on sphere numbers in A549CisR and H157CisR cells. The Matrigel‐based sphere formation assay was carried out with or without HIF inhibitor FM19G11 (0.5 μM). *P < 0.05; **P < 0.01.
Discussion
In this study, we showed that CSC stemness was enriched in A549CisR and H157CisR cells versus parental cells. This finding is consistent with the previous report by Barr et al.11 detailing that cisplatin‐resistant cells show stem cell‐like features. High numbers of CSCs should render high chemoresistance in tumor cells, as the recent in vitro and in vivo studies on several types of cancer indicate that CSCs have higher chemoresistance than non‐CSCs.26, 27, 28 However, we did not observe the CD133+ CSC‐like cell population increase in A549CisR and H157CisR cells compared to their parental cells in flow cytometric analyses (data not shown). We speculate that the IL‐6 effect on the increase of CSC‐like features in cisplatin‐resistant cells may not be due to a direct effect of increasing CSC numbers, but due to an increase in CSC growth and enrichment of stemness of existing CSCs, as suggested previously.29
In this study, we focused on the IL‐6 effect on increasing CSC stemness in cisplatin‐resistant cells. There may be several ways of triggering the IL‐6 effect on influencing cisplatin resistance. Our laboratory has previously shown that IL‐6 increases cisplatin resistance by upregulating molecules related to anti‐apoptosis and DNA repair.10 We speculate that the IL‐6 effect in increasing CSC stemness is another way of mediating cisplatin resistance in NSCLC. Interestingly, we detected IL‐6 level increases in parental cells after cisplatin treatment (Fig. 2d), but the IL‐6 levels in cisplatin‐resistant cells were lower than their parental cells (data not shown). This result suggests that the IL‐6 level increase is critical in the developmental process of cisplatin resistance, but not so critical after cisplatin resistance has been established.
We identified HIFs as the key IL‐6 downstream molecules responsible for the CSC stemness increase during the development of cisplatin resistance. The concept of hypoxia contributions in developing cisplatin resistance and CSC increase in chemoresistance are not new. Fischer et al.30 suggested the involvement of hypoxia in cisplatin resistance and Bar et al.31 showed the correlation between hypoxia and CSC increase. Recently, Samanta et al.32 reported that HIFs are required for chemotherapy resistance of CSCs in breast cancer. Although the concept of HIF involvement in cisplatin resistance and CSC increase is not totally novel, our discovery of the connection of IL‐6–HIF signaling in CSC increase during the cisplatin resistance process in lung cancer cells is a novel finding and clinically significant.
We showed the role of IL‐6 in upregulating HIFs after cisplatin treatment by two mechanisms: promoting HIF transcription, and blocking their degradation. The HIF regulation of IL‐6 gene expression in chondrocytes has been reported,33 but the regulation of IL‐6 on HIF expression at the transcriptional level has not been reported. Further investigation is necessary to conclude whether the IL‐6 effect on promoting HIF transcription (Fig. 4a,b) is a direct regulation or through modulation by the transcription factor regulated by IL‐6, such as Stat3. Xu et al.34 recently showed that targeting the Stat3 pathway, which is known to be downstream of IL‐6, blocked HIF1 expression in breast cancer cells.
We also showed IL‐6 regulation of HIFs in the ubiquitination pathway. We found that PHD levels were significantly upregulated in IL‐6 knockdown cells compared to sc cells on cisplatin treatment. The importance of PHD levels in HIF1α35, 36, 37 and HIF2α19 ubiquitination have also been suggested. Nevertheless, we did not observe significant difference in VHL levels in IL‐6si/sc cell sets following cisplatin treatment, and this observation is consistent with a recent report suggesting the VHL‐independent proteasomal degradation of HIF in colon cancer cells.38 We speculate that the suppressed PHDs in IL‐6‐expressing sc cells, in turn, suppressed HIF ubiquitination following cisplatin treatment, and consequently the HIF molecules may be protected from degradation. The IL‐6 regulation of PHDs in lung cancer cells after cisplatin treatment is our novel discovery. Whether IL‐6 also can trigger cisplatin resistance by regulating E3 ligase needs to be investigated in the future.
We observed that using the IL‐6 Ab or the HIF inhibitor both significantly reduced the CSC stemness in cisplatin‐resistant cells. In lung cancer therapeutics, the use of IL‐6 Ab has had some success in outcome and has already been reported.39 However, given the complexity of the physiological IL‐6 roles of both pro‐ and anti‐inflammatory,40 the therapeutic approach using IL‐6 Ab might result in complicated side effects. In this study, we showed that the use of HIF inhibitor reduced the cisplatin‐induced CSC increase to the level shown in IL‐6 knockdown cells, providing the potential to replace IL‐6 Ab therapy.
Several HIF1α inhibitors have recently been used in clinical trials to treat solid tumors. The pilot trial of EZN‐2968, an antisense oligonucleotide inhibitor of HIF1α, has been reported41 and the application of the mammalian target of rapamycin/HIF1α inhibitor temsirolimus combined with liposomal doxorubicin in a phase I trial has also been reported.42 An approach using PX‐478 as a HIF1α inhibitor has been attempted43 and the pilot trial of oral topotecan as a HIF1α inhibitor has been carried out to treat advanced solid tumors.44 However, no attempts have been made yet to block CSC increase during cisplatin treatment or to sensitize cisplatin‐resistant lung tumors to cisplatin. We suggest that using HIF inhibitor therapy can alternatively be used in targeting the IL‐6 signaling‐mediated CSC increase in cisplatin‐resistant tumors to overcome the IL‐6 therapy problem, however; further in vivo studies are necessary to prove the effectiveness of this strategy.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- ALDH
aldehyde dehydrogenase
- CisR
cisplatin‐resistance
- CSC
cancer stem cell
- HIF
hypoxia‐inducible factor
- HRE
hypoxia response element
- HRE‐luc
HRE–luciferase
- IHC
immunohistochemical
- IL‐6
interleukin‐6
- IL‐6si
IL‐6 knockdown
- NSCLC
non‐small cell lung cancer
- OD
optical density
- PHD
prolyl hydroxylase
- qPCR
quantitative real‐time PCR
- sc
scramble
- Stat3
signal transducer and activator of transcription 3
- VHL
von Hippel‐Lindau disease tumor suppressor
Acknowledgments
We thank Laura Finger for assistance with manuscript preparation. Grant support was provided by Meaghan's Hope.
Cancer Sci 107 (2016) 746–754
Funding Information
Meaghan's Hope.
References
- 1. Cersosimo RJ. Lung cancer: a review. Am J Health Syst Pharm 2002; 59: 611–42. [DOI] [PubMed] [Google Scholar]
- 2. Parsons A, Daley A, Begh R, Aveyard P. Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: systematic review of observational studies with meta‐analysis. BMJ 2010; 340: b5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer 2011; 71: 3–10. [DOI] [PubMed] [Google Scholar]
- 4. Galluzzi L, Vitale I, Michels J et al Systems biology of cisplatin resistance: past, present and future. Cell Death Dis 2014; 5: e1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Galluzzi L, Senovilla L, Vitale I et al Molecular mechanisms of cisplatin resistance. Oncogene 2012; 31: 1869–83. [DOI] [PubMed] [Google Scholar]
- 6. Lai SL, Hwang J, Perng RP, Whang‐Peng J. Modulation of cisplatin resistance in acquired‐resistant nonsmall cell lung cancer cells. Oncol Res 1995; 7: 31–8. [PubMed] [Google Scholar]
- 7. Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev 2002; 16: 2743–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yanagawa H, Sone S, Takahashi Y et al Serum levels of interleukin 6 in patients with lung cancer. Br J Cancer 1995; 71: 1095–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang CH, Hsiao CF, Yeh YM et al Circulating interleukin‐6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int J Cancer 2013; 132: 1977–85. [DOI] [PubMed] [Google Scholar]
- 10. Duan S, Tsai Y, Keng P, Chen Y, Lee SO, Chen Y. IL‐6 signaling contributes to cisplatin resistance in non‐small cell lung cancer via the up‐regulation of anti‐apoptotic and DNA repair associated molecules. Oncotarget 2015; 6: 27651–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barr MP, Gray SG, Hoffmann AC et al Generation and characterisation of cisplatin‐resistant non‐small cell lung cancer cell lines displaying a stem‐like signature. PLoS ONE 2013; 8: e54193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu CC, Lin JH, Hsu TW et al IL‐6 enriched lung cancer stem‐like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int J Cancer 2014; 136: 547–559. [DOI] [PubMed] [Google Scholar]
- 13. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia‐inducible factor 1 is a basic‐helix‐loop‐helix‐PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92: 5510–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gruber G, Greiner RH, Hlushchuk R et al Hypoxia‐inducible factor 1 alpha in high‐risk breast cancer: an independent prognostic parameter? Breast Cancer Res 2004; 6: R191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ranasinghe WK, Baldwin GS, Bolton D, Shulkes A, Ischia J, Patel O. HIF1alpha expression under normoxia in prostate cancer‐ which pathways to target? J Urol 2015; 193: 763–70. [DOI] [PubMed] [Google Scholar]
- 16. Ke Q, Costa M. Hypoxia‐inducible factor‐1 (HIF‐1). Mol Pharmacol 2006; 70: 1469–80. [DOI] [PubMed] [Google Scholar]
- 17. Song X, Liu X, Chi W et al Hypoxia‐induced resistance to cisplatin and doxorubicin in non‐small cell lung cancer is inhibited by silencing of HIF‐1alpha gene. Cancer Chemother Pharmacol 2006; 58: 776–84. [DOI] [PubMed] [Google Scholar]
- 18. Hernandez‐Luna MA, Rocha‐Zavaleta L, Vega MI, Huerta‐Yepez S. Hypoxia inducible factor‐1alpha induces chemoresistance phenotype in non‐Hodgkin lymphoma cell line via up‐regulation of Bcl‐xL. Leuk Lymphoma 2013; 54: 1048–55. [DOI] [PubMed] [Google Scholar]
- 19. Luo J, Lee SO, Cui Y, Yang R, Li L, Chang C. Infiltrating bone marrow mesenchymal stem cells (BM‐MSCs) increase prostate cancer cell invasion via altering the CCL5/HIF2alpha/androgen receptor signals. Oncotarget 2015; 6: 27555–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang YJ, Bailey JM, Rovira M, Leach SD. Sphere‐forming assays for assessment of benign and malignant pancreatic stem cells. Methods Mol Biol 2013; 980: 281–90. [DOI] [PubMed] [Google Scholar]
- 21. Cao L, Zhou Y, Zhai B et al Sphere‐forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol 2011; 11: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bertolini G, Roz L, Perego P et al Highly tumorigenic lung cancer CD133+ cells display stem‐like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA 2009; 106: 16281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang S, Xu ZY, Wang LF, Su W. CD133+ cancer stem cells in lung cancer. Front Biosci 2013; 18: 447–53. [DOI] [PubMed] [Google Scholar]
- 24. Sullivan JP, Spinola M, Dodge M et al Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res 2010; 70: 9937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen YC, Hsu HS, Chen YW et al Oct‐4 expression maintained cancer stem‐like properties in lung cancer‐derived CD133‐positive cells. PLoS ONE 2008; 3: e2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang XL, Jia Q, Lv L, Deng T, Gao J. Tumorspheres derived from HCC cells are enriched with cancer stem cell‐like cells and present high chemoresistance dependent on the Akt pathway. Anti‐Cancer Agents Med Chem 2015; 15: 755–63. [DOI] [PubMed] [Google Scholar]
- 27. Dobbin ZC, Katre AA, Steg AD et al Using heterogeneity of the patient‐derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 2014; 5: 8750–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu G, Yuan X, Zeng Z et al Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 2006; 5: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee SO, Yang X, Duan S et al IL‐6 promotes growth and epithelial‐mesenchymal transition of CD133+ cells of non‐small cell lung cancer. Oncotarget 2016; 7: 6626–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fischer C, Leithner K, Wohlkoenig C et al Panobinostat reduces hypoxia‐induced cisplatin resistance of non‐small cell lung carcinoma cells via HIF‐1alpha destabilization. Mol Cancer 2015; 14: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG. Hypoxia increases the expression of stem‐cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol 2010; 177: 1491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia‐inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA 2014; 111: E5429–38. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33. Ryu JH, Yang S, Shin Y, Rhee J, Chun CH, Chun JS. Interleukin‐6 plays an essential role in hypoxia‐inducible factor 2alpha‐induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum 2011; 63: 2732–43. [DOI] [PubMed] [Google Scholar]
- 34. Xu Q, Briggs J, Park S et al Targeting Stat3 blocks both HIF‐1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005; 24: 5552–60. [DOI] [PubMed] [Google Scholar]
- 35. Tan JT, Prosser HC, Vanags LZ, Monger SA, Ng MK, Bursill CA. High‐density lipoproteins augment hypoxia‐induced angiogenesis via regulation of post‐translational modulation of hypoxia‐inducible factor 1alpha. Faseb J 2014; 28: 206–17. [DOI] [PubMed] [Google Scholar]
- 36. Myllyharju J, Koivunen P. Hypoxia‐inducible factor prolyl 4‐hydroxylases: common and specific roles. Biol Chem 2013; 394: 435–48. [DOI] [PubMed] [Google Scholar]
- 37. Chintala S, Najrana T, Toth K et al Prolyl hydroxylase 2 dependent and Von‐Hippel‐Lindau independent degradation of Hypoxia‐inducible factor 1 and 2 alpha by selenium in clear cell renal cell carcinoma leads to tumor growth inhibition. BMC Cancer 2012; 12: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gariboldi MB, Taiana E, Bonzi MC et al The BH3‐mimetic obatoclax reduces HIF‐1alpha levels and HIF‐1 transcriptional activity and sensitizes hypoxic colon adenocarcinoma cells to 5‐fluorouracil. Cancer Lett 2015; 364: 156–64. [DOI] [PubMed] [Google Scholar]
- 39. Trikha M, Corringham R, Klein B, Rossi JF. Targeted anti‐interleukin‐6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res 2003; 9: 4653–65. [PMC free article] [PubMed] [Google Scholar]
- 40. Scheller J, Chalaris A, Schmidt‐Arras D, Rose‐John S. The pro‐ and anti‐inflammatory properties of the cytokine interleukin‐6. Biochim Biophys Acta 2011; 1813: 878–88. [DOI] [PubMed] [Google Scholar]
- 41. Jeong W, Rapisarda A, Park SR et al Pilot trial of EZN‐2968, an antisense oligonucleotide inhibitor of hypoxia‐inducible factor‐1 alpha (HIF‐1alpha), in patients with refractory solid tumors. Cancer Chemother Pharmacol 2014; 73: 343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moroney J, Fu S, Moulder S et al Phase I study of the antiangiogenic antibody bevacizumab and the mTOR/hypoxia‐inducible factor inhibitor temsirolimus combined with liposomal doxorubicin: tolerance and biological activity. Clin Cancer Res 2012; 18: 5796–805. [DOI] [PubMed] [Google Scholar]
- 43. Lee K, Kim HM. A novel approach to cancer therapy using PX‐478 as a HIF‐1alpha inhibitor. Arch Pharmacal Res 2011; 34: 1583–5. [DOI] [PubMed] [Google Scholar]
- 44. Kummar S, Raffeld M, Juwara L et al Multihistology, target‐driven pilot trial of oral topotecan as an inhibitor of hypoxia‐inducible factor‐1alpha in advanced solid tumors. Clin Cancer Res 2011; 17: 5123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]