

Abstract
Chronic myeloid leukemia (CML) therapy has markedly improved patient prognosis after introduction of imatinib mesylate for clinical use. However, a subset of patients develops resistance to imatinib and other tyrosine kinase inhibitors (TKIs), mainly due to point mutations in the region encoding the kinase domain of the fused BCR-ABL oncogene. To identify potential therapeutic targets in imatinib-resistant CML cells, we derived imatinib-resistant CML-T1 human cell line clone (CML-T1/IR) by prolonged exposure to imatinib in growth media. Mutational analysis revealed that the Y235H mutation in BCR-ABL is probably the main cause of CML-T1/IR resistance to imatinib. To identify alternative therapeutic targets for selective elimination of imatinib-resistant cells, we compared the proteome profiles of CML-T1 and CML-T1/IR cells using 2-DE-MS. We identified eight differentially expressed proteins, with strongly upregulated Na+/H+ exchanger regulatory factor 1 (NHERF1) in the resistant cells, suggesting that this protein may influence cytosolic pH, Ca2+ concentration or signaling pathways such as Wnt in CML-T1/IR cells. We tested several compounds including drugs in clinical use that interfere with the aforementioned processes and tested their relative toxicity to CML-T1 and CML-T1/IR cells. Calcium channel blockers, calcium signaling antagonists and modulators of calcium homeostasis, namely thapsigargin, ionomycin, verapamil, carboxyamidotriazole and immunosuppressive drugs cyclosporine A and tacrolimus (FK-506) were selectively toxic to CML-T1/IR cells. The putative cellular targets of these compounds in CML-T1/IR cells are postulated in this study. We propose that Ca2+ homeostasis can be a potential therapeutic target in CML cells resistant to TKIs. We demonstrate that a proteomic approach may be used to characterize a TKI-resistant population of CML cells enabling future individualized treatment options for patients.
Keywords: chronic myeloid leukemia, imatinib mesylate, drug resistance, proteomics, calcium homeostasis, endoplasmic reticulum, Wnt signaling, therapeutic targets
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disorder characterized by the presence of the Philadelphia (Ph) chromosome (1), encoding a chimeric protein Bcr-Abl with constitutive tyrosine kinase activity (2). Bcr-Abl kinase activates multiple downstream signaling pathways essential for survival and proliferation of CML cells (3,4). Imatinib is the first tyrosine kinase inhibitor (TKI) drug approved for CML therapy which blocks the ATP-binding site of Bcr-Abl and inhibits its activity (5). Unfortunately, not all patients with CML benefit from imatinib treatment in CML therapy. Primary refractoriness to imatinib is present in 13% of patients (6). Secondary (acquired) resistance, where the TKI treatment during therapy effectively selects drug-resistant clones, is a major complication that leads to disease progression in 30–40% of imatinib-treated patients (7). The most frequent causes of acquired resistance to CML therapy are point mutations in the kinase domain of Bcr-Abl, which prevent TKI drugs from effectively binding to Bcr-Abl (8). Other Bcr-Abl dependent mechanisms of resistance occur via amplification of the BCR-ABL gene and overexpression of the protein (9,10). Additional adaptive changes contributing to TKI resistance such as substitutive activation of the Src family of kinases (11), or activation of alternate cell signaling pathways such as PI3K/AKT/mTOR (12) have been documented.
Each causal change within a leukemic cell leading to TKI resistance is consequently accompanied by other adaptive molecular alterations resulting in a new drug-resistant cell phenotype. Proteomic characterization of such molecular alterations in resistant cancer cells enables identification of new molecular targets with therapeutic potential (13,14) and can be used for optimization of anticancer therapy (15). In order to identify the molecular alterations contributing to and associated with imatinib resistance in a model of CML, we performed proteomic analysis comparing imatinib-sensitive CML cells (CML-T1) with derived imatinib-resistant cells (CML-T1/IR). Among the most evident changes in the CML-T1/IR cells was upregulation of a multifunctional scaffolding protein Na+/H+ exchange regulatory factor 1 (NHERF1). Based on known NHERF1 functions, we evaluated the possible consequences of NHERF1 upregulation on the survival of the CML-T1/IR cells. We observed disrupted calcium homeostasis and demonstrated selective toxic effects of calcium transport and calcium signaling inhibitors in the imatinib-resistant CML cells. The pronounced toxicity of calcium homeostasis modulators emphasizes their therapeutic potential in CML therapy.
Materials and methods
Establishment of imatinib-resistant cells
CML-T1 cells (purchased from Leibniz Institut DSMZ, German Collection of Microorganisms and Cell Culture GmbH, Braunschweig, Germany) were grown in RPMI media in the presence of 10% fetal calf serum in a 37°C humidified atmosphere with 5% CO2. Resistant CML-T1 subclones, CML-T1/IR, were derived by prolonged cultivation in increasing concentrations of imatinib.
Mutation analysis in the kinase domain of BCR-ABL
Sanger sequencing was applied as previously described (16). Briefly, RNA was extracted from the CML-T1 and CML-T1/IR cells with TRIzol (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and the complementary DNA was synthesized by M-MLV reverse transcriptase (Promega, Madison, WI, USA) using random hexamer primers (Jena Bioscience GmbH, Jena, Germany). The cDNA region encoding the kinase domain of the fused BCR-ABL was amplified using nested PCR. The resulting 914-bp amplicon was sequenced from both strands. Based on the conclusive observation of mutated BCR-ABL transcripts, we explored the analysis using next-generation deep sequencing (NGS) with IRON-II BCR-ABL plates (IRON, International Robustness of Next-Generation sequencing) on a 454 GS Junior system (Roche Applied Science, Penzberg, Germany) to reveal the presence of mutations below the detection limit of Sanger sequencing. The protocol and algorithm previously established for NGS data evaluation (17) were followed.
Sample preparation for 2-DE
CML-T1 and CML-T1/IR cells (1×108) were harvested by centrifugation, washed twice with PBS and homogenized in a lysis buffer [7 M urea, 2 M thiourea, 4% CHAPS, 60 mM DTT and 1% ampholytes (IPG buffer pH 4.0–7.0; GE Healthcare Life Sciences, Little Chalfont, UK)] containing a protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland) for 20 min at room temperature. The lysates were cleared by centrifugation at 15,000 × g for 20 min at room temperature. Next, the supernatants were collected and the protein concentration was determined using the Bradford method (Bio-Rad, Inc., Hercules, CA, USA). The protein concentrations in all the samples were equalized to 7.3 mg/ml by dilution with the lysis buffer.
2-DE
Isoelectric focusing was performed with a Bio-Rad Protean IEF cell using 24 cm IPG strips (pH 4.0–7.0; GE Healthcare Life Sciences). Six technical replicates were run for each biological sample (6x CML-T1 and 6x CML-T1/IR). The strips were rehydrated overnight, each in 450 μl of sample, representing 3.3 mg of protein. Isoelectric focusing was performed for 60 kVh, with the maximum voltage not exceeding 5 kV, the current limited to 50 mA/strip and the temperature set to 18°C. The focused strips were equilibrated and reduced in equilibration buffer A (6 M urea, 50 mM Tris pH 8.8, 30% glycerol, 2% SDS and 450 mg DTT/50 ml of the buffer) for 15 min and then alkylated in equilibration buffer B (6 M urea, 50 mM Tris pH 8.8, 30% glycerol, 2% SDS and 1.125 mg iodacetamide/50 ml). The equilibrated strips were then placed on the top of 10% PAGE gel and secured in place by molten agarose. Electrophoresis was performed in a Tris-glycine-SDS system using a Protean Plus Dodeca Cell apparatus (Bio-Rad, Inc.) with buffer circulation and external cooling (20°C). The twelve gels were run at a constant voltage of 200 V for 6 h. Following electrophoresis, the gels were washed three times for 15 min in deionized water to remove the SDS. The washed gels were stained in CCB (SimplyBlue SafeStain, Invitrogen Life Technologies, Carlsbad, CA, USA) overnight, and then destained in deionized water.
Gel image analysis
The gels were scanned with a GS 800 calibrated densitometer (Bio-Rad, Inc.). Image analysis was performed with Phoretix 2D software (Nonlinear Dynamics, Newcastle upon Tyne, UK) in semi-manual mode with six gel replicates for one biological sample. Normalization of gel images was based on total spot density, and integrated spot density values (spot volumes) were then calculated after background subtraction. Average spot volume values (averages from all the six gels in the group) for each spot were compared between the groups. Protein spots were considered differentially expressed if they met both of the following criteria: i) the average difference of normalized spot volume was <1.5-fold and ii) the statistical significance of the change determined by the t-test was P<0.05.
MALDI MS, protein identification
The spots containing differentially expressed proteins were excised from the gels, cut into small pieces and washed three times with 25 mM ammonium bicarbonate in 50% acetonitrile (ACN). The gels were then dried in a SpeedVac Concentrator (Eppendorf, Hamburg, Germany). Sequencing grade modified trypsin (6 ng/μl) (Promega) in 25 mM ammonium bicarbonate in 5% ACN was added. Following overnight incubation at 37°C, the resulting peptides were extracted with 50% ACN. Peptide samples were spotted on a steel target plate (Bruker Daltonics, Bremen, Germany) and allowed to dry at room temperature. Matrix solution (3 mg α-cyano-4-hydroxycinnamic acid in 1 ml of 50% ACN containing 0.1% trifluoroacetic acid) was then added. MS was performed on an Autoflex II MALDI-TOF/TOF mass spectrometer (Bruker Daltonics) using a solid nitrogen laser (337 nm) and FlexControl software in reflectron mode with positive ion mass spectra detection. The mass spectrometer was externally calibrated with Peptide Calibration Standard II (Bruker Daltonics). Spectra were acquired in the mass range of 800–3,000 Da. The peak lists were generated using FlexAnalysis and searched against Swiss-Prot (2014 version) using the Mascot software. The peptide mass tolerance was set to 100 ppm, taxonomy Homo sapiens was selected, missed cleavage was set to 1, fixed modification for cysteine carbamidomethylation, and variable modifications for methionine oxidation and protein N-terminal acetylation were further settings selected. Proteins with a Mascot score over the threshold of 56 for P<0.05 calculated using the aforementioned settings were considered as identified.
Multidrug resistance (MDR) assay
The Vybrant™ Multidrug Resistance Assay kit (Thermo Fisher Scientific, Inc.) was used to measure drug efflux from the CML-T1 and the CML-T1/IR cells. The cells (5×104 cells/well) were grown in a 96-well plate for 24 h. Cells were then divided into two groups: the untreated group and the group treated with MDR drug efflux inhibitors cyclosporine A (CsA) and/or verapamil (at a final concentration ranging from 0.4 to 120 μg/ml). After 1 h, calcein AM was added to 100 μl of each examined cell suspension. After another 30 min, the cells in the plate were washed twice with 200 μl of cold RPMI-1640 culture medium, and the fluorescence of the retained calcein in both groups of cells was measured at a wavelength of λex = 485 nm and λem = 538 nm by FluoroMax-3 spectrofluorometer equipped with DataMax software (Jobin Yvon Horriba, Kyoto, Japan).
Cytosolic pH measurement
The assay was conducted as described previously by Kiedrowski (18). Cells were loaded with 1 μM BCECF-AM for 20 min at room temperature. To monitor the BCECF fluorescence, the cells were exposed every 5 sec to 488 and 440 nm excitation and the images of fluorescence emitted at >520 nm (F488 and F440) were measured by a FluoroMax-3 spectrofluorometer equipped with DataMax software (Jobin Yvon Horriba) and saved for offline analysis. In selected experiments, F488/F440 ratios were converted to cytosolic pH values based on in situ calibration performed at the end of the experiments as described in Kiedrowski (18).
Measurement of cytosolic Ca2+
Measurements of calcium concentration in the cytosol were performed as previously described (19). Briefly, the cells were washed in a modified HBSS buffer (140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 3 mM MgCl2, 10 mM HEPES, 50 mM glucose pH 7.4) and loaded with 3 μM Fura-2 acetoxymethyl ester (Fura-2/AM) for 30 min at 25°C in the dark, rinsed, and allowed to rest for 30 min prior to fluorescence measurements using a FluoroMax-3 spectrofluorometer equipped with DataMax software (Jobin Yvon Horriba, France). The fluorescence intensity of Fura-2 (excitation at 340 and 380 nm, and emission at 510 nm) was recorded every 15 sec, with an integration time of 3 sec. The concentration of free intracellular Ca2+ was determined as proportional to the ratio of fluorescence at 340/380 nm. The actual Ca2+ concentration was calculated with the Grynkiewicz equation (20). The Kd for Ca2+ binding to Fura-2 was measured to be 240 nM at the experimental temperature.
Wnt target gene microarray
Total RNA was isolated and purified from the CML-T1 and the CML-T1/IR cells with TRIzol reagent (Thermo Fisher Scientific, Inc.) and an RNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The RNA quality and quantity were determined at a 260/280 nm ratio on a Nanodrop ND-1000 (Thermo Fisher Scientific, Inc.). cDNA was synthesized by M-MLV reverse transcriptase (Promega) using random hexamer primers (Jena Bioscience GmbH).
The expression levels of the 92 genes involved in the Wnt signaling pathway within four control genes was analyzed on a TaqMan® array human Wnt Pathway Fast 96-well plate (Invitrogen Life Technologies) using a StepOnePlus™ Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). Analyses were performed three times for both the CMLT1 and the CMLT1/IR cells. The genes (n=27); genes for which the amplification signals were not observed in one or more replicates simultaneously for the CMLT1 control and the CMLT1/IR, were excluded from the analysis. Relative expression changes of target genes (n=65) were normalized to the expression of the housekeeping gene GUSB that is validated for routine molecular monitoring in CML cells (21). Relative expression levels of the genes were evaluated using the 2−ΔΔCq formula according to Livak et al (22) showing differential gene expression in the CMLT1/IR cells. For data control checking we re-analyzed differential relative expression using the GAPDH control gene, providing highly similar results as with GUSB.
BCR-ABL quantification was performed according to the method standardized in the EUTOS for CML project of ELN (www.eutos.org) and data were reported in the International Scale (IS). Primers and probes for BCR-ABL and GUSB were applied according to the European Partnership for Action Against Cancer recommendations and commercial plasmid standards were used to perform calibration curves (Ipsogen, Marseille, France).
Preparation of nuclear and cytoplasmic extracts
Cytoplasmic and nuclear extracts were prepared using a Nuclear and Cytoplasmic Extraction kit (NE-PER; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions with an additional modification in the final step of the nuclear protein extraction procedure, where the resulting pellets of the nuclear proteins were washed three times in ice cold PBS supplied with a protease inhibitor cocktail (Roche Diagnostics) and re-centrifuged at 16,000 × g to remove cytoplasmic protein contaminations.
Western blotting
Cell pellets were solubilized in lysis buffer (50 mM Tris pH 7.4; 1% Triton X-100, a protease inhibitor cocktail, 1 tablet/10 ml; Roche Diagnostics) on ice for 20 min. The cleared cell lysates (15,000 × g, 20 min) were collected and the protein concentration was determined by the Bradford method (Bio-Rad, Inc.). The samples containing 60 μg of protein were combined with an SDS loading buffer containing DTT, boiled for 5 min and resolved with SDS-PAGE using Novex precast 4–20% gradient gels (Thermo Fisher Scientific, Inc.). The separated proteins were transferred to PVDF membranes using the iBlot system according to manufacturer's instructions (Thermo Fisher Scientific, Inc.). The membranes were then blocked overnight in SuperBlock (PBS) blocking buffer (Thermo Fisher Scientific, Inc.). Then, the membranes were incubated with primary antibodies diluted to 1:1,000 in PBS containing 5% SuperBlock and 0.1% Tween-20. β-actin or Histone H2A (#4970 and #12349) were used as the loading controls; anti-NFAT1 rabbit mAb (#5862), anti-NHERF1 (#8601) and anti-MRP2/ABCC2 rabbit mAb (#12559; all from Cell Signaling Technology, Danvers, MA, USA) diluted 1:1,000 were used to detect the expression of NHERF1, MRP2 and NFAT. After thorough washing in PBS containing 0.1% Tween-20, a secondary anti-rabbit IgG, HRP-linked antibody (#7074; Cell Signaling Technology) was added (1:10,000). The signal was detected using enhanced chemiluminescence (ECL; GE Healthcare Life Sciences) assay, on X-ray film (Kodak, Rochester, NY, USA), developed, scanned and quantified by the Quantity One documentation system (Bio-Rad, Inc.).
Cell viability assays
Cells (1×104) were grown in a 24-well plate in 1 ml RPMI-1640 media (Thermo Fisher Scientific, Inc.) with increasing concentrations of the tested drugs for 3 days (72 h) at 37°C and a 5% CO2 humidified atmosphere. The toxicity of imatinib, amyloride, DCB, thapsigargin, ionomycin, verapamil, carboxyamidotriazole (CAI), FK-506 and CsA was measured using a Vybrant® MTT Cell Proliferation Assay kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Absorbance was detected at 570 nm using a microplate reader (Chameleon; Hidex, Turku, Finland).
Results
Development of imatinib-resistant CML-T1/IR subclones
We derived imatinib-resistant cells from an established model of the CML cell line CML-T1 (23). The CML-T1 cells express T-cell surface markers and carry the landmark BCR-ABL1 breakpoint cluster region translocation resulting in production of the p210 Bcr-Abl fusion protein (24). CML-T1 cells are sensitive to imatinib (IC50 0.45±0.015 μM, Fig. 1). The imatinib-resistant CML-T1/IR cells were derived by prolonged cultivation of CML-T1 in increasing concentrations of imatinib. The CML-T1/IR cells tolerated at least a 50-fold higher concentration of imatinib (Fig. 1).
Figure 1.

Viability of the CML-T1 and the CML-T1/IR cells in the presence of imatinib. CML-T1 and CML-T1/IR cells were grown in the presence of imatinib for 3 days. The cell viability was determined using an MTT assay on more than six biological replicates of the CML-T1 and CML-T1/IR cells, which were further seeded in three technical replicates onto the 96-well plates. The highest number of viable cells expressed by the maximal absorbance was set as 100%.
Mutation analysis reveals an imatinib-resistant Y253H mutation in the CML-T1/IR cells
The typical cause of resistance of CML cells to imatinib and other TKI inhibitors is a point mutation in the Abl kinase domain of the BCR-ABL fusion protein (25). We therefore performed mutational analysis of BCR-ABL in the CML-T1 and CML-T1/IR cells. We detected a Y253H mutation in the CML-T1/IR cells (69% of BCR-ABL transcripts), which is known to be a frequent causal mutation responsible for resistance to imatinib in human patients (26).
Proteomic analysis reveals upregulation of two NHERF1 variants in the CML-T1/IR cells
We hypothesized that besides the causal mutation in the BCR-ABL kinase domain there are additional, adaptive molecular changes in the CML-T1/IR cells, contributing to or associated with their survival in the presence of imatinib (27,28). These specific features may be exploitable as potential molecular targets for selective growth inhibition of imatinib-resistant cells for future therapies. To identify such alterations, we performed 2-DE proteomic analysis of the CML-T1 and CML-T1/IR cells (Fig. 2A). We identified eight differentially expressed proteins (Fig. 2A and Table I). The most marked change was the increased expression of the Na+/H+ exchange regulatory factor 1 (NHERF1 also known as SLC9A3R1). This protein was identified in two markedly upregulated variants of comparable molecular weight but of different isoelectric points (Table I, Fig. 2B). We further confirmed the upregulation of NHERF1 in the CML-T1/IR cells by western blot analysis (Fig. 2C). In addition to NHERF1, we detected upregulation of the endoplasmic reticulum (ER) calcium binding protein calreticulin, microtubule associated proteins and protein chaperones in the CML-T1/IR cells (Table I).
Figure 2.

2-DE analysis of CML-T1/IR and CML-T1 cells. (A) 2-DE gel from the CML-T1/IR cells with differentially expressed proteins indicated by arrows with numbers (↑ upregulated, ↓ downregulated). (B) Two protein variants of NHERF1 were detected as upregulated in the imatinib-resistant CML-T1/IR cells. (C) Verification of altered NHERF1 expression in the total cell lysates of CML-T1 and CML-T1/IR cells by western blot analysis revealed a 2.6-fold increase in the expression of NHERF1 in the CML-T1/IR cells, as quantified by Quantity One software (Bio-Rad, Inc.) based on more than three biological replicates, each performed in three technical replicates.
Table I.
Differentially expressed proteins in the CML-T1/IR cells.
| Spot no. | Uniprot | Protein name | Fold change | P-value | Sequence coverage (%) | Mascot score | MW (Da) | pI |
|---|---|---|---|---|---|---|---|---|
| Upregulated | ||||||||
| 1 | O14745 | Na+/H+ exchange regulatory cofactor NHERF1 | 2.84 | <0.009 | 21 | 57 | 39,130 | 5.55 |
| 2 | O14745 | Na+/H+ exchange regulatory cofactor NHERF1 | >10 | <0.001 | 32 | 84 | 39,130 | 5.55 |
| 3 | Q9Y230 | RuvB-like 2 | 6.8 | <0.015 | 29 | 81 | 51,296 | 5.49 |
| 4 | P27797 | Calreticulin | 1.8 | <0.023 | 24 | 71 | 48,283 | 4.29 |
| 5 | P00813 | Adenosine deaminase | 2.9 | <0.009 | 36 | 87 | 41,024 | 5.63 |
| 6 | P67936 | Tropomyosin α-4 chain | 1.56 | <0.036 | 63 | 198 | 28,619 | 4.67 |
| 7 | P04792 | Heat shock protein β-1 | >10 | <0.001 | 35 | 136 | 22,826 | 5.98 |
| Downregulated | ||||||||
| 8 | Q9Y230 | RuvB-like 2 | −2.17 | <0.02 | 38 | 149 | 51,296 | 5.49 |
| 9 | Q99536 | Synaptic vesicle membrane protein VAT-1 homolog | −3.7 | <0.039 | 34 | 60 | 42,122 | 5.88 |
| 10 | Q15691 | Microtubule-associated protein RP/EB family member 1 | −1.86 | <0.005 | 53 | 89 | 30,151 | 5.02 |
| 11 | P04792 | Heat shock protein β-1 | −2.2 | <0.001 | 44 | 75 | 22,826 | 5.98 |
Included in the table are the proteins with an expression difference of at least 1.5-fold and a statistical significance of change P<0.05. MW, molecular weight; pI, isoelectric point.
NHERF1: functional analysis
NHERF1 is a multifunctional scaffolding protein containing two PDZ domains (29). Via these domains NHERF1 interacts with various cellular proteins, mostly membrane receptors and transporters, modulating their expression, stability and activity (30). NHERF1 has been implicated in MDR in liver cancer by controlling the expression of MDR exporter MRP2 (31). NHERF1 negatively regulates the activity of sodium hydrogen ion exchanger SLC9A3 (NHE3), thus modulating the pH inside the cell (32). NHERF1 has also been demonstrated to influence cytosolic calcium concentration via transient receptor potential channel 5 protein (TRPC5) (33,34). Recently it was revealed that NHERF1 negatively regulates the canonical Wnt signaling pathway via direct interaction with a subset of Frizzled (Fzd) receptors (35).
Based on the functions aforementioned, we examined the possible effect of NHERF1 upregulation in the CML-T1/IR cells in order to identify a specific feature of the imatinib-resistant cells which may exhibit a 'molecular weakness' representing a potential therapeutic target. We first evaluated the potential connection of NHERF1 with MDR in the CML-T1/IR cells. Next we tested whether NHERF1 upregulation modulates H+ and Ca2+ concentrations in the cytosol of the CML-T1/IR cells and investigated whether NHERF1 upregulation and changes in ion homeostasis affect the activity of the Wnt signaling pathway.
NHERF1 upregulation does not contribute to MDR in the CML-T1/IR cells
We evaluated whether the upregulation of NHERF1 in the CML-T1/IR cells contributes to cell survival in high concentrations of imatinib by increasing the activity of the MDR protein pumps MRP2 as described in liver cancer (36). We performed an in vitro MDR assay based on the cellular efflux of the fluorescent probe calcein. This process was shown to be performed by the multidrug exporters MDR1 and MRP2 (37,38). Both the CML-T1 and the CML-T1/IR cells retained 100% of the incorporated calcein (no calcein efflux was detected). The addition of multidrug export inhibitors CsA and verapamil therefore had no effect on efflux. This suggests that these drug exporters are not present/active in CML-T1 and CML-T1/IR cells (Fig. 3A). Furthermore, while we were able to detect MRP2 by western blot in the lysates of several cell types including CML-derived K562 cells, expression of MRP2 in both the CML-T1 and CML-T1/IR cells was under our detection limit (Fig. 3B). We concluded that the activity of multidrug exporters in the CML-T1 and CML-T1/IR cells is negligible and that the increased NHERF1 expression does not affect the activity of MDR1 and MRP2 in the CML-T1/IR cells.
Figure 3.

Multidrug resistance (MDR) assay in CML-T1 and CML-T1/IR cells and immunodetection of MRP2 protein by western blot analysis in the CML-T1 and CML-T1/IR cells compared with other cell lysates. (A) Measurement of calcein retention as a surrogate of MDR exporter activity in the CML-T1 and CML-T1/IR cells was performed as described in 'Materials and methods'. Cyclosporine A (CsA) and verapamil were used as inhibitors of MDR. No MDR activity was detected in the CMLT1 and CMLT1/IR cells as reflected by 100% calcein retention in the cells. (B) Cell lysates of CML-T1 and CML-T1/IR were subjected to SDS-PAGE and western blot analysis for immunodetection of MRP2 together with the lysates of K562, HL-60, HeLa cells and mouse liver tissue, which were used as MRP2 positive controls.
Intracellular concentrations of H+ and Ca2+ ions differ in the CML-T1 and the CML-T1/IR cells
Based on the known interplay between NHERF1 and the Na+/H+ exchanger NHE3 with a consequent effect on cellular pH (39) we examined whether the NHERF1 upregulation in the CML-T1/IR cells affects the intracellular pH. We measured the intracellular pH and observed increased cytosolic pH in the CML-T1/IR cells (pH 7.25) compared to the control CML-T1 cells (pH 7.18), as shown in Fig. 4A.
Figure 4.
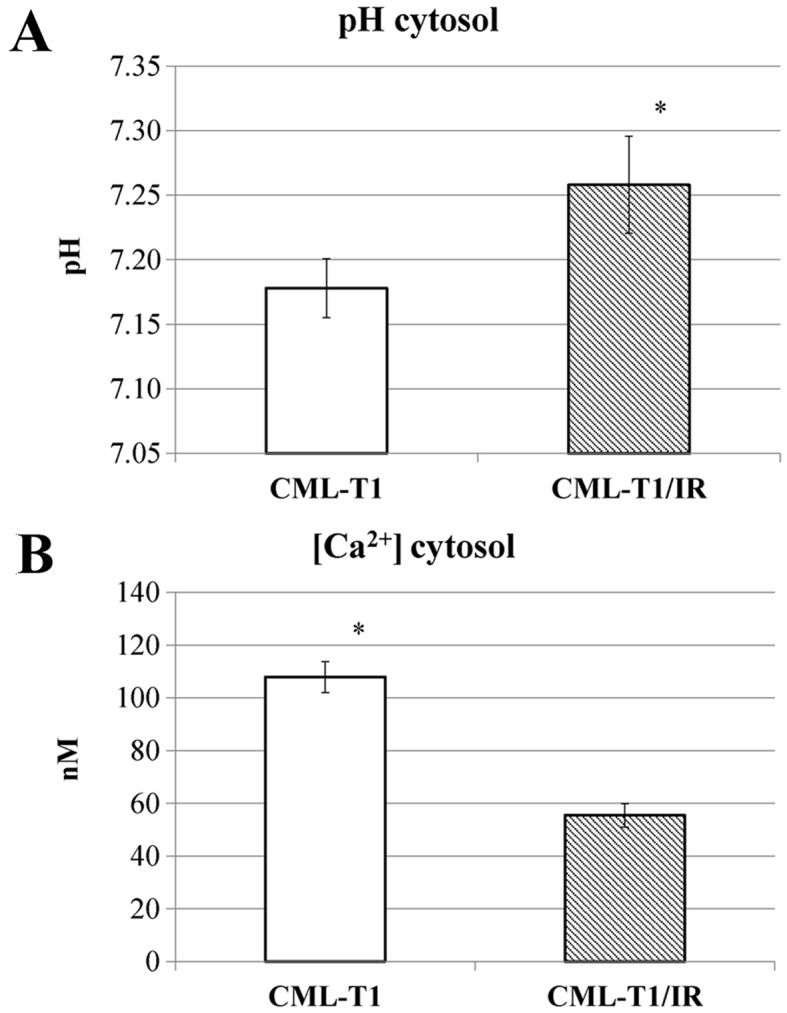
Cellular pH and calcium ion concentration measurements in CML-T1 and CML-T1/IR cells. (A) Intracellular pH was determined using BCECF fluorescence and (B) calcium ion concentration was measured using Fura-2 probe fluorescence measurements. Each graph shows the average values from three repeated measurements performed on three biological replicates.
Since NHERF1 was shown to regulate the activity of nonselective calcium permeable cation channels, namely TRPC4 and TRPC5 (34) we examined whether the presence of upregulated NHERF1 (or the increased cytosolic pH) in the CML-T1/IR cells also affects the cytosolic concentration of Ca2+ (40). Our measurements revealed a 50% decrease in cytosolic Ca2+ concentration in the CML-T1/IR cells (Fig. 4B).
In summary, ion homeostasis in the CML-T1/IR cytosol was altered, with an increased pH and a decreased cytosolic Ca2+ concentration compared to the original CMLT1 cells.
Calcium channel blockers and inhibitors of calcium signaling selectively inhibit the viability of CML-T1/IR cells
We hypothesized that the increased pH due to differential Na+/H+ exchange (41) and decreased Ca2+ concentration in the cytosol (42) contribute to CML-T1/IR survival in the presence of imatinib. To address whether the inhibition of Na+/H+ exchange selectively affects the growth of the CML-T1/IR cells, we targeted Na+/H+ exchange by amiloride. Since it is well established that a Na+/Ca2+ exchanger (NCX) may also contribute to pH and calcium concentration changes in the cells (43), we further inhibited Na+/Ca2+ exchange by 3′,4′-dichlorobenzamil hydrochloride (DCB) (44). We observed that none of the inhibitors had a selective effect on the viability of the CML-T1/IR cells. Both the CML-T1 and the CML-T1/IR cells tolerated comparable concentrations of inhibitors within IC50 values of 15 μM for amiloride and 7 μM for DCB (Fig. 5).
Figure 5.

Effects of selected inhibitors on the viability of CML-T1 and CML-T1/IR cells. Cell viability assay was determined using an MTT assay 3 days after addition of inhibitors to the cultivation media. Maximal absorbance (highest number of viable cells) of cells cultivated without imatinib was set as 100%. Each graph represents measurements of at least three biological samples, each further measured in three technical replicates. DCB, 2′,4′-dichlorobenzamil hydrochloride; CAI, carboxyamidotriazole.
To address whether shifted Ca2+ homeostasis is crucial for survival of the CML-T1/IR cells, we exposed the CML-T1 and CML-T1/IR cells to increasing concentrations of inhibitors of Ca2+ transport and Ca2+ signaling, namely thapsigargin, ionomycin, verapamil, and CAI. Thapsigargin inhibits the activity of sarco/ER Ca2+ ATPase (SERCA), preventing the uptake of cytosolic Ca2+ into the ER, thus increasing the cytosolic Ca2+ (45,46). Ionomycin increases intracellular Ca2+ by means of its increased entry across the plasma membrane and/or by depletion of intracellular Ca2+ stores such as ER (47). The clinically approved drug verapamil blocks voltage-dependent (L-type) Ca2+ channels (48). CAI inhibits non-voltage operated calcium channels and blocks both Ca2+ influx into the cells and Ca2+ release from the intracellular stores (49). Notably, CAI has been previously demonstrated to inhibit the growth of imatinib-resistant CML cell lines in vitro (50) and several studies have demonstrated a potential anticancer effect of CAI in vitro (51,52).
In a battery of in vitro cell viability assays we tested the aforementioned compounds for their toxicity to the CML-T1 and CML-T1/IR cells. All the tested agents were more toxic to the imatinib-resistant CML-T1/IR cells (Fig. 5). Among them, thapsigargin was the most potent in growth inhibition of CML-T1/IR cells, which were at least 16-fold more sensitive to this agent compared to the CML-T1 cells. The IC50 for the CML-T/IR cells was 75 nM while the IC50 in the CML-T1 cells was not reached at a 1.2 μM concentration of thapsigargin in the media. The CMLT1-IR cells were also 5-fold more sensitive to ionomycin (IC50values 200 nM for the CML-T1/IR cells and >1.6 μM for the CML-T1 cells). Verapamil was ~3-fold more toxic to the CML-T1/IR cells (IC50 30 nM for the CML-T1/IR cells and 90 nM for the CML-T1 cells) and CAI was 2.5-fold more toxic to the CML-T1/IR cells (IC50 200 nM for the CML-T1/IR cells and 1.6 μM for the CML-T1 cells).
Altered calcium homeostasis influences numerous intracellular processes including Wnt signaling. CsA and tacrolimus (FK-506) were shown to modulate calcium homeostasis (53,54) and inhibit Wnt-regulated pro-survival processes in imatinib-resistant cells (56). We, therefore, evaluated the effect of CsA and FK-506 on the proliferation of the CML-T1 and CML-T1/IR cells. In our assays both CsA and FK506 inhibited proliferation of the CML-T1/IR cells at significantly lower concentrations (3- and 1.8-fold, respectively) compared to the CML-T1 cells. The IC50 of CsA was 4 μM for the CML-T1/IR cells and 12 μM for the CML-T1 cells; the IC50 of FK-506 was 50 nM for the CML-T1/IR cells and 90 nM for the CML-T1 cells (Fig. 5).
In summary, we observed that a disruption of calcium homeostasis (and to a lesser extent also inhibition of Wnt signaling), but not the inhibition of Na+/H+ or Na+/Ca2+ exchange was selectively toxic to CML-T1/IR cells. The most effective growth inhibition of CML-T1/IR was achieved by using the agents causing depletion of the intracellular Ca2+ stores with an increase in the Ca2+ concentration in the cytosol (ionomycin, thapsigargin), but also calcium channel blockers (verapamil) and calcium signaling antagonist CAI. Low molecular weight antagonists of calcium homeostasis, calcium transport blockers and calcium signaling inhibitors may be used to selectively impair the growth of imatinib-resistant CML-T1/IR cells, thus suggesting their clinical potential in TKI-resistant CML.
The Wnt pathway is dysregulated in the CML-T1/IR cells
NHERF1 has been proven to negatively regulate Wnt signaling via its direct interaction with Fzd receptors (35). The potential of Wnt signaling serving as a therapeutic target is recently a subject of intensive studies in the field of cancer therapy and novel drug discovery (55). We therefore examined whether NHERF1 upregulation (accompanied with altered cytosolic ion concentration) in the CML-T1/IR cells is associated with altered Wnt signaling. We analyzed the activity of the Wnt pathway using RT-PCR Wnt microarray to determine the relative expression of Wnt target genes and regulatory molecules (Fig. 6A). In the CML-T1/IR cells, we observed a decreased expression of genes of the canonical β-catenin-dependent Wnt pathway, namely TLE1, TLE4, TCF7L2, LEF1, MYC and RHOU. Conversely, the expression of negative regulators of this pathway, KREMEN1 and SFRP5 was increased in the CML-T1/IR cells, indicating that the canonical β-catenin-dependent Wnt signaling was decreased in the CML-T1/IR cells.
Figure 6.

Changes in gene expression of the Wnt signaling pathway and immunodetection of NFAT by western blot analysis in CML-T1 and CML-T1/IR cells. (A) The heatmap shows genes whose mRNA expression was downregulated (blue) or upregulated (yellow) in the CML-T1/IR cells compared to the CML-T1 cells. (B) NFAT protein expression as determined by western blot analysis in the cell lysates and the nuclei of the CML-T1 and CML-T1/IR cells.
We also evaluated the expression of genes critical for the non-canonical CaMKII/Ca2+/NFAT Wnt pathway where the expression of mRNA encoding FZD8 receptor, essential for this pathway was decreased in the CML-T1/IR cells. This is contradictory to the results of Gregory et al (56), which showed that Wnt signaling contributed (via FZD8) to the pro-surviving effect of the CaMKII/Ca2+/NFAT Wnt pathway in imatinib-resistant cells. To obtain more information on the status of CaMKII/Ca2+/NFAT Wnt signaling in our cells, we determined the presence of transcription factor NFAT, the final effector of this pathway in the cytoplasm and the nuclei of the CML-T1 and CML-T1/IR cells. NFAT was not detected in the CML-T1/IR cells (Fig. 6B), while it was clearly detectable in the CML-T1 cells. The diminished expression of NFAT protein in the CML-T1/IR cells suggests that CaMKII/Ca2+/NFAT Wnt signaling is decreased in these cells and does not contribute to the survival against imatinib in our cell model.
All in all, our data suggest that imatinib resistance is accompanied by significant downregulation of both canonical and noncanonical CaMKII/Ca2+/NFAT Wnt signaling pathways in CML-T1/IR cells.
Discussion
The imatinib-resistant CML-T1/IR cells carry the causal mutation of Y253H in the kinase domain of the BCR-ABL gene. This mutation is undoubtedly the main driving force of resistance to imatinib. However, any resistant phenotype is a result of multiple molecular events including causative, contributing and adaptive cellular processes, enabling survival of the resistant cells. Detailed molecular analysis of therapy-resistant cells potentially opens the path to personalized therapies of drug-resistant malignancies. Using 2-DE analysis we revealed a strong upregulation of a multifunctional protein NHERF1. We demonstrated altered cytosolic pH and decreased calcium levels in the CML-T1/IR cells. While inhibition of Na+/H+ and Na+/Ca+ exchangers has no specific toxic effect in the resistant CML-T1/IR cells, modulators of cytosolic calcium concentration, calcium channels blockers and calcium signaling inhibitors were significantly more toxic to the CML-T1/IR cells compared to the CML-T1/IR cells. The most prominent toxic effect we observed using ionomycin and thapsigargin was presumably caused by toxic elevation of cytosolic Ca2+ and/or by the depletion of Ca2+ from ER, leading to ER stress, unfolded protein response and finally to apoptosis (57).
Altered Ca2+ concentration in the cytosol may affect the expression of many proteins and modulate signaling pathways such as CaMKII/Ca2+/NFAT Wnt. This was of particular interest to us since this calcium-dependent pathway was previously demonstrated to be critical for the survival of imatinib-resistant CML cells; NFAT inhibitor CsA effectively inhibited proliferation of the imatinib-resistant cells (56). Correspondingly, when we applied NFAT inhibitors, such as CsA or FK-506, in our experiments, we also observed their selective toxicity to CML-T1/IR. However, this growth inhibitory effect appeared to be NFAT-independent in our CML-T1/IR cells, because NFAT was markedly downregulated (if not absent) in the nuclei of the CML-T1/IR cells.
We, therefore, proposed that the selective toxicity of CsA and FK-506 in CML-T1/IR cells is due to their direct effect on calcium homeostasis. The established mechanism of ionomycin and thapsigargin toxicity (the inhibitors with the most pronounced toxic effect to the CML-T1/IR cells) is elevation of cytosolic Ca2+ and depletion of Ca2+ from the ER (47,45). This suggests that calcium levels in both the cytoplasm and the ER should be considered as critical for the survival of CML-T1/IR cells. Our hypothesis of altered calcium homeostasis was further supported by the results of our 2-DE analysis, where calreticulin was detected as upregulated in the CML-T1/IR cells (Table I). Calreticulin is an ER resident protein, which contributes to proper folding of nascent proteins and serves as a calcium binding buffer in the ER. If CML-T1/IR cells require increased expression of calreticulin to maintain ER Ca2+ homeostasis, blocking Ca2+ entry to the ER by thapsigargin or depleting Ca2+ from the ER by ionomycin can substantially reduce their viability. There is evidence that CsA and FK-506, in addition to their effect on Wnt signaling, may affect calcium homeostasis directly. CsA inhibits the activity of SERCA, an ER ATPase pump responsible for Ca2+ influx into the ER from the cytosol (58). FK-506 stimulates the activity of the ryanodine receptor (Ryr) which acts in opposition to SERCA and releases Ca2+ from the ER to the cytosol (59). Such a direct toxic effect of these clinically used immunosuppressants on calcium homeostasis may explain their selective cytotoxic effect on the CMLT1/IR cells in the absence of NFAT.
In addition to the selective toxicity of the experimental inhibitors, thapsigargin and ionomycin, three drugs already clinically established, CsA, tacrolimus (FK-506) and verapamil displayed similar selective toxicity to the imatinib-resistant CML-T1/IR cells. Verapamil and tacrolimus are known to inhibit multi-drug export in cancer cells (60,61). However, since there was no detectable multidrug-export activity in the CML-T1/IR cells, we propose that the observed cytotoxic effect of verapamil and tacrolimus was based on direct disruption of calcium homeostasis.
Our proteomic and functional analysis of imatinib resistance in CML cells provides a proof-of-concept and a vision for a future model of personalized TKI-resistant CML therapy, where the isolation of TKI-resistant cells can be combined with proteomic and functional analysis in order to identify potential therapeutic targets, which may be exploited for selective elimination of the drug-resistant population of cells.
Acknowledgments
This study was supported by the Czech Ministry of Health via the Project for Conceptual Development of Research Organizations grant no. 00023736, and via the 15-32961A grant, by the European Regional Development Fund via grants CZ.1.05/1.1.00/02.0109-BIOCEV, ERDF OPPK CZ.2.16/3.1.00/24001 and ERDF OPPK CZ.2.16/3.1.00/28007, by the Czech Science Foundation (www.gacr.cz) grant 15-14200S, and by the Ministry of Education, Youth and Sports of the Czech Republic (www.msmt.cz) via grants PRVOUK P24/LF1/3 and SVV 260 265/2016.
Abbreviations
- CML
chronic myeloid leukemia
- TKIs
tyrosine kinase inhibitors
- NHERF-1
Na+H+ exchanger regulatory factor 1
- MDR
multidrug resistance
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- PDZ
post synaptic density protein (PSD95)
- Dlg1
Discs, large (Drosophila) homolog 1
- Zo-1
zonula occludens-1 protein
- ABC
ATP-binding cassette transporters
- NFAT
nuclear factor of activated T cells
- DCB
3′,4′-dichlorobenzamil hydrochloride, TRPC, transient receptor potential calcium channel
- SERCA
sarco/endoplasmic reticulum Ca2+ ATPase
- CsA
cyclosporine A
- RyR
ryanodine receptor
References
- 1.Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960;25:85–109. [PubMed] [Google Scholar]
- 2.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247:1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 3.Deininger MW, Vieira S, Mendiola R, Schultheis B, Goldman JM, Melo JV. BCR-ABL tyrosine kinase activity regulates the expression of multiple genes implicated in the pathogenesis of chronic myeloid leukemia. Cancer Res. 2000;60:2049–2055. [PubMed] [Google Scholar]
- 4.Hazlehurst LA, Bewry NN, Nair RR, Pinilla-Ibarz J. Signaling networks associated with BCR-ABL-dependent transformation. Cancer Control. 2009;16:100–107. doi: 10.1177/107327480901600202. [DOI] [PubMed] [Google Scholar]
- 5.Waller CF. Imatinib mesylate. Recent Results Cancer Res. 2014;201:1–25. doi: 10.1007/978-3-642-54490-3_1. [DOI] [PubMed] [Google Scholar]
- 6.Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Laï JL, Philippe N, Facon T, Fenaux P, Preudhomme C. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–1018. doi: 10.1182/blood.V100.3.1014. [DOI] [PubMed] [Google Scholar]
- 7.Kantarjian HM, Talpaz M, O'Brien S, Giles F, Garcia-Manero G, Faderl S, Thomas D, Shan J, Rios MB, Cortes J. Dose escalation of imatinib mesylate can overcome resistance to standard-dose therapy in patients with chronic myelogenous leukemia. Blood. 2003;101:473–475. doi: 10.1182/blood-2002-05-1451. [DOI] [PubMed] [Google Scholar]
- 8.Ursan ID, Jiang R, Pickard EM, Lee TA, Ng D, Pickard AS. Emergence of BCR-ABL kinase domain mutations associated with newly diagnosed chronic myeloid leukemia: A meta-analysis of clinical trials of tyrosine kinase inhibitors. J Manag Care Spec Pharm. 2015;21:114–122. doi: 10.18553/jmcp.2015.21.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell LJ, Patsouris C, Rayeroux KC, Somana K, Januszewicz EH, Szer J. BCR/ABL amplification in chronic myelocytic leukemia blast crisis following imatinib mesylate administration. Cancer Genet Cytogenet. 2002;139:30–33. doi: 10.1016/S0165-4608(02)00615-5. [DOI] [PubMed] [Google Scholar]
- 10.Phan CL, Megat Baharuddin PJNB, Chin LP, Zakaria Z, Yegappan S, Sathar J, Tan S-M, Purushothaman V, Chang KM. Amplification of BCR-ABL and t(3;21) in a patient with blast crisis of chronic myelogenous leukemia. Cancer Genet Cytogenet. 2008;180:60–64. doi: 10.1016/j.cancergencyto.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 11.Roginskaya V, Zuo S, Caudell E, Nambudiri G, Kraker AJ, Corey SJ. Therapeutic targeting of Src-kinase Lyn in myeloid leukemic cell growth. Leukemia. 1999;13:855–861. doi: 10.1038/sj.leu.2401429. [DOI] [PubMed] [Google Scholar]
- 12.Quentmeier H, Eberth S, Romani J, Zaborski M, Drexler HG. BCR-ABL1-independent PI3Kinase activation causing imatinib-resistance. J Hematol Oncol. 2011;4:6. doi: 10.1186/1756-8722-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petrak J, Toman O, Simonova T, Halada P, Cmejla R, Klener P, Zivny J. Identification of molecular targets for selective elimination of TRAIL-resistant leukemia cells. From spots to in vitro assays using TOP15 charts. Proteomics. 2009;9:5006–5015. doi: 10.1002/pmic.200900335. [DOI] [PubMed] [Google Scholar]
- 14.Lorkova L, Scigelova M, Arrey TN, Vit O, Pospisilova J, Doktorova E, Klanova M, Alam M, Vockova P, Maswabi B, et al. Detailed functional and proteomic characterization of fludarabine resistance in mantle cell lymphoma cells. PLoS One. 2015;10:e0135314. doi: 10.1371/journal.pone.0135314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klanova M, Soukup T, Jaksa R, Molinsky J, Lateckova L, Maswabi BCL, Prukova D, Brezinova J, Michalova K, Vockova P, et al. Mouse models of mantle cell lymphoma, complex changes in gene expression and phenotype of engrafted MCL cells: Implications for preclinical research. Lab Invest. 2014;94:806–817. doi: 10.1038/labinvest.2014.61. [DOI] [PubMed] [Google Scholar]
- 16.Poláková KM, Polívková V, Rulcová J, Klamová H, Jurcek T, Dvoráková D, Zácková D, Pospísil Z, Mayer J, Moravcová J. Constant BCR-ABL transcript level ≥0.1% (IS) in patients with CML responding to imatinib with complete cytogenetic remission may indicate mutation analysis. Exp Hematol. 2010;38:20–26. doi: 10.1016/j.exphem.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 17.Machova Polakova K, Kulvait V, Benesova A, Linhartova J, Klamova H, Jaruskova M, de Benedittis C, Haferlach T, Baccarani M, Martinelli G, et al. Next-generation deep sequencing improves detection of BCR-ABL1 kinase domain mutations emerging under tyrosine kinase inhibitor treatment of chronic myeloid leukemia patients in chronic phase. J Cancer Res Clin Oncol. 2015;141:887–899. doi: 10.1007/s00432-014-1845-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiedrowski L. Cytosolic zinc release and clearance in hippocampal neurons exposed to glutamate - the role of pH and sodium. J Neurochem. 2011;117:231–243. doi: 10.1111/j.1471-4159.2011.07194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinsella BT, O'Mahony DJ, Fitzgerald GA. The human thromboxane A2 receptor α isoform (TPα) functionally couples to the G proteins Gq and G11 in vivo and is activated by the isoprostane 8-epi prostaglandin F2α. J Pharmacol Exp Ther. 1997;281:957–964. [PubMed] [Google Scholar]
- 20.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 21.White H, Deprez L, Corbisier P, Hall V, Lin F, Mazoua S, Trapmann S, Aggerholm A, Andrikovics H, Akiki S, et al. A certified plasmid reference material for the standardisation of BCR-ABL1 mRNA quantification by real-time quantitative PCR. Leukemia. 2015;29:369–376. doi: 10.1038/leu.2014.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Kuriyama K, Gale RP, Tomonaga M, Ikeda S, Yao E, Klisak I, Whelan K, Yakir H, Ichimaru M, Sparkes RS, et al. CML-T1: A cell line derived from T-lymphocyte acute phase of chronic myelogenous leukemia. Blood. 1989;74:1381–1387. [PubMed] [Google Scholar]
- 24.Drexler HG. Leukemia cell lines: In vitro models for the study of chronic myeloid leukemia. Leuk Res. 1994;18:919–927. doi: 10.1016/0145-2126(94)90103-1. [DOI] [PubMed] [Google Scholar]
- 25.Hochhaus A, Kreil S, Corbin AS, La Rosée P, Müller MC, Lahaye T, Hanfstein B, Schoch C, Cross NCP, Berger U, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16:2190–2196. doi: 10.1038/sj.leu.2402741. [DOI] [PubMed] [Google Scholar]
- 26.Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, Herrmann R, Lynch KP, Hughes TP. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–3475. doi: 10.1182/blood.V99.9.3472. [DOI] [PubMed] [Google Scholar]
- 27.Kominsky DJ, Klawitter J, Brown JL, Boros LG, Melo JV, Eckhardt SG, Serkova NJ. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clin Cancer Res. 2009;15:3442–3450. doi: 10.1158/1078-0432.CCR-08-3291. [DOI] [PubMed] [Google Scholar]
- 28.Adamia S, Pilarski PM, Bar-Natan M, Stone RM, Griffin JD. Alternative splicing in chronic myeloid leukemia (CML): A novel therapeutic target? Curr Cancer Drug Targets. 2013;13:735–748. doi: 10.2174/15680096113139990083. [DOI] [PubMed] [Google Scholar]
- 29.Reczek D, Berryman M, Bretscher A. Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J Cell Biol. 1997;139:169–179. doi: 10.1083/jcb.139.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ardura JA, Friedman PA. Regulation of G protein-coupled receptor function by Na+/H+ exchange regulatory factors. Pharmacol Rev. 2011;63:882–900. doi: 10.1124/pr.110.004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li M, Wang W, Soroka CJ, Mennone A, Harry K, Weinman EJ, Boyer JL. NHERF-1 binds to Mrp2 and regulates hepatic Mrp2 expression and function. J Biol Chem. 2010;285:19299–19307. doi: 10.1074/jbc.M109.096081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He P, Yun CC. Mechanisms of the regulation of the intestinal Na+/H+ exchanger NHE3. J Biomed Biotechnol. 2010;2010:238080. doi: 10.1155/2010/238080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Obukhov AG, Nowycky MC. TRPC5 activation kinetics are modulated by the scaffolding protein ezrin/radixin/moesin-binding phosphoprotein-50 (EBP50) J Cell Physiol. 2004;201:227–235. doi: 10.1002/jcp.20057. [DOI] [PubMed] [Google Scholar]
- 34.Tang Y, Tang J, Chen Z, Trost C, Flockerzi V, Li M, Ramesh V, Zhu MX. Association of mammalian Trp4 and phospholipase C isozymes with a PDZ domain-containing protein, NHERF. J Biol Chem. 2000;275:37559–37564. doi: 10.1074/jbc.M006635200. [DOI] [PubMed] [Google Scholar]
- 35.Wheeler DS, Barrick SR, Grubisha MJ, Brufsky AM, Friedman PA, Romero G. Direct interaction between NHERF1 and Frizzled regulates β-catenin signaling. Oncogene. 2011;30:32–42. doi: 10.1038/onc.2010.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karvar S, Suda J, Zhu L, Rockey DC. Distribution dynamics and functional importance of NHERF1 in regulation of Mrp-2 trafficking in hepatocytes. Am J Physiol Cell Physiol. 2014;307:C727–C737. doi: 10.1152/ajpcell.00011.2014. [DOI] [PubMed] [Google Scholar]
- 37.Bosch I, Croop J. P-glycoprotein multidrug resistance and cancer. Biochim Biophys Acta. 1996;1288:F37–F54. doi: 10.1016/0304-419x(96)00022-4. [DOI] [PubMed] [Google Scholar]
- 38.Canitrot Y, Lahmy S, Buquen JJ, Canitrot D, Lautier D. Functional study of multidrug resistance with fluorescent dyes. Limits of the assay for low levels of resistance and application in clinical samples. Cancer Lett. 1996;106:59–68. doi: 10.1016/0304-3835(96)04301-7. [DOI] [PubMed] [Google Scholar]
- 39.Donowitz M, Cha B, Zachos NC, Brett CL, Sharma A, Tse CM, Li X. NHERF family and NHE3 regulation. J Physiol. 2005;567:3–11. doi: 10.1113/jphysiol.2005.090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garciarena CD, Youm JB, Swietach P, Vaughan-Jones RD. H+-activated Na+ influx in the ventricular myocyte couples Ca2+-signalling to intracellular pH. J Mol Cell Cardiol. 2013;61:51–59. doi: 10.1016/j.yjmcc.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 41.Rich IN, Worthington-White D, Garden OA, Musk P. Apoptosis of leukemic cells accompanies reduction in intracellular pH after targeted inhibition of the Na+/H+ exchanger. Blood. 2000;95:1427–1434. [PubMed] [Google Scholar]
- 42.Pinton P, Ferrari D, Magalhães P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J Cell Biol. 2000;148:857–862. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Condrescu M, Opuni K, Hantash BM, Reeves JP. Cellular regulation of sodium-calcium exchange. Ann NY Acad Sci. 2002;976:214–223. doi: 10.1111/j.1749-6632.2002.tb04744.x. [DOI] [PubMed] [Google Scholar]
- 44.Iwamoto T, Watanabe Y, Kita S, Blaustein MP. Na+/Ca2+ exchange inhibitors: A new class of calcium regulators. Cardiovasc Hematol Disord Drug Targets. 2007;7:188–198. doi: 10.2174/187152907781745288. [DOI] [PubMed] [Google Scholar]
- 45.Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inesi G, Hua S, Xu C, Ma H, Seth M, Prasad AM, Sumbilla C. Studies of Ca2+ ATPase (SERCA) inhibition. J Bioenerg Biomembr. 2005;37:365–368. doi: 10.1007/s10863-005-9472-1. [DOI] [PubMed] [Google Scholar]
- 47.Beeler TJ, Jona I, Martonosi A. The effect of ionomycin on calcium fluxes in sarcoplasmic reticulum vesicles and liposomes. J Biol Chem. 1979;254:6229–6231. [PubMed] [Google Scholar]
- 48.Triggle DJ. L-type calcium channels. Curr Pharm Des. 2006;12:443–457. doi: 10.2174/138161206775474503. [DOI] [PubMed] [Google Scholar]
- 49.Hupe DJ, Boltz R, Cohen CJ, Felix J, Ham E, Miller D, Soderman D, Van Skiver D. The inhibition of receptor-mediated and voltage-dependent calcium entry by the antiproliferative L-651,582. J Biol Chem. 1991;266:10136–10142. [PubMed] [Google Scholar]
- 50.Alessandro R, Fontana S, Giordano M, Corrado C, Colomba P, Flugy AM, Santoro A, Kohn EC, De Leo G. Effects of carboxyamidotriazole on in vitro models of imatinib-resistant chronic myeloid leukemia. J Cell Physiol. 2008;215:111–121. doi: 10.1002/jcp.21290. [DOI] [PubMed] [Google Scholar]
- 51.Perabo FG, Wirger A, Kamp S, Lindner H, Schmidt DH, Müller SC, Kohn EC. Carboxyamido-triazole (CAI), a signal transduction inhibitor induces growth inhibition and apoptosis in bladder cancer cells by modulation of Bcl-2. Anticancer Res. 2004;24:2869–2877. [PubMed] [Google Scholar]
- 52.Guo L, Ye C, Chen W, Ye H, Zheng R, Li J, Yang H, Yu X, Zhang D. Anti-inflammatory and analgesic potency of carboxy-amidotriazole, a tumorostatic agent. J Pharmacol Exp Ther. 2008;325:10–16. doi: 10.1124/jpet.107.131888. [DOI] [PubMed] [Google Scholar]
- 53.Rovira P, Mascarell L, Truffa-Bachi P. The impact of immunosuppressive drugs on the analysis of T cell activation. Curr Med Chem. 2000;7:673–692. doi: 10.2174/0929867003374778. [DOI] [PubMed] [Google Scholar]
- 54.Maguire O, Tornatore KM, O'Loughlin KL, Venuto RC, Minderman H. Nuclear translocation of nuclear factor of activated T cells (NFAT) as a quantitative pharmacodynamic parameter for tacrolimus. Cytometry A. 2013;83:1096–1104. doi: 10.1002/cyto.a.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.An SM, Ding QP, Li LS. Stem cell signaling as a target for novel drug discovery: Recent progress in the WNT and Hedgehog pathways. Acta Pharmacol Sin. 2013;34:777–783. doi: 10.1038/aps.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gregory MA, Phang TL, Neviani P, Alvarez-Calderon F, Eide CA, O'Hare T, Zaberezhnyy V, Williams RT, Druker BJ, Perrotti D, et al. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell. 2010;18:74–87. doi: 10.1016/j.ccr.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krebs J, Agellon LB, Michalak M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem Biophys Res Commun. 2015;460:114–121. doi: 10.1016/j.bbrc.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 58.Bilmen JG, Wootton LL, Michelangeli F. The inhibition of the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase by macrocyclic lactones and cyclosporin A. Biochem J. 2002;366:255–263. doi: 10.1042/bj20020431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chelu MG, Danila CI, Gilman CP, Hamilton SL. Regulation of ryanodine receptors by FK506 binding proteins. Trends Cardiovasc Med. 2004;14:227–234. doi: 10.1016/j.tcm.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 60.Mahon FX, Belloc F, Lagarde V, Chollet C, Moreau-Gaudry F, Reiffers J, Goldman JM, Melo JV. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood. 2003;101:2368–2373. doi: 10.1182/blood.V101.6.2368. [DOI] [PubMed] [Google Scholar]
- 61.Nobili S, Landini I, Giglioni B, Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets. 2006;7:861–879. doi: 10.2174/138945006777709593. [DOI] [PubMed] [Google Scholar]