

Abstract
E. rhusiopathiae is the causative agent of erysipelas in animals and erysipeloid in humans, but its pathogenicity is poorly understood. To identify virulence factors associated with E. rhusiopathiae and screen engineered vaccine candidates, we used proteomics and transcriptomics to compare the highly virulent strain HX130709 with an isogenic avirulent derivative, HX130709a. 1,299 proteins and 1,673 transcribed genes were identified and 1,292 of the proteins could be associated with genes. In a comparison between HX130907 and HX130709a, 168 proteins and 475 genes exhibited differences in regulation level. Among these, levels for 61 proteins and transcripts were positively or negatively correlated. Gene Ontology (GO) analysis suggests that many of the down-regulated proteins in the attenuated strain have catalytic or binding functions. Potential protein-protein interactions suggest that some of the down-regulated proteins may regulate PTS, GMP synthase and ribosomal proteins. Morphological results showed that HX130709 and HX130709a have similar colony and capsule morphology. Growth curves and pyruvate measurements suggest that TCA cycle and saccharide phosphorylation levels were decreased and gluconeogenesis was increased in HX130709a. Our study confirms that SpaA and neuraminidase, but not hyaluronidase and capsule, are associated with virulence in E. rhusiopathiae. We conclude that the virulence of E. rhusiopathiae may be associated with slow reactions of the TCA cycle and down-regulation of selected proteins.
Introduction
Erysipelothrix rhusiopathiae (E. rhusiopathiae) is a small gram-positive, slender, and straight rod-shaped bacterium that can cause erysipelas in swine and other animals, including sheep, fish, reptiles and birds. E. rhusiopathiae is also an important pathogen in humans and is the causative agent of erysipeloid, a skin disease [1]. The bacterium can be isolated from sick and healthy animals (pork, seafood, chicken) and from environment in which they live [2, 3]. E. rhusiopathiae belong to the genus Erysipelothrix along with E. tonsillarum and two other unnamed species [4]. Among the 15 serotypes of E. rhusiopathiae, serotypes 1a and 2 have the greatest impact on the swine industry [1, 5–9]. Swine erysipelas has caused serious losses in the swine industries of North America, Europe, Asia, and Australia. Swine erysipelas occurs in three forms: acute, subacute, and chronic. The characteristics of acute erysipelas in swine are sudden death or general signs of septicemia. Sub-acute erysipelas causes urticarial or diamond-skin lesions that appear as early as the second or third day after infection. The chronic form of infection can develop from acute or subacute disease, and can be characterized by localized arthritis or proliferative pathological changes in the heart (endocarditis) [10].
E. rhusiopathiae infection in humans is mostly occupationally related, those most prone include butchers, abattoir workers, veterinarians, farmers, fishermen, fish-handlers and housewives [11]. Clinical manifestations in humans are highly similar to those in swine and occur in a localized cutaneous form (erysipeloid), a generalized cutaneous form, and a septicemic form associated with endocarditis [10].
The virulence of E. rhusiopathiae varies considerably among different serotypes. Though the mechanisms of pathogenicity are poorly understood, and no toxin has been identified in this organism, several candidate virulence factors have been identified, including neuraminidase [12], capsular antigens [13], RspA and RspB[14], and the 64-66kDa antigen[15]. However, because these studies compared highly virulent with moderate or low virulence strains, the complete virulence repertoire may not have been revealed. In the present study, a highly virulent strain (HX130709) was attenuated by 70 passages on agar medium containing a gradually increasing concentration (0.0025% to 0.03%) of acriflavine dye, with attenuation confirmed by mouse pathogenicity. Proteomic and transcriptomic analyses were then used to identify differences between the attenuated strain HX130709a and its parent, HX130709, to reveal the virulence repertoire of E. rhusiopathiae.
Materials and Methods
Reagents
Reagents were obtained from the following sources: 2-D Quant Kit (GE Healthcare), TEAB (Applied Biosystems, Milan Italy), Trypsin Gold (Promega, Madison WI, USA), iTRAQ Reagents -8plex Chemistry (Applied Biosystems), TruSeq SBS KIT-HS, TruSeq PE Cluster Kit and TruSeq SR Cluster Kit (Illumina), TRIzol (Invitrogen), CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega), mouse GAPDH antibody (GeneTex), DNase I (Ambion), Terminator 5΄-phosphate-dependent exonuclease (Epicentre).
Bacterial strains and culture conditions
HX130709, a highly virulent strain isolated from a case of septicemia, has been confirmed by PCR and is pathogenic in mice. To generate HX130709a, HX130709 was attenuated by 70 passages on BHI-T80 agar containing an acriflavine dye (0.0025% to 0.03%). The reduced pathogenicity of the attenuated strain was confirmed in mice[16]. Animal experiments were approved by the Institutional Animal Care and Ethics Committee of Nanjing Agricultural University (Approval No. IACECNAU20100902).
Sample preparation
Brain Heart infusion agar and broth supplemented with 0.1% Tween 80 were used for bacterial cultivation. Pure cultures of strains HX130709 and HX130709a were grown in 500 ml of BHI broth for 4 h and 10 h, respectively. Cultures were harvested by centrifugation at 4000 rpm for 15 min at 4°C and washed three times in 1 × PBS buffer. Washed cells were collected in sterile tubes, flash frozen in liquid nitrogen, and submitted to BGI tech for proteomic and transcriptomic analyses. Three biological replicates were prepared independently for each sample.
Quantitative transcriptomics (RNA-seq)
RNA isolation and mRNA purification
Total RNA was isolated with TRIzol reagent using the standard protocol, and dissolved in 200 μL RNase-free water. The concentration of total RNA was determined using a NanoDrop spectrophotometer (Thermo Scientific, USA), and the RNA integrity value (RIN) was determined using the RNA 6000 Pico LabChip and an Agilent 2100 Bioanalyzer (Agilent, USA). Total RNA was incubated with 10 U DNase I at 37°C for 1 h, and then nuclease-free water was added to bring the sample volume to 250 μL. Messenger RNA was further purified by digesting ribosomal RNA and tRNA with Terminator 5΄-phosphate-dependent exonuclease. The resulting RNA samples were quantified using a DU800 spectrophotometer (Beckman Coulter, USA) and mixed with fragmentation buffer to generate short mRNA fragments.
cDNA synthesis and Illumina sequencing
cDNA was synthesized using the mRNA fragments as templates. The short cDNA fragments were purified and resuspended in EB buffer for end repair and single nucleotide A (adenine) addition, then ligated to adapters. After agarose gel electrophoresis, appropriately sized products were selected as templates for PCR amplification. During the QC steps, the Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System were used to monitor sample library quantity and quality. The library was sequenced using the Illumina HiSeq™ 2000 high-throughput sequencing system.
Bioinformatics analysis
Sequencing reads were mapped to the reference genome Fujisawa (NC_015601) using BLASTN with a threshold e value of 0.00001 and the “-F F” parameter [17], which allowed alignments with up to two mismatches. Reads that mapped to rRNA genes or that failed to align using these parameters were excluded from further analysis. The read totals were expressed as RPKM (Reads Per Kilo bases per Million reads) [18] for each gene, and then differently regulated genes were identified using the DEGseq package and the MARS (MA-plot-based method with Random Sampling model) method [19]. We used FDR ≤ 0.001 and an absolute value of log2Ratio ≥ 1 as thresholds to judge the significance of differences in gene expression.
Quantitative proteomics
Protein preparation
Harvested bacteria were washed three times with ice-cold phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10.1 mM Na2HPO4, 1.8mM KH2PO4, pH 7.4). The supernatant was discarded after the final centrifugation at 12,000 × g for 30 min. The pellets were extracted with lysis buffer (7 M Urea, 2 M Thiourea, 4% CHAPS, 40 mM Tris-HCl, pH 8.5) with a final concentration of 1mM PMSF and 2mM EDTA. After 5 min of vortexing, DTT was added to the samples to a final concentration of 10 mM. The samples were sonicated at 200 W for 15 min and then centrifuged at 4°C, 30,000 × g for 15 min. The samples were mixed well with 5× volume of ice-cold acetone containing 10% (v/v) TCA and incubated at –20°C overnight. After centrifugation at 4°C, 30,000 × g, the supernatant was discarded. The precipitate was washed with ice-cold acetone three times. The pellet was vacuum-dried and dissolved in lysis buffer (7M urea, 2 M thiourea, 4% NP40, 20mM Tris-HCl, pH 8.0–8.5). The samples were sonicated at 200 W for 15 min and centrifuged at 4°C, 30,000 × g for 15 min, and then the supernatant was transferred to a new tube. To reduce disulfide bonds in proteins, 10 mM DTT (final concentration) was added and samples were incubated at 56°C for 1 h. Subsequently, 55 mM IAM (final concentration) was added to block the cysteines, and samples were incubated for 1 h in the dark. The supernatant was mixed well with 5× volume of ice-cold acetone for 2 h at –20°C to precipitate proteins. After centrifugation at 4°C, 30,000 × g, the supernatant was discarded and the pellet was vacuum-dried for 5 min. The samples were then dissolved in 500 μL 0.5 M TEAB and sonicated at 200 W for 15 min. Finally, samples were pelleted at 4°C, 30,000 × g for 15 min. The supernatant was transferred to a new tube and protein content quantified. Protein preparations were stored at –80°C for later analysis.
iTRAQ (Isobaric tag for relative and absolute quantitation) labeling and SCX fractionation
100 μg of total protein was withdrawn from each sample and then digested at 37°C for 16 hours with Trypsin Gold, at a protein: trypsin ratio of 30: 1. After digestion, peptides were vacuum-dried and re-dissolved in 0.5M TEAB, then processed with 8-plex iTRAQ reagent, according to the manufacturer’s protocol. Briefly, one unit of iTRAQ reagent was thawed and mixed with 24 μL isopropanol. The peptides labeled with the isobaric tags were pooled then dried by vacuum centrifugation.
SCX chromatography was performed using a LC-20AB HPLC Pump system (Shimadzu, Kyoto, Japan). The peptide mixtures labeled with isobaric tags were reconstituted in 4 mL buffer A (25 mM NaH2PO4 in 25% ACN, pH 2.7) and loaded onto a 4.6×250 mm Ultremex SCX column containing 5-μm particles (Phenomenex). The peptides were eluted with a gradient of buffer A for 10 min, at a flow rate of 1 mL/min, then eluted with 5–60% buffer B (25mM NaH2PO4, 1 M KCl in 25% ACN, pH 2.7) for 27 min, and then with 60–100% buffer B for 1 min. Prior to the next sample injection, the system was maintained in 100% buffer B for 1 min, then equilibrated with buffer A for 10 min. Elution was monitored and fractions were pooled every 1 min. The eluted peptides were grouped into 20 fractions and desalted with a Strata X C18 column (Phenomenex) then vacuum-dried.
LC-ESI-MS/MS analysis based on Q EXACTIVE
Each fraction was re-dissolved in buffer A containing 2% ACN and 0.1% FA and centrifuged at 20000 × g for 10 min. The final concentration of peptide was about 0.5 μg/μL. 10 μL supernatant was loaded by autosampler onto a 2 cm C18 trap column for analysis in a LC-20AD nanoHPLC (Shimadzu, Kyoto, Japan). The peptides were eluted onto an analytical C18 column (inner diameter 75 μm) packed in-house. The samples were loaded at 8 μL/min for 5 min, then the column was run with a gradient from 2% to 35% in buffer B (98% ACN, 0.1% FA) at 300 nL/min for 35 min. A linear gradient to 80% was run through the column for 2 min, 80% buffer B was maintained for 4 min then returned to 5% for 1 min.
The peptides subjected to nanoelectrospray ionization were analyzed by tandem mass spectrometry (MS/MS) in a Q EXACTIVE instrument (Thermo Fisher Scientific, San Jose, CA) coupled online with the HPLC. Orbitrap was used to detect intact peptides at a resolution of 70,000. MS/MS was used to select peptides in high-energy collision dissociation (HCD) operating mode with a normalized collision energy setting of 27.0 and Orbitrap was set at a resolution of 17,500. The 15 most abundant precursor ions above a threshold ion count of 20,000 in the MS scan with a subsequent Dynamic Exclusion duration of 15 s were analyzed with a data-dependent procedure that alternated between one MS scan followed by 15 MS/MS scans. The electrospray voltage was set as 1.6 kV. Automatic gain control (AGC) was applied to optimize the spectra generated by the Orbitrap. The AGC target for full MS was 3e6 and 1e5 for MS2. For MS scans, the m/z scan range was between 350 and 2,000 Da. For MS2 scans, the m/z scan range was between 100 and 1,800.
Data analysis
Raw data files obtained from the Orbitrap were converted into MGF format using Proteome Discoverer 1.2 (PD 1.2, Thermo), [5600 msconverter]. Proteins were identified using the Mascot search engine (Matrix Science, London, UK; version 2.3.02) against a database (downloaded from NCBI http://www.ncbi.nlm.nih.gov/protein/?term=txid1648[Organism:exp]) containing 3,409 sequences. For protein identification, a mass tolerance of 20 Da (ppm) was used for intact peptide masses and 0.05 Da for fragmented ions, and one missing trypsin cleavage was permitted in the trypsin digests. Gln->pyro-Glu (N-term Q), Oxidation (M), and Deamidated (NQ) were specified as potential modifications, and Carbamidomethyl (C), iTRAQ8plex (N-term), and iTRAQ8plex (K) were set as invariable modifications. Peptide charge states were set to +2 and +3. We included an automatic decoy database search as part of the analysis. In this option, whenever a protein sequence from the target database is tested, a random decoy sequence of the same length is also tested. To reduce false positives, proteins required support from at least one unique peptide, and only peptides that exceeded “identity” at 99% confidence were accepted.
For protein quantitation, at least two unique peptides were required. The median ratio by Mascot was used to weight and normalize quantitative protein ratios. Proteins with p-values < 0.05 and fold changes >1.5 were considered to be significantly different between samples.
Functional annotation
The functional annotation of proteins was conducted using the Blast2GO application and the non-redundant (NR) protein database at NCBI. The KEGG database (http://www.genome.jp/kegg/) and the COG protein database (http://www.ncbi.nlm.nih.gov/COG/) were used to classify and group the identified proteins.
Growth curves
A single colony of HX130709 (F0) and HX130709a (F70) was used to inoculate overnight cultures in BHI medium. 300 μL overnight culture was inoculated into 30 mL BHI medium and incubated at 200 rpm. 500 μL samples were collected in triplicate after 1 h and centrifuged at 12,000 rpm for 3 min. The supernatant was discarded, pelleted bacteria were re-suspended in an equal volume of PBS, and optical density (OD600 nm) was measured.
Morphological observations
HX130709 and the attenuated derivative strain HX130709a were streaked onto BHI agar containing 0.1% Tween 80, incubated overnight, and colony morphology observed. Bacteria cultured to log phase were pelleted and fixed with 2.5% glutaraldehyde and then sectioned. Thin sections were stained with uranyl acetate and lead citrate and examined with a Hitachi H-7500 transmission electron microscope.
Lactate dehydrogenase activity
Bacteria were cultured to mid-logarithmic phase in BHI medium at 37°C. Triplicate samples were collected for each of 3 time points for the virulent strain HX130709 (5h, 6h, 7h) and the attenuated strain HX13079a (8h, 9h, 10h). Each 10 mL sample was centrifuged at 6,000 rpm for 10 min, then washed in PBS 3 times. Pelleted bacteria were resuspended in 500 μL PBS and lysed by sonication. The lysate was centrifuged at 12,000 rpm for 1 min and supernatant collected. 50 μL supernatant was added to wells in 96-well plates, lactate dehydrogenase (LDH) substrate was added, and the plates were incubated at 37°C for 30 min, following the protocol accompanying the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit. Color development was halted by the addition of stop solution and optical density at 490 nm (OD490) was recorded using an ELISA plate reader (BioTek, USA). In parallel, sampled bacteria were plated at a final dilution of 10−7 on BHI agar plates to determine bacterial concentration (colony forming units, CFU). Results were expressed as LDH/cfu.
Results and Discussion
Summary of RNA-seq and iTRAQ data
Transcriptome data were obtained by high-throughput RNA-sequencing as described in materials and methods. Three RNA samples were analyzed from HX130709 and its attenuated derivative HX130709a (six samples total). After excluding low-scoring sequence reads, the average read length was 90 bp and the total reads obtained were 3,675,074, 4,692,139 and 3,839,099 for samples HX130709-1, -2, -3 respectively, and 3,251,469, 4,755,689 and 5,151,752 for samples HX130709a-1, -2, -3, respectively. Correlation analysis among the replicates of HX130709, and among the replicates of HX130709a, generated Pearson’s correlation coefficients over 0.95 except in two cases. Because the coefficients for HX130709a-1 vs. HX130709a-2 (0.206) and HX130709a-1 vs. HX130709a-3 (0.2008) were unacceptably low, the HX130709a-1 data set was discarded. In spite of this loss, the mean sequencing coverage for both strains was 193–259 fold. Reads were mapped to the E. rhusiopathiae genome sequence (Fujisawa) using Soap (v2.01). The results indicated that the transcriptional percentages for ORFs encoded by the Fujisawa strain were 96.37% and 84.58% for HX130709 and HX130709a, respectively. Surprisingly, no SNPs were found in HX130709 or HX130709a. Gene differential regulation was analyzed using the DEGseq software package. Out of 1,673 genes examined, 197 genes exhibited increased transcription in HX130709a and 278 genes exhibited decreased transcription, relative to the levels in HX130709 (S1 Table).
Three biological replicates for each strain were included in the iTRAQ experiment comparing virulent HX130709 and avirulent HX130709a. After trypsinization and labeling with distinct isobaric tags, protein fragments in the six samples were separated and identified by LC-ESI-MS/MS. Because the error distribution analysis among the replicates of HX130709, and among the replicates of HX130709a, showed that the mean errors for HX130709a-1 with replicas -2 and -3 were 1.37 and 1.43, respectively, the data obtained from HX130709a-1 was again set aside. In the five remaining samples, a total of 312,350 mass spectra were generated. After excluding low-scoring spectra, 79,681 unique spectra that matched to specific peptides were obtained. Mascot analysis identified 12,154 peptides, 11,655 unique peptides, and 1,299 proteins in total, in the five samples (S1 File). Covariance distribution analysis for these samples yielded a mean covariance of 0.063 (S1 Fig), indicating good reproducibility between biological replicates in each group. A protein with ≥ 1.5-fold difference and p-value ≤ 0.05 was regarded as being differentially regulated. Relative to protein levels in the virulent strain HX130709, the abundance of 101 proteins increased, and 67 proteins decreased, in HX130709a (Table 1).
Table 1. Differentially regulated proteins identified in proteome and transcriptome (HX130709a/HX130709).
Protein level differences with p-values < 0.05, and fold changes of >1.5 were considered significant.
| Accession | Description | Cov | Unique Peptide | ratio | functions | mRNA expression | Gene ID |
|---|---|---|---|---|---|---|---|
| Up-regulation proteins (101) | |||||||
| gi|336065729 | hypothetical protein | 33 | 6 | 2.005 | Unknown | ||
| gi|322464469 | polysaccharide deacetylase | 57.2 | 23 | 1.563 | Predicted xylanase/chitin deacetylase | ||
| gi|322462985 | LytTr DNA-binding domain protein | 35.9 | 4 | 1.502 | Response regulator of the LytR/AlgR family | ||
| gi|336065511 | two-component system response regulator | 62.2 | 13 | 1.949 | Response regulator of the LytR/AlgR family | up | ERH_0269 |
| gi|322463071 | amino acid permease | 2.1 | 1 | 1.606 | Amino acid transporters | up | ERH_0305 |
| gi|336066470 | hypothetical protein | 74.3 | 4 | 1.718 | Unknown | down | ERH_1233 |
| gi|336065272 | ferric uptake regulator family protein | 58.3 | 7 | 1.56 | Fe2+/Zn2+ uptake regulation proteins | ||
| gi|322463447 | ABC transporter, substrate-binding protein, QAT family | 44.1 | 10 | 6.58 | Periplasmic glycine betaine/choline-binding (lipo)protein of an ABC-type transport system (osmoprotectant binding protein) | up | ERH_1627 |
| gi|336065597 | glutaredoxin-like protein NrdH | 75 | 5 | 2.394 | Thiol-disulfide isomerase and thioredoxins | down | ERH_0356 |
| gi|336066593 | peptidase, M23B family | 3.2 | 2 | 1.565 | Membrane proteins related to metalloendopeptidases | ||
| gi|336066734 | spermidine/putrescine ABC transporter ATP-binding protein | 70.3 | 16 | 2.416 | ABC-type spermidine/putrescine transport systems, ATPase components | up | ERH_1498 |
| gi|336066419 | hypothetical protein | 7.1 | 2 | 1.666 | Unknown | ||
| gi|322464148 | hypothetical protein HMPREF0357_10136 | 17.5 | 5 | 1.512 | Predicted nucleotidyltransferase | ||
| gi|336066664 | LPXTG-motif cell wall anchor domain-containing protein | 9 | 13 | 1.535 | Unknown | ||
| gi|489869940 | signal peptidase | 37.9 | 2 | 2.437 | Unknown | down | ERH_0786 |
| gi|489872223 | PTS cellobiose transporter subunit IIC | 6.8 | 3 | 6.19 | Phosphotransferase system cellobiose-specific component IIC | up | ERH_0219 |
| gi|322464339 | biotin/lipoate A/B protein ligase family protein | 40.9 | 7 | 1.554 | Lipoate-protein ligase A | down | ERH_0787 |
| gi|322464475 | helix-turn-helix protein, YlxM/p13 family | 27.8 | 3 | 1.646 | Uncharacterized protein conserved in bacteria | ||
| gi|322463067 | response regulator receiver domain protein | 32.5 | 6 | 1.514 | Response regulators consisting of a CheY-like receiver domain and a winged-helix DNA-binding domain | ||
| gi|489869961 | carbamate kinase | 30.9 | 7 | 1.809 | Carbamate kinase | down | ERH_0797 |
| gi|336066177 | ABC transporter ATP-binding protein | 49.2 | 8 | 1.662 | ABC-type multidrug transport system, ATPase component | up | ERH_0939 |
| gi|336066253 | formamidopyrimidine-DNA glycosylase | 23.4 | 4 | 1.916 | Formamidopyrimidine-DNA glycosylase | ||
| gi|336066573 | copper chaperone | 71.4 | 4 | 1.805 | Unknown | ||
| gi|336065993 | oxaloacetate decarboxylase subunit alpha | 38.5 | 13 | 4.116 | Pyruvate/oxaloacetate carboxyltransferase | ||
| gi|336066878 | hypothetical protein | 13.6 | 1 | 2.808 | Unknown | ||
| gi|336066225 | hypothetical protein | 8.5 | 2 | 1.665 | ABC-type uncharacterized transport system, permease component | ||
| gi|489870987 | thioredoxin | 69.6 | 5 | 1.964 | Thiol-disulfide isomerase and thioredoxins | down | ERH_1500 |
| gi|322463512 | hypothetical protein HMPREF0357_10813 | 52.4 | 5 | 1.99 | Unknown | ||
| gi|336065984 | triphosphoribosyl-dephospho-CoA synthase | 19.6 | 4 | 2.114 | Triphosphoribosyl-dephospho-CoA synthetase | up | ERH_0746 |
| gi|322464227 | CAAX amino terminal protease family protein | 6.2 | 2 | 1.789 | Predicted metal-dependent membrane protease | ||
| gi|336066870 | hypothetical protein | 36.1 | 4 | 2.983 | Unknown | ||
| gi|336066016 | hypothetical protein | 57.3 | 7 | 2.395 | Uncharacterized conserved protein | ||
| gi|322463205 | glycerophosphodiester phosphodiesterase family protein | 18.6 | 10 | 1.705 | Glycerophosphoryl diester phosphodiesterase | ||
| gi|336066010 | prolyl aminopeptidase | 30 | 7 | 1.914 | Predicted hydrolases or acyltransferases (alpha/beta hydrolase superfamily) | up | ERH_0772 |
| gi|322463949 | hypothetical protein HMPREF0357_11250 | 57.6 | 6 | 1.981 | Predicted DNA-binding protein with PD1-like DNA-binding motif | ||
| gi|336066178 | hypothetical protein | 9.7 | 5 | 1.987 | Unknown | ||
| gi|336066448 | TetR family transcriptional regulator | 34.8 | 6 | 1.53 | Transcriptional regulator | ||
| gi|336065561 | Crp/Fnr family transcriptional regulator | 12.1 | 2 | 2.196 | Predicted transcriptional regulators | ||
| gi|336065842 | hypothetical protein | 40 | 2 | 2.474 | Unknown | down | ERH_0602 |
| gi|336065758 | tyrosine recombinase XerC | 35.1 | 8 | 1.617 | Site-specific recombinase XerD | ||
| gi|336065996 | oxaloacetate decarboxylase subunit beta | 10.8 | 4 | 2.757 | Na+-transporting methylmalonyl-CoA/oxaloacetate decarboxylase, beta subunit | ||
| gi|336065500 | endonuclease/exonuclease/phosphatase family protein | 11 | 3 | 1.852 | Unknown | ||
| gi|489872418 | methionine sulfoxide reductase A | 55.2 | 5 | 1.982 | Peptide methionine sulfoxide reductase | ||
| gi|336066213 | putative metalloprotease | 25.9 | 3 | 1.62 | Predicted metal-dependent hydrolase | ||
| gi|489871071 | peptide ABC transporter ATP-binding protein | 16.8 | 4 | 2.263 | ABC-type antimicrobial peptide transport system, ATPase component | ||
| gi|336065450 | D-alanine—poly(phosphoribitol) ligase subunit 2 | 31.6 | 2 | 1.769 | Unknown | down | ERH_0208 |
| gi|489871733 | ABC transporter | 11.6 | 1 | 4.118 | ATPase components of ABC transporters with duplicated ATPase domains | ||
| gi|322463931 | BadF/BadG/BcrA/BcrD ATPase family protein | 31.6 | 7 | 1.671 | Predicted N-acetylglucosamine kinase | ||
| gi|336065340 | acid phosphatase/vanadium-dependent haloperoxidase related protein | 18.2 | 2 | 1.864 | Uncharacterized protein conserved in bacteria | up | ERH_0098 |
| gi|322463944 | acetyltransferase, GNAT family | 30.6 | 4 | 1.693 | Histone acetyltransferase HPA2 and related acetyltransferases | ||
| gi|336066251 | replication initiation and membrane attachment protein | 35.8 | 13 | 1.775 | Replication initiation/membrane attachment protein | ||
| gi|322463437 | HD domain protein | 24.4 | 8 | 1.55 | HD superfamily phosphohydrolases | ||
| gi|336066440 | hypothetical protein | 67.2 | 6 | 1.878 | Unknown | down | ERH_1203 |
| gi|336065559 | N-acetyltransferase GCN5 | 14.6 | 2 | 1.858 | Acetyltransferases, including N-acetylases of ribosomal proteins | ||
| gi|336066518 | hypothetical protein | 10.3 | 1 | 2.093 | Unknown | down | ERH_1282 |
| gi|489869871 | citrate lyase | 34 | 5 | 1.838 | Phosphoribosyl-dephospho-CoA transferase (holo-ACP synthetase) | ||
| gi|336065544 | YeeE/YedE family integral membrane protein | 21.4 | 8 | 1.68 | Predicted transporter component | up | ERH_0303 |
| gi|336065463 | glycoside hydrolase | 23.6 | 11 | 4.251 | Beta-glucanase/Beta-glucan synthetase | up | ERH_0221 |
| gi|336065828 | hypothetical protein | 34.1 | 3 | 1.767 | Unknown | down | ERH_0588 |
| gi|489869873 | citrate lyase subunit gamma | 27.6 | 2 | 3.173 | Citrate lyase, gamma subunit | ||
| gi|336066517 | copper chaperone | 17.3 | 1 | 2.156 | Copper chaperone | ||
| gi|322463327 | ABC 3 transport family protein | 10.3 | 4 | 2.911 | ABC-type Mn2+/Zn2+ transport systems, permease components | ||
| gi|336065783 | MarR family transcriptional regulator | 17.7 | 2 | 2.505 | Unknown | ||
| gi|336065994 | putative oxaloacetate decarboxylase subunit gamma | 18 | 1 | 3.612 | Transcriptional regulators | ||
| gi|322463480 | transcriptional regulator, LysR family | 16.8 | 4 | 1.506 | Unknown | ||
| gi|29603463 | rhusiopathiae surface protein B | 4.7 | 3 | 3.364 | Transcriptional regulator | ||
| gi|336065820 | host cell surface-exposed lipoprotein | 17.6 | 2 | 1.502 | Unknown | ||
| gi|336066586 | D-methionine ABC transporter ATP-binding protein | 61.8 | 3 | 2.205 | Unknown | ||
| gi|336065271 | hypothetical protein | 51 | 2 | 1.635 | ABC-type metal ion transport system, ATPase component | up | ERH_0029 |
| gi|322463387 | LPXTG-motif cell wall anchor domain protein | 16.6 | 1 | 2.225 | Uncharacterized conserved protein | up | ERH_1687 |
| gi|322463680 | ABC transporter, permease protein | 2.3 | 1 | 17.319 | Unknown | down | ERH_1245 |
| gi|336066678 | glycosyltransferase family protein | 32.8 | 11 | 1.543 | ABC-type sugar transport systems, permease components | down | ERH_1442 |
| gi|336065843 | hypothetical protein (Glycosyltransferases) | 15.4 | 2 | 1.706 | Glycosyltransferases involved in cell wall biogenesis | ||
| gi|336066252 | dephospho-CoA kinase | 25.6 | 4 | 2.513 | Predicted membrane protein | ||
| gi|322464309 | Biotin-requiring enzyme | 19.6 | 2 | 6.095 | Dephospho-CoA kinase | up | ERH_0757 |
| gi|322463815 | LPXTG-motif cell wall anchor domain protein | 13.5 | 11 | 2.184 | Biotin carboxyl carrier protein | ||
| gi|336065715 | hypothetical protein | 18.1 | 5 | 1.712 | Unknown | up | ERH_0475 |
| gi|322464470 | ATPase/histidine kinase/DNA gyrase B/HSP90 domain protein | 35.2 | 12 | 2.296 | Predicted periplasmic solute-binding protein | ||
| gi|322464365 | hypothetical protein HMPREF0357_10353 | 5.8 | 1 | 3.662 | Signal transduction histidine kinase | ||
| gi|322463326 | ABC 3 transport family protein | 8.3 | 3 | 2.436 | Unknown | down | ERH_0047 |
| gi|336066379 | XRE family transcriptional regulator | 54.4 | 3 | 2.366 | ABC-type Mn2+/Zn2+ transport systems, permease components | ||
| gi|509078903 | ABC transporter, permease protein | 6.6 | 2 | 1.617 | Predicted transcriptional regulators | down | ERH_1244 |
| gi|336066105 | ACT domain-containing protein | 30.3 | 3 | 1.705 | ABC-type sugar transport system, permease component | up | ERH_0867 |
| gi|322464145 | putative endoribonuclease L-PSP | 27.1 | 3 | 1.806 | ACT domain-containing protein | ||
| gi|336065349 | DNA binding helix-turn helix protein | 52.9 | 4 | 1.579 | Putative translation initiation inhibitor, yjgF family | ||
| gi|336066891 | MutT/NUDIX family protein | 48.6 | 7 | 3.258 | Predicted transcriptional regulator | ||
| gi|336066864 | glycine betaine/carnitine/choline ABC transporter permease | 21.1 | 3 | 8.34 | NTP pyrophosphohydrolases including oxidative damage repair enzymes | up | ERH_1629 |
| gi|322463363 | DNA replication and repair protein RecF | 9.4 | 3 | 1.701 | ABC-type proline/glycine betaine transport systems, permease component | up | ERH_0007 |
| gi|336066579 | purine nucleosidase | 50.5 | 13 | 1.543 | Recombinational DNA repair ATPase (RecF pathway) | ||
| gi|336066735 | XRE family transcriptional regulator | 42.9 | 5 | 2.366 | Inosine-uridine nucleoside N-ribohydrolase | ||
| gi|336066556 | N-acetyltransferase GCN5 | 2.1 | 1 | 1.665 | Predicted transcriptional regulators | ||
| gi|489871234 | PTS glucose transporter subunit IIBC | 15.3 | 5 | 2.049 | Acetyltransferases, including N-acetylases of ribosomal proteins | down | ERH_1399 |
| gi|336065655 | ABC transporter permease | 6.1 | 2 | 1.522 | Phosphotransferase system IIC components, glucose/maltose/N-acetylglucosamine-specific | up | ERH_0414 |
| gi|336065992 | citrate-sodium symporter | 9.7 | 4 | 2.961 | ABC-type sugar transport systems, permease components | ||
| gi|336065983 | GntR family transcriptional regulator | 37.6 | 8 | 1.991 | Na+/citrate symporter | ||
| gi|336066469 | hypothetical protein | 16.9 | 2 | 1.753 | Transcriptional regulators | ||
| gi|336066654 | MutT/NUDIX family protein | 8.8 | 1 | 1.759 | Unknown | ||
| gi|336065454 | two-component system sensor histidine kinase | 25.8 | 8 | 2.469 | ADP-ribose pyrophosphatase | ||
| gi|336066363 | ABC transporter ATP-binding protein | 19.7 | 3 | 1.696 | Signal transduction histidine kinase | ||
| gi|489870514 | beta-carotene 15,15~-monooxygenase | 12.3 | 3 | 1.533 | ATPase components of ABC transporters with duplicated ATPase domains | up | ERH_0064 |
| gi|336066325 | hypothetical protein ERH_1087 | 16.8 | 3 | 1.858 | Uncharacterized conserved protein | up | ERH_1087 |
| Down-regulation proteins (67) | |||||||
| gi|336066742 | hypothetical protein | 25.5 | 4 | 0.52 | Unknown | ||
| gi|336066589 | aspartate—ammonia ligase | 32.6 | 9 | 0.407 | Asparagine synthetase A | down | ERH_1353 |
| gi|336066753 | amino acid ABC transporter amino acid-binding protein | 17.9 | 4 | 0.294 | ABC-type amino acid transport/signal transduction systems, periplasmic component/domain | down | ERH_1517 |
| gi|336065791 | methionine adenosyltransferase | 50.8 | 15 | 0.553 | S-adenosylmethionine synthetase | ||
| gi|336065393 | putative D-alanyl-D-alanine carboxypeptidase | 13.1 | 5 | 0.552 | D-alanyl-D-alanine carboxypeptidase | ||
| gi|489869850 | bacteriocin ABC transporter ATP-binding protein | 33.8 | 4 | 0.55 | ABC-type antimicrobial peptide transport system, ATPase component | ||
| gi|336066913 | hypothetical protein | 24.9 | 18 | 0.277 | Unknown | down | ERH_1678 |
| gi|322463568 | cell envelope-like function transcriptional attenuator common domain protein | 14.3 | 7 | 0.617 | Transcriptional regulator | ||
| gi|336065737 | putative SUF system FeS cluster assembly protein SufD | 37.9 | 8 | 0.637 | ABC-type transport system involved in Fe-S cluster assembly, permease component | ||
| gi|322463946 | putative Na/Pi-cotransporter II-like protein | 21.6 | 10 | 0.584 | Na+/phosphate symporter | ||
| gi|105303396 | surface protective antigen SpaA | 41.5 | 1 | 0.248 | FOG: Glucan-binding domain (YG repeat) | down | ERH_0094 |
| gi|336065502 | subtilase familycell-envelope associated proteinase | 4.5 | 4 | 0.503 | Subtilisin-like serine proteases | ||
| gi|336065501 | cold-shock protein | 74.2 | 4 | 0.503 | Cold shock proteins | ||
| gi|336065445 | Na+ efflux pump ABC transporter permease | 27 | 10 | 0.245 | ABC-type Na+ efflux pump, permease component | down | ERH_0203 |
| gi|336066925 | guanosine monophosphate reductase 2 | 58.3 | 16 | 0.558 | IMP dehydrogenase/GMP reductase | down | ERH_1690 |
| gi|336065366 | type III pantothenate kinase | 22.3 | 4 | 0.571 | Putative transcriptional regulator, homolog of Bvg accessory factor | down | ERH_0124 |
| gi|336066690 | LPXTG-motif cell wall anchor domain-containing protein | 27.7 | 6 | 0.179 | Unknown | down | ERH_1454 |
| gi|336065396 | hypothetical protein | 54.7 | 19 | 0.196 | Unknown | down | ERH_0154 |
| gi|18146962 | Tet(M) | 44.6 | 21 | 0.246 | Translation elongation factors (GTPases) | ||
| gi|322463820 | hypothetical protein HMPREF0357_11121 | 15.6 | 4 | 0.485 | Unknown | down | ERH_1253 |
| gi|489869985 | hypothetical protein | 8.3 | 1 | 0.493 | Unknown | down | ERH_0811 |
| gi|322463107 | transcriptional regulator, TetR family | 24.7 | 4 | 0.581 | Unknown | ||
| gi|489871386 | dipeptidase | 58.5 | 9 | 0.339 | Peptidase E | down | ERH_1312 |
| gi|322464532 | response regulator receiver domain protein | 24.6 | 5 | 0.444 | Response regulators consisting of a CheY-like receiver domain and a winged-helix DNA-binding domain | down | ERH_0982 |
| gi|336066596 | ABC transporter permease | 34.2 | 12 | 0.54 | ABC-type antimicrobial peptide transport system, permease component | ||
| gi|336065388 | inositol monophosphatase family protein | 45.5 | 9 | 0.159 | Archaeal fructose-1,6-bisphosphatase and related enzymes of inositol monophosphatase family | ||
| gi|336066608 | ABC transporter ATP-binding protein | 35.2 | 6 | 0.508 | ABC-type multidrug transport system, ATPase component | ||
| gi|336066551 | peptidase, M42 family | 46.4 | 11 | 0.536 | Cellulase M and related proteins | down | ERH_1315 |
| gi|489871941 | pyrrolidone-carboxylate peptidase | 59.2 | 11 | 0.393 | Pyrrolidone-carboxylate peptidase (N-terminal pyroglutamyl peptidase) | down | ERH_0382 |
| gi|336066606 | enterochelin ABC transporter substrate-binding protein | 39.1 | 9 | 0.48 | ABC-type enterochelin transport system, periplasmic component | ||
| gi|336066243 | basic membrane lipoprotein | 42.1 | 10 | 0.558 | Uncharacterized ABC-type transport system, periplasmic component/surface lipoprotein | ||
| gi|336065553 | two-component system response regulator | 34.2 | 6 | 0.537 | Response regulators consisting of a CheY-like receiver domain and a winged-helix DNA-binding domain | ||
| gi|336065967 | leucine-rich repeat protein | 13.5 | 5 | 0.64 | Leucine-rich repeat (LRR) protein | down | ERH_0728 |
| gi|322463725 | PASTA domain protein | 31.8 | 13 | 0.51 | Unknown | down | ERH_1362 |
| gi|322463102 | hypothetical protein HMPREF0357_11450 | 7.8 | 1 | 0.531 | Unknown | ||
| gi|503619403 | hypothetical protein | 37.4 | 4 | 0.629 | Unknown | ||
| gi|336066550 | hypothetical protein | 30.6 | 3 | 0.602 | Unknown | ||
| gi|336066068 | CobQ/CobB/MinD/ParA nucleotide binding domain-containing protein | 26.9 | 8 | 0.402 | Flp pilus assembly protein, ATPase CpaE | ||
| gi|336066221 | phosphate starvation-inducible protein PhoH | 64.2 | 14 | 0.238 | Phosphate starvation-inducible protein PhoH, predicted ATPase | down | ERH_0983 |
| gi|322463351 | PTS system glucoside-specific EIICBA component family protein | 23 | 9 | 0.666 | Phosphotransferase system IIC components, glucose/maltose/N-acetylglucosamine-specific | ||
| gi|489872217 | PTS glucose transporter subunit IIABC | 15.7 | 6 | 0.208 | Phosphotransferase system IIC components, glucose/maltose/N-acetylglucosamine-specific | down | ERH_0222 |
| gi|336065624 | hypothetical protein | 8.8 | 2 | 0.467 | Predicted membrane protein | down | ERH_0383 |
| gi|336066461 | 2-dehydro-3-deoxygluconokinase | 28.5 | 9 | 0.653 | Sugar kinases, ribokinase family | ||
| gi|322464377 | Flp/Fap pilin component | 14.3 | 1 | 0.304 | Flp pilus assembly protein, pilin Flp | down | ERH_0825 |
| gi|489869981 | ABC transporter ATP-binding protein | 47.4 | 7 | 0.186 | ABC-type multidrug transport system, ATPase component | down | ERH_0807 |
| gi|336066791 | peptide chain release factor 2 | 40.9 | 11 | 0.634 | Protein chain release factor B | ||
| gi|336065318 | N-acetyltransferase GCN5 | 22.8 | 5 | 0.642 | Histone acetyltransferase HPA2 and related acetyltransferases | ||
| gi|336066001 | hypothetical protein | 30 | 4 | 0.658 | FOG: CBS domain | ||
| gi|489872308 | flavin reductase | 37.2 | 5 | 0.628 | Predicted flavoprotein | ||
| gi|489871309 | multidrug ABC transporter ATP-binding protein | 25.2 | 6 | 0.593 | ABC-type antimicrobial peptide transport system, ATPase component | down | ERH_1361 |
| gi|489871187 | ABC transporter | 40.7 | 17 | 0.57 | ABC-type multidrug transport system, ATPase and permease components | ||
| gi|489872323 | inositol monophosphatase | 48.6 | 11 | 0.599 | Archaeal fructose-1,6-bisphosphatase and related enzymes of inositol monophosphatase family | ||
| gi|336066429 | DNA polymerase IV | 12.1 | 3 | 0.645 | Nucleotidyltransferase/DNA polymerase involved in DNA repair | ||
| gi|322464283 | hypothetical protein HMPREF0357_10271 | 6.4 | 1 | 0.555 | Unknown | down | ERH_0732 |
| gi|322463718 | ABC transporter, ATP-binding protein | 3.5 | 1 | 0.497 | ABC-type enterochelin transport system, ATPase component | ||
| gi|322463306 | hypothetical protein HMPREF0357_10606 | 5.1 | 1 | 0.649 | Unknown | ||
| gi|322464381 | Flp pilus assembly protein CpaB | 35.3 | 7 | 0.327 | Flp pilus assembly protein CpaB | ||
| gi|489870985 | diacylglycerol kinase | 27.1 | 7 | 0.655 | Sphingosine kinase and enzymes related to eukaryotic diacylglycerol kinase | ||
| gi|336065298 | sulfatase family protein | 31.7 | 15 | 0.446 | Phosphoglycerol transferase and related proteins, alkaline phosphatase superfamily | ||
| gi|336066345 | alpha/beta hydrolase domain-containing protein | 18.7 | 4 | 0.326 | Esterase/lipase | ||
| gi|322464343 | ABC transporter, ATP-binding protein | 11.2 | 6 | 0.659 | ABC-type transport system involved in cytochrome bd biosynthesis, ATPase and permease components | ||
| gi|336066648 | ABC transporter permease/ATP-binding protein | 23.6 | 14 | 0.644 | ABC-type multidrug transport system, ATPase and permease components | ||
| gi|322463081 | NMT1/THI5-like protein | 37.7 | 1 | 0.485 | ABC-type nitrate/sulfonate/bicarbonate transport systems, periplasmic components | ||
| gi|336065736 | Fe-S cluster assembly ATP-binding protein | 62 | 15 | 0.533 | ABC-type transport system involved in Fe-S cluster assembly, ATPase component | ||
| gi|336066238 | peptidase M16 domain-containing protein | 44.9 | 7 | 0.577 | Predicted Zn-dependent peptidases | ||
| gi|489871578 | PTS sugar transporter | 71.5 | 6 | 0.409 | Phosphotransferase system, mannose/fructose-specific component IIA | ||
| gi|336065707 | hemolysin-like protein | 28.7 | 10 | 0.396 | Hemolysins and related proteins containing CBS domains | ||
Functional annotation of the 1299 proteins was based on the three principal classifications developed by the gene ontology (http://www.geneontol.ogy.org). With respect to biological processes, most proteins were associated with metabolic processes (32.18%) and cellular processes (26.68%). When classified according to cellular component, 30.76% of proteins were described as located in the cell, 30.76% in cell parts, 13.14% in membranes, and 9.45% in membrane parts. Classification by molecular function showed that most proteins had catalytic (46.97%) and binding (36.54%) activities (Fig 1). Overall, the gene ontology classifications for the differentially expressed proteins are very similar to those obtained for all 1,299 proteins identified by Mascot. However, when the differentially expressed proteins are classified by molecular function, catalytic activity (50.75% vs. 44.07%) and binding (34.33% vs. 29.66%) are overrepresented.
Fig 1. Protein classification based on functional annotations using the Gene Ontology resource.
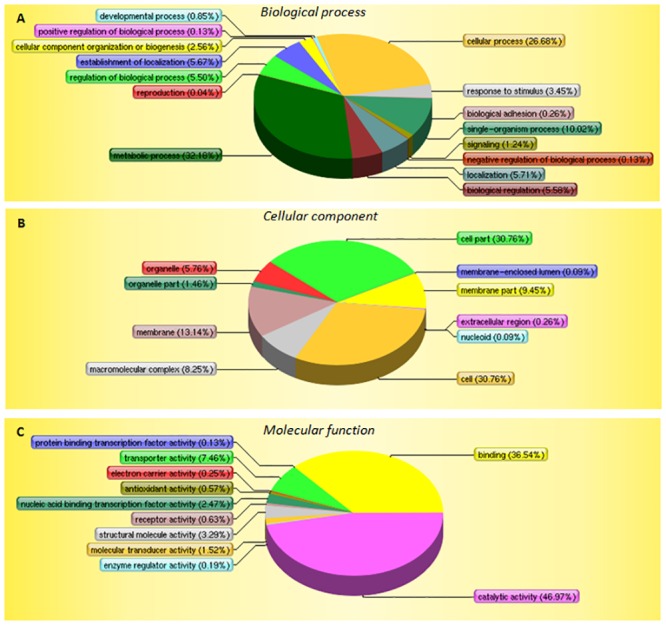
(A) GO biological processes; (B) GO molecular functions; and (C) GO cellular components. p-values were calculated using tools at the Life Science Website.
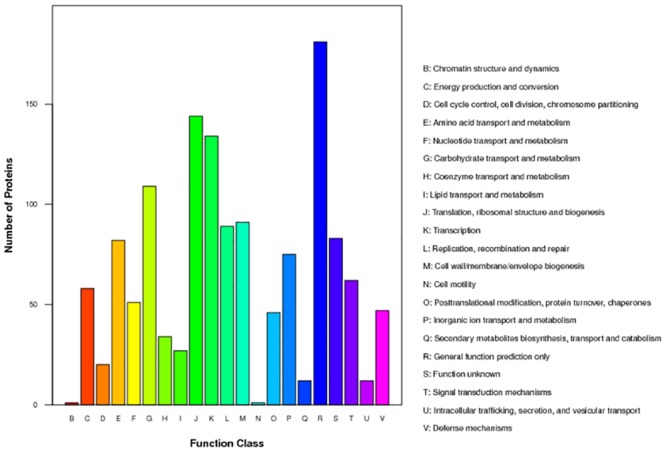
The COG database (http://www.ncbi.nlm.nih.gov/COG) was used to classify and group the identified proteins (Fig 2). The ten most common categories were as follows: [R] General function prediction (~13.93%), [J] Translation, ribosomal structure and biogenesis (~11.08%), [K] Transcription (~10.31%), [G] Carbohydrate transport and metabolism (~8.39%), [M] Cell wall/membrane/envelope biogenesis (~7.00%), [L] Replication, recombination and repair (~6.85%), [S] Function unknown (~6.39%), [E] Amino acid transport and metabolism (~6.31%), [P] Inorganic ion transport and metabolism (~5.77%), and [T] Signal transduction mechanisms (~4.77%). As expected, the majority of proteins are involved in basic cellular functions, such as replication, transcription, translation and metabolism.
Fig 2. Classification of protein functions based on COGs.
Correlation between mRNA and protein expression profiles
A coupled transcriptomics-proteomics project provides a unique opportunity to investigate whether protein regulation is correlated with gene transcription. We investigated the correlation between mRNA and protein profiles and found that the levels of 1219 proteins could be correlated, either negatively or positively, with mRNA levels. Among the 168 differentially regulated proteins, only 61 could be correlated with mRNA level variations. These proteins could be clustered into four groups based on the pattern of changes in mRNA and protein levels: Group I, the mRNA and protein levels are positively correlated (45 proteins); Group II, the mRNA level remains almost unchanged while the protein level is decreased (43 proteins); Group III, the mRNA level remains almost unchanged but the protein level is increased (64 proteins); Group IV, the mRNA level is decreased but the protein level is increased (16 proteins) (Table 1). Group I includes two subgroups: both the mRNA and protein levels are increased synchronously (21 proteins); both the mRNA and protein levels are decreased synchronously (24 proteins) (Table 1).
Protein-protein interaction analysis
Protein-protein interactions play an important role in bacterial pathogenicity and metabolism. We therefore examined the 67 down-regulated proteins for potential protein interactions using the STRING database [20]. Associations were predicted to exist among these proteins, and especially between them and components of the phosphotransferase system, GMP synthases and ribosomal proteins (Fig 3). These interactions may function in reducing nucleotide and protein synthesis and saccharide phosphorylation.
Fig 3. Potential protein–protein interaction network among down-regulated proteins as analyzed by STRING.
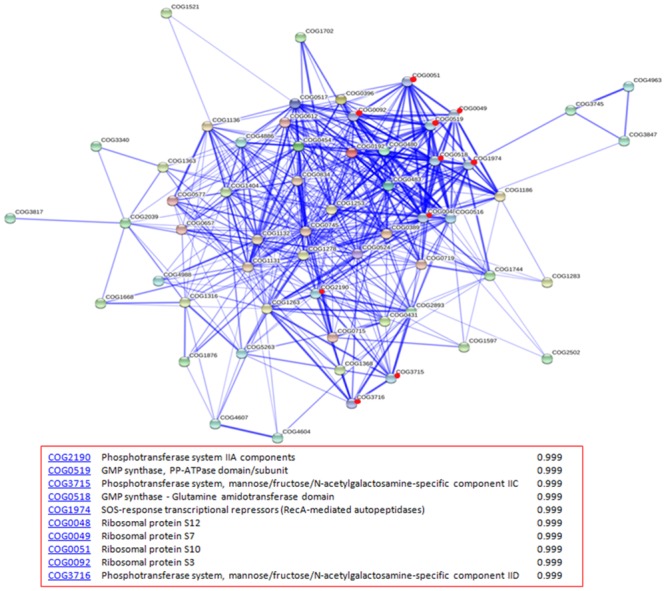
Stronger associations are represented by thicker lines. Red dots indicate predicted proteins that have strong associations with the down-regulated proteins.
Phosphotransferase system (PTS)
The phosphoenolpyruvate sugar phosphotransferase system (PTS) is widespread among microorganisms. The PTS couples carbon source uptake and substrate phosphorylation, generating intracellular sugar-phosphate [21]. Pathway analysis suggests that the phosphorylation levels of glucose, maltose, D-glucosamine, N-acetylgalatosamine and galactosamine were down-regulated (1.6–5 fold) in HX130709a, resulting in reduced glucose 6-phosphate and glyceraldehydes-3-phophate synthesis, consistent with the up-regulation of gluconeogenesis (Fig 4).
Fig 4. Pathway analysis of up- and down-regulated proteins associated with the TCA cycle.
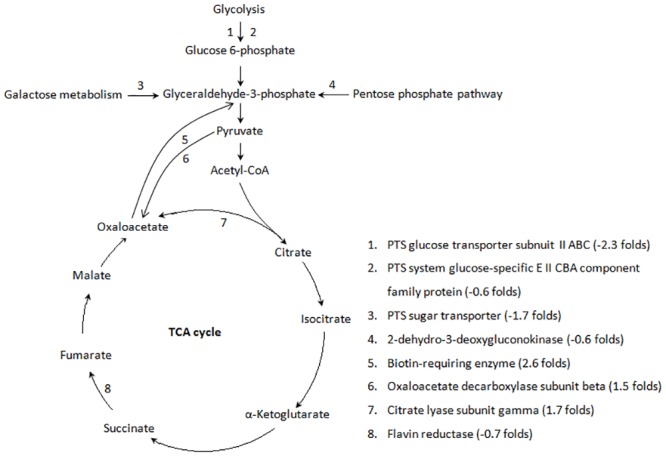
TCA Cycle
A pathway analysis focused on the TCA cycle showed that the pathway components responsible for the conversion of pyruvate and citrate to oxaloacetate were up-regulated in HX130709a relative to HX130709. In contrast, components involved in the metabolism of succinate to fumarate were down-regulated. We hypothesize that the lower levels of medium-supplied carbohydrates were used by the attenuated E. rhusiopathiae, but the cells compensated by up-regulating production of oxaloacetate and shunting excess oxaloacetate into the gluconeogenesis pathway. However, pathway components involved in the conversion of pyruvate to acetyl-CoA were not changed. Combining this fact with the down-regulation of components involved in the metabolism of succinate to fumarate, we suggest that the TCA cycle as a whole is down-regulated in HX130709a (Fig 4). Growth curves also showed that HX130709 grows more rapidly than HX130709a (Fig 5). Compared with HX130709, pyruvate levels in HX130709a may be slightly higher, but the difference is not significant (Fig 6). The colony morphology of HX130709a was convex and irregular, similar to that of HX130709. Electron microscopy showed capsule material as an electron-dense layer outside the outer membrane and no significant morphological difference between the virulent and attenuated strains (Fig 7), which is inconsistent with the result reported by Yoshihiro et al. [13, 22]. The role of the capsule in virulence has been clearly demonstrated using isogenic mutants with defined mutations. However, differences in growth curves and pyruvate metabolism between parent strains and their avirulent derivatives have not been reported, nor have differences in mRNA and protein levels. Our results demonstrate that capsule is not closely associated with virulence in E. rhusiopathiae.
Fig 5. Growth curves for HX130709 and its attenuated derivative HX130907a.
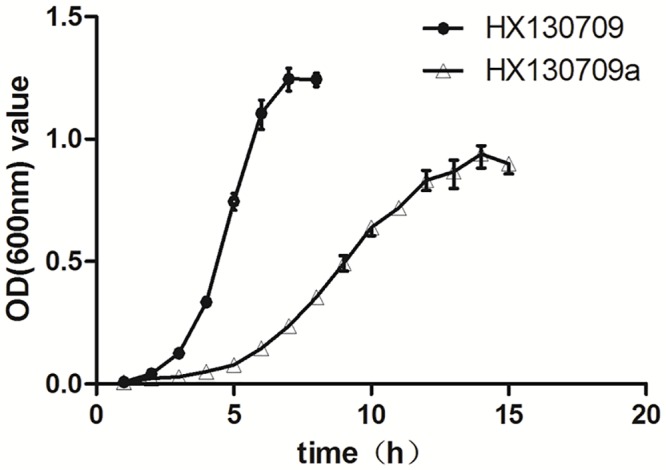
HX130709 and HX130709a were cultured and triplicate samples were collected at 1 h intervals. The samples were centrifuged and pellets suspended in an equal volume of PBS. Values shown are optical densities (600 nm).
Fig 6. Pyruvate detection with LDH assay.
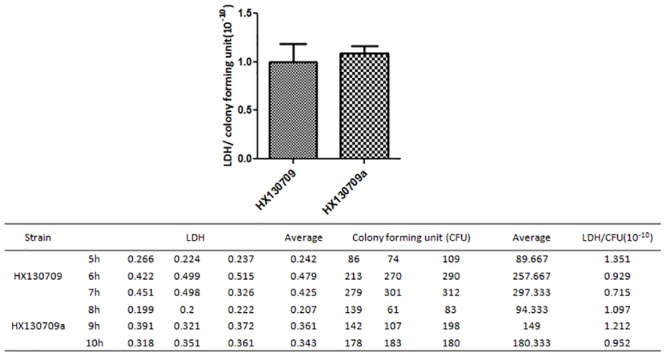
Bacteria were cultured to mid-logarithmic phase in BHI medium and triplicate samples were collected at 3 time points for the virulent strain HX130709 (5 h, 6 h, 7 h) and the attenuated strain HX13079a (8 h, 9 h, 10 h). Lactate dehydrogenase levels and bacterial counts (colony forming units; CFU) were measured as described in materials and methods. Results are expressed as LDH/cfu. Pyruvate levels are assumed to correlate positively with LDH activity.
Fig 7. Morphological observation of HX130709 and HX130709a.
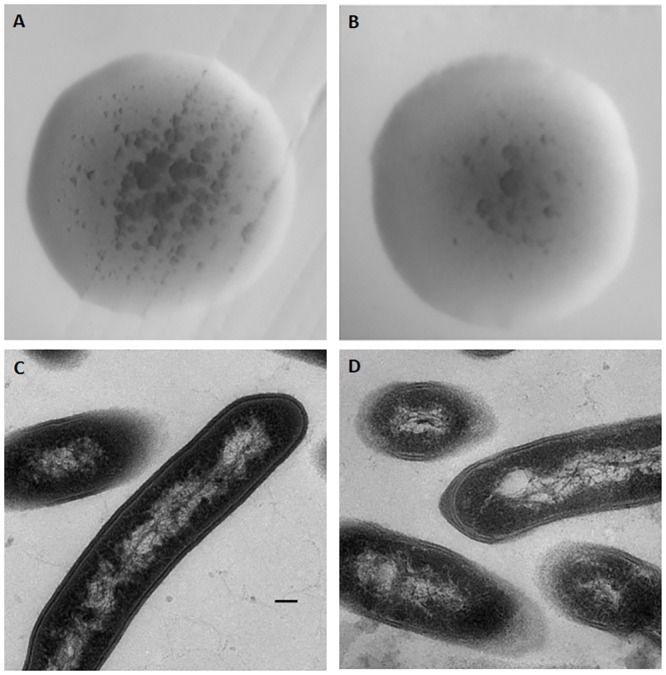
Colony morphology and electron micrographs of E. rhusiopathiae. Colony morphology of (A) HX130709 and (B) HX130709a. Electron micrographs of (C) HX130709 and (D) HX130709a. The scale bar is 100 nm.
Virulence factors
In a previous study, Kwok and Li sequenced the genome of a virulent strain of E. rhusiopathiae (SY1027) and used BLASTP and VFDB to identify 37 potential virulence factors [23]. Using a similar strategy, we examined the proteins and genes identified by proteomic and transcriptomic analyses in our study and found 13 virulence factor candidates. To our surprise, compared with HX130709, no potential virulence factors were differentially expressed in HX130709a. However, it is possible that these factors are not associated with pathogenicity in E. rhusiopathiae.
Virulence factors such as neuraminidase, hyaluronidase, RspA/RspB, SpaA, and hemolysin have been suggested to play a role in E. rhusiopathiae pathogenicity. Among the proteins and genes identified in our transcriptomic and proteomic analyses, transcriptional activity was detected for neuraminidase, hyaluronidase and spaA, which were decreased 2.7, 2.35 and 15.8 fold in HX130709a vs. HX130709, respectively. Unexpectedly, neuraminidase was not detected as a protein product, nor were transcripts detected that corresponded to the rspA/rspB genes. Since pelleted bacteria were used in the proteomic and transcriptomic analyses, it is possible that neuraminidase was secreted into the culture medium and that the mRNAs for rspA/rspB were unstable. Protein levels for hyaluronidase and RspA were stable, but protein levels for RspB and SpaA were increased and decreased 3.364 and 4 fold, respectively. Hemolysin was not detected in mRNA or protein forms, but a hemolysin-like protein was identified and its protein level was down-regulated.
Neuraminidase is an enzyme responsible for cleavage of sialic acids from sialo-glycoconjugates such as glycoproteins, glycolipids, and oligo- and polysaccharides. The down-regulation of neuraminidase has been associated with decreased virulence in E. rhusiopathiae [10]. We found that the mRNA level of neuraminidase was decreased 2.7 fold in attenuated strain HX130907a, which is consistent with this result. Shimoji et al. conducted transposon mutagenesis with Tn916 to construct mutants defective in hyaluronidase production and reported that hyaluronidase was not associated with virulence in E. rhusiopathiae [24]. Although mRNA levels for hyaluronidase decreased 2.35 fold in our study, protein expression were essentially identical in HX130709 and HX130907a, providing additional support that hyaluronidase is not associated with virulence in E. rhusiopathiae. RspA expression was also identical in the two strains, but RspB was increased in the attenuated strain HX130907a, suggesting that RspA and RspB are not associated with virulence in E. rhusiopathiae. Finally, Borrathybay et al. deleted the spaA gene from wild-type E. rhusiopathiae strain C43065 and found that the virulence of the ΔspaA mutant decreased more than 76 fold [25], but they did not compare growth curves for the wild-type and mutant strains. We also found that spaA transcription and SpaA protein levels decreased in HX130907a, supporting the hypothesis that SpaA is associated with virulence in E. rhusiopathiae.
In summary, the transcriptomes and proteomes of an attenuated E. rhusiopathiae and its parent strain were compared. 475 genes and 168 proteins were found to be up- or down-regulated and the levels for 61 proteins could be correlated with gene transcription levels. The growth of the attenuated strain is slower than its parent strain, but pyruvate metabolism appears to be unaffected. Our data are consistent with other studies showing that SpaA and neuraminidase, but not hyaluronidase and capsule, are associated with virulence in E. rhusiopathiae. We conclude that the down-regulation of the TCA cycle and the down-regulation of several proteins are associated with virulence in this organism.
Supporting Information
The difference was plotted versus the percentage of the proteins identified. Approximately 82.4% of proteins had cv differences less than 0.1, and more than 99.7% of the proteins had cv errors less than 0.5.
(PNG)
{kind=link}
(TIF)
(TIF)
(XLS)
(DOCX)
Acknowledgments
This manuscript was modified by Ms. Elizabeth G. Wills.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was supported by public welfare Grants from the Ministry of Agriculture, the People’s Republic of China (201203039) and the Priority Academic Program for the Development of Jiangsu Higher Education Institutions (PAPD).
References
- 1.Wood RL. Erysipelas Diseases of swine, 8th ed (Straw BE, D'Allaire S, Mengeling WL, et al), Iowa State University Press, Ames: 1999:419–30. [Google Scholar]
- 2.Bender JS, Shen HG, Irwin CK, Schwartz KJ, Opriessnig T. Characterization of Erysipelothrix species isolates from clinically affected pigs, environmental samples, and vaccine strains from six recent swine erysipelas outbreaks in the United States. Clinical and vaccine immunology: CVI. 2010;17(10):1605–11. Epub 2010/08/20. 10.1128/CVI.00206-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brooke CJ, Riley TV. Erysipelothrix rhusiopathiae: bacteriology, epidemiology and clinical manifestations of an occupational pathogen. Journal of medical microbiology. 1999;48(9):789–99. Epub 1999/09/11. . [DOI] [PubMed] [Google Scholar]
- 4.Shimoji Y. Pathogenicity of Erysipelothrix rhusiopathiae: virulence factors and protective immunity. Microbes and infection / Institut Pasteur. 2000;2(8):965–72. Epub 2000/08/30. . [DOI] [PubMed] [Google Scholar]
- 5.Bender JS, Irwin CK, Shen H-G, Schwartz KJ, Opriessnig T. Erysipelothrix spp. genotypes, serotypes, and surface protective antigen types associated with abattoir condemnations. Journal of Veterinary Diagnostic Investigation. 2011;23(1):139–42. WOS:000286953200025. [DOI] [PubMed] [Google Scholar]
- 6.Eamens GJ, Forbes WA, Djordjevic SP. Characterisation of Erysipelothrix rhusiopathiae isolates from pigs associated with vaccine breakdowns. Veterinary microbiology. 2006;115(4):329–38. Epub 2006/04/20. 10.1016/j.vetmic.2006.02.015 . [DOI] [PubMed] [Google Scholar]
- 7.Imada Y, Takase A, Kikuma R, Iwamaru Y, Akachi S, Hayakawa Y. Serotyping of 800 strains of Erysipelothrix isolated from pigs affected with erysipelas and discrimination of attenuated live vaccine strain by genotyping. Journal of Clinical Microbiology. 2004;42(5):2121–6. 10.1128/Jcm.42.5.2121-2126.2004. ISI:000221424100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozawa M, Yamamoto K, Kojima A, Takagi M, Takahashi T. Etiological and Biological Characteristics of Erysipelothrix rhusiopathiae Isolated between 1994 and 2001 from Pigs with Swine Erysipelas in Japan. Journal of Veterinary Medical Science. 2009;71(6):697–702. ISI:000267775600001. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi T, Nagamine N, Kijima M, Suzuki S, Takagi M, Tamura Y, et al. Serovars of Erysipelothrix strains isolated from pigs affected with erysipelas in Japan. Journal of Veterinary Medical Science. 1996;58(6):587–9. WOS:A1996UU68500021. [DOI] [PubMed] [Google Scholar]
- 10.Wang Q, Chang BJ, Riley TV. Erysipelothrix rhusiopathiae. Veterinary microbiology. 2010;140(3–4):405–17. Epub 2009/09/08. 10.1016/j.vetmic.2009.08.012 . [DOI] [PubMed] [Google Scholar]
- 11.Reboli AC, Farrar WE. Erysipelothrix rhusiopathiae: an occupational pathogen. Clinical microbiology reviews. 1989;2(4):354–9. Epub 1989/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Q, Chang BJ, Mee BJ, Riley TV. Neuraminidase production by Erysipelothrix rhusiopathiae. Veterinary microbiology. 2005;107(3–4):265–72. Epub 2005/05/03. 10.1016/j.vetmic.2005.01.022 . [DOI] [PubMed] [Google Scholar]
- 13.Shimoji Y, Yokomizo Y, Sekizaki T, Mori Y, Kubo M. Presence of a capsule in Erysipelothrix rhusiopathiae and its relationship to virulence for mice. Infection and immunity. 1994;62(7):2806–10. Epub 1994/07/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimoji Y, Ogawa Y, Osaki M, Kabeya H, Maruyama S, Mikami T, et al. Adhesive surface proteins of Erysipelothrix rhusiopathiae bind to polystyrene, fibronectin, and type I and IV collagens. Journal of bacteriology. 2003;185(9):2739–48. Epub 2003/04/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galan JE, Timoney JF. Cloning and expression in Escherichia coli of a protective antigen of Erysipelothrix rhusiopathiae. Infection and immunity. 1990;58(9):3116–21. Epub 1990/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou Y, Zhu X, Muhammad HM, Jiang P, Li Y. Characterization of Erysipelothrix rhusiopathiae strains isolated from acute swine erysipelas outbreaks in Eastern China. The Journal of veterinary medical science / the Japanese Society of Veterinary Science. 2015;77(6):653–60. Epub 2015/02/05. 10.1292/jvms.14-0589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoder-Himes DR, Chain PS, Zhu Y, Wurtzel O, Rubin EM, Tiedje JM, et al. Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(10):3976–81. Epub 2009/02/24. 10.1073/pnas.0813403106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008;5(7):621–8. Epub 2008/06/03. 10.1038/nmeth.1226 . [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Feng Z, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–8. Epub 2009/10/27. 10.1093/bioinformatics/btp612 . [DOI] [PubMed] [Google Scholar]
- 20.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, et al. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic acids research. 2009;37(Database issue):D412–6. Epub 2008/10/23. 10.1093/nar/gkn760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaigalat L, Schluter JP, Hartmann M, Mormann S, Tauch A, Puhler A, et al. The DeoR-type transcriptional regulator SugR acts as a repressor for genes encoding the phosphoenolpyruvate:sugar phosphotransferase system (PTS) in Corynebacterium glutamicum. BMC molecular biology. 2007;8:104 Epub 2007/11/17. 10.1186/1471-2199-8-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi F, Harada T, Ogawa Y, Ono H, Ohnishi-Kameyama M, Miyamoto T, et al. Capsular polysaccharide of Erysipelothrix rhusiopathiae, the causative agent of swine erysipelas, and its modification with phosphorylcholine. Infection and immunity. 2012;80(11):3993–4003. Epub 2012/09/06. 10.1128/IAI.00635-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwok AH, Li Y, Jiang J, Jiang P, Leung FC. Complete genome assembly and characterization of an outbreak strain of the causative agent of swine erysipelas—Erysipelothrix rhusiopathiae SY1027. BMC microbiology. 2014;14:176 Epub 2014/07/06. 10.1186/1471-2180-14-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimoji Y, Asato H, Sekizaki T, Mori Y, Yokomizo Y. Hyaluronidase is not essential for the lethality of Erysipelothrix rhusiopathiae infection in mice. The Journal of veterinary medical science / the Japanese Society of Veterinary Science. 2002;64(2):173–6. Epub 2002/03/27. . [DOI] [PubMed] [Google Scholar]
- 25.Borrathybay E, Gong FJ, Zhang L, Nazierbieke W. Role of Surface Protective Antigen A in the Pathogenesis of Erysipelothrix rhusiopathiae strain C43065. Journal of microbiology and biotechnology. 2014. Epub 2014/09/17. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The difference was plotted versus the percentage of the proteins identified. Approximately 82.4% of proteins had cv differences less than 0.1, and more than 99.7% of the proteins had cv errors less than 0.5.
(PNG)
(TIF)
(TIF)
(XLS)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.