

Abstract
“Click chemistry” was explored to synthesize two series of 2-(1,2,3-triazolyl)adenosine derivatives (1–14). Binding affinity at the human A1, A2A, and A3ARs (adenosine receptors) and relative efficacy at the A3AR were determined. Some triazol-1-yl analogues showed A3AR affinity in the low nanomolar range, a high ratio of A3/A2A selectivity, and a moderate-to-high A3/A1 ratio. The 1,2,3-triazol-4-yl regiomers typically showed decreased A3AR affinity. Sterically demanding groups at the adenine C2 position tended to reduce relative A3AR efficacy. Thus, several 5′-OH derivatives appeared to be selective A3AR antagonists, i.e., 10, with 260-fold binding selectivity in comparison to the A1AR and displaying a characteristic docking mode in an A3AR model. The corresponding 5′-ethyluronamide analogues generally showed increased A3AR affinity and behaved as full agonists, i.e., 17, with 910-fold A3/A1 selectivity. Thus, N6-substituted 2-(1,2,3-triazolyl)-adenosine analogues constitute a novel class of highly potent and selective nucleoside-based A3AR antagonists, partial agonists, and agonists.
Introduction
Adenosine receptors (AR) are G-protein-coupled receptors and consist of four subtypes classified as A1, A2A, A2B, and A3. Among the four AR subtypes, the A3AR is the most recently identified.1 The distribution of A3AR is species-dependent, and in humans this subtype occurs in the lungs, liver, heart, kidneys, and brain.2–4 Activation of this receptor subtype inhibits adenylyl cyclase activity, increases phosphatidylinositol-specific phospholipase C activity, and stimulates Ca2+ mobilization.3 Adenosine A3 receptor stimulation induces cardioprotection through the activation of KATP channels4 and is also involved in neuroprotection, suggesting the possibility of using A3AR agonists to treat cardial and cerebral ischemia.5 A3AR agonists also exhibit systemic anticancer and chemoprotective effects.6 A3AR modulators have been proposed as antiinflammatory and antiasthmatic drugs.7,8 Selective A3AR antagonists promise to be useful in the regulation of cell growth8,9 and as cerebroprotective agents.10,11 They also seem to enhance anticancer treatment by counteracting P-glycoprotein efflux in multidrug resistance.12 A3AR antagonists are also proposed as potential therapeutics for the treatment of glaucoma; application of A3-AR antagonists externally to the eye substantially lowers intraocular pressure in mice and monkeys.13–15
Although diverse in structure, most AR antagonists share some common structural features. In general, they are planar, aromatic, or π-electron-rich and nitrogen-containing heterocycles. Additionally, most AR antagonists lack the ribose moiety, which seems essential for agonist activity.16 Various heterocyclic classes have been identified as promising leads for A3AR antagonists, among them 1,4-dihydropyridines, pyridines, deazaadenines, pyrazolopyrimidines, adenines, and 1,2,4-triazolo[ 4,3-a]quinoxalin-1-ones.4,7,17,18
However, the A3AR, more than other AR subtypes, is amenable to the design of nucleoside-based antagonists. The efficacy of nucleoside derivatives in activation of the A3AR is particularly sensitive to molecular substitution of the ligand.19 A wide range of adenosine derivatives have been shown to antagonize this receptor, including the highly potent A1AR agonist 2-chloro-N6-cyclopentyladenosine. N6-Benzyl groups are associated with reduced A3AR efficacy, leading to partial agonists and antagonists. However, many of the nucleosides so far demonstrated to be antagonists of the A3AR are not highly subtype-selective.20 Nucleoside-based A3AR antagonists maintaining an intact ribose moiety were reported by Volpini et al.,21 with a series of 8-alkynyladenosine derivatives that exhibited A3AR selectivity, but suffered from weak A3AR affinity. A spirolactam derivative, in which the 5′-alkyluronamide group was cyclized onto the 4′ carbon, was found to potently and selectively antagonize the A3AR.15,19 An advantage of nucleoside-based A3AR antagonists over other heterocyclic antagonists is the ability to achieve high affinity at murine species.
Recently, researchers from CV Therapeutics described a series of 2-pyrazolyladenosine analogues.22 Several representative compounds containing an N6-methyl substituent proved to display high affinity and selectivity for the A3AR. This study confirms the former finding23 that introduction of a methyl group into the N6 position increases the affinity for the human A3AR and enhances the selectivity versus A1 and A2AARs. On the basis of these results, we explored the versatile “click chemistry” approach24 to synthesize two series of N6-methyl-2-(1,2,3-triazolyl)adenosine derivatives and evaluated their affinity, selectivity, and efficacy at the A3AR. Although a number of 1,2,3-triazole nucleoside derivatives have been described,25 most involve base replacement with 1,2,3-triazole or introduction of 1,2,3-triazole at C8 or at the sugar moiety.
Results and Discussion
Chemistry
The synthesis of the 1,2,3-triazol-1-yladenosine derivatives is depicted in Scheme 1. 2-Iodo-N6-methyladenosine 2223 was prepared by reacting 2126 with 2.0 M CH3NH2 in THF. Since the reaction conditions27 for a Cu(I)-catalyzed nucleophilic substitution are very similar to those used in the “click” variant of Huisgen’s 1,3-dipolar cycloaddition, we initially attempted to perform a one-pot conversion of 22 to the desired 1,4-disubstituted 1,2,3-triazoles. The 2-azido derivative 23 was isolated as the main reaction product, and only a minor amount of the appropriate triazole was formed. This event forced us to perform the reaction in two steps. First the azido intermediate 23 was prepared in 66% yield from 22. 1H and 13C NMR in DMSO-d6 proved the presence of a tautomeric fused tetrazole form (17%) of the 2-azidoadenosine derivative 23, due to a spontaneous cyclization (Scheme 2). Such azido/tetrazole tautomerism has been previously reported for 2-azidoadenine and for 2-azidoadenosine.28,29 Next we applied a Cu(I)-catalyzed 1,3-cycloaddition reaction of azide 23 with the appropriate alkyne to generate the triazole analogues 1–11 (Scheme 1).30 Generally, the use of a water/butanol mixture as a solvent for the 1,3-cycloaddition allowed simple isolation of the desired compounds, which precipitated from the reaction medium.
Scheme 1.

Synthesis of 1,2,3-Triazol-1-yl Analogues of N6-Methyladenosine 1–11a
a Reagents and conditions: (a) CH3NH2 in THF, 2 days; (b) CuSO4·5H2O, sodium ascorbate, L-proline, Na2CO3, NaN3, H2O/tBuOH 1:1, 60 °C; (c) CuSO4·5H2O, sodium ascorbate, alkyne, H2O/tBuOH 3:1, room temp.
Scheme 2.

Azido/Tetrazole Tautomerism of a 2-Substituted Adenosine Derivative 23
Similarly, the 1,2,3-triazol-4-yl analogues 12–14 (Scheme 3) were prepared by a Cu+-catalyzed Huisgen 1,3-dipolar cycloaddition reaction of 2-ethynyl-N6-methyladenosine (25) with the appropriate azide.
Scheme 3.

Synthesis of 1,2,3-Triazol-4-yl Analogues of N6-Methyladenosine 12–14a
a Reagents and conditions: (a) trimethylsilylacetylene, CuI, (Ph3P)3PdCl2, DMF; (b) 7 N NH3 in MeOH, 0 °C; (c) CuSO4·5H2O, sodium ascorbate, alkyne, H2O/tBuOH 3:1, room temp.
The synthesis of the 5′-N-ethylcarbamoyl 2-(1,2,3)-triazol-1-yladenosine analogues 15–19 was carried out starting from 2-iodo-9-(2′,3′-O-isopropylidene-β-D-ribofuranosyl)-N6-methyladenine (26). After permanganate oxidation, carboxylic acid 27 was converted into its p-nitrophenyl ester 28, which upon treatment with ethylamine gave uronamide 29. Deprotection with 80% trifluoroacetic acid yielded 5′-ethylcarbamoyl-N6-methyl-2-iodoadenosine (30).30 The conversion of this 2-iodo derivative into the azido intermediate 31 was performed in 79% yield. The presence of a tautomeric fused tetrazole form (20%) of the 2-azidoadenosine derivative 31, due to a spontaneous cyclization, was here also observed in the NMR spectrum. Finally we applied the Cu(I)-catalyzed 1,3-cycloaddition reaction of azide 31 with the appropriate alkyne to generate the triazole analogues 15–19 (Scheme 4).
Scheme 4.

Synthesis of 1,2,3-Triazol-1-yl Analogues of N6-Methyladenosine-5′-N-ethyluronamide 15–19a
a Reagents and conditions: (a) KMnO4, KOH, room temp, 20 h; (b) p-nitrophenol, EDCI, DMF, room temp; (c) ethylamine; (d) 80% TFA/H2O; (e) CuSO4·5H2O, sodium ascorbate, L-proline, Na2CO3, NaN3, H2O/tBuOH 1:1, 60 °C; (f) CuSO4·5H2O, sodium ascorbate, alkyne, H2O/tBuOH 1:1, room temp.
2-Azido-N6-(5-chloro-2-methoxybenzyl)-2-(4-cyclopentylmethyl-1,2,3-triazol-1-yl)-9-(β-D-ribofuranosyl)adenine (20) was prepared in three steps starting from intermediate 21, as depicted in Scheme 5.
Scheme 5.

Synthesis of Compound 20, 2-(4-Cyclopentylmethyl-1,2,3-triazol-1-yl)-N6-(2-chloro-5-methoxybenzyl)adenosinea
a Reagents and conditions: (a) 2-chloro-5-methoxybenzylammonium chloride, Et3N, EtOH, reflux; (b) CuSO4·5H2O, sodium ascorbate, L-proline, Na2CO3, NaN3, H2O/tBuOH 1:1, 60 °C; (c) CuSO4·5H2O, sodium ascorbate, alkyne, H2O/tBuOH 1:1, room temp.
Biological Evaluation
The binding affinities of the newly synthesized adenosine derivatives were measured at the hA1, hA2A, and hA3ARs expressed in CHO (Chinese hamster ovary) cells as previously described,20 and their relative efficacy in the activation of the A3AR was determined (Table 1). The binding affinity of more potent compounds at the ARs was evaluated with full competition curves, while the weaker compounds at the hA1 and hA2A ARs were measured at a fixed concentration of 10 μM. Several compounds showed affinity for the A3AR in the low nanomolar range, a very high ratio of A3/A2A selectivity, and a moderate-to-high A3/A1 selectivity ratio. A functional assay of A3AR activation consisted of the ability of a single, high concentration of the nucleoside (10 μM) to inhibit forskolin-stimulated adenylyl cyclase measured by the method of Nordstet and Fredholm,32 in comparison to the full agonist NECA (10 μM). Cl-IB-MECA (2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine) was also a full agonist (100%) in this assay.19 The range of efficacies observed depended on the nature of the groups at the 2 and N6 positions.
Table 1.
Binding Affinities of Adenosine Derivatives at Human A1, A2A, and A3ARs Expressed in CHO Cells and Relative Efficacy at the A3ARa
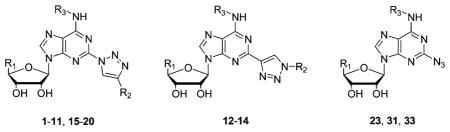
| |||||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 |
Ki (nM) or % inhibition (in parentheses) at 10 μM
|
% efficacyb hA3 |
|||
| hA1 | hA2A | hA3 | |||||
| 1 | CH2OH | H | CH3 | 1000 ± 30 | (13 ± 3) | 10.4 ± 0.2 | 41 ± 6 |
| 2 | CH2OH | ethyl | CH3 | 2920 ± 910 | (18) | 13.8 ± 3.3 | 23c |
| 3 | CH2OH | butyl | CH3 | 848 ± 76 | (23) | 11.7 ± 3.1 | 3 ± 8 |
| 4 | CH2OH | 2-hydroxyethyl | CH3 | 1270 ± 260 | (14) | 45.0 ± 4.4 | 25c |
| 5 | CH2OH | dimethylaminomethyl | CH3 | 3800 ± 600 | (6) | 117 ± 25 | 8c |
| 6 | CH2OH | phenyl | CH3 | (36) | (5) | 14.9 ± 1.7 | 14 ± 8 |
| 7 | CH2OH | pyridin-2-yl | CH3 | 1970 ± 210 | (40) | 10.3 ± 1.5 | 11 ± 4 |
| 8 | CH2OH | 4-propoxyphenyl | CH3 | (49) | (14) | 25.2 ± 2.6 | 31c |
| 9 | CH2OH | benzyl | CH3 | 589 ± 55 | (20) | 9.5 ± 0.7 | −1 ± 5 |
| 10d | CH2OH | cyclopentylmethyl | CH3 | 335 ± 13 | (39) | 1.3 ± 0.4 | −5 ± 7 |
| 11 | CH2OH | cyclohexylmethyl | CH3 | 1430 ± 60 | (16 ± 1) | 21.3 ± 8.1 | 2 ± 5 |
| 12 | CH2OH | benzyl | CH3 | 770 ± 210 | (21 ± 5) | 53.9 ± 6.6 | 2 ± 3 |
| 13 | CH2OH | 3-methoxybenzyl | CH3 | 957 ± 65 | (43 ± 10) | 86.1 ± 3.8 | −1 ± 3 |
| 14 | CH2OH | 3-Cl-benzyl | CH3 | 956 ± 6 | (39 ± 10) | 81.1 ± 5.0 | 0 ± 5 |
| 15d | C2H5NHCO | H | CH3 | 590 ± 70 | (18 ± 3) | 2.1 ± 0.1 | 102 ± 5 |
| 16 | C2H5NHCO | butyl | CH3 | 750 ± 110 | (43 ± 1) | 5.6 ± 0.2 | 89 ± 3 |
| 17d | C2H5NHCO | pyridin-2-yl | CH3 | 1640 ± 90 | (45 ± 12) | 1.8 ± 0.6 | 90 ± 7 |
| 18d | C2H5NHCO | benzyl | CH3 | 510 ± 50 | (33 ± 2) | 2.8 ± 1.3 | 86 ± 5 |
| 19 | C2H5NHCO | cyclopentylmethyl | CH3 | 1250 ± 150 | (36 ± 7) | 11.5 ± 1.4 | 83c |
| 20 | CH2OH | cyclopentylmethyl | 2-Cl–5-MeO–Bn | 830 ± 40 | 6000 | 18 ± 11 | −6 ± 3 |
| 23 | CH2OH | CH3 | 230 ± 10 | (23) | 10.8 ± 3.1 | 84 ± 9 | |
| 31 | C2H5NHCO | CH3 | 429 ± 55 | (18 ± 3) | 11.4 ± 4.2 | 112c | |
| 33 | CH2OH | 2-Cl–5-MeO–Bn | 60 ± 10 | 1800 ± 500 | 1.4 ± 0.1 | 44 ± 5 | |
All binding experiments were performed using cells stably transfected with cDNA encoding one of the human ARs. Binding at human A1, A2A, and A3ARs in this study was carried out as described in Experimental Section using [3H]CCPA, [3H]CGS 21680, or [125I]IAB-MECA as a radioligand. Values from the present study are expressed as Ki values (mean ± SEM, n = 3, unless otherwise noted) or as percent displacement of radioligand.
% activation at 10 μM, relative to cyclic AMP inhibitory effect of 10 μM NECA (=100%). Cl-IB-MECA was also a full agonist (100%) in this assay.
n = 2.
10, LC 153; 15, LC 260; 17, LC 257; 18, LC 259.
The 2-azido precursor 23 showed high binding affinity at the A3AR (Ki = 10.8 nM) and modest selectivity in comparison to the A1AR. The 1,2,3-triazol-1-yl derivatives obtained by 1,3-dipolar cycloaddition of azide 23 with acetylene (1), butyne (2), and hexyne (3) maintained high affinity for the A3AR and increased selectivity. They displayed Ki values of 10.4, 13.8, and 11.7 nM, respectively. Also, aromatic triazole substituents (6, 7, 9) resulted in similar Ki values of about 10 nM and even greater selectivity. Introducing nitrogen or oxygen including substituents at position 4 of the 1,2,3-triazole ring (4, 5, and 8) reduced the A3AR affinity. Among the investigated analogues, the 4-cyclopentylmethyl derivative 10 exhibited the highest affinity for the A3AR (Ki = 1.3 nM) and 260-fold selectivity in comparison to the A1AR. Replacement of the cyclopentyl ring with a phenyl (9) or cyclohexyl (11) moiety adversely affected A3AR affinity. Remarkably, the 1,2,3-triazol-4-yl regiomers (12–14) showed decreased affinity for the A3AR in comparison to similar 1,2,3-triazol-1-yl regiomers. In particular, a comparison of homologous compounds 12 and 9 indicated a 6-fold loss of affinity at the A3AR for the 4-yl isomer, approximately the same affinity at the A1AR, and no significant measurable gain in affinity at the A2AAR.
Replacement of the ribose 4′-hydroxymethyl moiety of the 2-azido derivative 23 by a 5′-N-ethyluronamide did not appreciably affect affinity at any of the AR subtypes. However, a similar substitution in the 2-(1,2,3-triazol-1-yl)-substituted series provided a modest (2- to 5-fold) increase in A3AR affinity and small or no changes in A1AR affinity, as demonstrated for the unsubstituted, 4-butyl, 4-pyridin-2-yl, and 4-benzyl substituted 1,2,3-triazol-1-yl combinations (15, 16, 17, and 18, in comparison to 1, 3, 7, and 9, respectively). Curiously, one 5′-uronamide, compound 19, exhibited decreased A3AR affinity (Ki = 11.5 nM) compared to its potent 5′-OH analogue 10 (Ki = 1.3 nM).
Replacement of the N6-methyl substituent of the 2-azido precursor 23 by a sterically demanding 2-chloro-5-methoxybenzyl group yielded 33, which manifested very high A3AR affinity (Ki = 1.4 nM). A similar replacement of the N6-methyl group of an analogue 10, also having a bulky 2-position substituent, to yield 20 reduced A3AR affinity but not appreciably. This was in accordance with previous observations that a simultaneous substitution at the 6 and 2 positions did not improve A3AR affinity.22,33 Thus, the effects of substitution at the 2 and N6 positions were not independent; however, it was possible to retain considerable A3AR selectivity (46-fold in compound 20). This was not representative of findings in a previous study in which double substitution greatly diminished the affinity and selectivity at the human A3AR.22
Whereas some previously synthesized 2-substituted adenosine derivatives22,23 displayed selective A3AR agonist activity, all 2-triazol-1-yl-N6-methyladenosine analogues synthesized with an unmodified ribose moiety (1–11 and 20) behaved as antagonists or weak partial agonists. Similar findings were reported for 2-ester derivatives of adenosine, in which a combination of 2 and N6 substitution reduced efficacy.34 Direct ring substitution at the 4-position of the 1,2,3-triazole with alkyl or aryl groups resulted in weak partial agonists (1, 2, 4–8), but subtle changes of structure resulted in a loss of efficacy, e.g., the 4-butyl derivative 3. 2-Triazol-1-yl-N6-methyladenosine analogues with a methylene spacer between the 1,2,3-triazole moiety and a ring system yielded full A3AR antagonists (9–11, 20), since they bound to the receptor but did not activate it. The 2-triazol-4-yl-N6-methyladenosine derivatives (12–14) also behaved as full A3AR antagonists. Thus, the 5′-OH derivatives 3, 9–14, and 20 appeared to be A3AR antagonists with the following order of decreasing selectivity for the A3AR in comparison to the A1AR: 4-cyclopentylmethyl-N6-methyl 10 (260-fold) > 4-butyl-N6-methyl 3 (72-fold), 4-cyclohexylmethyl-N6-methyl 11 (67-fold) > 4-cyclopentylmethyl-N6-(5-chloro-2-methoxybenzyl) 20.
Interestingly, the 5′-N-ethyluronamide modification was able to reestablish the A3AR agonist activity in analogues with sterically bulky substitution at the 2 position. This is consistent with previous findings that similar 5′-uronamides overcome the efficacy-reducing activity of substitution at the adenine 2 and N6 positions but not at the ribose 3′ position.19,35,36 Indeed, all 5′-N-ethyluronamide analogues studied here (15–19) proved to be full agonists at the A3AR. Among them are highly selective A3AR agonists, N6-methyladenosine-5′-N-ethyluronamide 2-(1,2,3-triazol-1-yl) derivatives: pyridin-2-yl 17 (910-fold) > unsubstituted 15 (280-fold) > benzyl 18 (180-fold). The 2-azido-N6-methyl precursors 23 and 31 also showed full agonist activity, whereas azide 33 having a bulky N6 group showed partial agonist activity.
Selected potent agonists in this series were measured in a functional assay of the human A2BAR. At 10 μM, compounds 3–7, 15, and 16 did not significantly stimulate adenylyl cyclase in human A2BAR-expressing CHO cells (<10% of the effect of 10 μM NECA, as a full agonist). Compounds 2, 8–14, 17, 19, and 23 at 10 μM stimulated adenylyl cyclase by <50%. Compounds 18 and 31 produced approximately 50% stimulation at 10 μM. Thus, selectivity for the A3AR was demonstrated; these nucleosides that activate the A3AR at low nanomolar concentrations activated the A2BAR only at substantial micromolar concentrations.
On the basis of previous findings, it is predicted that only the analogues containing the substituted N6-benzyl group (5-chloro-2-methoxy), i.e., full agonist 20 and partial agonist 33, would be expected to bind in the nanomolar range to the rat A3AR. Small alkyl groups at the N6 position, such as methyl and ethyl, although conducive to high affinity at the human A3-AR, led to negligible affinity at the rat homologue of the receptor. Selected compounds were measured in binding to the rat A3AR expressed in CHO cell membranes using [125I]I-AB-MECA. The Ki values determined were as follows: compounds 5, 7, and 8, Ki > 10 μM; compound 9, Ki = 1.79 μM; compound 10, Ki = 0.312 μM.
Molecular Modeling
To explain the structural basis for the high binding affinity of the nucleoside 2-(4-cyclopentylmethyl-1,2,3-triazol-1-yl)-N6-methyl-9-(β-D-ribofuranosyl)adenine 10 at hA3AR, we performed a computational study of ligand docking in a previously derived A3AR model based on the high-resolution structure of bovine rhodopsin.37,38 Various bound conformations of the C2-substituent and χ1 angles for the adenine ring were generated for an energetically favorable binding location and orientation, and the resulting conformations were compared energetically in the putative binding site.
The result of docking 10 in the putative binding site of the A3AR is shown in Figure 1A. The purine ring was surrounded by a hydrophobic pocket, defined by L91 (3.33) and L246 (6.51). In addition, the H-bonds formed between the exocyclic amine and the hydroxyl group of S247 (6.52) and between the purine N1 atom and the side chain of N250 (6.55). The 2′-OH group of the ribose moiety formed a H-bond with the side chain of Q167 (EL2), and the 3′-OH group formed an intramolecular H-bond with the 5′-OH group. Unlike the previously reported docking models of N6-substituted adenosines,37 here the 5′-OH group H-bonded with the side chain of H272 (7.43) and the backbone carbonyl group of S271 (7.42). The cyclopentyl moiety interacted with aliphatic hydrophobic residues, M177 (5.38) and V178 (5.39), through a hydrophobic interaction and were situated in proximity to F168 (EL2) F182 (5.43), consistent with the optimized binding affinity of compound 10.
Figure 1.

(A) Docking complexes of compound 10, 2-(4-cyclopentylmethyl-1,2,3-triazol-1-yl)-N6-methyladenosine. (B) Superimposition of Cl-IB-MECA in red and compound 10 in color by atom type. Residues that were within 5 Å to the ligand in this putative binding site were L91 (3.33), T94 (3.36), H95 (3.37), Q167 (EL2), F168 (EL2), M172 (EL2), S181 (5.42), M177 (5.38), V178 (5.39), F182 (5.43), W243 (6.48), L246 (6.51), S247 (6.52), N250 (6.55), C251 (6.56), I268 (7.39), S271 (7.42), and H272 (7.43). The ligand is represented by a ball-and-stick model. The H-bonds are indicated with yellow dots. By use of the MOLCAD ribbon surface program, the A3AR is shown in a ribbon model with different colors for each TM (TM1, red; TM2, orange; TM3, yellow; TM4, green; TM5, cyan; TM6, blue; TM7, purple; H8, violet).
A comparison of the docking modes of Cl-IB-MECA and compound 10 in the putative binding domain showed considerable overlap of the ribose rings and of the adenine moieties, although in compound 10 both were situated a little closer to extracellular loop 2 (Figure 1B). Previously, it was noted that 5′-uronamide analogues, typically of derivatives having bulky N6-subsitituents, generally gain affinity in comparison to the corresponding 5′-hydroxyl analogue. Here, the 5′-uronamide analogue 19 (agonist) of the most potent 5′-hydroxyl analogue 10 (antagonist) displayed a lower binding affinity, which could be explained by the shift of the ribose position in adenosine analogue having a bulky 2-(4-cyclopentylmethyl-1,2,3-triazol-1-yl) substituent in comparison to those having N6 bulky substituents. The binding of the cyclopentylmethyl group in 10 was directed more toward the upper part of TM5, partially overlapping with the binding site of the 3-iodophenyl ring in Cl-IB-MECA. Curiously, other closely related triazolo derivatives displayed a higher potency of the 5′-uronamide analogues; thus, compound 10 must bind to the receptor in a very distinct manner. There was a subtle difference in orientation between the 2-cyclopentyl group and bulkier groups like benzyl (9) or cyclohexylmethyl (11), which were associated with unfavorable van der Waals interactions and resulted in a decrease of binding affinity of 7- and 16-fold, respectively. In addition, the same preferred χ1 angles of the energetically favorable bound conformation, common to Cl-IB-MECA and compound 10, were consistent with the empirical finding that the combination of bulky N6 and C2 substituents was unfavorable for A3AR selectivity because of competitive interaction of these bulky substituents. Thus, the modeling has demonstrated that the human A3AR preference of 2-(4-cyclopentylmethyl-1,2,3-triazol-1-yl) derivatives in the 5′-OH series might be explained by optimal van der Waals interactions.
Conclusions
Several 2-(1,2,3-triazol-1-yl)-N6-methyl-substituted adenosine derivatives described in the present study displayed A3AR affinities in the low nanomolar range, showed very high A3/A2A, and a moderate to high A3/A1 selectivity. Contrary to what we expected, the 2-triazole analogues with an unmodified ribose moiety (1–14) showed antagonist or weak partial agonist activity at the A3AR. A 2-(4-cyclopentylmethyl-(1,2,3-triazol-1-yl))-N6-methyl derivative 10 was 260-fold selective in binding in comparison to the A1AR. The binding of the 4-cyclopentylmethyl group in 10, in distinction to the binding of closely related bulky groups pendent on the triazole ring, was directed more toward the upper part of TM5 partially overlapping with the binding site of the 3-iodophenyl ring in Cl-IB-MECA. The 5′-N-ethyluronamide modification was dominant over the efficacy reducing effects at the 2 position and was capable of fully re-establishing the A3AR agonist activity, resulting in highly potent and selective A3AR agonists 15–19. The most selective agonist derivative was compound 17, 9-(5-ethylcarbamoyl-β-D-ribofuranosyl)-N6-methyl-2-(4-pyridin-2-yl-1,2,3-triazol-1-yl)adenine, which was 910-fold selective in binding to the A3AR in comparison to the A1AR. The retention of high human A3AR affinity in compound 20 was not typical of previous findings that double bulky substitution at the 2 and N6 positions tended to reduce A3AR affinity markedly. Thus, the 2-triazol-1-yl-N6-methyladenosine analogues 1–11 constitute a novel class of highly potent and selective nucleoside-based A3AR partial agonists and antagonists (all of which maintain an intact ribose in the molecular structure) and agonists. Since the reported analogues show excellent affinity for the A3AR and span the full intrinsic activity range, they might be useful as pharmacological tools or as leads for further optimization.
Experimental Section
All reagents were from standard commercial sources and of analytic grade, except for the benzylic azides, which were prepared by treating the corresponding benzylic bromides with NaN3 in DMF. Precoated Merck silica gel F254 plates were used for TLC, and spots were examined under UV light at 254 nm and further visualized by sulfuric acid–anisaldehyde spray. Column chromatography was performed on ICN silica gel (63–200 μm, 60 Å, ICN Biochemicals, Eschwege, Germany). NMR spectra were obtained with a Varian Mercury 300 MHz spectrometer. Chemical shifts are given in ppm (δ) relative to the residual solvent signals, which in the case of DMSO-d6 were 2.54 ppm for 1H and 40.5 ppm for 13C. Structural assignment was confirmed with COSY and DEPT. All signals assigned to hydroxyl groups were exchangeable with D2O. Exact mass measurements were performed on a quadrupole/orthogonal-acceleration time-of-flight (Q/oaTOF) tandem mass spectrometer (qToF 2, Micromass, Manchester, U.K.) equipped with a standard electrospray ionization (ESI) interface. Samples were infused in a 2-propanol/water (1:1) mixture at 3 μL/min. For the compounds that precipitated from the reaction medium, the yields were calculated from the amount obtained after filtration and are lower than the actual yields, since in most cases a considerable amount remained in solution.
N6-Methyl-9-(β-D-ribofuranosyl)-2-(1,2,3-triazol-1-yl)adenine (1)
In a pressure tube was added 23 (165 mg, 0.51 mmol), trimethylsilylacetylene (292 μL, 2.05 mmol), and 4 mL of DMF. The mixture was stirred at 105 °C for 15 h. After solvent evaporation, the yellowish residue was dissolved in 2 mL of a 1.0 M solution of tetrabutylammonium fluoride in THF and stirred for 5 h. The reaction was monitored by NMR. After evaporation of the solvent, the residue was dissolved in ethyl acetate. Water was added, and the triazole product was precipitated in the water layer. After overnight cooling and filtration, the precipitate was further purified on a silica gel column (CH2Cl2/MeOH, 92:8) and yielded compound 1 as a white solid (82 mg, 46%). 1H NMR (300 MHz, DMSO-d6): δ 3.06 (d, 3H, J = 4.5 Hz, N6-CH3), 3.54–3.61 (m, 1H, H-5′A), 3.65–3.72 (m, 1H, H-5′B), 3.96 (dd, 1H, J = 3.8 and 7.9 Hz, H-4′), 4.20 (dd, 1H, J = 4.7 and 8.2 Hz, H-3′), 4.65 (app q, J = 5.9 Hz, H-2′), 4.98 (t, 1H, J = 5.6 Hz, 5′-OH), 5.23 (d, 1H, J = 5.0 Hz, 3′-OH), 5.48 (d, 1H, J = 6.2 Hz, 2′-OH), 5.95 (d, 1H, J = 5.9 Hz, H-1′), 7.92 (d, 1H, J = 1.2 Hz, H-4′′), 8.37 (d, 1H, J = 4.6 Hz, N6-H), 8.46 (s, 1H, H-8), 8.82 (br s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 27.84 (N6-CH3), 62.19 (C-5′), 71.13 (C-3′), 74.30 (C-2′), 86.42 (C-4′), 88.01 (C-1′), 119.75 (C-5), 124.67 (C-5′′), 134.25 (C-4′′), 141.13 (C-8), 149.56 and 149.85 (C-2 and C-4), 156.082 (C-6). HRMS (ESI-MS) C13H17N8O4 [M + H]+: 349.1367 found; 349.1372 calcd. Anal. (C13H16N8O4·½H2O) C, H, N.
2-(4-Ethyl-1,2,3-triazol-1-yl)-N6-methyl-9-(β-D-ribofuranosyl)-adenine (2)
Compound 23 (70 mg, 0.217 mmol), sodium ascorbate (8.6 mg, 0.043 mmol), and CuSO4·5H2O (2.2 mg, 0.009 mmol) were suspended in 20 mL of H2O/tBuOH (3:1). The mixture was saturated with 1-butyne and stirred for 4 days at room temperature in a Parr apparatus. Purification on a preparative TLC plate (CH2Cl2/MeOH, 90:10) resulted in compound 2 as a white solid in 40% yield. 1H NMR (300 MHz, DMSO-d6): δ 1.27 (t, 3H, J = 7.62 Hz, CH3), 2.75 (q, 2H, J = 7.6 Hz, CH2), 3.05 (d, 3H, J = 4.4 Hz, N6-CH3), 3.51–3.59 (m, 1H, H-5′A), 3.61–3.70 (m, 1H, H-5′B), 3.94 (dd, 1H, J = 4.0 and 7.6 Hz, H-4′), 4.15 (dd, 1H, J = 4.4 and 7.9 Hz, H-3′), 4.60 (app q, J = 5.6 Hz, H-2′), 4.97 (t, 1H, J = 5.6 Hz, 5′-OH), 5.22 (d, 1H, J = 4.7 Hz, 3′-OH), 5.47 (d, 1H, J = 6.2 Hz, 2′-OH), 5.93 (d, 1H, J = 6.2 Hz, H-1′), 8.34 (d, 1H, J = 4.4 Hz, N6-H), 8.45 (s, 1H, H-8), 8.55 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 14.32 (CH3), 19.08 (CH2), 27.84 (N6-CH3), 62.13 (C-5′), 71.15 (C-3′), 74.34 (C-2′), 86.43 (C-4′), 87.84 (C-1′), 119.57 (C-5), 120.962 (C-5′′), 140.92 (C-8), 149.31, 149.63, 149.88 (C-2, C-4, and C-4′′), 156.05 (C-6). HRMS (ESI-MS) C15H21N8O4 [M + H]+: 377.1682 found; 377.1685 calcd. Anal. (C15H20N8O4) C, H, N.
General Procedure for the Synthesis of 4′′-Substituted 2-(1,2,3-Triazol-1-yl)adenosine Derivatives 3–11
Compound 23 (70 mg, 0.217 mmol), sodium ascorbate (8.6 mg, 0.043 mmol), and CuSO4·5H2O (2.2 mg, 0.009 mmol) were suspended in 2 mL of H2O/tBuOH (3:1). The appropriate alkyne (2 equiv) was subsequently added, and the mixture was stirred overnight at room temperature. The 2-triazol-1-yl compounds (generally) precipitated from this reaction medium and were isolated by filtration with water.
2-(4-Butyl-1,2,3-triazol-1-yl)-N6-methyl-9-(β-D-ribofuranosyl)-adenine (3)
The reaction of 23 (70 mg, 0.217 mmol) with 1-hexyne (50 μL, 0.435 mmol) gave compound 3 in 59% yield. 1H NMR (300 MHz, DMSO-d6): δ 0.93 (t, 3H, J = 7.3 Hz, CH3), 1.38 (m, 2H, CH2), 1.66 (m, 2H, CH2), 2.71 (t, 2H, J = 7.2 Hz, C4′′-CH2), 3.05 (d, 3H, J = 4.0 Hz, N6-CH3), 3.52–3.62 (m, 1H, H-5′A), 3.62–3.72 (m, 1H, H-5′B), 3.95 (dd, 1H, J = 3.6 and 7.2 Hz, H-4′), 4.18 (dd, 1H, J = 4.8 and 8.1 Hz, H-3′), 4.62 (app q, 1H, J = 5.7 Hz, H-2′), 5.01 (t, 1H, J = 5.2 Hz, 5′-OH), 5.29 (d, 1H, J = 4.0 Hz, 3′-OH), 5.54 (d, 1H, J = 5.6 Hz, 2′-OH), 5.94 (d, 1H, J = 5.9 Hz, H-1′), 8.36 (d, 1H, J = 4.1 Hz, N6-H), 8.45 (s, 1H, H-8), 8.56 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 14.37 (CH3), 22.37 (CH2), 25.21 (CH2), 27.83 (N6-CH3), 31.68 (CH2), 62.14 (C-5′), 71.15 (C-3′), 74.36 (C-2′), 86.43 (C-4′), 87.88 (C-1′), 119.56 (C-5), 121.33 (C-5′′), 140.96 (C-8), 147.88 and 149.90 (C-2, C-4 and C-4′′), 156.07 (C-6). HRMS (ESI-MS) C17H25N8O4 [M + H]+: 405.1992 found, 405.1998 calcd. Anal. (C17H24N8O4) C, H, N.
2-[4-(2-Hydroxyethyl)-1,2,3-triazol-1-yl]-N6-methyl-9-(β-D-ribofuranosyl) adenine (4)
The reaction of 23 (70 mg, 0.217 mmol) with 3-butyn-1-ol (33 μL, 0.435 mmol) afforded compound 4 without precipitation. After solvent evaporation, the mixture was purified on a silica gel column (90:10 CH2Cl2/MeOH + 1% 7 N NH3 in MeOH), yielding compound 11 as a white solid in 68% yield. 1H NMR (300 MHz, DMSO-d6): δ 2.85 (t, 2H, J = 6.9 Hz, CH2), 3.03 (d, 3H, J = 4.7 Hz, N6-CH3), 3.52–3.59 (m, 1H, H-5′A), 3.63–3.72 (m, 3H, H-5′B and CH2), 3.94 (dd, 1H, J = 3.5 and 7.3 Hz, H-4′), 4.16 (dd, 1H, J = 4.8 and 8.1 Hz, H-3′), 4.59 (app q, 1H, J = 5.4 Hz, H-2′), 4.74 (t, 1H, J = 5.8 Hz, CH2–OH), 4.97 (app t, 1H, J = 5.6 Hz, 5′-OH), 5.24 (d, 1H, J = 5.0 Hz, 3′-OH), 5.50 (d, 1H, J = 6.2 Hz, 2′-OH), 5.93 (d, 1H, J = 6.2 Hz, H-1′), 8.31 (d, 1H, J = 4.7 Hz, N6-H), 8.43 (s, 1H, H-8), 8.54 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 27.83 (N6-CH3), 29.70 (CH2), 60.85 (CH2–OH), 62.13 (C-5′), 71.15 (C-3′), 74.35 (C-2′), 86.43 (C-4′), 87.81 (C-1′), 119.56 (C-5), 122.02 (C-5′′), 140.49 (C-8), 145.49, 149.88, and 149.65 (C-2, C-4, and C-4′′), 156.05 (C-6). HRMS (ESI-MS) C15H21N8O5 [M + H]+: 393.1630 found, 393.1634 calcd. Anal. (C15H20N8O5·H2O) C, H, N.
2-(4-Dimethylaminomethyl-1,2,3-triazol-1-yl)-N6-methyl-9-(β-D-ribofuranosyl)adenine (5)
The reaction of 23 (70 mg, 0.217 mmol) with 1-dimethylamino-2-propyne (47 μL, 0.435 mmol) gave compound 5 without precipitation. The volatiles were removed under reduced pressure, and the residue was purified on a silica gel column (80:20 CH2Cl2/MeOH + 1% 7 N NH3 in MeOH). Compound 5 was obtained as a white solid in 67% yield. 1H NMR (300 MHz, DMSO-d6): δ 2.20 (s, 6H, 2 × CH3), 3.05 (d, 3H, J = 4.4 Hz, N6-CH3), 3.54–3.62 (m, 3H, H-5′A and CH2), 3.65–3.72 (m, 1H, H-5′B), 3.95 (dd, 1H, J = 4.1 and 7.6 Hz, H-4′), 4.18 (dd, 1H, J = 5.0 and 8.2 Hz, H-3′), 4.62 (app q, 1H, J = 5.4 Hz, H-2′), 4.98 (t, 1H, J = 5.6 Hz, 5′-OH), 5.23 (d, 1H, J = 5.0 Hz, 3′-OH), 5.49 (d, 1H, J = 6.2 Hz, 2′-OH), 5.95 (d, 1H, J = 6.2 Hz, H-1′), 8.38 (d, 1H, J = 4.4 Hz, N6-H), 8.45 (s, 1H, H-8), 8.64 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 27.85 (N6-CH3), 45.18 (N(CH3)2), 53.85 (CH2), 62.10 (C-5′), 71.12 (C-3′), 74.38 (C-2′), 86.41 (C-4′), 87.84 (C-1′), 119.64 (C-5), 123.328 (C-5′′), 140.97 (C-8), 144.41, 149.59, 149.79 (C-2, C-4, and C-4′′), 156.05 (C-6). HRMS (ESI-MS) C16H24N9O4 [M + H]+: 406.1944 found, 406.1951 calcd. Anal. (C16H23N9O4) C, H, N. N calcd, 31.09; found, 30.41.
N6-Methyl-2-(4-phenyl-1,2,3-triazol-1-yl)-9-(β-D-ribofuranosyl) adenine (6)
The reaction of 23 (70 mg, 0.217 mmol) with phenylacetylene (48 μL, 0.435 mmol) yielded compound 6 (54%) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 3.11 (d, 3H, J = 4.5 Hz, N6-CH3), 3.55–3.63 (m, 1H, H-5′A), 3.67–3.74 (m, 1H, H-5′B), 3.97 (dd, J = 3.6 and 7.2 Hz, H-4′), 4.21 (dd, J = 4.8 and 8.1 Hz, H-3′), 4.66 (app q, J = 5.4 Hz, H-2′), 5.03 (app t, 1H, J = 5.6 Hz, 5′-OH), 5.29 (d, 1H, J = 5.0 Hz, 3′-OH), 5.54 (d, 1H, J = 6.2 Hz, 2′-OH), 5.99 (d, 1H, J = 5.9 Hz, H-1′), 7.4 (t, 1H, J = 7.3 Hz, Ph), 7.50 (t, 2H, J = 7.5 Hz, Ph), 8.06 (d, 2H, J = 7.3 Hz, Ph), 8.44 (d, 1H, J = 4.5 Hz, N6-H), 8.49 (s, 1H, H-8), 9.31 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 27.92 (N6-CH3), 62.16 (C-5′), 71.17 (C-3′), 74,39 (C-2′), 86.48 (C-4′), 87.94 (C-1′), 119.85 (C-5), 120.68 (C-5′′), 126.27, 128.95, 129.61, and 130.85 (Ph), 141.082 (C-8), 147.02 and 149.77 (C-2, C-4, and C-4′′), 156.10 (C-6). HRMS (ESI-MS) C19H21N8O4 [M + H]+: 425.1689 found, 425.1685 calcd. Anal. (C19H20N8O4) C, H, N.
N6-Methyl-2-[4-pyridin-2-yl-1,2,3-triazol-1-yl]-9-(β-D-ribofuranosyl) adenine (7)
The reaction of 23 (70 mg, 0.217 mmol) with 2-ethynylpyridine (44 μL, 0.435 mmol) afforded compound 7 as a white solid in 55% yield. 1H NMR (300 MHz, DMSO-d6): δ 3.09 (d, 3H, J = 4.1 Hz, N6-CH3), 3.57–3.66 (m, 1H, H-5′A), 3.67–3.76 (m, 1H, H-5′B), 3.97 (dd, 1H, J = 3.9 and 7.5 Hz, H-4′), 4.19 (dd, 1H, J = 4.8 and 8.1 Hz, H-3′), 4.64 (app q, 1H, J = 6.0 Hz, H-2′), 4.99 (t, 1H, J = 5.6 Hz, 5′-OH), 5.26 (d, 1H, J = 5.0 Hz, 3′-OH), 5.54 (d, 1H, J = 6.2 Hz, 2′-OH), 5.99 (d, 1H, J = 5.9 Hz, H-1′), 7.42 (m, 1H, pyridin-2-yl), 7.96 (m, 1H, pyridin-2-yl), 8.16 (d, 1H, J = 7.3 Hz, pyridin-2-yl), 8.42 (d, 1H, J = 4.1 Hz, 1H, N6-H), 8.49 (s, 1H, H-8), 8.68 (d, 1H, J = 4.1 Hz, pyridin-2-yl), 9.16 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 27.92 (N6-CH3), 62.13 (C-5′), 71.14 (C-3′), 74.49 (C-2′), 86.47 (C-4′), 88.16 (C-1′), 119.92 (C-5), 120.53 (C-5′′), 121.07 (pyridin-2-yl), 122.25 (pyridin-2-yl), 137.79 (pyridin-2-yl), 141.28 (C-8), 148.47, 150.45 and 150.56 (C-2, C-4, C-4′′, and pyridin-2-yl), 156.28 (C-6). HRMS (ESI-MS) C18H19N9O4Na [M + Na]+: 448.1458 found, 448.1457 calcd. Anal. (C18H19N9O4) C, H, N.
N6-Methyl-2-[4-(4-propoxyphenyl)-1,2,3-triazol-1-yl]-9-(β-D-ribofuranosyl) adenine (8)
The reaction of 23 (70 mg, 0.217 mmol) with 1-eth-1-ynyl-4-propoxybenzene (57 μL, 0.435 mmol) afforded compound 8 as a white solid in 36% yield. 1H NMR (300 MHz, DMSO-d6): δ 0.99 (t, 3H, J = 7.3 Hz, CH3), 1.71–1.78 (m, 2H, CH2), 3.10 (d, 3H, J = 4.1 Hz, N6-CH3), 3.54–3.62 (m, 1H, H-5′A), 3.64–3.72 (m, 1H, H-5′B), 3.98 (m, 3H, H-4′ and CH2), 4.18 (dd, 1H, J = 4.8 and 8.1 Hz, H-3′), 4.64 (app q, 1H, J = 5.4 Hz, H-2′), 4.99 (t, 1H, J = 5, 7 Hz, 5′-OH), 5.25 (d, 1H, J = 4.1 Hz, 3′-OH), 5.50 (d, 1H, J = 5.9 Hz, 2′-OH), 5.97 (d, 1H, J = 6.2 Hz, H-1′), 7.03 (d, 2H, J = 8.8 Hz, Ph), 7.94 (d, 2H, J = 8.8 Hz, Ph), 8.36 (d, 1H, J = 4.1 Hz, 1H, N6-H), 8.45 (s, 1H, H-8), 9.15 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 11.08 (CH3), 22.73 (CH2), 27.90 (N6-CH3), 62.16 (C-5′), 69.71 (OCH2), 71.18 (C-3′), 74.38 (C-2′), 86.46 (C-4′), 87.91 (C-1′), 115.50 (Ph), 119.60 (C-5), 123.28 (C-5′′), 127.66 (Ph), 140.01 (C-8), 147.01, 149.82, 150.45 (C-4, C-2, and C-4′′), 156.10 (C-6). HRMS (ESI-MS) C22H27N8O5 [M + H]+: 483.2109 found, 483.2104 calcd. Anal. (C22H26N8O5) C, H, N.
N6-Methyl-2-(4-benzyl-1,2,3-triazol-1-yl)-9-(β-D-ribofuranosyl) adenine (9)
The reaction of 23 (70 mg, 0.217 mmol) with 3-phenyl-1-propyne (54 μL, 0.435 mmol) gave compound 9 as a white solid in 43% yield. 1H NMR (300 MHz, DMSO-d6): δ 3.03 (d, 3H, J = 3.8 Hz, N6-CH3), 3.53–3.62 (m, 1H, H-5′A), 3.64–3.71 (m, 1H, H-5′B), 3.95 (dd, 1H, J = 3.7 and 7.2 Hz, H-4′), 4.11 (s, 2H, CH2), 4.17 (dd, 1H, J = 4.8 and 8.1 Hz, H-3′), 4.61 (app q, 1H, J = 5.7 Hz, H-2′), 4.97 (app t, 1H, J = 5.3 Hz, 5′-OH), 5.22 (d, 1H, J = 4.7 Hz, 3′-OH), 5.47 (d, 1H, J = 5.9 Hz, 2′-OH), 5.94 (d, 1H, J = 5.9 Hz, H-1′), 7.23 (m, 1H, Ph), 7.32 (d, 4H, J = 4.4 Hz, Ph), 8.34 (d, 1H, J = 3.8 Hz, N6-H), 8.45 (s, 1H, H-8), 8.59 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 25.32 (N6-CH3), 32.51 (CH2), 62.11 (C-5′), 71.13 (C-3′), 74.43 (C-2′), 86.41 (C-4′), 87.96 (C-1′), 121.42 (C-5), 121.97 (C-5′′), 126.91, 129.12, 129.21, and 140.02 (Ph), 140.88 (C-8), 147.42, 149.58, and 149.81 (C-4, C-2, and C-4′′), 156.09 (C-6). HRMS (ESI-MS) C20H23N8O4 [M + H]+: 439.1846 found, 439.1842 calcd. Anal. (C20H22N8O4) C, H, N.
2-(4-Cyclopentylmethyl-1,2,3-triazol-1-yl)-N6-methyl-9-(β-D-ribofuranosyl) adenine (10)
The reaction of 23 (70 mg, 0.217 mmol) with 3-cyclopentyl-1-propyne (57 μL, 0.435 mmol) yielded compound 10 (32%) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 1.23–1.28 (m, 2H, cyclopentyl), 1.48–1.62 (m, 4H, cyclopentyl), 1.71–1.75 (m, 2H, cyclopentyl), 2.19–2.25 (m, 1H, cyclopentyl), 2.72 (d, 2H, J = 7.3 Hz, CH2-cyclopentyl), 3.05 (d, 3H, J = 3.9 Hz, N6-CH3), 3.53–3.61 (m, 1H, H-5′A), 3.63–3.72 (m, 1H, H-5′B), 3.96 (dd, 1H, J = 3.6 and 7.2 Hz, H-4′), 4.19 (dd, 1H, J = 4.8 and 8.1 Hz, H-3′), 4.62 (app q, 1H, J = 5.9 Hz, H-2′), 4.98 (t, 1H, J = 5.3 Hz, 5′-OH), 5.23 (d, 1H, J = 4.7 Hz, 3′-OH), 5.49 (d, 1H, J = 6.5 Hz, 2′-OH), 5.95 (d, 1H, J = 5.9 Hz, H-1′), 8.34 (d, 1H, J = 3.9 Hz, N6-H), 8.45 (s, 1H, H-8), 8.55 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 25.37 (cyclopentyl), 27.83 (N6-CH3), 31.56 (CH2), 32.55 (cyclopentyl), 62.15 (C-5′), 71.15 (C-3′), 74.34 (C-2′), 86.43 (C-4′), 87.87 (C-1′), 119.60 (C-5), 121.58 (C-5′′), 140.92 (C-8), 147.33, 149.89 (C-4, C-2, and C4′′), 156.07 (C-6). HRMS (ESI-MS) C19H27N8O4 [M + H]+: 431.2153 found, 431.2155 calcd. Anal. (C19H26N8O4) C, H, N.
2-(4-Cyclohexylmethyl-1,2,3-triazol-1-yl)-N6-methyl-9-(β-D-ribofuranosyl) adenine (11)
The reaction of 23 (70 mg, 0.217 mmol) with cyclohexyl-1-propyne (63 μL, 0.435 mmol) gave compound 11 in 82% yield. 1H NMR (300 MHz, DMSO-d6): δ 0.86–1.28 (br m, 6H, cyclohexyl), 1.54–1.72 (br m, 5H, cylcohexyl), 2.58 (d, 2H, J = 6.9 Hz, CH2), 3.03 (d, 3H, J = 3.9 Hz, N6-CH3), 3.51–3.59 (m, 1H, H-5′A), 3.63–3.70 (m, 1H, H-5′B), 3.94 (dd, 1H, J = 4.2 and 7.5 Hz, H-4′), 4.16 (dd, 1H, J = 4.8 and 8.1 Hz, H-3′), 4.59 (app q, 1H, J = 6.3 Hz, H-2′), 4.97 (t, 1H, J = 5.5 Hz, 5′-OH), 5.22 (d, 1H, J = 4.8 Hz, 3′-OH), 5.47 (d, 1H, J = 6.3 Hz, 2′-OH), 5.93 (d, 1H, J = 6.0 Hz, H-1′), 8.30 (d, 1H, J = 3.9 Hz, N6-H), 8.43 (s, 1H, H-8), 8.51 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 26.31 (cyclohexyl), 26.66 (cyclohexyl), 27.84 (N6-CH3), 33.16 (cyclohexyl), 38.22 (CH2), 62.14 (C-5′), 71.15 (C-3′), 74.35 (C-2′), 86.44 (C-4′), 87.86 (C-1′), 119.60 (C-5), 121.93 (C-5′′), 140.92 (C-8), 146.45, 149.92 (C-4, C-2, and C-4′′), 153.51 (C-6). HRMS (ESI-MS) C20H29N8O4 [M + H]+: 445.2305 found, 445.2311 calcd. Anal. (C20H28N8O4) C, H, N.
General Procedure for the Synthesis of 4′′-Substituted 2-(1,2,3-Triazol-4-yl)adenosine Derivatives 12–14
Compound 25 (100 mg, 0.32 mmol), sodium ascorbate (13 mg, 0.06 mmol mmol), and CuSO4·5H2O (3 mg, 0. 013 mmol) were suspended in 30 mL of H2O/tBuOH (3:1). The appropriate azide (2 equiv) was subsequently added, and the mixture was stirred overnight at room temperature. The 2-triazol-4-yl compounds (generally) precipitated from this reaction medium and were isolated by filtration with water.
2-(1-Benzyl-1,2,3-triazol-4-yl)-N6-methyl-9-(β-D-ribofuranosyl) adenine (12)
The reaction of 25 (100 mg, 0.32 mmol) with 85 mg (0.64 mmol) of benzylazide gave compound 12 in 78% yield (110 mg). 1H NMR (300 MHz, DMSO-d6): δ 3.03 (br s, 3H, N6-CH3), 3.52–3.58 (m, 1H, H-5′A), 3.60–3.66 (m, 1H, H-5′B), 3.92 (app d, H-4′, J = 2.9 Hz, H-4′), 4.15 (dd, 1H, J = 4.7 and 7.6 Hz, H-3′), 4.60 (app q, 1H, J = 5.9 Hz, H-2′), 5.08 (t, 1H, J = 5.4 Hz, 5′-OH), 5.19 (d, 1H, J = 3.5 Hz, 3′-OH), 5.44 (d, 1H, J = 5.6 Hz, 2′-OH), 5.68 (s, 2H, CH2), 5.97 (d, 6.2 Hz, H-1′), 7.38 (br s, 5H, Ph), 7.82 (br s, 1H, N6-H), 8.36 (s, 1H, H-8), 8.66 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 27.54 (N6-CH3), 53.62 (CH2), 62.29 (C-5′), 71.30 (C-3′), 74.30 (C-2′), 86.41 (C-4′), 87.77 (C-1′), 119.52 (C-5), 126.41 (C-5′′), 128.63, 128.86, 129.48 and 136.71 (Ph), 140.27 (C-8), 148.22, 153.56, 153.72 (C-2, C-4, and C-4′), 155.69 (C-6). HRMS (ESI-MS) C20H23N8O4 [M + H]+: 439.1834 found, 439.1842 calcd. Anal. (C20H22N8O4) C, H, N.
2-[1-(3-Methoxybenzyl)-1,2,3-triazol-4-yl)-N6-methyl-9-(β-D-ribofuranosyl) adenine (13)
The reaction of 25 (100 mg, 0.32 mmol) with 104 mg (0.64 mmol) of 3-methoxybenzylazide gave compound 13 in 80% yield (120 mg). 1H NMR (300 MHz, DMSO-d6): δ 3.03 (br s, 3H, N6-CH3), 3.52–3.60 (m, 1H, H-5′A), 3.64–3.72 (m, 1H, H-5′B), 3.95 (app d, H-4′, J = 2.9 Hz, H-4′), 4.15 (dd, 1H, J = 4.7 and 7.6 Hz, H-3′), 4.60 (app q, 1H, J = 6.4 Hz, H-2′), 5.07 (t, 1H, J = 5.1 Hz, 5′-OH), 5.21 (d, 1H, J = 4.2 Hz, 3′-OH), 5.46 (d, 1H, J = 6.3 Hz, 2′-OH), 5.62 (s, 2H, CH2), 5.95 (d, 1H, J = 6.6 Hz, H-1′), 6.91 (m, 3H, Ph), 7.29 (t, 1H, J = 7.9 Hz, Ph), 7.79 (br s, 1H, N6-H), 8.34 (s, 1H, H-8), 8.63 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 27.59 (N6-CH3), 53.45 (CH2), 55.82 (OCH3), 62.30 (C-5′), 71.33 (C-3′), 74.25 (C-2′), 86.42 (C-4′), 87.70 (C-1′), 114.44 and 114.20 (Ph), 119.51 (C-5), 120.72 (Ph), 130.65 (C-5′′), 138.21 (C-8), 148.22, 153.56, 153.64 (C-2, C-4, and C-4′), 155.69 (C-6), 160.14 (Ph). HRMS (ESI-MS) C21H25N8O5 [M + H]+: 469.1938 found, 469.1947 calcd. Anal. (C21H24N8O5) C, H, N.
2-[1-(3-Chlorobenzyl)-1,2,3-triazol-4-yl)-N6-methyl-9-(β-D-ribofuranosyl) adenine (14)
The reaction of 25 (100 mg, 0.32 mmol) with 107 mg (0.64 mmol) of 3-chlorobenzylazide gave compound 14 in 73% yield (110 mg). 1H NMR (300 MHz, DMSO-d6): δ 3.04 (br s, 3H, N6-CH3), 3.53–3.60 (m, 1H, H-5′A), 3.64–3.71 (m, 1H, H-5′B), 3.96 (app d, H-4′, J = 2.9 Hz, H-4′), 4.17 (dd, 1H, J = 4.7 and 7.6 Hz, H-3′), 4.64 (app q, 1H, J = 5.8 Hz, H-2′), 5.09 (t, 1H, J = 5.3 Hz, 5′-OH), 5.20 (d, 1H, J = 4.4 Hz, 3′-OH), 5.45 (d, 1H, J = 6.2 Hz, 2′-OH), 5.97 (d, 1H, J = 6.2 Hz, H-1′), 7.31–7.36 (m, 1H, Ph), 4.41–7.47 (m, 3H, Ph), 7.83 (br s, 1H, N6-H), 8.37 (s, 1H, H-8), 8.72 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 27.54 (N6-CH3), 52.80 (CH2), 62.31 (C-5′), 71.33 (C-3′), 74.27 (C-2′), 86.43 (C-4′), 87.69 (C-1′), 119.56 (C-5), 126.63 (C-5′′), 127.53, 128.53, 128.84, 131.42, 133.98, and 139.15 (Ph), 140.36 (C-8), 148.22, 150.03, and 153.43 (C-2, C-4, and C-4′), 155.78 (C-6). HRMS (ESI-MS) C20H22N8O4Cl [M + H]+: 473.1452 found, 473.1452 calcd. Anal. (C20H21N8O4Cl) C, H, N.
9-(5-Ethylcarbamoyl-β-D-ribofuranosyl)-N6-methyl-2-(1,2,3-triazol-1-yl)adenine (15)
In a pressure tube was added 31 (110 mg, 0.30 mmol), trimethylsilylacetylene (259 μL, 1.81 mmol), and 4 mL of DMF. The mixture was stirred at 105 °C for 15 h. Solvent evaporation yielded a yellowish solid that was dissolved 6 mL of a 1.0 solution of tetrabutylammonium fluoride in THF and stirred for 5 h. After solvent evaporation, the residue was dissolved in ethyl acetate. Water was added, and the triazole precipitated in the water layer. After overnight cooling and filtration, the precipitate was further purified on a silica gel column (CH2Cl2/MeOH, 93:7) and yielded compound 15 as a white solid (49 mg, 42%). 1H NMR (300 MHz, DMSO-d6): δ 0.90 (t, 3H, CH3), 3.05–3.21 (m, 2H, N-CH2), 3.05 (d, 3H, J = 4.2 Hz, N6-CH3), 4.26 (m, 1H, H-3′), 4.33 (d, 1H, J = 2.1 Hz, H-4′), 5.61 (d, 1H, J = 6.2 Hz, 3′-OH), 5.71 (d, 1H, J = 4.7 Hz, 2′-OH), 6.04 (d, 1H, J = 7.2 Hz, H-1′), 7.91 (d, 1H, J = 1.2 Hz, H-4′′), 8.07 (t, 1H, J = 5.3 Hz, NHCO), 8.41 (d, 1H, J = 4.2 Hz, N6-H), 8.54 (s, 1H, H-8), 8.82 (br s, 1H, H-5′′). 13CMR (300 MHz, DMSO-d6): δ 15.16 (CH3), 27.85 (N6-CH3), 34.07 (CH2), 73.64 (C-2′ and C-3′), 84.98 (C-4′), 88.02 (C-1′), 119.91 (C-5), 124.70 (C-5′′), 134.29 (C-4′′), 141.59 (C-8), 149.69 and 149.90 (C-2 and C-4), 156.14 (C-6), 169.73 (C═O). HRMS (ESI-MS) C15H20N9O4 [M + H]+: 390.1676 found, 390.1683 calcd. Anal. (C15H19N9O4) C, H, N.
General Procedure for the Synthesis of 4′′-Substituted 2-(1,2,3-Triazol-1-yl)adenosine Derivatives 16–18
To a mixture of 31 (100 mg, 0.28 mmol), CuI (5 mg, 0.03 mmol), and triethylamine (40 μL, 0.28 mmol) in water/acetonitrile (1:1), the appropriate alkyne (2 equiv) was added. The mixture was stirred for 5 days at room temperature. The reaction was monitored by 1H NMR. The product was precipitated with water and cooled overnight. After filtration, the yellowish solid was purified on a silica gel column (CH2Cl2/MeOH, 90:10) to obtain the 1,2,3-triazol-1-yladenosine derivative as a white solid.
2-(4-Butyl-1,2,3-triazol-1-yl)-9-(5-ethylcarbamoyl-β-D-ribofuranosyl)-N6-methyladenine (16)
The reaction of compound 31 (100 mg, 0.28 mmol) with 1-hexyne (64μL, 0.56 mmol) gave 65 mg (53%) of compound 16 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 0.88–0.95 (m, 6H, 2 × CH3), 1.31–1.43 (m, 2H, CH2), 1.61–1.71 (m, 2H, CH2), 2.72 (t, 2H, J = 7.8 Hz, C4′′-CH2), 3.05–3.22 (m, 2H, N-CH2), 3.05 (d, 3H, J = 4.1 Hz, N6-CH3), 4.25 (m, 1H, H-3′), 4.33 (d, 1H, J = 2.1 Hz, H-4′), 4.73 (m, 1H, H-2′), 5.59 (d, 1H, J = 6.3 Hz, 3′-OH), 5.69 (d, 1H, J = 4.5 Hz, 2′-OH), 6.04 (d, 1H, J = 6.9 Hz, H-1′), 8.09 (t, 1H, J = 5.4 Hz), 8.37 (d, 1H, J = 4.1 Hz, N6-H), 8.53 (s, 1H, H-8), 8.56 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): 14.36 (CH3), 15.17 (CH3), 22.35 (CH2), 25.21 (CH2), 27.84 (N6-CH3), 31.70 (CH2), 34.07 (CH2), 73.60 and 73.64 (C-2′ and C-3′), 84.99 (C-4′′), 87.91 (C-1′), 119.79 (C-5), 121.45 (C-5′′), 141.49 (C-8), 147.97, 149.72, and 149.93 (C-2, C-4, and C-4′′), 156.11 (C-6), 169.73 (C═O). HRMS (ESI-MS) C19H28N9O4 [M + H]+: 446.2256 found, 446.2264 calcd. Anal. (C19H27N9O4) C, H, N.
9-(5-Ethylcarbamoyl-β-D-ribofuranosyl)-N6-methyl-2-(4-pyridin-2-yl-1,2,3-triazol-1-yl)adenine (17)
The reaction of compound 31 (50 mg, 0.14 mmol) with 2-ethynylpyridine (56 μL, 0.56 mmol) gave 35 mg (54%) of compound 17 as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 0.93 (t, 3H, J = 7.2 Hz, CH3), 3.09–3.21 (m, 2H, N-CH2), 3.10 (d, 3H, J = 4.2 Hz, N6-CH3), 4.25 (m, 1H, H-3′), 4.35 (d, 1H, J = 2.1 Hz, H-4′), 4.74 (m, 1H, H-2′), 5.68 (d, 1H, J = 6.6 Hz, 2′-OH), 5.75 (d, 1H, J = 4.5 Hz, 3′-OH), 6.08 (d, 1H, J = 6.6 Hz, H-1′), 7.41 (m, 1H, pyridin-2-yl), 7.96 (m, 1H, pyridin-2-yl), 8.10 (t, 1H, J = 6.0 Hz, NHCO), 8.16 (d, 1H, J = 8.1 Hz, pyridin-2-yl), 8.48 (d, 1H, J = 4.1 Hz, 1H, N6-H), 8.60 (s, 1H, H-8), 8.67 (d, H, J = 5.1 Hz, pyridin-2-yl), 9.17 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 15.18 (CH3), 27.94 (N6-CH3), 34.11 (N-CH2), 73.69 and 73.78 (C-2′ and C-3′), 84.95 (C-4′), 87.98 (C-1′), 120.04 (C-5), 120.65 (C-5′′), 122.20 and 124.11 (pyridin-2-yl), 138.027 (pyridin-2-yl), 141.64 (C-8), 147.95, 149,64, 149.67, and 149.98 (C-2, C-4, C-4′′, and pyridin-2-yl), 150.49 (pyridin-2-yl), 156.13 (C-6), 169.76 (C═O). HRMS (ESI-MS) C20H23N10O4 [M + H]+: 467.1899 found, 467.1903 calcd. Anal. (C20H22N10O4) C, H, N. N calcd, 30.03; found, 29.55.
9-(5-Ethylcarbamoyl-β-D-ribofuranosyl)-N6-methyl-2-(4-benzyl-1,2,3-triazol-1-yl)adenine (18)
The reaction of compound 31 (70 mg, 0.19 mmol) with 3-phenyl-1-propyne (49μL, 0.56 mmol) gave 35 mg (38%) of compound 18 as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 0.87 (t, 3H, J = 7.5 Hz, CH3), 3.03–3.20 (m, 2H, N-CH2), 3.03 (d, 3H, 4.5 Hz, N6-CH3), 4.11 (s, 2H, CH–Ph), 4.24 (m, 1H, H-3′), 4.32 (d, 1H, J = 2.1 Hz, H4′), 4.72 (m, 1H, H-2′), 5.59 (d, 1H, J = 6.6 Hz, 3′-OH), 5.69 (d, 1H, J = 4.5 Hz, 2′-OH), 6.03 (d, 1H, J = 6.9 Hz, H1′), 7.22 (m, 1H, Ph), 7.32 (d, 4H, J = 4.2 Hz, Ph), 8.06 (t, 1H, J = 5.7 Hz, NHCO), 8.38 (d, 1H, J = 4.1 Hz, N6-H), 8.54 (s, 1H, H-8), 8.60 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 15.16 (CH3), 27.84 (N6-CH3), 31.72 (CH2), 34.07 (N-CH2), 73.64 (C-2′ and C-3′), 84.98 (C-4′), 87.87 (C-1′), 119.91 (C-5), 122.11 (C-5′′), 126.93, 129.13, 129.22, and 140.07 (Ph), 140.48 (C-8), 147.05, 149.87 (C-4, C-2, and C-4′′), 156.12 (C-6), 169.71 (C═O). HRMS (ESI-MS) C22H26N9O4 [M + H]+: 480.2098 found, 480.2107 calcd. Anal. (C22H25N9O4) C, H, N.
2-(4-Cyclopentylmethyl-1,2,3-triazol-1-yl)-9-(5-ethylcarbamoyl-β-D-ribofuranosyl)-N6-methyladenine (19)
Compound 31 (60 mg, 0.17 mmol), sodium ascorbate (13 mg, 0.066 mmol) and CuSO4·5H2O (3.5 mg, 0.013 mmol) were suspended in 4 mL of tBuOH/H2O (1:1). 3-Cyclopentyl-1-propyne (58 μL, 0.44 mmol) was subsequently added, and the mixture was stirred for 2 days at room temperature. The 2-triazol-1-yl compound precipitated from the reaction medium. Water was added, and the mixture was cooled overnight. The precipitate was filtered off and washed with water and hexane to obtain 19 as a white solid in 33% yield. 1H NMR (300 MHz, DMSO-d6): δ 0.89 (t, 3H, CH3), 1.19–1.28 (m, 2H, cyclopentyl), 1.45–1.75 (m, 6H, cyclopentyl), 2.15–2.25 (m, 1H, CH, cyclopentyl), 2.72 (d, 2H, J = 7.2 Hz, CH2), 3.06 (d, 3H, J = 4.5 Hz, N6-CH3), 3.09–3.22 (m, 2H, N-CH2), 4.24 (dt, 1H, J = 1.5 and 4.8 Hz, H-3′), 4.33 (d, 1H, J = 2.1 Hz, H-4′), 4.71–7.76 (app q, 1H, J = 6.6 Hz, H-2′), 5.61 (d, 1H, J = 6.3 Hz, 2′-OH), 5.71 (d, 1H, J = 4.8 Hz, 3′-OH), 6.03 (d, 1H, J = 6.9 Hz, H-1′), 8.10 (t, 1H, J = 5.7 Hz, NHCO), 8.40 (d, 1H, J = 5.1 Hz, N6-H), 8.54 (s, 1H, H-8), 8.56 (s, 1H, H-5′′). 13C NMR (300 MHz, DMSO-d6): δ 15.17 (CH3), 25.36 (cyclopentyl), 27.85 (N6-CH3), 31.53 (cyclopentyl), 31.51 (CH2), 34.07 (CH2), 73.63 and 73.56 (C-2′ and C-3′), 84.99 (C-4′), 87.93 (C-1′), 119.77 (C-5), 121.69 (C-5′′), 141.51 (C-8), 147.48 and 149.92 (C-2, C-4, and C-4′′), 156.10 (C-6), 169.74 (C═O). HRMS (ESI-MS) C21H30N9O4 [M + H]+: 472.2415 found, 472.2420 calcd. Anal. (C21H29N9O4) C, H, N.
N6-(5-Chloro-2-methoxybenzyl)-2-(4-cyclopentylmethyl-1,2,3-triazol-1-yl)-9-(β-D-ribofuranosyl)adenine (20)
Compound 33 (100 mg, 0.22 mmol), sodium ascorbate (17 mg, 0.086 mmol), and CuSO4·5H2O (3.5 mg, 0.017 mmol) were suspended in 4 mL of H2O/tBuOH (1:1). 3-Cyclopentyl-1-propyne (29 μL, 0.44 mmol) was subsequently added, and the mixture was stirred for 2 days at room temperature. The 2-triazol-1-yl compound precipitated from the reaction medium. Water was added, and the mixture was cooled overnight. The precipitate was filtered off and washed with water and hexane to obtain 20 as a white solid in 59% yield. 1H NMR (300 MHz, DMSO-d6): δ 1.23–1.28 (m, 2H, cyclopentyl), 1.48–1.61 (m, 4H, cyclopentyl), 1.66–1.73 (m, 2H, cyclopentyl), 2.15–2.25 (m, 1H, cyclopentyl), 2.72 (d, 2H, J = 7.2 Hz, CH2-cyclopentyl), 3.56–3.61 (m, 1H, H-5′A), 3.66–3.71 (m, 1H, H-5′B), 3.86 (s, 3H, OCH3), 3.97 (m, 1H, H-4′), 4.20 (m, 1H, H-3′), 4.64 (m, 1H, H-2′), 4.73 (br s, 2H, N6-CH2), 4.96 (t, 1H, J = 6.0 Hz, 5′-OH), 5.22 (d, 1H, J = 4.8 Hz, 3′-OH), 5.48 (d, 1H, J = 5.7 Hz, 2′-OH), 5.95 (d, 1H, J = 6.3 Hz, H-1′), 7.04 (d, 1H, J = 9.0 Hz, Ph), 7.25–7.29 (m, 2H, Ph), 8.40 (s, 1H, H-8), 8.81 (s, 1H, H-5′′), 8.81 (br s, 1H, N6-H). 13C NMR (300 MHz, DMSO-d6): δ 25.36 (cyclopentyl), 31.54 (CH2), 32.54 (cyclopentyl), 38.86 (CH2), 56.57 (OCH3), 62.13 (C-5′), 71.15 (C-3′), 74.37 (C-2′), 86.46 (C-4′), 88.07 (C-1′), 113.15 (Ph), 119.58 (C-5), 121.37 (C-5′′), 124.66, 128.23, and 129.93 (Ph), 141.37 (C-8), 147.40, 149.66, and 150.12 (C-2, C-4, and C-4′′), 155.51 (Ph), 156.33 (C-6). HRMS (ESI-MS) C26H32N8O5Cl [M + H]+: 571.2184 found, 571.2184 calcd. Anal. (C26H31N8O5Cl) C, H, N.
2-Azido-N6-methyl-9-(β-D-ribofuranosyl)adenine (23)
Sodium ascorbate (19.4 mg, 0.098 mmol) and CuSO4·5H2O (12.2 mg, 0.049 mmol) were added to a mixture of 22 (200 mg, 0.491 mmol), sodium azide (38,3 mg, 0.589 mmol), L-proline (11,3 mg, 0.098 mmol), and sodium carbonate (10.4 mg, 0.098 mmol) in 10 mL of H2O/tBuOH (1:1). The mixture was stirred overnight at 65 °C and was monitored by 1H NMR. Then 50 mL of dilute NH4OH was added and the crude mixture extracted with ethyl acetate (3 × 60 mL). The organic layer was washed with brine (60 mL), dried over MgSO4, and purified on a silica gel column (CH2Cl2/MeOH, 95:5) to afford compound 23 as a slightly yellow solid in 66% yield. 1H NMR (300 MHz, DMSO-d6): δ 2.91 (d, 3H, J = 4.4 Hz, N6-CH3), 3.48–3.55 (m, 1H, H-5′A), 3.58–3.66 (m, 1H, H-5′B), 3.90 (dd, 1H, J = 3.8 and 7.3 Hz, H-4′), 4.10 (dd, 1H, J = 4.7 and 9.7 Hz, H3′), 4.53 (app q, 1H, J = 5.9 Hz, H-2′), 5.04 (dd, 1H, J = 5.2 and 6.2 Hz, 5′-OH), 5.19 (d, 1H, J = 5.0 Hz, 3′OH), 5.43 (d, 1H, J = 6.2 Hz, 2′-OH), 5.78 (d, 1H, J = 6.2 Hz, H-1′), 8.12 (d, J = 4.4 Hz, N6-H), 8.27 (s, 1H, H-8). Small peaks from 1/6 tetrazole tautomeric form: δ 3.15 (d, 3H, J = 5.0 Hz, N6-CH3), 3.95 (d, H-4′), 4.15 (d, H-3′), 5.51 (d, 2′-OH), 5.94 (d, H-1′), 8.51 (s, H-8). 13C NMR (300 MHz, DMSO-d6): δ 27.54 (N6-CH3), 62.21 (C-5′), 71.17 (C-3′), 74.12 (C-2′), 86.35 (C-4′), 88.01 (C-1′), 118.07 (C-5), 139.99 (C-8), 156.06 and 156.20 (C-2 and C-6). Small peaks from 1/6 tetrazole tautomeric form: δ 31.89 (N6-CH3), 61.79 (C-5′), 70.77 (C-3′), 74.37 (C-4′), 112.30 (C-12), 142.91 (C-11), 147.60 (C-6). HRMS (ESI-MS) C11H15N8O4 [M + H]+: 323.1208 found, 323.1216 calcd. Anal. (C11H14N8O4) C, H, N.
N6-Methyl-9-(β-D-ribofuranosyl)-2-[2-trimethylsilylethyn-1-yl]adenine (24)
Compound 22 (500 mg, 1.23 mmol), CuI (12 mg, 0.062 mmol), and (Ph3P)3PCl2 were dissolved in 9 mL of DMF. Triethylamine (205 μL, 1.47 mmol) and trimethylsilylacetylene (210 mg, 1.47 mmol) were added, and the reaction mixture was stirred overnight. After solvent evaporation, the residue was dissolved in CH2Cl2 and filtered through a pad of Celite. Purification on a silica gel column (CH2Cl2/MeOH, 95:5) yielded 305 mg (66%) of compound 23. 1H NMR (300 MHz, DMSO-d6): δ 0.00 (9H, s, (CH3)3Si), 2.7 (3H, N6-CH3), 3.26–3.34 (m, 1H, H-5′A), 3.37–3.44 (m, 1H, H-5′B), 3.68 (dd, 1H, J = 3.5 Hz, H-4′), 3.86 (dd, 1H, J = 3.4 and 8.2 Hz, H-3′), 4.23 (app q, 1H, J = 5.9 Hz, H-2′), 4.87 (t, 1H, J = 5.0, 5′-OH), 4.94 (d, 1H, J = 4.99 Hz, 3′-OH), 5.21 (d, 1H, J = 6.2 Hz, 2′-OH), 5.63 (d, 1H, J = 6.2 Hz, H-1′), 7.69 (br s, 1H, N6-H), 8.19 (s, 1H, H-8). HRMS (ESI-MS) C16H24N5O4Si: [M + H]+: 378.1586 found; 378.1597 calcd.
2-Ethynyl-N6-methyl-9-(β-D-ribofuranosyl)purine (25)
An amount of 300 mg (0.8 mmol) of compound 24 was dissolved in 7 N ammonia in methanol and stirred for 2 h at 0 °C. After solvent evaporation, the residue was purified by silica gel chromatography (CH2Cl2/MeOH, 95:5) to obtain 160 mg (65%) of derivative 25. 1H NMR (300 MHz, DMSO-d6): δ 2.91 (s, 3H, N6-CH3), 3.48–3.56 (m, 1H, H-5′A), 3.61–3.68 (m, 1H, H-5′B), 3.92 (m, 1H, H-4′), 4.02 (s, 1H, C≡CH), 4.11 (dd, 1H, J = 5.0 and 8.2 Hz, H-3′), 4.53 (app q, 1H, J = 5.9 Hz, H-2′), 5.15 (m, 2H, 3′-OH and 5′-OH), 5.44 (d, 1H, J = 6.15 Hz, 2′-OH), 5.84 (d, 1H, J = 6.16 Hz, H-1′), 7.95 (br s, 1H, N6-H), 8.40 (s, 1H, H-8). HRMS (ESI-MS) C13H16N5O4 [ M + H]+: 306.1197 found; 306.1202 calcd.
1-Deoxy-1-(6-methylamino-2-iodo-9H-purin-9-yl)-2,3-O-isopropylidene-β-D-ribufuranuronic Acid (27)
To a stirred solution of 3.8 g (8.5 mmol) of 26 in 560 mL of H2O were added 1.4 g of KOH and, dropwise, a solution of 4.03 g (25.5 mmol) of KMnO4 in 110 mL of H2O. The mixture was stirred in the dark for 20 h, cooled to 0 °C, and quenched with 30 mL of 7% H2O2. The mixture was filtered through Celite. The filtrate was concentrated in vacuo and then acidified to pH 4 with 3 N HCl. The resulting precipitate was filtered off and successively washed with water and ether to give 2.98 g (76%) of 27 as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 1.36 (s, 3H, CH3), 1.51 (s, 3H, CH3), 2.89 (d, 3H, J = 3.3 Hz, N6-CH3), 4.68 (d, 1H, J = 1.8 Hz, H-4′), 5.40 (d, 1H, J = 6.0 Hz, H-2′), 5.47 (dd, 1H, J = 6.0 and 1.8 Hz, H-3′), 6.28 (s, 1H, H-1′), 8.08 (d, 1H, J = 3.3 Hz, N6-H), 8.16 (s, 1H, H-8). HRMS (ESI-MS) C14H16N5O5I [M + H]+: 462.0273 found; 462.0276 calcd.
9-(5-Ethylcarbamoyl-β-D-ribofuranosyl)-2-iodo-N6-methyladenine (30)
p-Nitrophenol (402 mg, 2.89 mmol) and 1-[3-(dimethylamino) propyl]-3-ethylcarbodiimide hydrochloride (506 mg, 2.65 mmol) were added to a solution of 27 (1.11 g, 2.41 mmol) in 10 mL of dry DMF. The reaction mixture was stirred for 3 h at room temperature and cooled to 0 °C, and 1.6 mL (24.1 mmol) of ethylamine was added. The solution turned yellow immediately and was further stirred for 1 h at room temperature. After evaporation of the volatiles, the residue was partitioned between ethyl acetate (3 × 100 mL) and H2O (100 mL). The organic layer was washed with brine (100 mL), dried over MgSO4, and concentrated to dryness. The residue was dissolved in 80% aqueous TFA (20 mL) and stirred for 2 h at room temperature. The mixture was concentrated in vacuo, coevaporated several times with EtOH, and purified by silica gel chromatography (CH2Cl2/MeOH, 96:4). Compound 30 was obtained as a white solid in 78% yield (840 mg). 1H NMR (300 MHz, DMSO-d6): δ 1.05 (t, 3H, J = 7.2 Hz, CH3), 2.91 (d, 3H, J = 4.4 Hz, N6-CH3), 3.19–3.29 (m, 2H, N-CH2), 4.16 (dt, 1H, J = 2.1 and 4.4 Hz, H-3′), 4.31 (d, 1H, J = 2.1 Hz, H-4′), 4.58 (app q, 1H, J = 5.6 Hz, H-2′), 5.59 (d, 1H, J = 6.5 Hz, 2′-OH), 5.71 (d, 1H, J = 4.7 Hz, 3′-OH), 5.92 (d, 1H, J = 7.1 Hz, H-1′), 8.12 (t, 1H, J = 5.5 Hz, NHCO), 8.19 (d, 1H, J = 4.4 Hz, N6-H), 8.38 (s, 1H, H-8). HRMS (ESI-MS) C13H18N6O4I [M + H]+: 449.0429 found, 449.0436 calcd.
2-Azido-9-(5-ethylcarbamoyl-β-D-ribofuranosyl)-N6-methyladenine (31)
Sodium ascorbate (69 mg, 0.34 mmol) and CuSO4· 5H2O (5.6 mg, 0.17 mmol) were added to a mixture of 30 (780 mg, 1.74 mmol), sodium azide (226 mg, 3.48 mmol), L-proline (40 mg, 0.35 mmol), and sodium carbonate (37 mg, 0.35 mmol) in 20 mL of H2O/tBuOH (1:1). The mixture was stirred for 2 days at 65 °C and monitored by 1H NMR. An amount of 100 mL of dilute NH4OH was added, and the crude mixture was extracted with ethyl acetate (5 × 150 mL) and washed with brine (150 mL). The organic layer was dried over MgSO4 and purified on a silica gel column (CH2Cl2/MeOH, 96:4) to afford compound 31 as a white solid in 79% yield (500 mg). 1H NMR (300 MHz, DMSO-d6): δ 1.06 (t, 3H, J = 7.2 Hz, CH3), 2.96 (d, 3H, J = 4.0 Hz, N6-CH3), 3.16–3.29 (m, 2H, N-CH2), 4.14 (dt, 1H, J = 1.8 and 4.8 Hz, H-3′), 4.29 (d, 1h, J = 1.8, H-4′), 4.53–4.59 (app q, 1H, J = 6.3 Hz, H-2′), 5.52 (d, 1H, J = 6.3 Hz, 2′-OH), 5.68 (d, 1H, J = 4.8 Hz, 3′-OH), 5.90 (d, 1H, J = 7.2 Hz, H-1′), 8.20 (d, 1H, J = 4.0 Hz, N6-H), 8.35 (s, 1H, H-8), 8.49 (t, 1H, J = 6.0 Hz, NHCO). Small peaks from 1/5 tetrazole tautomeric form: δ 1.08–1.12 (t, 3H, J = 7.2 Hz, CH3), 4.34 (d, 1H, J = 2.1 Hz, H-4′), 4.67–4.73 (app q, 1H, J = 7.2 Hz, H-2′), 5.54 (d, 1H, J = 4.5 Hz, 2′-OH) 6.00 (d, 1H, J = 7.2 Hz, H-1′). 13C NMR (300 MHz, DMSO-d6): δ 15.48 (CH3), 27.52 (N6-CH3), 33.94 (N-CH2), 73.06 and 73.75 (C-2′ and C-3′), 85.15 (C-4′), 88.09 (C-1′), 118.36 (C-5), 140.69 (C-8), 156.032 and 156.143 (C-2 and C-4), 169.79 (C-6). HRMS (ESI-MS) C13H18N9O4 [M + H]+: 364.1473 found; 364.1481 calcd. Anal. (C13H17N9O4) C, H, N.
N6-(5-Chloro-2-methoxybenzyl)-2-iodo-9-(β-D-ribofuranosyl)-adenine (32)
Compound 21 (1 g, 1.86 mmol) was dissolved in EtOH (30 mL). 5-Chloro-2-methoxybenzylammonium chloride (580 mg, 2.79 mmol) and Et3N (392 μL, 2.79 mmol) were added, and the solution was refluxed overnight. The mixture was concentrated to dryness, dissolved in 7 N NH3 in methanol, and stirred at room temperature for 2 h to deprotect the 2′-hydroxyl group. The volatiles were removed under reduced pressure, and the residue was purified by silica gel column (CH2Cl2/MeOH, 97:3). The product, compound 32, was realized in 80% yield. 1H NMR (300 MHz, DMSO-d6): δ 3.51–3.58 (m, 1H, H-5′A), 3.63–3.68 (m, 1H, H-5′B), 3.85 (s, 3H, OCH3), 3.94 (m, 1H, H-4′), 4.13 (m, 1H, H-3′), 4.52–4.59 (m, 3H, N6-CH2 and H-2′), 5.03 (t, 1H, J = 5.6 Hz, 5′-OH), 5.21 (d, 1H, J = 5.0 Hz, 3′-OH), 5.48 (d, 1H, J = 5.8 Hz, 2′-OH), 5.83 (d, 1H, J = 6.2 Hz, H-1′), 7.03 (d, 1H, J = 8.8 Hz, Ph), 7.16 (d, 1H, J = 2.7 Hz, Ph), 7.29 (dd, 1H, J = 2.7 and 8.8 Hz, Ph), 8.35 (s, 1H, H-8), 8.62 (br s, 1H, N6-H). HRMS (ESI-MS) C18H20N5O5-ICl [M + H]+: 548.0204 found; 548.0199 calcd.
2-Azido-N6-(5-chloro-2-methoxybenzyl)-9-(β-D-ribofuranosyl) adenine (33)
Sodium ascorbate (14 mg, 0.073 mmol) and CuSO4· 5H2O (9 mg, 0.037 mmol) were added to a mixture of 32 (200 mg, 0.365 mmol), sodium azide (47 mg, 0.73 mmol), L-proline (8 mg, 0.073 mmol), and sodium carbonate (8 mg, 0.073 mmol) in 4 mL of H2O/tBuOH (1:1). The mixture was stirred for 2 days at 65 °C and monitored by 1H NMR. An amount of 10 mL of dilute NH4-OH was added, and the crude mixture was extracted with ethyl acetate (5 × 15 mL) and washed with brine (15 mL). The organic layer was dried over MgSO4 and purified on a silica gel column (CH2Cl2/MeOH, 97:3) to afford compound 33 as a white solid in 82% yield. 1H NMR (300 MHz, DMSO-d6): δ 3.51–3.56 (m, 1H, H5′-A), 3.60–3.66 (m, 1H, H5′-B), 3.83 (s, 3H, OCH3), 3.93 (m, 1H, H 4′), 4.13 (m, 1H, H-3′), 4.56–4.62 (m, 3H, N6-CH2 and H-2′), 5.03 (t, 1H, J = 5.4 Hz, 5′-OH), 5.17 (d, 1H, J = 4.8 Hz, 3′-OH), 5.41 (d, 1H, J = 6.3 Hz, 2′-OH), 5.81 (d, 1H, J = 6.0 Hz, H-1′), 7.02 (d, 1H, J = 8.7 Hz, Ph), 7.15 (d, 1H, J = 3.0 Hz, Ph), 7.28 (dd, 1H, J = 2.7 and 8.7 Hz, Ph), 8.34 (s, 1H, H-8), 8.63 (br s, 1H, N6-H). 13C NMR (300 MHz, DMSO-d6): δ 38.59 (CH2), 56.49 (OCH3), 62.19 (C-5′), 71.15 (C-3′), 74.13 (C-2′), 86.36 (C-4′), 88.13 (C-1′), 113.02 (Ph), 119.58 (C-5), 124.29 (Ph), 128.10 (Ph), 129.93 (Ph), 140.43 (C-8), 150.69 (C-4), 155.51 (Ph and C-2), 156.33 (C-6). HRMS (ESI-MS) C18H20N8O5Cl [M + H]+: 463.1248 found, 463.1245 calcd. Anal. (C18H19N8O5Cl) C, H, N.
Cell Culture and Membrane Preparation
CHO cells expressing recombinant human ARs or the rat A3AR were cultured in DMEM (Dulbecco’s modified Eagle’s medium) and F12 (1:1) supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 μmol/mL glutamine. After harvest and homogenization, the cells were centrifuged at 500g for 10 min. The pellet was resuspended in 50 mM Tris-HCl buffer (pH 7.4) containing 10 mM MgCl2 and 1 mM EDTA. The suspension was homogenized with an electric homogenizer for 10 s and was then recentrifuged at 20000g for 20 min at 4 °C. The resulting pellets were resuspended in buffer containing 3 units/mL of adenosine deaminase, and the suspension was stored at −80 °C prior to the binding experiments. The protein concentration was measured using the Bradford assay.39
Radioligand Binding Studies
For the A3AR binding experiments, the procedures were similar to those previously described.19 Briefly, each tube contained 100 μL of membrane suspension, 50 μL of [125I]I-AB-MECA ([125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide, final concentration of 0.5 nM), and 50 μL of increasing concentrations of compounds in Tris-HCl buffer (50 mM, pH 7.4) containing 10 mM MgCl2 and 1 mM EDTA. Nonspecific binding was determined using 10 μM NECA (adenosine-5′-N-ethyluronamide). The mixtures were incubated at 25 °C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester (Brandel, Gaithersburg, MD). Filters were washed three times with ice-cold buffer. Radioactivity was determined in a Beckman 5500B γ-counter. The binding of [3H]CCPA (2-chloro-N6-cyclopentyladenosine) to the recombinant hA1AR and the binding of [3H]CGS21680 (2-[p-(2-carboxyethyl)phenylethylamino]-5′-N-ethylcarboxamidoadenosine) to the recombinant hA2AAR was performed as previously described.20,44
Cyclic AMP Accumulation Assay
Intracellular levels of 3′,5′-cyclic AMP were measured by the competitive protein binding method.32 CHO cells expressing recombinant human40 ARs were harvested by trypsinization. After resuspension in the medium, cells were plated in 24-well plates in 0.5 mL of medium/well. After 24 h the medium was removed and cells were washed three times with 1 mL/well of DMEM containing 50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.4. Cells were then treated with agonists and/or test compounds in the presence of rolipram (10 μM) and adenosine deaminase (3 units/mL) and incubated at 37 °C. For the A3AR, after 45 min forskolin (10 μM) was added to the medium, and incubation was continued for an additional 15 min. The reaction was terminated upon removal of the medium, and the cells were lysed with 200 μL/well of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20 °C. For determination of cyclic AMP production, protein kinase A (PKA) was incubated with [3H]cyclic AMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μL of the cell lysate, and 30 μL of 0.1 M HCl. Bound radioactivity was separated by rapid filtration through Whatman GF/C filters under reduced pressure and washed once with cold buffer. Bound radioactivity was subsequently measured by scintillation spectrometry. Calculation of the relative maximal efficacy at the A3AR was determined at a fixed concentration of the nucleoside analogue (10 μM) and expressed as a relative percent of the effect of 10 μM NECA determined in each experiment, which typically reached ×50% inhibition of the forskolin stimulated cyclase.
Molecular Modeling
All calculations were performed on a Silicon Graphics Octane2 workstation (600 MHz IP30 processor, MIPS R14000). Compound 10, 2-(4-cyclopentylmethyl-1,2,3-triazole)-N6-methyladenosine, was constructed with the use of the Sketch Molecule of SYBYL 7.1 (Tripos Inc., 1699 South Hanley Road, St. Louis, MO 63144). A grid search was performed in which flexible bonds were rotated by 0° and 180° for t1 (C5–C6–N6–CMe) at the N6 position, t2 (4′O–4′C–5′C–5′OH) at the 5′-position, and t3 (N3–C2–N1′–N2′) and by 60°, 180°, and −60° for t4 (N3′–C4′–CMe–CCyc) and t5 (C4′–CMe–CCyc–CCyc) at the C2 position. The low-energy conformers from the grid search were reoptimized, removing all torsional constraints. Merck molecular force field (MMFF)41 and charges were applied with the use of distance-dependent dielectric constants and the conjugate gradient method until the gradient reached 0.05 kcal·mol−1·Å−1. After the low-energy conformers from the result of the grid search were clustered, the representative ones from all groups were reoptimized by semiempirical molecular orbital calculations with the PM3 method in the MOPAC 6.0 package.42
A human A3AR model (PDB code 1OEA) constructed by homology to the X-ray structure of bovine rhodopsin with 2.8 Å resolution (PDB code 1F88)38 was used for the docking study. All atom types were assigned by the Amber7_FF99 force field.43 Amber charges for protein and MMFF charges for ligand were calculated. The starting geometry of the ligand conformation was chosen from the human A3AR complex model with Cl-IB-MECA,19 which was already validated by point mutation. The ribose binding position was fixed, using an atom-by-atom fitting method for the carbon atoms of the ribose ring. To determine the binding region of the 2-(4-cyclopentylmethyl-1,2,3-triazole) moiety at the adenine 2 position, the flexible bond defining a χ1 (O–C1′–N9–C4) angle was searched while docked within the putative binding cavity through various low-energy conformers with diverse t1–t5 angles, rotating by −60°, −110°, and −160°, assuming an anti conformation. Several conformations without any steric bump were selected for further optimization. The initial structures of all complexes were optimized using the Amber force field with a fixed dielectric constant of 4.0 and a terminating gradient of 0.05 kcal·mol−1·Å−1.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Supporting Information Available: Elemental analysis data for compounds 1–20, 23, 31, and 33. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Müller CE. Adenosine receptor ligands. Recent developments. Part I. Agonists. Curr Top Med Chem. 2000;7:1269–1288. doi: 10.2174/0929867003374101. [DOI] [PubMed] [Google Scholar]
- 2.Linden J. Cloned adenosine A3 receptors: pharmacological properties, species differences and receptor functions. Trends Pharmacol Sci. 1994;15:298–306. doi: 10.1016/0165-6147(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 3.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J International Union of Pharmacology XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobson KA, Gao Z-G. Adenosine receptors as therapeutic targets. Nat Rev Drug Discovery. 2006;5:247–246. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fishman P, Bar-Yehuda S. Pharmacology and therapeutic applications of A3 receptor subtype. Curr Top Med Chem. 2003;3:463–469. doi: 10.2174/1568026033392147. [DOI] [PubMed] [Google Scholar]
- 6.Fishman P. The A3 adenosine receptor as a new target for cancer therapy and chemoprotection. Exp Cell Res. 2001;269:230–236. doi: 10.1006/excr.2001.5327. [DOI] [PubMed] [Google Scholar]
- 7.Müller CE. Medicinal chemistry of adenosine A3 receptor ligands. Curr Top Med Chem. 2003;3:445–462. doi: 10.2174/1568026033392174. [DOI] [PubMed] [Google Scholar]
- 8.Jacobson KA, Moro S, Kim YC, Li AH. A3 adenosine receptors: Protective vs. damaging effects identified using novel agonists and antagonists. Drug Dev Res. 1998;45:113–124. doi: 10.1002/(SICI)1098-2299(199811/12)45:3/4<113::AID-DDR5>3.0.CO;2-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brambilla R, Cattabeni F, Ceruti S, Barbieri D, Franceshi C, Kim Y, Jacobson KA, Klotz KN, Lohse MJ, Abbracchio MP. Activation of the A3 adenosine receptor effects cell cycle progression and cell growth. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;361:225–234. doi: 10.1007/s002109900186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Lubitz DK, Lin RC, Popik P, Carter MF, Jacobson KA. Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol. 1994;263:59–67. doi: 10.1016/0014-2999(94)90523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Lubitz DK, Carter MF, Deutsch SI, Lin RC, Mastrapaolo J, Meshulam Y, Jacobson KA. The effects of adenosine A3 adenosine receptor stimulation on seizures in mice. Eur J Pharmacol. 1995;275:23–29. doi: 10.1016/0014-2999(94)00734-o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borea PA, Baraldi PG, Chen SF, Leung E. Enhancing treatment of MDR cancer with adenosine A3 antagonists. 2004000224. PCT Int Appl WO. 2003
- 13.Civan MM, Macknight ADC. The ins and outs of aqueous humour secretion. Exp Eye Res. 2004;78:625–631. doi: 10.1016/j.exer.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 14.Okamura T, Kurogi Y, Hashimoto K, Sato S, Nishikawa H, Kiryu K, Nagao Y. Structure–activity relationships of adenosine A3 receptor ligands: New potential therapy for the treatment of glaucoma. Bioorg Med Chem Lett. 2004;14:3775–3779. doi: 10.1016/j.bmcl.2004.04.099. [DOI] [PubMed] [Google Scholar]
- 15.Yang H, Avila MY, Peterson-Yantorno K, Coca-Prados M, Stone RA, Jacobson KA, Civan MM. The cross-species A3 adenosine-receptor antagonist MRS 1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure. Curr Eye Res. 2005;30:747–754. doi: 10.1080/02713680590953147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poulsen SA, Quinn RJ. Adenosine receptors: New opportunities for future drugs. Bioorg Med Chem. 1998;6:619–641. doi: 10.1016/s0968-0896(98)00038-8. [DOI] [PubMed] [Google Scholar]
- 17.Perreira M, Jiang JK, Klutz AM, Gao ZG, Shainberg A, Lu C, Thomas CJ, Jacobson KA. “Reversine” and its 2-substituted adenine derivatives as potent and selective A3 adenosine receptor antagonists. J Med Chem. 2005;48:4910–4918. doi: 10.1021/jm050221l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lenzi O, Colotta V, Catarzi D, Varano F, Filacchioni G, Martini C, Trincavelli L, Ciampi O, Varani K, Marighetti F, Morizzo E, Moro S. 4-Amido-2-aryl-1,2,4-triazolo[4,3-a]quinoxalin-1-ones as new potent and selective human A3 adenosine receptor antagonists. Synthesis, pharmacological evaluation, and ligandreceptor modeling studies. J Med Chem. 2006;49:3916–3925. doi: 10.1021/jm060373w. [DOI] [PubMed] [Google Scholar]
- 19.Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. Structural determinants of A3 adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J Med Chem. 2002;45:4471–4484. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao ZG, Blaustein JB, Gross AS, Melman N, Jacobson KA. N6-Substituted adenosine derivatives: selectivity, efficacy and species differences at A3 adenosine receptors. Biochem Pharmacol. 2003;65:1675–1684. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Volpini R, Constanzi S, Lambertucci C, Vittori S, Klotz KN, Lorenzen A, Cristalli G. Introduction of alkynyl chains on C-8 of adenosine led to very selective antagonists of the A3 adenosine aeceptor. Bioorg Med Chem Lett. 2001;11:1931–1934. doi: 10.1016/s0960-894x(01)00347-x. [DOI] [PubMed] [Google Scholar]
- 22.Elzein E, Palle V, Wu Y, Maa T, Zeng D, Zablocki J. 2-Pyrazolyl-N6-substituted adenosine derivatives as high affinity and selective adenosine A3 receptor agonists. J Med Chem. 2004;47:4766–4773. doi: 10.1021/jm049682h. [DOI] [PubMed] [Google Scholar]
- 23.Volpini R, Constanzi S, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli G. N6-Alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3 receptor and a starting point for searching A2B ligands. J Med Chem. 2002;45:3271–3279. doi: 10.1021/jm0109762. [DOI] [PubMed] [Google Scholar]
- 24.Lewis WG, Green LG, Grynszpan F, Radić Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Click chemistry in situ: acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew Chem, Int Ed. 2002;41:1053–1057. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 25.Alvarez R, Velázquez S, San-Félix A, Aquaro S, De Clercq E, Perno CF, Karlsson A, Balzarini J, Camarassa MJ. 1,2,3-Triazol-[2′,5′-bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]-3′-spiro-5′′-(4′′-amino-1,2′′-oxothiole 2′′,2′′-dioxide) (TSAO) analogues: synthesis and anti-HIV-1 activity. J. Med. Chem. 1994; 37: 4185–4198. (b) Gunji, H., Vasella, A. Oligonucleotides with a nucleobase-including backbone. Part 4. A convergent synthesis of ethylenediyl-linked adenosine tetramers Helv. Chim. Acta 2000; 83: 3229–3245. (c) Epple, R., Kudirka, R., Greenberg, W. A. Solidphase synthesis of nucleoside analogs. J. Comb. Chem. 2003; 5: 292–310. (d) O’Mahony, G., Ehrman, E., Grøtli, M. Synthesis of adenosine-based fluorosides containing a novel heterocyclic ring system. Tetrahedron Lett. 2005; 46: 4745–6748. (e) Moukha-Chafiq, O., Taha, M. L., Lazrek, H. B., Pannecouque, C., Witvrouw, M., De Clercq, E., Barascut, L., Imbach, J. L. Synthesis and biological activity of 4-substituted 1-[1-(2-hydroxymethoxy)-methyl-1,2,3-triazol-( 4 & 5)-ylmethyl]-1H-pyrazolo-[3,4-d]pyrimidines. Nucleosides, Nucleotides Nucleic Acids 2001; 20: 1797–1810. (f) Moukha-Chafiq, O., Taha, M. L., Lazrek, H. B., Vasseur, J. J., Pannecouque, C., Witvrouw, M., De Clercq, E. Synthesis and biological evaluation of some 4-substituted 1-[1-(2-hydroxybutyl)-1,2,3-triazol-(4 & 5)-ylmethyl]-1H-pyrazolo[3,4-d]pyrimidines. Nucleosides, Nucleotides Nucleic Acids 2001; 20: 1811–1821. (g) Wigerinck, P., Van Aerschot, A., Claes, P., Balzani, J., Declercq, E., Herdewijn, P. 3′-(1,2,3-Triazol-1-yl)-2′,3′-dideoxythymidine and 3′-(1,2,3-triazol-1-yl)-2′,3′-dideoxyuridine. J Heterocycl Chem. 1989;26:1635–1642. [Google Scholar]
- 26.Matsuda A, Shinozaki M, Yamaguchi T, Homma H, Nomoto R, Miyasaka T, Watanabe Y, Abiru T. Nucleosides and nucleotides. 103. 2-Alkynyladenosines: a novel class of selective adenosine A2 receptor agonists with potent antihypertensive effects. J Med Chem. 1992;35:241–252. doi: 10.1021/jm00080a007. [DOI] [PubMed] [Google Scholar]
- 27.Feldman AK, Colasson B, Fokin VV. One-pot synthesis of 1,4-disubstituted 1,2,3-triazoles from in situ generated azides. Org Lett. 2004;6:3897–3899. doi: 10.1021/ol048859z. [DOI] [PubMed] [Google Scholar]
- 28.Temple C, Jr, Kussner CL, Montgommery JA. Studies on the azidomethine–tetrazole equilibrium. V. 2- and 6-azidopurines. J Org Chem. 1966;31:2210–2215. [Google Scholar]
- 29.Lioux T, Gosselin G, Mathé C. Azido/tetrazole tautomerism in 2-azidoadenine beta-D-pentofuranonucleoside derivatives. Eur J Org Chem. 2003;20:3997–4002. [Google Scholar]
- 30.Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. Copper(I)-catalysed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J Am Chem Soc. 2005;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 31.Umino T, Yoshioka K, Saitoh Y, Minakawa N, Nakata H, Matsuda A. Nucleosides and Nucleotides. 200. Reinvestigation of 5′-N-ethylcarboxamidoadenosine derivatives: structure–activity relationship for P3 purinoceptor-like proteins. J Med Chem. 2001;44:208–214. doi: 10.1021/jm000150k. [DOI] [PubMed] [Google Scholar]
- 32.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body-fluid. Anal Biochem. 1990;189:234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 33.Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Molecular-cloning and characterisation of the human-A3 adenosine receptor. Proc Natl Acad Sci US A. 1993;90:10365–10369. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohno M, Gao ZG, Van Rompaey P, Tchilibon S, Kim SK, Harris BA, Blaustein J, Gross AS, Duong HT, Van Calenbergh S, Jacobson KA. Modulation of adenosine receptor affinity and intrinsic efficacy in nucleosides substituted at the 2-position. Biooorg Med Chem. 2004;12:2995–3007. doi: 10.1016/j.bmc.2004.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cosyn L, Gao ZG, Van Rompaey P, Lu C, Jacobson KA, Van Calenbergh S. Synthesis of hypermodified adenosine derivatives as selective adenosine A3 receptor ligands. Bioorg Med Chem. 2006;14:1403–1412. doi: 10.1016/j.bmc.2005.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao ZG, Jeong LS, Moon HR, Kim HO, Choi WJ, Shin DH, Elhalem E, Comin MJ, Melman N, Mamedova L, Gross AS, Rodriguez JB, Jacobson KA. Structural determinants of efficacy at A3 adenosine receptors: Modification of the ribose moiety. Biochem Pharmacol. 2004;67:893–901. doi: 10.1016/j.bcp.2003.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tchilibon S, Kim SK, Gao ZG, Harris BA, Blaustein J, Gross AS, Melman N, Jacobson KA. Exploring distal regions of the A3 adenosine receptor binding site: Sterically-constrained N6-(2-phenylethyl)adenosine derivatives as potent ligands. Bioorg Med Chem. 2004;12:2021–2034. doi: 10.1016/j.bmc.2004.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp TE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 39.Bradford MM. Rapid and sensitive method for quantification of microgram quantities of protein utilizing principle of protein–dye binding. Anal Biochem. 1976;72:248. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 40.Baraldi PG, Cacciari B, Pineda de las Infantas MJ, Romagnoli R, Spalluto G, Volpini R, Constanzi S, Vittori S, Cristalli G, Melman N, Park K, Jacobson KA. Synthesis and biological activity of a new series of N6-arylcarbamoyl, 2-(ar)-alkynyl-N6-arylcarbamoyl, and N6-carboxamido derivatives of adenosine-5′-N-ethyluronamide as A1 and A3 adenosine receptor agonists. J Med Chem. 1998;41:3174–3185. doi: 10.1021/jm980147p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halgren TA. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J Comput Chem. 1999;20:730–748. doi: 10.1002/(SICI)1096-987X(199905)20:7<730::AID-JCC8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 42.Stewart JJP. MOPAC: A semiempirical molecular orbital program. J Comput-Aided Mol Des. 1990;4:1–105. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 43.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins nucleic acids and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 44.Hutchison AJ, Williams M, de-Jesus R, Yokoyama R, Oei HH, Ghai GR, Webb RL, Zoganas HC, Stone GA, Jarvis MF. 2-(Arylalkylamino)adenosine-5′-uronamides. A new class of highly selective adenosine A2 receptor ligands. J Med Chem. 1990;33:1919–1924. doi: 10.1021/jm00169a015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.