

ABSTRACT
Proliferation and migration of epidermal keratinocytes are essential for proper cutaneous wound closure after injury. αv integrins and several of their ligands—vitronectin, TGFβ and thrombospondin—are up-regulated in healing wounds. However, the role of αv integrins in wound re-epithelialization is unknown. Here, we show that genetic depletion or antibody-mediated blockade of pan-integrin αv, or the specific heterodimer αvβ6, in keratinocytes limited epidermal proliferation at the wound edge and prevented re-epithelialization of wounded human organotypic skin both in vivo and in vitro. While we did not observe a migration defect upon αv blockade in vivo, αv was necessary for keratinocyte migration over longer distances in organotypic skin. Integrin αv is required for local activation of latent TGFβ, and the wound healing defect in the setting of integrin αv loss was rescued by exogenous, active TGFβ, indicating that the αv-TGFβ signaling axis is a critical component of the normal epidermal wound healing program. As chronic wounds are associated with decreased TGFβ signaling, restoration of TGFβ activity may have therapeutic utility in some clinical settings.
KEYWORDS: integrin αv, organotypic models, skin, TGFβ, wound healing
Introduction
Cutaneous wounds heal through 3 major processes: re-epithelialization, deposition of granulation tissue (connective tissue) and contraction.1 Upon injury, fibroblasts migrate into the wound space where they both proliferate and contribute to the formation of granulation tissue through the secretion extracellular matrix proteins including fibronectin, vitronectin and collagen.2,3 New blood vessels form in the granulating areas (angiogenesis), and leukocytes migrate rapidly to the wound site to prevent infection.4 Epidermal keratinocytes then migrate over the granulation tissue and proliferate to form full-thickness epidermis.2 Both keratinocytes and fibroblasts synthesize key extracellular matrix components to re-form the basement membrane beneath the wound. Some contraction due to fibroblast pulling on the extracellular matrix also facilitates human wound closure, where the associated traction forces contribute to scarring.4,5
Integrin αβ heterodimers in the keratinocyte plasma membrane bind to newly-synthesized extracellular matrix proteins to initiate signal transduction pathways that regulate cellular responses essential for wound healing. There are 16 different α subunits and 8 different β subunits, which together form 24 αβ heterodimers — many of which are expressed in human keratinocytes.6 One of these heterodimers, α9β1, is essential for keratinocyte re-epithelialization during wound healing, while another, α3β1, inhibits keratinocyte migration during wound healing.7,8 The αv class of integrins (specifically, αvβ6) and their ligands—vitronectin, fibronectin, TGFβ, thrombospondin and osteopontin—are upregulated during wound healing in humans.9,10 However, specific roles for αv integrins in wound re-epithelialization are unclear.11
αv integrins activate one of their ligands—transforming growth factor β (TGFβ)—which is secreted into the matrix as an inactive latent form, and contributes to wound healing.12,13 TGFβ isoforms bind to heterodimers of TGFβRI and TGFβRII, which form serine/threonine kinase receptors. Ligand binding leads to phosphorylation of both Smad2 and Smad3, which then bind Smad4 to form transcription factor complexes that translocate to the nucleus to promote transcription of genes that enhance motile, mesenchymal traits through up-regulation of Snail and Slug transcription factors.14,15 Furthermore, several integrin genes are direct transcriptional targets of TGFβ-Smad signaling. Thus, the integrin-TGFβ interaction serves as a feed-forward mechanism to potentiate integrin signaling.
TGFβ stimulates migration of fibroblasts and endothelial cells into granulating wound tissue and promotes deposition and activation of ECM proteins integrins and Focal Adhesion Kinase (FAK), all of which have pro-proliferative and migratory roles.9,10,16-19 However, the specific TGFβ role in keratinocytes during re-epithelialization is unclear, as genetic ablation of TGFβ signaling pathway elements accelerates wound re-epithelialization in some settings.20-24
We previously showed that integrin αv is necessary for keratinocyte proliferation in organotypic skin, and thus we hypothesized that αv integrins are essential for wound re-epithelialization.6 Here, we develop a novel, human organotypic wound re-epithelialization assay and show, both in vivo and in vitro, that αv integrin function in keratinocytes is required for keratinocyte proliferation and efficient wound re-epithelialization. In in vivo xenograft wounds, human skin tissue does not contract rapidly upon wounding, as it does in mouse skin, and thus re-epithelialization can be directly visualized over time.25-27 We show that integrin αv blockade resulted in reduced TGFβ signaling, and significantly compromised wound re-epithelialization that was rescued upon TGFβ restoration. Thus, the αv-TGFβ signaling axis is required for epidermal keratinocyte proliferation during human wound healing.
Results
αv integrins are necessary for organotypic wound re-epithelialization
We utilized human organotypic skin to study epidermal generation and proliferation. To establish these tissues, primary keratinocytes were seeded into a native human dermal matrix, and allowed to proliferate and stratify for 10 days at the air-liquid interface.6,28-30 We have previously used this approach to show that human keratinocytes require integrin αv to maintain the capacity to both proliferate in culture, and to regenerate stratified epidermis.6 However, inducible genetic αv depletion in established mature epidermis has no obvious deleterious effect.6 This suggested that αv may be required for wound re-epithelialization, but may be dispensable for normal epidermal tissue maintenance. To test this idea, we first used a complementary antibody-mediated approach to antagonize integrin αv activity. The validated integrin αv blocking antibody (L230) recognizes all αv heterodimers.31,32 Consistent with this, 7 µg/mL of blocking antibody led to efficient decoration of organotypic skin, primarily in the basal epidermal cells (Figure S1C). As we observed previously with shRNA-mediated αv depletion, exposure to L230 at the time of initial seeding led to a marked reduction in tissue thickness (Figure S1A,B). However, L230 treatment at day 5 after the epidermis was stratified did not alter tissue thickness (Figure S1A,B).
To determine whether integrin αv is required for re-epithelialization during wound healing, we next developed a novel organotypic re-epithelialization assay utilizing a 3D-printed runway platform (Fig. 1A). In this approach, keratinocytes were seeded onto the left half of the runway, and were blocked from migrating to the right half using a 3D-printed physical barrier (Fig. 1A). Upon removal of the barrier cover, normal keratinocytes spontaneously migrated to completely re-epithelialize the right half of the runway (Fig. 1A). Dermal fibroblasts were excluded from these runway platforms. Utilizing these runway platforms, we began antibody treatment (at a dose of 7 µg/mL) 5 days after seeding keratinocytes, at the same time the barriers were removed (Fig. 1B). L230 blocking antibody markedly inhibited re-epithelialization compared to control antibody treatment (Fig. 1C, D). Additionally, the L230 treated tissue on the left side of the runway appeared histologically distinct with some areas of acantholysis that were not seen in control tissues, despite their similarity in tissue thickness (Figure S1D). This was associated with a partial differentiation defect as indicated by diminished levels of Loricrin and Filaggrin in suprabasal layers (Figure S2A,B).
Figure 1.
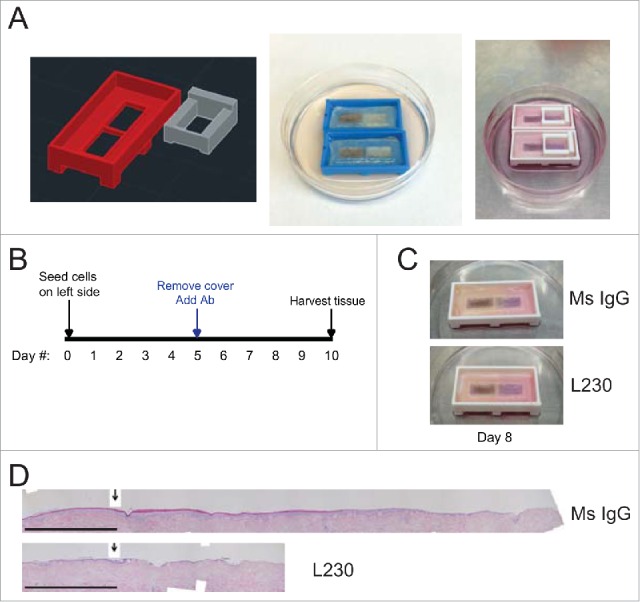
Blocking antibody targeting integrin αv blocks re-epithelialization in an organotypic wound healing model. (A) Images of 3D printed runway for organotypic re-epithelialization assay. (B) Timecourse for organotypic re-epithelialization assay. Keratinocytes are seeded onto the left side of the runway on day 0, and the right side of the runway is blocked until day 5. On day 5, blocking antibodies are added, and keratinocytes migrate over the course of 5 days, until the tissue is harvested on day 10. (C) Visualization of re-epithelialization assay upon treatment with control (Ms IgG) antibody or L230 antibody. (D) Representative Hematoxylin & Eosin (H&E) stain for tissue shown in C, harvested at day 10. Arrow indicates the start of re-epithelialization. Scale bar = 1mm. Tissues shown are representative of 2 independent experiments, each performed in triplicate.
Integrin αv is required for proliferation during human wound healing in vivo
We next utilized the L230 αv blocking antibody to examine the role of integrin αv in epidermal wound healing in vivo. We first grafted human skin onto SCID mice and injected L230 into the grafted skin. The L230 antibody only stained human epidermis, but not mouse epidermis, indicating that this blocking antibody is specific for human αv (Figure S3A). While L230 efficiently decorated the human epidermis, it did not alter the thickness or overall histological appearance of the tissue, compared to control antibody treatment (Figure S3B,C). This is in contrast to the L230 treatment in organotypic culture, which led to some relative skin fragility (Figure S1D).
We next questioned whether L230 mediated integrin αv inhibition would inhibit normal cutaneous wound healing in human skin. Human skin grafted onto mice resists contraction upon wounding.25 We tested this by wounding human skin xenografts on SCID mice using a 2mm punch biopsy, which removed both epidermis and dermis, without altering the fascia or panniculus carnosus layers (Figure S4). We monitored these wounds over time and determined that these wounds re-epithelialized without any appreciable contraction (Figure S4). We next examined wound healing at day 2 or day 4 in xenografted mice treated with L230 αv-blocking antibody (200 µg administered every day beginning one day prior to skin wounding), or a mouse IgG (or IgG1 isotype) control antibody (Fig. 2). At day 2, none of the wounds had healed completely. However, control tissues displayed much higher epidermal proliferation and tissue thickness at the wound edge compared to L230 treated tissues (Fig. 2A, C, D, E). Epidermis that was distant to the active wound edge did not show any difference in proliferation or epidermal tissue thickness in response to L230 (Fig. 2B, D, E). In contrast to the proliferation defects, L230 treated wounds did not display any significant difference in the absolute distance that basal keratinocytes had migrated into the wound bed (Fig. 2F). At day 4, all wounds had almost completely re-epithelialized, though the L230-treated wounds continued to display reduced epidermal proliferation and epidermal thickness compared to mouse IgG1-treated epidermis (Fig. 2D-F). These data indicate that αv integrins are necessary for the normal hyperproliferative response in the acute wound environment, but are dispensable for normal tissue homeostasis.
Figure 2.
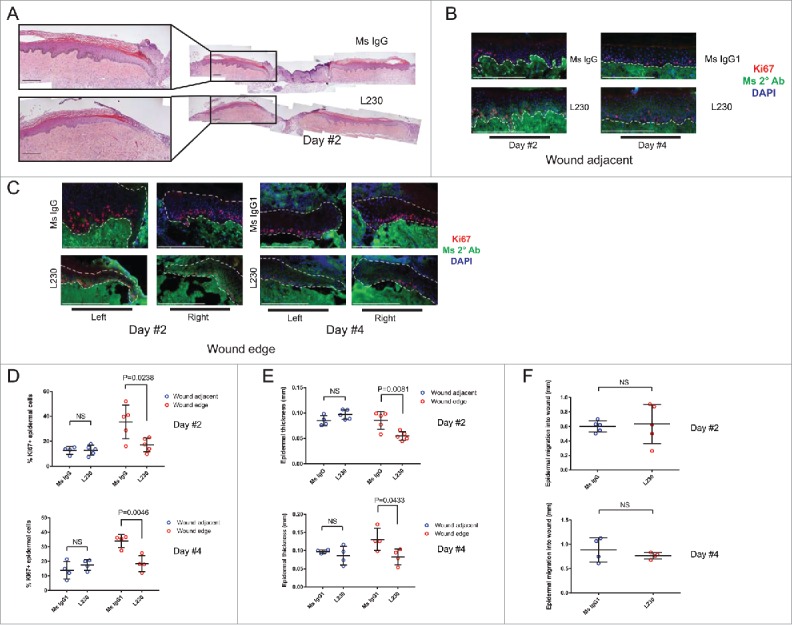
Integrin αv is necessary for proliferation during wound healing of human skin in vivo. (A) Representative Hematoxylin and Eosin (H&E) stains for wounds from mice treated with a control mouse IgG antibody or an L230 antibody. Wounds were harvested 2 days after wounding. B,C. Representative images of Ki67 and mouse secondary antibody labeling of wound adjacent tissue (B) or wound edge tissue (C). Dotted lines indicate the basement membrane zone. (D, E) Quantification of %Ki67+ epidermal cells (D) or epidermal thickness (E) at the wound edge and in wound adjacent tissue. Wound edge was considered within 0.25mm of wound. (F) Quantification of epidermal migration into the wound in mm. N = 4 mice per group for day 2 and 5 mice per group for day 4. NS = not statistically significant. P values were calculated using a student's t-test. Scale bar = 200 µm.
Integrin αv's role in re-epithelialization depends on TGFβ signaling
We next questioned whether αv's regulation of TGFβ signaling in keratinocytes is the mechanism through which αv supports cutaneous wound healing. We thus performed re-epithelialization assays using our organotypic runways using a pan-αv blocking antibody (L230) or an αvβ6 specific blocking antibody (10D5), with or without exogenous, active TGFβ1. The αvβ6 subunit was targeted specifically, as previous work indicated that it is expressed in basal epidermis, and is required for maintenance of keratinocyte proliferation in culture.6 The αv integrins promote the activation of latent, extracellular TGFβ, and we therefore hypothesized that addition of active TGFβ1 would rescue the signaling pathway in the absence of αv activity. As expected, αv knock down reduced TGFβ cleavage from the latent form into the mature, active form (Figure S5). Furthermore, antibody-mediated blockade of pan-αv or αvβ6 reduced levels of the TGFβ signaling pathway intermediate phospho-Smad3 (Figure S6). We found that both L230 and 10D5 significantly reduced epidermal re-epithelialization (Fig. 3A-C). This defect was nearly completely rescued by the concurrent addition of 100 pM TGFβ1 (Fig. 3B, C). We also examined proliferation at the wound edge in these organotypic tissues. We found that L230 or 10D5 treatment reduced the percentage of PCNA+ cells in the wound edge in these tissues (Fig. 3D, E). This was also largely rescued by TGFβ (Fig. 3D, E).
Figure 3.

αv's role in re-epithelialization is partially dependent on TGFβ signaling. (A) Representative Hematoxylin & Eosin stain for runway re-epithelialization assay utilizing the indicated antibodies and TGFβ1 treatments. Arrows indicate the starting edge for the re-epithelialization assay. Scale bar= 1 mm. (B) Quantification of tissues from (A), in terms of epidermal thickness. Epidermal thickness was measured at various distances away from the starting edge of the re-epithelialization assay. N = 9 for Ms IgG, L230, and 10D5, N = 6 for L230 + 100 pM TGFβ and 10D5 + 100 pM TGFβ. p<0.005 (L230) and p<0.05 (10D5) by 2-way ANOVA. C. Quantification of cell layers from (A). Cell layers were counted at various distances away from the starting edge of the re-epithelialization assay. N = 9 for Ms IgG, L230, and 10D5, N = 6 for L230 + 100 pM TGFβ and 10D5 + 100 pM TGFβ. p < 0.001 (L230) and p < 0.001 (10D5) by 2-way ANOVA. D. Representative PCNA staining for tissues represented in panel (A). E. Quantification of the % of PCNA+ cells from panel (D) at the migratory edge. p < 0.05 using one-way ANOVA. *= p < 0.05, **= p < 0.005, and ***= p<0.0005 using Tukey's HSD post-hoc test. Scale bar = 100 µm.
We also specifically examined epidermal migration in the 3-D printed runway platform by labeling keratinocytes with dsRed and observing their ability to migrate across the tissue over the course of 5 days (Fig. 4A). We found that L230 and 10D5 treatment greatly impaired the ability of these cells to migrate (Fig. 4A, B). However, this defect was also completely rescued by addition of TGFβ (Fig. 4A, B). This migration defect contrasts with the results seen in the setting of αv blockade in vivo in which keratinocyte migration into the wound was not impaired. This is likely a reflection of the migration distance in each assay. In the organotypic runway assay, keratinocytes are allowed to migrate up to 6mm, while in the in vivo wound healing assay, wound closure requires only 1mm of keratinocyte migration. It is also likely that some active TGFβ secreted by dermal fibroblasts in vivo is sufficient partially rescue the re-epithelialization defect seen in vitro, restoring migration, but not proliferation.
Figure 4.

Epidermal migration is dependent on an αv-TGFβ signaling axis. (A) Representative fluorescent images of runway tissue seeded with keratinocytes labeled with a dsRed reporter. Tissues were treated with the indicated antibodies and recombinant proteins, which were added 5 days after seeding, and imaged every day. TGFβ1-containing media was replaced every day. (B) Quantification of the images in (A), normalized to the tissues at day 5, prior to treatments. N=3 tissues per group. p<0.0001 by 2-way ANOVA. **=p<0.005, ***=p<0.0005, calculated using Bonferroni post-hoc test. Comparisons are statistically significant between L230 vs. Ms IgG, L230 + 100 pM TGFβ and 10D5 + 100 pM TGFβ, and between 10D5 vs. Ms IgG, L230 + 100 pM TGFβ and 10D5 + 100 pM TGFβ.
We verified these results obtained with the blocking antibody using a genetic knock down approach. We transduced keratinocytes with a doxycycline-inducible integrin αv shRNA. Inducible αv loss in organotypic tissue also resulted in impaired epidermal re-epithelialization (Figure S6), a defect that was partially rescued by TGFβ1 (Fig. S7A,B). Thus, using both blocking antibodies and shRNA, we demonstrated that keratinocyte αv integrins are required for human wound re-epithelialization in a TGFβ-dependent manner.
Discussion
αv integrins and several of their ligands, including TGFβ, thrombospondin and vitronectin, are up-regulated in the epidermis of healing wounds.33 However, the functional significance of this altered expression in human skin wounds was previously unclear. Previous work in murine systems suggested that integrin β5 may have no significant role in wound healing, and integrin β6 may be most important in the skin of aged mice.34-36 However, these results may not be directly applicable to human wound healing because mouse wounds exhibit rapid contraction from the panniculus carnosus muscle layer beneath the skin.27 With the exception of the head and neck region, humans lack this muscle layer and heal primarily via re-epithelialization with minimal wound contraction.27 Here, we show, through specific blockade of human epidermal integrin αv, that keratinocyte αv is necessary for human cutaneous wound healing. Furthermore, utilizing a novel organotypic wound re-epithelialization assay, we show that a critical role of αv in re-epithelialization is to support TGFβ signaling. Understanding how keratinocytes respond to changes in the extracellular matrix to stimulate skin re-epithelialization would help advance efforts to optimize wound healing in the setting of acute injury or chronic disease. Dysregulation at the level of inflammation, ECM deposition, ECM degradation or keratinocyte proliferation and migration can result in chronic wounds,37 which are a significant health burden in the United States, with over 6.5 million patients affected.38 Currently, there are limited treatment options for chronic wounds, with only 2 FDA-approved therapies: platelet derived growth factor (PDGF) and a human skin equivalent.39 These treatments have limited clinical success, and there are no approved treatments that promote keratinocyte re-epithelialization.
Current strategies in development for treatment of chronic wounds target the processes of re-epithelialization, angiogenesis and/or tissue granulation,39 and include plasminogen administration, MMP-9 inhibition, keratinocyte growth factor (KGF) and granulocyte-macrophage colony stimulating factor (GM-CSF).40-43
Chronic wounds are associated with decreased TGF-β activity and our results suggest that restoration of TGFβ in certain clinical settings may have therapeutic utility.44 In this regard, the strategy of using RGD-based peptides (which activate αv integrins) for treatment of chronic wounds is logical, and has shown some success both in vitro and in the clinic.45-47 Additionally, agonistic antibodies specific to αvβ6 may promote keratinocyte proliferation in non-healing wounds. However, caution is warranted here, as β6 over-expression in the mouse results in chronic wound formation.48
This newly discovered role for αv integrins in epidermal wound healing may extend to other epithelial tissues. Much of the research on αv integrins has focused on mesenchymal tissues and the haematopoietic system. Deletion of αv integrins in myeloid lineages causes depletion of Tregs (specifically, Th17 T helper cells), enhanced T cell activation and failure of macrophages and dendritic cells to properly remove apoptotic cells.49,50 These phenotypes are largely attributed to dysregulation of TGFβ signaling in response to αv loss. Depletion of αv in myofibroblasts results in reduced susceptibility to hepatic, pulmonary and renal fibrosis, resulting from reduced TGFβ activation.51 In the brain, αv loss in neural cells results in defective association between vessels and brain parenchyma, resulting in hemorrhage.52,53 The roles that αv plays in various tissues may be largely dependent on the roles of TGFβ signaling in that particular tissue. The roles for αv in other epithelial tissues with proliferative basal cells, such as breast, colon, intestine, ovary, cervix, esophagus, and many others, are still unknown. Given our newly-defined role for αv in re-epithelialization during cutaneous wound healing, this integrin may also contribute to mucosal wound healing in settings such as inflammatory bowel disease, thus opening up new therapeutic opportunities.54
Despite our evidence for a pro-proliferative role for integrin αv during wound healing, there are several reports for an anti-proliferative role of αv integrins in other epithelial tissues. Deletion of αv in the eyelid and conjunctiva results in spontaneous squamous cell carcinoma formation, potentially due to dysregulation of TGFβ signaling.55 Deletion of αv in the mouse epidermis in the absence of p53 leads to accelerated tumor formation (potentially due to Akt activation), but subsequently, slowed tumor growth, potentially due to decreased immune cell infiltration, vascularization of the tumors, or defective keratinocyte proliferation.11 Integrin β6 deficient mice show enhanced proliferation in hair follicles after depilation, also in a TGFβ-dependent manner.56 Some of these differences in phenotypes may be the result of the dose-dependent effects of TGFβ signaling, as partial αv integrin blockade may have different effects than complete genetic loss. Further, TGFβ signaling may have somewhat different effects in mouse and human skin.
We show that αv's supports cutaneous re-epithelialization largely through maintenance of TGFβ signaling. This is in contrast to literature which shows that genetic ablation of TGFβ signaling pathway components results in accelerated wound re-epithelialization, potentially due to release of TGFβ's anti-proliferative effects.16,20,24 These differences may lie in the mechanisms of TGFβ antagonism. Integrin αv regulates the direct activation of latent TGFβ in the extracellular matrix. Upon genetic ablation of TGFβ receptors or Smads, there is still TGFβ ligand present, which can induce non-canonical TGFβ signaling to activate ERK or Akt signaling pathways, which may be responsible for accelerated wound healing or tumorigenesis in the absence of canonical TGFβ signaling. Consistent with our findings, TGFβ has been shown to promote epithelial proliferation in a corneal wound model, where ablation of TGFβRII delayed wound healing, as well as p38 MAPK activation.57 Also consistent with this, TGFβ was shown to promote keratinocyte proliferation in an in vivo wound healing model through upregulation of miR-132, indicating that TGFβ can have pleiotropic effects.58 In addition, the expression and activity of several integrin heterodimers can be affected by ERK and AKT signaling. ERK signaling is known to up-regulate p38 activity, which can promote α3β1 mediated spreading and migration on laminin-332.59 MEK1/ERK signaling also increases expression of α2 and α3 integrins which promote keratinocyte migration.60 AKT signaling increases integrin α4 expression in other cell types,61 while PI3K signaling results in displacement of α6β4 from hemidesmosomes. This facilitates migration by loosening adhesion to basement membrane.59 We show that αv's regulation of TGFβ in keratinocytes is necessary for epidermal re-epithelialization. The effects of TGFβ on re-epithelialization in different tissues or organisms may be dose-dependent and highly localized.
Materials and methods
Cell culture and reagents
Primary human keratinocytes were isolated from neonatal foreskins obtained from the Hospital of the University of Pennsylvania. Foreskins were incubated in 50:50 dispase (Fisher):DMEM (Dulbecco modified Eagle medium, high glucose 4.5g/L) + 5% FBS (fetal bovine serum, Invitrogen) mixture overnight at 4°C. The epidermis was carefully peeled from the underlying dermis and incubated in trypsin for 10 minutes at 37°C. The trypsin was neutralized with DMEM + 5% FBS and 1% antibiotic/antimycotic (Gibco) and spun at 300 g for 5 minutes. The supernatant was removed, and the pellet was plated in keratinocyte media containing 50% Gibco Keratinocyte-SFM + L-glutamine + EGF and BPE, 50% Gibco Cascade Biologics 154 medium for keratinocytes and 1% penicillin/streptomycin (100U/mL, Gibco) for keratinocyte culture. HEK293T cells were purchased from ATCC and also cultured in DMEM + 5% FBS + 1% antibiotic/antimycotic. Recombinant human TGFβ1 was purchased from R&D Systems. Lentivirus production and transduction, hybridoma culture, and fluorescence activated cell sorting are described in the Supplementary Methods.
3D printed Runway Platform
Organotypic runways were printed using ABSplus plasstin on a Statasys uPrint SE Plus 3D printer at the University of Pennsylvania Biomedical library. The growing space consists of a 5×20mm channel, separated by a 0.5mm bar in the center. The stereolithography files containing detailed specifications for the 3D runway platform and cover are attached as supplementary material.
Organotypic culture and runway re-epithelialization
Split-thickness human skin was obtained and washed in PBS containing penicillin/streptomycin, and incubated at 37°C for 7–10 days. PBS was changed every 2 days. The epidermis was separated from the dermis and subsequently discarded. The dermis was washed and incubated in PBS at 4°C for 6–12 weeks. PBS was changed every 2–3 days. For assembly of organotypic tissue, the dermis was cut into 1×2 cm2 pieces. The dermis was elevated to a 3D printed runway platform in a manner such that the basement membrane was oriented up. Several drops of BD matrigel were placed on the bottom of the dermis to create a seal. Keratinocyte growth media (KGM) was added to the lower chamber: KGM (3:1 mixture of DMEM:Ham's F12 supplemented with 10% FBS, adenine (1.8x 10–4M), hydrocrtisone (0.4 µg/mL), insulin (5 µg/mL), cholera toxin (1x 10–10M), EGF (10ng/mL), transferrin (5 µg/mL), and triido-L-thyronine (1.36ng/mL)). Epithelial cells were seeded onto one half of the runway platform on the BM side at density of 5.0×105 keratinocytes, in a total volume of 20 µL of KGM. The upper chamber was kept dry and exposed only to air while the media on the lower chamber was changed every other day. Organotypic skin tissue was harvested at 10–14 days. Organotypic tissues were treated with a polyclonal mouse IgG antibody, an L230 αv- blocking antibody or a 10D5 αvβ6- blocking antibody at a dose of 7 µg/mL. Media was replaced every day during antibody treatments. Imaging of fluorescently labeled cells was performed using a Leica MZ16 F stereomicroscope with a Leica DFC7000 T 2.8 MP color microscope camera and the accompanying Leica Application Suite v4.6 software.
Human skin xenografts
Organotypic human skin was assembled (as described previously) in a 1cm2 square format, and incubated at 37°C for 3–4 days before being grafted onto SCID mice. For skin grafting, SCID mice were anesthetized in an isoflurane chamber, and 1 cm2 of epidermis was removed on the dorsal region of the mouse, down to fascia. Reconstituted human skin was sewn onto the mouse dorsal region with individual interrupted stitches using 6–0 nylon sutures. Mice were dressed with Bactroban ointment, Adaptic, Telfa pad, and Coban wrap. Mice were unwrapped 2 weeks after grafting.
In vivo wound healing
Human skin xenografts were wounded approximately 6 weeks after grafting. Wounding was performed using a 2mm punch biopsy. Human skin and dermis was removed, while the fascia and muscle layer remained intact. Wounds were dressed with Bactroban ointment, Adaptic, Telfa pad, and Coban wrap, and monitored every day. For antibody treatments during wound healing experiments, 200 µg of polyclonal mouse IgG or L230 was administered every day, starting one day prior to wounding.
Western blotting
Adherent cells were washed with PBS and then lysed with RIPA Lite lysis buffer: 50mM Tris pH 7.5, 150mM NaCl, 1mM EDTA, 1% NP-40 containing protease inhibitors (Roche) and phosphatase inhibitors (Roche). Lysates were quantified using Bradford assay, and reduced in Laemmli sample buffer containing β-mercaptoethanol (BioRad). Cell lysates were subjected to SDS gel electrophoresis in 4–15% Tris-Glycine precast polyacrylamide gels (BioRad) in running buffer (25mM Tris, 192mM glycine, 0.1% SDS, pH 8.3). Protein was transferred to PVDF membrane (Millipore) using a Trans-blot Semi-Dry Transfer Cell (BioRad) in semi-dry transfer buffer. Membranes were blocked with 5% milk in TBST or 5% BSA in TBST and incubated in primary antibodies at 4°C overnight recognizing β-actin (Cell Signaling #3700, 1:5000), phospho-FGFR (Tyr653/654, Cell Signaling 55H2, 1:1000), FGFR1 (Cell Signaling D8E4, 1:1000), integrin αv (BD Transduction Laboratories #611012, 1:1000), Smad3 (Cell Signaling #9523, 1:1000), phospho-Smad3 (Cell Signaling #9520, 1:1000), TGF-β (Cell Signaling 56E4, 1:1000) or vinculin (Millipore # MAB3574, 1:2000). After incubation with HRP secondary antibody (cell signaling) for 30 minutes-1 hour at 4°C, proteins were detected using ECL Western Blotting Detection Reagents (GE-Amersham Biosciences) or Luminata Crescendo Western HRP Substrate (Millipore).
Quantification and statistical analysis
Tissue thickness and re-epithelialization were quantified using ImageJ software. For experiments with 2 groups, statistical significance was measured using a student's t-test. For experiments with >2 groups, one-way ANOVA was used to measure statistical significance. For experiments in which ANOVA showed significance, Tukey's HSD (honest significance difference) test was performed. For experiments in which there were 2 factors, 2-way ANOVA followed by Tukey's HSD was performed. *= p < 0.05, **= p < 0.005, ***= p < 0.0005, and NS = not statistically significant.
Supplementary Material
Abbreviations
- ECM
extracellular matrix
- FAK
focal adhesion kinase
- GM-CSF
granulocyte macrophage colony-stimulating factor
- KGF
keratinocyte growth factor
- MAPK
mitogen-activated protein kinase
- MMP-9
matrix metallopeptidase 9
- PCNA
proliferating cell nuclear antigen
- TGFβ
transforming growth factor β
- TGFβRI
transforming growth factor β receptor I
- TGFβRII
transforming growth factor β receptor II
- SCID
severe combined immunodeficiency
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Steve Prouty for his assistance in tissue processing and histology. We also thank Dr. Thomas Leung for critical pre-submission review.
Funding
T.W.R. is supported by a grant from the NIH/NCI (RO1 CA163566), a Penn/Wistar Institute N.I.H. SPORE (P50CA174523), and the Melanoma Research Alliance. E.K.D. received support from an NIH/NIAMS training grant (T32 AR0007465-30), an NIH/NCI F31 NRSA Individual Fellowship (F31 CA186446) and the Patel Family Scholar Award. C.A.N was supported by an NIH/NIAMS training grant (T32 AR0007465-32).
References
- [1].Grinnell F. Fibroblasts, myofibroblasts, and wound contraction. J Cell Biol [Internet] 1994. [cited 2015October8]; 124:401-4. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2119916&tool=pmcentrez&rendertype=abstract; PMID:8106541; http://dx.doi.org/ 10.1083/jcb.124.4.401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nat Rev Cancer [Internet] 2012. [cited 2015July29]; 12:170-80. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22362215; PMID:22362215; http://dx.doi.org/ 10.1038/nrc3217 [DOI] [PubMed] [Google Scholar]
- [3].Schäfer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol [Internet] 2008. [cited 2015September15]; 9:628-38. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18628784; ; http://dx.doi.org/ 10.1038/nrm2455 [DOI] [PubMed] [Google Scholar]
- [4].Shaw TJ, Martin P. Wound repair at a glance. J Cell Sci [Internet] 2009. [cited 2014November1]; 122:3209-13. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2736861&tool=pmcentrez&rendertype=abstract; PMID:19726630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Penn JW, Grobbelaar AO, Rolfe KJ. The role of the TGF-β family in wound healing, burns and scarring: a review. Int J Burns Trauma [Internet] 2012. [cited 2015October8]; 2:18-28. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3415964&tool=pmcentrez&rendertype=abstract; PMID:22928164 [PMC free article] [PubMed] [Google Scholar]
- [6].Duperret EK, Dahal A, Ridky TW. Focal adhesion-independent integrin αv regulation of FAK and c-myc is necessary for 3D skin formation and tumor invasion. J Cell Sci [Internet] 2015. [cited 2015September14]; 128:3887-4013. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26359297; http://dx.doi.org/ 10.1242/jcs.175539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Margadant C, Raymond K, Kreft M, Sachs N, Janssen H, Sonnenberg A. Integrin alpha3beta1 inhibits directional migration and wound re-epithelialization in the skin. J Cell Sci [Internet] 2009. [cited 2013March12]; 122:278-88. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19118220; PMID:19118220 [DOI] [PubMed] [Google Scholar]
- [8].Singh P, Chen C, Pal-Ghosh S, Stepp MA, Sheppard D, Van De Water L. Loss of integrin alpha9beta1 results in defects in proliferation, causing poor re-epithelialization during cutaneous wound healing. J Invest Dermatol [Internet] 2009. [cited 2013January14]; 129:217-28. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18633440; PMID:18633440; http://dx.doi.org/ 10.1038/jid.2008.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cavani A, Zambruno G, Marconi A, Manca V, Marchetti M, Giannetti A. Distinctive Integrin Expression in the Newly Forming Epidermis During Wound Healing in Humans. J Invest Dermatol [Internet] 1993. [cited 2014December12]; 101:600-4. Available from: http://dx.doi.org/10.1111/1523-1747.ep12366057; PMID:8409530 [DOI] [PubMed] [Google Scholar]
- [10].Clark RA, Ashcroft GS, Spencer MJ, Larjava H, Ferguson MW. Re-epithelialization of normal human excisional wounds is associated with a switch from α v β 5 to α v β 6 integrins. Br J Dermatol [Internet] 1996. [cited 2015June3]; 135:46-51. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8776358; PMID:8776358; http://dx.doi.org/ 10.1111/j.1365-2133.1996.tb03606.x [DOI] [PubMed] [Google Scholar]
- [11].Savar A, Acin S, Gonzalez CL, El-Sawy T, Mejia O, Li Z, Esmaeli B, Lacy-Hulbert A, El-Naggar AK, McCarty JH, et al.. Loss of epithelial p53 and αv integrin cooperate through Akt to induce squamous cell carcinoma yet prevent remodeling of the tumor microenvironment. Oncogene [Internet] 2014. [cited 2014December12]; 34:516-24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24469034; PMID:24469034; http://dx.doi.org/ 10.1038/onc.2013.585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci [Internet] 2003. [cited 2015October8]; 116:217-24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12482908; PMID:12482908; http://dx.doi.org/ 10.1242/jcs.00229 [DOI] [PubMed] [Google Scholar]
- [13].Mamuya FA, Duncan MK. αV Integrins and TGF-β Induced EMT; a Circle of Regulation. J Cell Mol Med 2013; 16:445-55; http://dx.doi.org/ 10.1111/j.1582-4934.2011.01419.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Massagué J. TGFbeta in Cancer. Cell [Internet] 2008. [cited 2015February5]; 134:215-30. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3512574&tool=pmcentrez&rendertype=abstract; http://dx.doi.org/ 10.1016/j.cell.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gordon KJ, Blobe GC. Role of transforming growth factor-β superfamily signaling pathways in human disease. Biochim Biophys Acta [Internet] 2008. [cited 2015July9]; 1782:197-228. Available from: http://www.sciencedirect.com/science/article/pii/S092544390800029X; PMID:18313409; http://dx.doi.org/ 10.1016/j.bbadis.2008.01.006 [DOI] [PubMed] [Google Scholar]
- [16].Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E. Loss of TGFβ signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell [Internet] 2007. [cited 2013February16]; 12:313-27. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2424201&tool=pmcentrez&rendertype=abstract; PMID:17936557; http://dx.doi.org/ 10.1016/j.ccr.2007.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Essayem S, Kovacic-Milivojevic B, Baumbusch C, McDonagh S, Dolganov G, Howerton K, Larocque N, Mauro T, Ramirez A, Ramos DM, et al.. Hair cycle and wound healing in mice with a keratinocyte-restricted deletion of FAK. Oncogene [Internet] 2006. [cited 2013March2]; 25:1081-9. Available from: www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2710133&tool=pmcentrez&rendertype=abstract; http://dx.doi.org/ 10.1038/sj.onc.1209130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol [Internet] 2005. [cited 2013February27]; 6:56-68. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15688067; PMID:15688067; http://dx.doi.org/ 10.1038/nrm1549 [DOI] [PubMed] [Google Scholar]
- [19].Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer [Internet] 2014. [cited 2014August8]; 14:598-610. Available from: http://dx.doi.org/10.1038/nrc3792; PMID:25098269; http://dx.doi.org/ 10.1038/nrc3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chan T, Ghahary A, Demare J, Yang L, Iwashina T, Scott PG, Tredget EE. Development, characterization, and wound healing of the keratin 14 promoted transforming growth factor-beta1 transgenic mouse. Wound Repair Regen [Internet] 2002. [cited 2015October8]; 10:177-87. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12100379; PMID:12100379; http://dx.doi.org/ 10.1046/j.1524-475X.2002.11101.x [DOI] [PubMed] [Google Scholar]
- [21].Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev [Internet] 2003. [cited 2015September14]; 83:835-70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12843410; PMID:12843410 [DOI] [PubMed] [Google Scholar]
- [22].Falanga V, Schrayer D, Cha J, Butmarc J, Carson P, Roberts AB, Kim SJ. Full-thickness wounding of the mouse tail as a model for delayed wound healing: accelerated wound closure in Smad3 knock-out mice. Wound Repair Regen [Internet] 2004. [cited 2015October8]; 12:320-6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15225210; PMID:15225210; http://dx.doi.org/ 10.1111/j.1067-1927.2004.012316.x [DOI] [PubMed] [Google Scholar]
- [23].Amendt C, Mann A, Schirmacher P, Blessing M. Resistance of keratinocytes to TGFbeta-mediated growth restriction and apoptosis induction accelerates re-epithelialization in skin wounds. J Cell Sci [Internet] 2002. [cited 2015October8]; 115:2189-98. Available from: http://jcs.biologists.org/content/115/10/2189.abstract; PMID:11973359 [DOI] [PubMed] [Google Scholar]
- [24].Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, et al.. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol [Internet] 1999. [cited 2015October8]; 1:260-6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10559937; PMID:10559937; http://dx.doi.org/ 10.1038/12971 [DOI] [PubMed] [Google Scholar]
- [25].Escámez MJ, García M, Larcher F, Meana A, Muñoz E, Jorcano JL, Del Río M. An in vivo model of wound healing in genetically modified skin-humanized mice. J Invest Dermatol [Internet] 2004. [cited 2015October8]; 123:1182-91. Available from: http://dx.doi.org/10.1111/j.0022-202X.2004.23473.x; http://dx.doi.org/ 10.1111/j.0022-202X.2004.23473.x [DOI] [PubMed] [Google Scholar]
- [26].Truong A-TN, Kowal-Vern A, Latenser BA, Wiley DE, Walter RJ. Comparison of dermal substitutes in wound healing utilizing a nude mouse model. J Burns Wounds [Internet] 2005. [cited 2015September2]; 4:e4. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1501115&tool=pmcentrez&rendertype=abstract; PMID:16921409 [PMC free article] [PubMed] [Google Scholar]
- [27].Wong VW, Sorkin M, Glotzbach JP, Longaker MT, Gurtner GC. Surgical approaches to create murine models of human wound healing. J Biomed Biotechnol [Internet] 2011. [cited 2015October8]; 2011:1-8. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2995912&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Monteleon CL, McNeal A, Duperret EK, Oh SJ, Schapira E, Ridky TW. IQGAP1 and IQGAP3 Serve Individually Essential Roles in Normal Epidermal Homeostasis and Tumor Progression. J Invest Dermatol [Internet] 2015; 135(9):2258-65:[cited 2015June4]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/25848980; PMID:25848980; http://dx.doi.org/ 10.1038/jid.2015.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].McNeal AS, Liu K, Nakhate V, Natale CA, Duperret EK, Capell BC, Dentchev T, Berger SL, Herlyn M, Seykora JT, et al.. CDKN2B Loss Promotes Progression from Benign Melanocytic Nevus to Melanoma. Cancer Discov [Internet] 2015. [cited 2015October8]; 5:1072-85. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26183406; PMID:26183406; http://dx.doi.org/ 10.1158/2159-8290.CD-15-0196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ridky TW, Chow JM, Wong DJ, Khavari PA. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat Med [Internet] 2010. [cited 2012November15]; 16:1450-5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21102459; http://dx.doi.org/ 10.1038/nm.2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Goswami RK, Bajjuri KM, Forsyth JS, Das S, Hassenpflug W, Huang ZZ, Lerner RA, Felding-Habermann B, Sinha SC. Chemically Programmed Antibodies Targeting Multiple Alpha(v) Integrins and Their Effects on Tumor-Related Functions in Vitro. Bioconjug Chem [Internet] 2011. [cited 2015October19]; 22:1535-44. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3277862&tool=pmcentrez&rendertype=abstract; PMID:21774545; http://dx.doi.org/ 10.1021/bc2000879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Weinacker A, Chen A, Agrez M, Cone RI, Nishimura S, Wayner E, Pytela R, Sheppard D. Role of the integrin α v β 6 in cell attachment to fibronectin. Heterologous expression of intact and secreted forms of the receptor. J Biol Chem [Internet] 1994. [cited 2015October20]; 269:6940-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8120056; PMID:8120056 [PubMed] [Google Scholar]
- [33].Longmate WM, Dipersio CM. Integrin Regulation of Epidermal Functions in Wounds. Adv Wound Care [Internet] 2014. [cited 2015October18]; 3:229-46. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3955963&tool=pmcentrez&rendertype=abstract; http://dx.doi.org/ 10.1089/wound.2013.0516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang X, Griffiths M, Wu J, Farese RV, Sheppard D. Normal development, wound healing, and adenovirus susceptibility in beta5-deficient mice. Mol Cell Biol [Internet] 2000. [cited 2013April21]; 20:755-9. Available from: http://mcb.asm.org/content/20/3/755.short; PMID:10629031; http://dx.doi.org/ 10.1128/MCB.20.3.755-759.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Huang X, Wu J, Cass D, Erle D. Inactivation of the integrin β 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol [Internet] 1996. [cited 2013May6]; 133:921-8. Available from: http://jcb.rupress.org/content/133/4/921.abstract; PMID:8666675; http://dx.doi.org/ 10.1083/jcb.133.4.921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].AlDahlawi S, Eslami A, Häkkinen L, Larjava HS. The alphavbeta6 integrin plays a role in compromised epidermal wound healing. Wound Repair Regen [Internet] 2006. [cited 2015June1]; 14:289-97. Available from: http://proxy.library.upenn.edu:2077/doi/10.1111/j.1743-6109.2006.00123.x/full; PMID:16808807; http://dx.doi.org/ 10.1111/j.1743-6109.2006.00123.x [DOI] [PubMed] [Google Scholar]
- [37].Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res [Internet] 2012. [cited 2015August10]; 49:35-43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22797712; PMID:22797712 [DOI] [PubMed] [Google Scholar]
- [38].Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, Longaker MT. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen [Internet] 2009. [cited 2015February15]; 17:763-71. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2810192&tool=pmcentrez&rendertype=abstract; PMID:19903300; http://dx.doi.org/ 10.1111/j.1524-475X.2009.00543.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mustoe T. Understanding chronic wounds: a unifying hypothesis on their pathogenesis and implications for therapy. Am J Surg [Internet] 2004. [cited 2015June30]; 187:S65-70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15147994; http://dx.doi.org/ 10.1016/S0002-9610(03)00306-4 [DOI] [PubMed] [Google Scholar]
- [40].Shen Y, Guo Y, Mikus P, Sulniute R, Wilczynska M, Ny T, Li J. Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood [Internet] 2012. [cited 2015October18]; 119:5879-87. Available from: http://www.bloodjournal.org/content/119/24/5879.abstract; PMID:22563086; http://dx.doi.org/ 10.1182/blood-2012-01-407825 [DOI] [PubMed] [Google Scholar]
- [41].Gooyit M, Peng Z, Wolter WR, Pi H, Ding D, Hesek D, Lee M, Boggess B, Champion MM, Suckow MA, et al.. A Chemical Biological Strategy to Facilitate Diabetic Wound Healing. ACS Chem Biol [Internet] 2014. [cited 2015October18]; 9:105-10. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3947039&tool=pmcentrez&rendertype=abstract; PMID:24053680; http://dx.doi.org/ 10.1021/cb4005468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Koria P, Yagi H, Kitagawa Y, Megeed Z, Nahmias Y, Sheridan R, Yarmush ML. Self-assembling elastin-like peptides growth factor chimeric nanoparticles for the treatment of chronic wounds. Proc Natl Acad Sci U S A [Internet] 2011. [cited 2015October18]; 108:1034-9. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3024670&tool=pmcentrez&rendertype=abstract; PMID:21193639; http://dx.doi.org/ 10.1073/pnas.1009881108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Marques da Costa R, Jesus FM, Aniceto C, Mendes M. Double-blind randomized placebo-controlled trial of the use of granulocyte-macrophage colony-stimulating factor in chronic leg ulcers. Am J Surg [Internet] 1997. [cited 2015October18]; 173:165-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9124619; PMID:9124619; http://dx.doi.org/ 10.1016/S0002-9610(97)89589-X [DOI] [PubMed] [Google Scholar]
- [44].Pastar I, Stojadinovic O, Krzyzanowska A, Barrientos S, Stuelten C, Zimmerman K, Blumenberg M, Brem H, Tomic-Canic M. Attenuation of the transforming growth factor β-signaling pathway in chronic venous ulcers. Mol Med [Internet] 2010. [cited 2015November24]; 16:92-101. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2804290&tool=pmcentrez&rendertype=abstract; PMID:20069132; http://dx.doi.org/ 10.2119/molmed.2009.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Steed DL, Ricotta JJ, Prendergast JJ, Kaplan RJ, Webster MW, McGill JB, Schwartz SL. Promotion and Acceleration of Diabetic Ulcer Healing by Arginine-Glycine-Aspartic Acid (RGD) Peptide Matrix. Diabetes Care [Internet] 1995. [cited 2016January25]; 18:39-46. Available from: http://care.diabetesjournals.org/content/18/1/39.abstract; PMID:7698046; http://dx.doi.org/ 10.2337/diacare.18.1.39 [DOI] [PubMed] [Google Scholar]
- [46].Mertz PM, Davis SC, Franzen L, Uchima FD, Pickett MP, Pierschbacher MD, Polarek JW. Effects of an arginine-glycine-aspartic acid peptide-containing artificial matrix on epithelial migration in vitro and experimental second-degree burn wound healing in vivo. J Burn Care Rehabil [Internet] [cited 2016January25]; 17:199-206. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8736364; PMID:8736364; http://dx.doi.org/ 10.1097/00004630-199605000-00004 [DOI] [PubMed] [Google Scholar]
- [47].Fong E, Tzlil S, Tirrell DA. Boundary crossing in epithelial wound healing. Proc Natl Acad Sci U S A [Internet] 2010. [cited 2016January25]; 107:19302-7. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2984212&tool=pmcentrez&rendertype=abstract; PMID:20974917; http://dx.doi.org/ 10.1073/pnas.1008291107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Häkkinen L, Koivisto L, Gardner H, Saarialho-Kere U, Carroll JM, Lakso M, Rauvala H, Laato M, Heino J, Larjava H. Increased expression of beta6-integrin in skin leads to spontaneous development of chronic wounds. Am J Pathol [Internet] 2004. [cited 2015June1]; 164:229-42. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1602209&tool=pmcentrez&rendertype=abstract; http://dx.doi.org/ 10.1016/S0002-9440(10)63113-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Acharya M, Mukhopadhyay S, Païdassi H, Jamil T, Chow C, Kissler S, Stuart LM, Hynes RO, Lacy-Hulbert A. αv Integrin expression by DCs is required for Th17 cell differentiation and development of experimental autoimmune encephalomyelitis in mice. J Clin Invest [Internet] 2010. [cited 2015October19]; 120:4445-52. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2993596&tool=pmcentrez&rendertype=abstract; PMID:21099114; http://dx.doi.org/ 10.1172/JCI43796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lacy-Hulbert A, Smith AM, Tissire H, Barry M, Crowley D, Bronson RT, Roes JT, Savill JS, Hynes RO. Ulcerative colitis and autoimmunity induced by loss of myeloid v integrins. Proc Natl Acad Sci [Internet] 2007. [cited 2015October19]; 104:15823-8. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1994135&tool=pmcentrez&rendertype=abstract; PMID:17895374; http://dx.doi.org/ 10.1073/pnas.0707421104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, et al.. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med [Internet] 2013. [cited 2015June1]; 19:1617-24. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3855865&tool=pmcentrez&rendertype=abstract; PMID:24216753; http://dx.doi.org/ 10.1038/nm.3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].McCarty JH, Monahan-Earley RA, Brown LF, Keller M, Gerhardt H, Rubin K, Shani M, Dvorak HF, Wolburg H, Bader BL, et al.. Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Mol Cell Biol [Internet] 2002. [cited 2015October19]; 22:7667-77. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=135679&tool=pmcentrez&rendertype=abstract; PMID:12370313; http://dx.doi.org/ 10.1128/MCB.22.21.7667-7677.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].McCarty JH, Lacy-Hulbert A, Charest A, Bronson RT, Crowley D, Housman D, Savill J, Roes J, Hynes RO. Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development [Internet] 2005. [cited 2015October19]; 132:165-76. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15576410; PMID:15576410; http://dx.doi.org/ 10.1242/dev.01551 [DOI] [PubMed] [Google Scholar]
- [54].Neurath MF. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol [Internet] 2014. [cited 2015October19]; 7:6-19. Available from: http://dx.doi.org/10.1038/mi.2013.73; PMID:24084775; http://dx.doi.org/ 10.1038/mi.2013.73 [DOI] [PubMed] [Google Scholar]
- [55].McCarty JH, Barry M, Crowley D, Bronson RT, Lacy-Hulbert A, Hynes RO. Genetic ablation of alphav integrins in epithelial cells of the eyelid skin and conjunctiva leads to squamous cell carcinoma. Am J Pathol [Internet] 2008. [cited 2014November26]; 172:1740-7. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2408432&tool=pmcentrez&rendertype=abstract; PMID:18467691; http://dx.doi.org/ 10.2353/ajpath.2008.070700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Xie Y, McElwee KJ, Owen GR, Häkkinen L, Larjava HS. Integrin β6-deficient mice show enhanced keratinocyte proliferation and retarded hair follicle regression after depilation. J Invest Dermatol [Internet] 2012. [cited 2012November13]; 132:547-55. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22113470; PMID:22113470; http://dx.doi.org/ 10.1038/jid.2011.381 [DOI] [PubMed] [Google Scholar]
- [57].Terai K, Call MK, Liu H, Saika S, Liu CY, Hayashi Y, Chikama T, Zhang J, Terai N, Kao CW-C, et al.. Crosstalk between TGF-β and MAPK signaling during corneal wound healing. Invest Ophthalmol Vis Sci [Internet] 2011. [cited 2015October21]; 52:8208-15. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3208026&tool=pmcentrez&rendertype=abstract; PMID:21917935; http://dx.doi.org/ 10.1167/iovs.11-8017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Li D, Wang A, Liu X, Meisgen F, Grünler J, Botusan IR, Narayanan S, Erikci E, Li X, Blomqvist L, et al.. MicroRNA-132 enhances transition from inflammation to proliferation during wound healing. J Clin Invest [Internet] 2015. [cited 2015October18]; 125:3008-26. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26121747; PMID:26121747; http://dx.doi.org/ 10.1172/JCI79052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Santoro MM, Gaudino G, Marchisio PC. The MSP receptor regulates alpha6beta4 and alpha3beta1 integrins via 14-3-3 proteins in keratinocyte migration. Dev Cell [Internet] 2003. [cited 2016May20]; 5:257-71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12919677; PMID:12919677; http://dx.doi.org/ 10.1016/S1534-5807(03)00201-6 [DOI] [PubMed] [Google Scholar]
- [60].Chernyavsky AI, Arredondo J, Karlsson E, Wessler I, Grando SA. The Ras/Raf-1/MEK1/ERK signaling pathway coupled to integrin expression mediates cholinergic regulation of keratinocyte directional migration. J Biol Chem [Internet] 2005. [cited 2016May20]; 280:39220-8. Available from: http://www.jbc.org/content/280/47/39220.full; PMID:16150734; http://dx.doi.org/ 10.1074/jbc.M504407200 [DOI] [PubMed] [Google Scholar]
- [61].Su Y, Zheng L, Wang Q, Bao J, Cai Z, Liu A. The PI3K/Akt pathway upregulates Id1 and integrin α4 to enhance recruitment of human ovarian cancer endothelial progenitor cells. BMC Cancer [Internet] 2010. [cited 2016May20]; 10:459. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2940800&tool=pmcentrez&rendertype=abstract; PMID:20796276; http://dx.doi.org/ 10.1186/1471-2407-10-459 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.