

Abstract
The Big Data era in Biomedical research has resulted in large-cohort data repositories such as The Cancer Genome Atlas (TCGA). These repositories routinely contain hundreds of matched patient samples for genomic, proteomic, imaging, and clinical data modalities, enabling holistic and multi-modal integrative analysis of human disease. Using TCGA renal and ovarian cancer data, we conducted a novel investigation of multi-modal data integration by combining histopathological image and RNA-seq data. We compared the performances of two integrative prediction methods: majority vote and stacked generalization. Results indicate that integration of multiple data modalities improves prediction of cancer grade and outcome. Specifically, stacked generalization, a method that integrates multiple data modalities to produce a single prediction result, outperforms both single-data-modality prediction and majority vote. Moreover, stacked generalization reveals the contribution of each data modality (and specific features within each data modality) to the final prediction result and may provide biological insights to explain prediction performance.
I. Introduction
Rapid advances in biomedical research alongside the dawn of the Bid Data era have resulted in large biomedical data repositories such as the Cancer Genome Atlas (TCGA). These repositories have emerged in response to a critical need for improved prediction of cancer patient prognosis and response to treatment, which remain difficult due to the heterogeneity and molecular complexity of cancer [1]. Despite a large body of research investigating biomarkers for cancer endpoints such as grading and patient survival, it is unclear which data modalities (e.g., histopathological imaging, genomic, or clinical) are most valuable or useful. Though evidence suggests that the choice of data modality affects prediction performance, few studies have directly compared the performance of prediction models derived from different data modalities. Despite cross-platform and cross-batch variance, the combination of multiple datasets of similar modality can improve biomarker detection and prediction of cancer endpoints, suggesting that information in these datasets are complementary [2].
Among available integrative data research, few studies have focused on prediction modeling, much less combination of prediction models. Furthermore, few studies have highlighted the importance of combining comprehensive histopathological image features with –omics data [3]. Existing integrative imaging and genomic studies focus primarily on radiology [4, 5] or imaging meta-data [6]. We use TCGA renal (KIRC) and ovarian (OV) cancer that includes histopathological whole-slide imaging and RNA-seq (i.e., gene, isoform, exon, and junction expression) modalities. We then use two methods, majority voting and stacked generalization, to assess the effect of combining multiple data modalities on prediction of cancer grade and patient survival. Although stacked generalization was initially developed as a generalized variant of cross validation for classifier model selection, it has not been documented as a method for combining multiple data modalities [7, 8] and has rarely been used for biomedical applications [9].
We aim to address the following questions: (1) How does the choice of biomedical data modality affect the prediction of cancer endpoints? (2) Does the combination of multiple data modalities improve prediction performance? (3) In terms of biology, how can we interpret a prediction model that includes multiple data modalities?
II. Methods
A. Datasets
We use large-cohort KIRC and OV datasets from The Cancer Genome Atlas (TCGA) to predict two cancer endpoints: grade and patient survival. We use four genomic data modalities, all derived from RNA-seq, and one imaging data modality from histopathological whole-slide imaging (WSI). Clinical information pertaining to cancer grade and patient survival is only available for a subset of samples. Table 1 lists the total number of samples available for prediction modeling for each cancer and prediction endpoint. Samples are divided into training and validation sets in order to evaluate prediction modeling.
Table 1. Datasets and Prediction Endpoints for Renal, Ovarian, and Pancreatic Cancer.
| Cancer Grade | Patient Survival | |||||
|---|---|---|---|---|---|---|
| Renal Cancer | Training | Validation | Training | Validation | ||
| Grade 1 or 2 | 95 | 94 | Known Survival <5 Years | 62 | 61 | |
| Grade 3 or 4 | 115 | 114 | Known Survival >= 5 Years | 49 | 48 | |
|
| ||||||
| Total Samples | 210 | 208 | Total Samples | 111 | 109 | |
|
| ||||||
| Ovarian Cancer | Training | Validation | Training | Validation | ||
| Grade 1 or 2 | 14 | 14 | Known Survival <5 Years | 63 | 62 | |
| Grade 3 or 4 | 111 | 111 | Known Survival >= 5 Years | 18 | 17 | |
|
| ||||||
| Total Samples | 125 | 125 | Total Samples | 81 | 79 | |
B. RNA-Seq Data Preparation
RNA-seq expression data obtained from TCGA were processed using the SeqWare engine [10]. Briefly, MapSplice was used to align RNA-seq reads to the human genome (hg19, GRCh37) [11]. Subsequently, RSEM was used to quantify all genes and isoforms with respect to the genome annotation file, also obtained from TCGA [12]. Genome alignments were used to quantify exon and junction reads. Table 2 lists the total number of features for each modality.
Table 2. Dimensionality of Data Modalities.
| Data Modality | # of Features |
|---|---|
| RNA-Seq Gene | 20531 |
| RNA-Seq Isoform | 73599 |
| RNA-Seq Exon | 239322 |
| RNA-Seq Junction | 249567 |
| Histopathological WSIs | 4610 |
We apply the Trimmed Mean of M-values (TMM) method to normalize all modalities of RNA-seq genomic data [13]. We estimate TMM scaling factors using the edgeR package in R [14]. The TMM method requires a reference sample, so we preselect a set of reference samples for normalization that are not used for prediction modeling. The reference samples are not usable for a particular cancer and endpoint. Table 3 lists the total number of reference samples used for each cancer and endpoint. The TMM method trims extreme M-values and A-values (upper and lower 30% for M-values and upper and lower 5% for A-values), and then computes the weighted average of M-values as the scaling factor. The normalized expression estimates for genes, isoforms, exons, and junctions are the raw read count, divided by the product of total read count and TMM scaling factor, multiplied by the average total read count across all sequencing samples. We add one to the normalized expression estimates and then log2 transform the data to yield the final expression estimates.
Table 3. Reference Samples Used for Normalization.
| Cancer | Endpoint | Data Modality | # of Normalization Reference Samples |
|---|---|---|---|
| Renal Cancer | Cancer Grade | Genomic | 51 |
| Imaging | 33 | ||
| Patient Survival | Genomic | 249 | |
| Imaging | 231 | ||
| Ovarian Cancer | Cancer Grade | Genomic | 12 |
| Imaging | 313 | ||
| Patient Survival | Genomic | 102 | |
| Imaging | 403 |
C. Image Data Preparation
TCGA contains hematoxylin and eosin (H&E) stained whole-slide image (WSI) tissue samples. We begin with WSIs of 1,092 tumor samples from 563 OV patients and 906 tumor samples from 451 KIRC patients. We reduce this initial set to the matched set of patients indicated in Table 1.
We identify and remove WSI regions that are not informative for cancer diagnosis but may influence analysis. As described by Kothari et al. in [15], a typical TCGA WSI contains non-tissue regions such as large white regions representing blank, tissue-less portions of the slide and bluish-green regions representing pen marks used by pathologists to annotate the slide. We remove these non-tissue regions using HSV-color space thresholding and morphological analysis. Tissue fold artifacts are then detected as described by Kothari et al [15]. Finally, we crop the WSI into a matrix of 512×512-pixel non-overlapping tiles. Finally, we select tiles with greater than 50% tissue and less than 10% tissue fold artifacts for feature extraction.
We extract 461 image features from each tile. These features capture color, texture, and morphological properties [16]. We combine the features extracted from all tiles for a patient into a single feature vector. To do this, we represent each feature of a patient as a histogram with a fixed number of bins. In other words, we quantize each feature into B bins. For each WSI, we then estimate the percent of its tiles that fall into each bin. Finally, all B percentages for all features represent a single patient profile. Quantization values for a feature are estimated using the number of bins B=10, and a feature-dependent dynamic range: lower limit Li and upper limit Ui, calculated based on the distribution Di of the feature i across all tiles of all patients in the reference set. The number of reference images available for each cancer and endpoint is listed in Table 3. Mathematically, the limits are:
where the function Qp(D) returns the pth percentile of distribution D, and IQD is the interquartile distance.
D. Single Data Modality Prediction Modeling
We use nested cross validation for prediction modeling of each individual data modality, as described by Parry et al. [17] and based on the guidelines for prediction modeling developed by MAQC-II [18]. We perform 5-fold nested cross validation using the training dataset to optimize the feature size and classifier. Using the minimum redundancy, maximum relevance (mRMR) feature selection, we choose an optimal feature size between 1 to 100 features [19]. We select the optimal classifier from four: Bayesian (i.e., nearest centroid, LDA, diagonal LDA), K-nearest neighbors, logistic regression, and SVM. The optimal prediction model is then applied to the validation set to obtain the final prediction performance, measured as AUC.
E. Integrating Data Modalities with Stacked Generalization and Majority Voting
Stacked generalization was originally formulated as a method for combining multiple prediction models to obtain a single prediction result [7]. We expand the definition of stacked generalization to combine multiple prediction models derived from different data modalities. As described in Table 1, each cancer endpoint dataset is partitioned into a training set and an independent validation, or testing, set. Using 5-fold cross validation, as described for the single data modality prediction modeling, we identify optimal prediction models for each data modality. Modality decision values are then used to derive the prediction model by solving using linear regression. Level-1 testing data are derived from the prediction models by training using the entire set of Level-0 training data and calculating decision values using the Level-0 testing data. The Level-1 testing data are then used to calculate the final decision values for stacked generalization.
The majority voting method can be viewed as a simplification of stacked generalization that equally weights all constituent prediction models, calculating an average decision value across all modalities for each sample.
III. Results
A. Single Data Modality Prediction Performance is Dependent on Clinical Endpoint and Data Modality
Using only a single data modality, prediction performance of KIRC and OV grade and patient survival is highly dependent on the cancer type and data modality. The dependence on cancer type is concordant with results observed in the MAQC-II study [18]. Cancer type contributes 57.3% of the variance in prediction performance. For most data modalities, we can observe in, that prediction performance of KIRC endpoints (AUC around 0.70) is much higher than that of ovarian cancer endpoints. However, prediction performance for each individual data modality varies for each cancer type and endpoint. The accuracy of renal cancer grading varies from 0.6 to over 0.7 AUC, with RNA-seq junction expression resulting in the lowest external validation performance (cyan ‘x’). Variance in prediction of renal cancer patient survival is higher due to the very low performance of the image data modality. Similarly, prediction of ovarian cancer grade varies from as low as 0.35 to 0.7 AUC, with RNA-seq junction expression resulting in the lowest external validation performance and image data modality resulting in the highest performance. Prediction of ovarian cancer patient survival is low (around 0.5 AUC) regardless of data modality. Analysis of variance shows that data modality is a statistically significant (p=6e-5) source of variance in prediction performance. Although it only contributes 4.1% to total variance, in combination with cancer type or prediction endpoint, it contributes 10.7% and 18.1%, respectively, both statistically significant.
B. Stacked Generalization Improves Prediction
Compared to most individual data modalities, stacked generalization improves prediction performance (Figure 2a). For KIRC grade, stacked generalization performs better than the image, isoform, and junction data modalities, statistically similar to exon data modality. However, stacked generalization significantly underperforms compared to the individual gene expression modality. For KIRC survival, stacked generalization significantly outperforms the gene expression and image data modalities while performing statistically similar to the junction and isoform modalities. The exon data modality individually outperforms stacked generalization for renal cancer survival. For OV grade, stacked generalization significantly outperforms all data modalities except for the image data modality, which outperforms stacked generalization. For the OV survival endpoint, stacked generalization significantly outperforms all data modalities except for the isoform data modality, which performs statistically similar to stacked generalization.
Figure 2. Stacked Generalization Improves Prediction Performance.
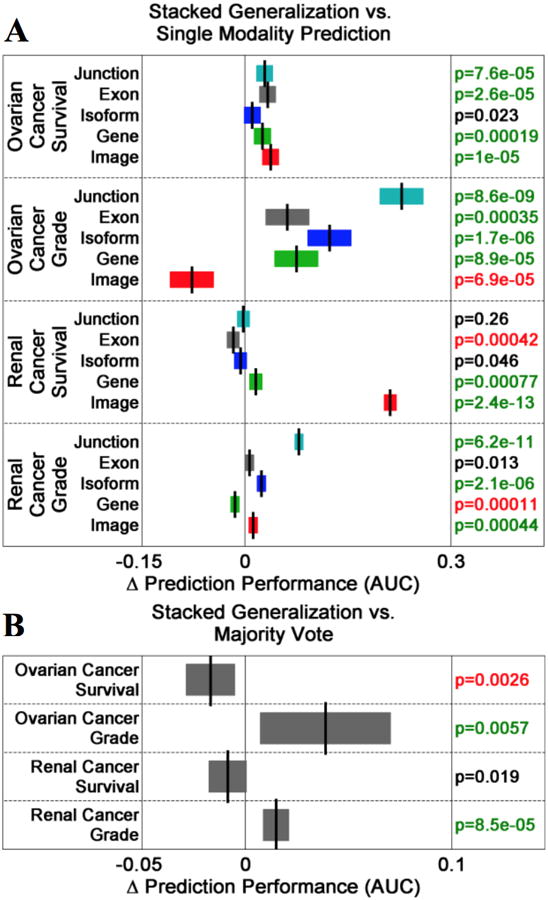
(A) Comparisons of stacked generalization to single data modalities for each cancer endpoint. Box plots indicate differences in prediction performance. Green/red-highlighted p-values indicate that the change in performance is positive/negative and statistically significant. (B) Stacked generalization out-performs the majority vote method of prediction modeling for the OV grade and KIRC grade endpoints. Both methods perform similarly for the KIRC survival endpoint. However, majority vote out-performs stacked generalization for the OV survival endpoint.
Stacked generalization significantly outperforms majority voting for the KIRC and OV grading endpoints (Figure 2b). Stacked generalization and majority voting perform statistically similar for the renal cancer survival endpoint. However, majority voting outperforms stacked generalization for the ovarian cancer survival endpoint (p=0.0026).
C. Stacked Generalization Prediction Models May Reveal Biological Insight
Level-1 prediction models, derived from linear regression, can be interpreted as weights for each data modality. Gene expression is the dominant data modality for the renal cancer grade, renal cancer patient survival, and ovarian cancer grade endpoints, contributing 56%, 48%, and 41%, respectively. For renal cancer survival, imaging is the dominant data modality, contributing 40%. A close inspection of the dominant features and data modalities for each endpoint may lead to biological or experimental insights for renal and ovarian cancer. For example, the specific image features contributing to the ovarian cancer survival prediction model may be concordant with morphological cellular properties interpretable by pathologists [20]. Furthermore, the overall low contribution of exon, junction, and isoform data modalities to the final prediction models may be a result of data quality, as we discuss in the following sections.
IV. Discussion
Although we have used five TCGA data modalities (histopathological whole-slide images and four levels of RNA-seq expression [gene, exon, isoform, and junction]), inclusion or exclusion of some modalities or improving the quality of data may improve prediction performance. Some data modalities, such as gene and isoform expression, may be highly correlated, limiting the benefit of combination. TCGA histopathological WSI data is difficult to handle because of data artifacts and biological heterogeneity [21].
In addition, potential limiting factors for individual data modality prediction modeling and level-1 prediction modeling in stacked generalization warrant further investigation. We have used a mixture of linear and non-linear classifiers for level-0 (i.e., individual data modality) prediction modeling as well as a sophisticated feature selection method (e.g. mRMR) that identifies optimal groups of features. More sophisticated feature selection methods may be better able to identify optimal features and produce better single-modality-data prediction models.
V. Conclusions
The emergence of large-cohort data repositories, such as TCGA, that host multiple biomedical data modalities have enabled integrative analysis that can potentially lead to improved diagnosis or prognosis of cancer endpoints. Our results indicate that a simple data integration method, stacked generalization, can improve prediction performance and provide biological insights. However, the results of this study must be considered in light of some limiting factors in terms of data and prediction modeling methods.
We have used histopathological image data with minimal processing to remove artifacts; further processing of the images to select biological regions-of-interest may improve performance. Finally, we have used linear regression for the level-1 model of stacked generalization, which simply computes a weighted combination of prediction decision values from each data modality. In future work, it may be beneficial to investigate non-linear prediction modeling for stacked generalization.
Figure 1. Multi-Modal Integrative Prediction Modeling with Stacked Generalization.
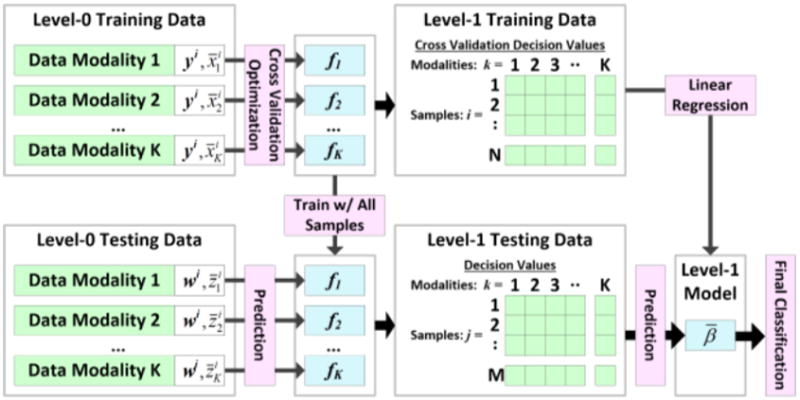
Classifiers from all modalities produce a single prediction model.
Contributor Information
John H. Phan, Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332 USA
Ryan Hoffman, Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332 USA.
Sonal Kothari, Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332 USA.
Po-Yen Wu, School of Electrical and Computer Engineering, Georgia Institute of Technology, Atlanta, GA 30332 USA.
May D. Wang, Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332 USA
References
- 1.Chin L, Andersen JN, Futreal PA. Cancer genomics: from discovery science to personalized medicine. Nature medicine. 2011;17:297–303. doi: 10.1038/nm.2323. [DOI] [PubMed] [Google Scholar]
- 2.Phan JH, Young AN, Wang MD. Robust Microarray Meta-Analysis Identifies Differentially Expressed Genes for Clinical Prediction. The Scientific World Journal. 2012;2012 doi: 10.1100/2012/989637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutman DA, Cobb J, Somanna D, Park Y, Wang F, Kurc T, et al. Cancer Digital Slide Archive: an informatics resource to support integrated in silico analysis of TCGA pathology data. Journal of the Am Med Informatics Association. 2013;20:1091–1098. doi: 10.1136/amiajnl-2012-001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gevaert O, Mitchell LA, Achrol AS, Xu J, Echegaray S, Steinberg GK, et al. Glioblastoma Multiforme: Exploratory Radiogenomic Analysis by Using Quantitative Image Features. Radiology. 2014 doi: 10.1148/radiol.14131731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutman DA, Cooper LA, Hwang SN, Holder CA, Gao J, Aurora TD, et al. MR imaging predictors of molecular profile and survival: multi-institutional study of the TCGA glioblastoma data set. Radiology. 2013;267:560–569. doi: 10.1148/radiol.13120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ping Z, Siegal GP, Almeida JS, Schnitt SJ, Shen D. Mining genome sequencing data to identify the genomic features linked to breast cancer histopathology. J of Pathology Informatics. 2014;5 doi: 10.4103/2153-3539.126147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolpert DH. Stacked generalization. Neural networks. 1992;5:241–259. [Google Scholar]
- 8.Ting KM, Witten IH. Issues in Stacked Generalization. Journal of Artificial Intelligence Research. 1999;10:271–289. [Google Scholar]
- 9.He L, Yang Z, Zhao Z, Lin H, Li Y. Extracting Drug-Drug Interaction from the Biomedical Literature Using a Stacked Generalization-Based Approach. PloS one. 2013;8:e65814. doi: 10.1371/journal.pone.0065814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Connor B, Merriman B, Nelson S. SeqWare Query Engine: storing and searching sequence data in the cloud. BMC bioinformatics. 2010;11:S2. doi: 10.1186/1471-2105-11-S12-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic acids re. 2010;38:e178–e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kothari S, Phan JH, Wang MD. Eliminating tissue-fold artifacts in histopathological whole-slide images for improved image-based prediction of cancer grade. J of pathology informatics. 2013 doi: 10.4103/2153-3539.117448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kothari S, Osunkoya AO, Phan JH, Wang MD. Biological interpretation of morphological patterns in histopathological whole-slide images; Proceedings of the ACM Conference on Bioinformatics, Comp Biology and Biomedicine; 2012. pp. 218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parry R, Jones W, Stokes T, Phan J, Moffitt R, Fang H, et al. k-Nearest neighbor models for microarray gene expression analysis and clinical outcome prediction. The pharmacogenomics journal. 2010;10:292–309. doi: 10.1038/tpj.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nature biotechnology. 2010;28:827–838. doi: 10.1038/nbt.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding C, Peng H. Minimum redundancy feature selection from microarray gene expression data. Journal of bioinformatics and computational biology. 2005;3:185–205. doi: 10.1142/s0219720005001004. [DOI] [PubMed] [Google Scholar]
- 20.Kothari S, Phan JH, Young AN, Wang MD. Histological Image Feature Mining Reveals Emergent Diagnostic Properties for Renal Cancer. 2011 doi: 10.1109/BIBM.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kothari S, Phan JH, Stokes TH, Wang MD. Pathology imaging informatics for quantitative analysis of whole-slide images. Journal of the American Medical Informatics Association. 2013;20:1099–1108. doi: 10.1136/amiajnl-2012-001540. [DOI] [PMC free article] [PubMed] [Google Scholar]