

Abstract
In addition to the previously reported (+)-araguspongine A, (+)-araguspongine C, (+)-araguspongine D, (−)-araguspongine E, and (+)-xestospongin B, two new N-oxide araguspongines, (+)-araguspongine K and (+)-araguspongine L, are described here. Their structures were established on the basis of spectral analyses including 1H–15N HMBC. The promising in vitro antimalarial and antituberculosis activities of araguspongine C are reported.
The family of araguspongine/xestospongin alkaloids contains 13 members.1–6 Chemically, they are dimeric 2,9-disubstituted 1-oxaquinolizidines. Conformational aspects of the 1-oxaquinolizidine ring system have been studied via molecular modeling, NMR, and X-ray crystallographic analyses.1,7 Stereochemical complexities, cis- versus trans-decaline-like conformers and the relative configuration of variously substituted 1-oxaquinolizidines, have also been studied and explained.7
In these studies, it was concluded that trans-2,9-dialky-lated 1-oxaquinolizidine rings reside predominantly in the trans-decaline-like conformer, while cis-dialkylated rings reside in the cis-decaline-like conformer. Moreover, the chemical shifts for H-10 in these cis-disubstituted compounds were typically in the range δ 3.8–4.2 and resonate as broad doublets with J ≈ 3 Hz. This is nearly 1 ppm downfield to the shift for H-10 in the C-9 epimeric trans-disubstituted compounds, which typically appear as doublets with J ≈ 8 Hz. These results were further confirmed via calculating isomer distribution obtained from alumina-catalyzed isomerization of araguspongines B, D, and E.1
The 2,9-trans substituents in araguspongine D, having a trans-decaline-like conformation, are both of equatorial orientation, while the 2,9-cis substituents in araguspongine B, having a cis-decaline-like conformation, are also both of equatorial orientation. Thus, it was presumed that the isomerization of C-9 accompanied with inversion of a nitrogen lone pair of electrons must occur by alumina treatment of the dimeric 2,9-disubstituted 1-oxaquinolizi-dine moiety. In other words, if both C-9 and C-9′ substituents in araguspongine D are isomerized by alumina treatment, the conformation of the resulting product, araguspongine B, will change to the thermodynamically stable cis-decaline-like conformation having equatorially oriented substituents at C-9 and C-9′.1
Members of this family have been isolated principally from Xestospongia exigua (Kirkpatrick) and have been shown to possess a variety of pharmacological activities. Araguspongines A,8 C,8 D,6 and E6 were isolated from Haliclona exigua (the correct taxonomic identity is Xesto-spongia exigua), and araguspongines A, D, and E and xestospongin B were obtained from Australian specimens of Xestospongia exigua.3 An undescribed species of Xesto-spongia4 yielded araguspongines A–H and J.4 Araguspongine A has been reported from an undescribed species of Niphates11 from Singapore.
Araguspongines possess diverse important biological activities. For example, araguspongines A and C were shown to inhibit rat brain nitric oxide synthase activity in vitro.8 Araguspongines A, C, and J1,4 and xestospongin B3 exhibited stronger vasodilation activity than papaverine in the perfused model experiment using isolated mesenteric artery of SD-rat. They were also shown to be equally potent inhibitors of both inositol 1,4,5-triphosphate receptor-mediated Ca2+ release and endoplasmic reticulum-calcium pump.9,10
Furthermore, several araguspongines were reported to possess other biological activities, including cytotoxicity,11 antifungal activity,2 and somatostatin and vasoactive intestinal peptide inhibition activity.12
As part of an ongoing research program at the Department of Pharmacognosy, College of Pharmacy, King Saud University, aimed at the discovery of bioactive diverse compounds from marine macrofauna and flora, it was decided to screen Xestospongia exigua (Kirkpatrick) for activity. As far as the original objectives were concerned, the study culminated in the isolation of the known (+)-araguspongine A (1), (+)-araguspongine C (2), (+)-araguspongine D, (−)-araguspongine E, and (+)-xestospongin B, in addition to two novel N-oxide araguspongines, (+)-araguspongine K (3) and (+)-araguspongine L (4), whose isolation, structure elucidation, and antimalarial and antituberculosis activities are the subject of this paper.
Results and Discussion
The frozen coarsely minced X. exigua sponge was percolated several times using 95% ethanol. The dried ethanol extract was partitioned between aqueous acetonitrile and hexane presaturated with each other. The acetonitrile fraction was chromatographed several times using flash silica gel to afford the known araguspongines A (1), C (2), D, and E and xestospongin B, in addition to the new araguspongines K (3) and L (4).
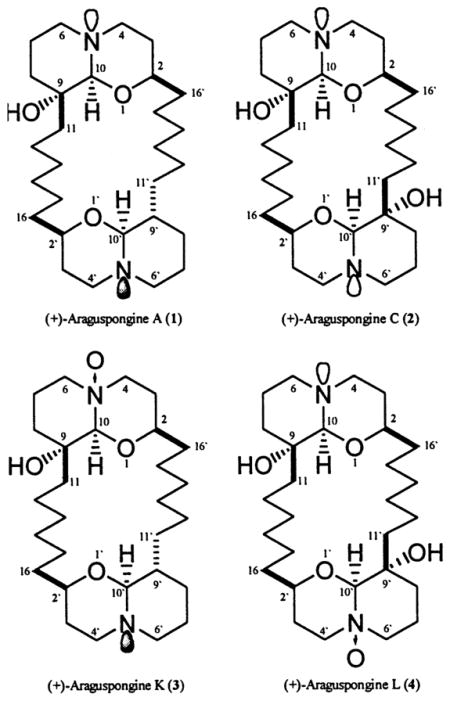
The identity of the known compounds was established by direct comparison of their spectral data with those previously reported for (+)-araguspongine A,3 (+)-araguspongine C,4 (+)-araguspongine D,1 (−)-araguspongine E,1 and (+)-xestospongin B.3 Full NMR data of (+)-araguspongine A (Table 1) are reported here for the first time.
Table 1.
NMR Data of Araguspongines A (1) and K (3)a
| position | 1
|
3
|
|||
|---|---|---|---|---|---|
| δH (m, J Hz) | δC, m | δH (m, J Hz) | δC, m | HMBCc | |
| 2 | 3.44 (dd, 10.7, 10.7) | 76.2, db | 3.75 (dd, 10.7, 10.7) | 77.2, d | 10d |
| 3 | 1.50 (m) | 26.0, t | 1.32 (m) | 31.9, t | 4 |
| 1.61 (m) | 1.55 (m) | ||||
| 4 | 2.89 (m) | 52.4, t | 2.83 (br d, 9.6) | 57.3, t | 3, 6, 10 |
| 2.97 (br d, 8.5) | 3.66 (ddd, 11.8, 11.8, 2.6) | ||||
| 6 | 2.22 (br d, 8.4) | 44.3, t | 3.40 (br dd, 13.4, 3.0) | 68.3, t | 10 |
| 2.91 (m) | 3.51 (ddd, 13.4, 13.4, 4.3) | ||||
| 7 | 1.21 (m) | 20.8, t | 1.54 (m) | 16.5, t | |
| 1.66 (m) | 1.55 (m) | ||||
| 8 | 1.30 (m) | 22.6, t | 1.32 (m) | 22.9, t | 10 |
| 1.40 (m) | 1.52 (m) | ||||
| 9 | 70.7, s | 72.5, s | 10, 11 | ||
| 10 | 3.93 (s) | 91.5, d | 4.24 (s) | 98.4, d | 6 |
| 11 | 1.50 (m) | 38.5, t | 1.57 (m) | 39.7, t | |
| 1.54 (m) | 1.58 (m) | ||||
| 12 | 1.21 (m) | 29.4, t | 1.28 (m) | 29.5, t | |
| 1.51 (m) | 1.58 (m) | ||||
| 13 | 1.25 (m) | 32.2, t | 1.52 (m) | 31.6, t | |
| 1.42 (m) | 1.72 (m) | ||||
| 14 | 1.20 (m) | 25.3, t | 1.30 (m) | 25.6, t | |
| 1.52 (m) | 1.50 (m) | ||||
| 15 | 1.30 (m) | 24.8, t | 1.30 (m) | 25.1, t | |
| 1.60 (m) | 1.52 (m) | ||||
| 16 | 1.48 (m) | 35.5, t | 1.32 (m) | 35.0, t | |
| 1.57 (m) | 1.57 (m) | ||||
| 2′ | 3.23 (dd, 10.7, 10.7) | 75.2, d | 3.33 (dd, 10.7, 10.7) | 75.4, d | 3′, 4′, 10′ |
| 3′ | 1.48 (m) | 32.3, t | 1.48 (m) | 32.4, t | |
| 1.55 (m) | 1.66 (m) | ||||
| 4′ | 2.65 (br d, 10.5) | 54.2, t | 2.72 (ddd, 11.1, 11.1, 4.2) | 54.4, t | |
| 2.85 (br d, 14.2) | 2.92 (ddd, 11.1, 2.9, 2.0) | ||||
| 6′ | 1.88 (ddd, 11.4, 11.4, 2.8) | 54.0, t | 1.96 (ddd, 11.7, 11.7, 2.8) | 54.3, t | 9′, 10′ |
| 2.07 (ddd, 11.4, 2.8, 2.8) | 2.18 (ddd, 11.7, 2.9, 2.9) | ||||
| 7′ | 1.49 (m) | 24.7, t | 1.58 (m) | 24.8, t | 9′ |
| 1.68 (m) | 1.62 (m) | ||||
| 8′ | 1.28 (m) | 28.9, t | 1.28 (m) | 29.0, t | 10′ |
| 1.52 (m) | 1.78 (m) | ||||
| 9′ | 1.54 (m) | 40.3, d | 1.63 (m) | 40.5, d | |
| 10′ | 3.09 (d, 8.6) | 95.5, d | 3.06 (d, 8.6) | 95.6, d | 4′ |
| 11′ | 1.23 (m) | 31.1, t | 1.32 (m) | 31.3, t | |
| 1.42 (m) | 1.60 (m) | ||||
| 12′ | 1.28 (m) | 28.8, t | 1.28 (m) | 29.0, t | |
| 1.52 (m) | 1.78 (m) | ||||
| 13′ | 1.21 (m) | 31.5, t | 1.17 (m) | 30.1, t | |
| 1.43 (m) | 1.37 (m) | ||||
| 14′ | 1.20 (m) | 31.6, t | 1.32 (m) | 31.8, t | |
| 1.41 (m) | 1.52 (m) | ||||
| 15′ | 1.30 (m) | 24.7, t | 1.30 (m) | 25.1, t | |
| 1.61 (m) | 1.52 (m) | ||||
| 16′ | 1.48 (m) | 36.0, t | 1.29 (m) | 35.6, t | |
| 1.57 (m) | 1.51 (m) | ||||
Spectra recorded in CDCl3.
13C multiplicities were determined by DEPT 135°.
Two-bond and three-bond correlations.
Proton number.
(+)-Araguspongine K (3) was isolated as colorless tiny needles in 0.0007% yield. It was shown to possess the molecular formula C28H50O4N2 as derived from the presence of an ion peak at m/z 478.3770 [M]+ and NMR data. The IR spectrum showed an absorption band at 3410 cm−1, indicative of the presence of a hydroxyl function, in addition to a Bohlmann band (2752 cm−1). Therefore, the identity of 3 was concluded to be hydroxylated bis-1-oxaquinolizidine, with at least one oxaquinolizidine ring adopting a trans-decaline-like conformation. The 13C NMR spectrum showed 28 carbon resonances distributed as 22 triplets, five doublets, and a singlet. Half of these resonances were similar to those of the trans-oxaquinolizidine half of araguspongine A (1) (Table 1).
Moreover, compound 3 is an N-oxide araguspongine as derived from 1H–15N HMBC spectra, which showed an oxygenated nitrogen (N-5) resonating at δN 117.1. This nitrogen showed two two-bond correlations (1H–15N) with a sharp singlet proton resonating at δH 4.24 (H-10), and H-6axial, which resonated at δH 3.51 as a ddd (J = 13.4, 13.4, 4.3 Hz). H-10′ resonated at δH 3.06 as a doublet (J = 8.6 Hz), a chemical shift and a coupling pattern and constant identical to those expected for a trans-decaline-like conformation of disubstituted oxaquinolizidines.7 The large coupling constant of H-10′ (J = 8.6 Hz) indicated that H-9′, resonating at δH 1.63, is axial and therefore with β disposition. On the other hand, H-2′ resonated at δH 3.33 as a dd (J = 10.7, 10.7 Hz), and hence it must also be axial, however, with α disposition. These data were in full agreement with the previously mentioned observations about the relative stereochemistry of the 2,9-disubstituted trans-decaline-like conformation of oxaquinolizidines.3,7 These data along with key HMBC correlations (Table 1) suggested that compound 3 is araguspongine A N-oxide.
(+)-Araguspongine L (4) was also isolated as colorless tiny needles in 0.005% yield. High-resolution electrospray ionization mass spectroscopy (HRESIMS) of 4 gave a characteristic ion peak at m/z 494.3719 [M]+ and was consistent with the molecular formula C28H50O5N2. The IR spectrum showed an absorption band for the hydroxyl group (3390 cm−1) and was devoid of Bohlmann bands, indicating the relative stereochemistry of 4 as bis-cis-1-oxaquinolizidine. From the 13C NMR data it was deduced that 4 has 28 carbon resonances, including 22 triplets, four doublets, and two singlets. Half of these resonances were ascribable to araguspongine C (2) (Table 2). The 1H–15N HMBC spectrum showed two nitrogen atoms, one resonating at δN 43.1 (N-5) and the other resonating at δN 117.0 (N-5′), and hence, N-5′ was concluded to be an N-oxide. This conclusion was further confirmed, from the HMBC (1H–15N) spectra, due to the presence of 2JH–N cross-peaks between this nitrogen (N-5′) and a sharp singlet proton resonating at δH 4.23 (H-10′), which correlated to a carbon resonating as a doublet at δC 98.6 (C-10′), and two protons resonating each as a ddd at δH 3.40 (Heq-6′) and δH 3.51 (Hax-6′) and correlated to a carbon resonating as a triplet at δC 68.4 (C-6′).
Table 2.
NMR Data of Araguspongines C (2) and L (4)a
| position | 2
|
4
|
||
|---|---|---|---|---|
| δH (m, J Hz) | δC, m | δH (m, J Hz) | δC, m | |
| 2 | 3.56 (dd, 11.0, 11.0) | 76.8, db | 3.74 (dd, 10.8, 10.8) | 76.7, d |
| 3 | 1.05 (m) | 26.4, t | 1.10 (m) | 26.3, t |
| 1.76 (m) | 1.75 (m) | |||
| 4 | 2.97 (ddd, 13.5, 3.5, 3.5) | 52.9, t | 2.95 (ddd, 13.5, 3.8, 3.8) | 52.9, t |
| 3.11 (ddd, 13.5, 13.5, 3.5) | 3.09 (ddd, 13.5, 13.5, 3.0) | |||
| 6 | 2.34 (br dd, 12.0, 3.0) | 44.6, t | 2.32 (br dd, 11.7, 2.4) | 44.6, t |
| 3.03 (ddd, 12.0, 12.0, 3.0) | 3.00 (ddd, 11.7, 11.7, 2.6) | |||
| 7 | 1.52 (m) | 21.3, t | 1.52 (m) | 21.3, t |
| 1.74 (m) | 1.74 (m) | |||
| 8 | 1.30 (m) | 23.0, t | 1.30 (m) | 22.9, t |
| 1.42 (m) | 1.40 (m) | |||
| 9 | 71.1, s | 71.1, s | ||
| 10 | 4.06 (s) | 90.7, d | 4.03 (s) | 90.6, d |
| 11 | 1.30 (m) | 39.0, t | 1.29 (m) | 40.0, t |
| 1.60 (m) | 1.60 (m) | |||
| 12 | 1.38 (m) | 30.0, t | 1.35 (m) | 29.9, t |
| 1.52 (m) | 1.55 (m) | |||
| 13 | 1.26 (m) | 32.7, t | 1.76 (m) | 32.2, t |
| 1.30 (m) | 1.82 (m) | |||
| 14 | 1.30 (m) | 31.9, t | 1.32 (m) | 31.8, t |
| 1.51 (m) | 1.52 (m) | |||
| 15 | 1.32 (m) | 25.3, t | 1.30 (m) | 25.3, t |
| 1.50 (m) | 1.60 (m) | |||
| 16 | 1.31 (m) | 36.7, t | 1.29 (m) | 36.5, t |
| 1.55 (m) | 1.51 (m) | |||
| 2′ | 3.53 (dd, 11.7, 11.7) | 77.7, d | ||
| 3′ | 1.32 (m) | 30.3, t | ||
| 1.55 (m) | ||||
| 4′ | 2.84 (br d, 12.4) | 57.4, t | ||
| 3.65 (ddd, 12.4, 12.4, 2.8) | ||||
| 6′ | 3.40 (ddd, 13.5, 3.2, 3.2) | 68.4, t | ||
| 3.51 (ddd, 13.5, 13.5, 4.2) | ||||
| 7′ | 2.65 (m) | 16.7, t | ||
| 2.68 (m) | ||||
| 8′ | 1.30 (m) | 23.1, t | ||
| 1.40 (m) | ||||
| 9′ | 72.7, s | |||
| 10′ | 4.23 (s) | 98.6, d | ||
| 11′ | 1.31 (m) | 38.9, t | ||
| 1.58 (m) | ||||
| 12′ | 1.35 (m) | 29.9, t | ||
| 1.55 (m) | ||||
| 13′ | 1.76 (m) | 32.7, t | ||
| 1.82 (m) | ||||
| 14′ | 1.31 (m) | 31.9, t | ||
| 1.49 (m) | ||||
| 15′ | 1.30 (m) | 25.4, t | ||
| 1.60 (m) | ||||
| 16′ | 1.32 (m) | 35.5, t | ||
| 1.52 (m) | ||||
Spectra recorded in CDCl3.
13C multiplicities were determined by DEPT 135°.
The presence of the N-oxide moiety in this half of the molecule made its carbon resonances very similar to those of the N-oxide-containing half of compound 3. Especially noteworthy are downfield shifts for C-4′ (4.5 ppm), C-6′ (23.8 ppm), and C-10′ (8 ppm) when compared to carbons 4, 6, and 10, respectively, which indicated whatever moiety was added, it should have had a profound magnetic and electronic effect on this part of the molecule. Therefore, the identity of araguspongine L was established as araguspongine C N-oxide.
During the in vitro antimalarial activity test, araguspongines K and L were found to be inactive, unlike araguspongine C, which was active against Plasmodium falciparum African (D6) clone with IC50 670 ng/mL and selectivity index >7.1 and against P. falciparum Indochina (W2) clone with IC50 280 ng/mL and selectivity index >17.0, while those of chloroquine were 7 and 220 ng/mL and those of artemisinin were 6 and 7 ng/mL against D6 and W2 clones, respectively. On the other hand, araguspongine C was submitted for antituberculosis activity evaluation and was shown to possess high activity against the H37Rv strain of Mycobacterium tuberculosis with MIC = 3.94 μM, while that of the positive control, rifampin, was 0.61 μM.
Experimental Section
General Experimental Procedure
Optical rotations were measured on a Jasco DIP-370 digital polarimeter. The IR spectra were recorded on a ATI Mattson Genesis Series FTIR spectrophotometer. The 1H and 13C NMR spectra were obtained on a Bruker DRX-400 spectrometer operating at 400 and 100 MHz, respectively. Both 1H and 13C NMR spectra were recorded in CDCl3, and the chemical shift values are expressed in δ (ppm) relative to the internal standard TMS. For the 13C NMR spectra, spectral editing was determined by DEPT. 2D NMR data were obtained using the standard pulse sequence of the Bruker DRX-400 for COSY, HMQC, HMBC, and NOESY. The HRMS spectra were measured on a Bioapex FTMS with electrospray ionization.
15N NMR
Inverse-detected 15N NMR spectra were recorded using a Bruker DRX-500 spectrometer equipped with a 3 mm inverse-detected gradient probe. A gradient HMBC pulse sequence with 1 ms Gaussian Z-axis gradient pulses (70:30: 50) was used, and a 100 ms delay for long-range coupling was used. Referencing of the indirectly detected 15N dimension was accomplished using nitromethane as an external standard. A GHMBC experiment was performed on nitromethane, and the 15N correlation was calibrated to 380.2 so that the chemical shifts are relative to liquid ammonia. This same calibration value was then used for araguspongines. The acquisition time was 24 h for each compound.
Sample Material
The sponge was collected from vertical reef slopes at −17 m at Bayadha, 4 miles north of Jeddah, on the Saudi Arabian Red Sea coast, in November 1999. The specimens are very similar to Xestospongia exigua (Kirkpatrick, 1900) (order Haplosclerida, family Petrosiidae), characterized in life by a dark reddish brown external coloration and cream interior, delicate skeletal architecture, and relatively small spicules. The sponge is massively encrusting (10 cm long × 10 cm wide × 5 cm thick) and the texture relatively crumbly and very sticky to the touch. The surface is irregular with small oscules on mounds on the surface. The skeleton is a delicate round-meshed reticulation of small oxea spicules ranging in length from 63 to 120 μm. A voucher specimen has been deposited in the Natural History Museum, London (BMNH 2002.5.1.1).
Extraction and Isolation
The frozen, coarsely minced sponge (930 g) was percolated at room temperature with 95% EtOH (1 L × 3), and the extract was evaporated in vacuo to leave 58 g of residue. This residue was partitioned between 15% aqueous acetonitrile (1 L) and hexane (1 L × 3) presaturated with each other. The acetonitrile fraction was chromatographed, after evaporation, over flash Si gel (800 g, 10 × 30 cm), using increasing concentrations of MeOH in EtOAc as eluent, to yield partially pure compounds, which upon further chromatography yielded (+)-araguspongine K (7 mg) and (+)-araguspongine L (47 mg), in addition to the previously known araguspongines A (3 mg), C (257 mg), D (5 mg), and E (21 mg) and xestospongin B (2 mg).
(+)-Araguspongine K (3): colorless tiny needles: [α]D25 +2.60° (c 0.35, CHCl3); IR neat (NaCl) νmax 3410, 2927, 2752 cm−1; 1H NMR (CDCl3, 400 MHz), see Table 1; 13C NMR (CDCl3, 100 MHz), see Table 1; HRESIMS m/z 478.3770 [M]+ (calcd for C28H50O4N2 478.3765).
(+)-Araguspongine L (4): colorless tiny needles: [α]D25 +1.68° (c 0.36, CHCl3); IR neat (NaCl) νmax 3390, 2928 cm−1; 1H NMR (CDCl3, 400 MHz), see Table 2; 13C NMR (CDCl3, 100 MHz), see Table 2; HRESIMS m/z 494.3719 [M]+ (calcd for C28H50O5N2 494.3714).
Bioassays
Araguspongines C, K, and L were submitted to the National Center for the Development of Natural Products, School of Pharmacy, the University of Mississippi, MS 38677, to evaluate their in vitro antimalarial activity against African (D6) and Indochina (W2) clones of Plasmodium falciparum using the previously reported method.13 Chloroquine and artemisinin were used as positive controls.
On the other hand, in vitro quantitative antituberculosis activity was evaluated at the Institute for Tuberculosis Research, College of Pharmacy, University of Illinois at Chicago, Chicago, IL 60612, using the microplate-based Alamar Blue assay (MABA) protocol.14 It is a colorimetric oxidation–reduction assay that involves the addition of Alamar Blue dye as an indicator. It evaluates the metabolic activity of the microorganisms. The activity of the evaluated drug is expressed as minimum inhibitory concentration (μM). Araguspongine C was tested for its activity against Mycobacterium tuberculosis (H37Rv). Rifampin was used as a positive control.
Acknowledgments
S. G. Franzblau of the Institute for Tuberculosis Research, College of Pharmacy, University of Illinois at Chicago, is greatly acknowledged for evaluating the antimycobacterial activity. For financial support we acknowledge NIH, grant numbers R01A136596 and K02A101502, and the Research Center, College of Pharmacy, King Saud University, grant number C.P.R.C. 79.
References and Notes
- 1.Kobayashi M, Miyamoto Y, Aoki S, Murakami N, Kitagawa I, In Y, Ishida T. Heterocycles. 1998;47:195–203. [Google Scholar]
- 2.Moon S, MacMillan J, Olmstead M, Ta T, Pessah I, Molinski T. J Nat Prod. 2002;65:249–254. doi: 10.1021/np010427z. [DOI] [PubMed] [Google Scholar]
- 3.Nakagawa M, Endo M. Tetrahedron Lett. 1984;25:3227–3230. [Google Scholar]
- 4.Kobayashi M, Kawazoe K, Kitagawa I. Chem Pharm Bull. 1989;37:1676–1678. doi: 10.1248/cpb.37.1676. [DOI] [PubMed] [Google Scholar]
- 5.Reddy M, Faulkner D. Nat Prod Lett. 1997;11:53–59. [Google Scholar]
- 6.Venkateswarlu Y, Reddy M, Rao J. J Nat Prod. 1994;57:1283–1285. [Google Scholar]
- 7.Hoye T, North J, Yao L. J Org Chem. 1994;59:6904–6910. [Google Scholar]
- 8.Rao J, Desaiah D, Vig P, Venkateswarlu Y. Toxicology. 1998;129:103–112. doi: 10.1016/s0300-483x(98)00067-5. [DOI] [PubMed] [Google Scholar]
- 9.Gafni J, Munsch J, Lam T, Catlin M, Costa L, Molinski T, Pessah I. Neuron. 1997;19:723–733. doi: 10.1016/s0896-6273(00)80384-0. [DOI] [PubMed] [Google Scholar]
- 10.De Smet P, Parys J, Callewaert G, Weidema A, Hill E, De Smedt H, Erneux C, Sorrentino V, Missiaen L. Cell Calcium. 1999;26:9–13. doi: 10.1054/ceca.1999.0047. [DOI] [PubMed] [Google Scholar]
- 11.Pettit G, Orr B, Herald D, Doubek D, Tackett L, Schmidt J, Boyd M, Pettit R, Hooper J. Bioorg Med Chem Lett. 1996;6:1313–1318. [Google Scholar]
- 12.Vassas A, Bourdy G, Paillard J, Lavayre J, Pais M, Quirion J, Debitus C. Planta Med. 1996;62:28–30. doi: 10.1055/s-2006-957790. [DOI] [PubMed] [Google Scholar]
- 13.El Sayed KA, Dunbar DC, Goins DK, Cordova CR, Perry TL, Wesson KJ, Sanders SC, Janus SA, Hamann MT. J Nat Toxins. 1996;5:261–285. [Google Scholar]
- 14.Franzblau SG, Witzig RS, Mclaughlin JC, Torres PM, Guillermo HA, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, Gilman RH. J Clin Microbiol. 1998;36:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]