

Abstract
The P2Y1 receptor is a G protein-coupled receptor (GPCR) and is stimulated by extracellular ADP and ATP. Site-directed mutagenesis of the three extracellular loops (ELs) of the human P2Y1 receptor indicates the existence of two essential disulfide bridges (Cys124 in EL1 and Cys202 in EL2; Cys42 in the N-terminal segment and Cys296 in EL3) and several specific ionic and H-bonding interactions (involving Glu209 and Arg287). Through molecular modeling and molecular dynamics simulations, an energetically sound conformational hypothesis for the receptor has been calculated that includes transmembrane (TM) domains (using the electron density map of rhodopsin as a template), extracellular loops, and a truncated N-terminal region. ATP may be docked in the receptor, both within the previously defined TM cleft and within two other regions of the receptor, termed meta-binding sites, defined by the extracellular loops. The first meta-binding site is located outside of the TM bundle, between EL2 and EL3, and the second higher energy site is positioned immediately underneath EL2. Binding at both the principal TM binding site and the lower energy meta-binding sites potentially affects the observed ligand potency. In meta-binding site I, the side chain of Glu209 (EL2) is within hydrogen-bonding distance (2.8 Å) of the ribose O3′, and Arg287 (EL3) coordinates both α- and β-phosphates of the triphosphate chain, consistent with the insensitivity in potency of the 5′-monophosphate agonist, HT-AMP, to mutation of Arg287 to Lys. Moreover, the selective reduction in potency of 3′NH2-ATP in activating the E209R mutant receptor is consistent with the hypothesis of direct contact between EL2 and nucleotide ligands. Our findings support ATP binding to at least two distinct domains of the P2Y1 receptor, both outside and within the TM core. The two disulfide bridges present in the human P2Y1 receptor play a major role in the structure and stability of the receptor, to constrain the loops within the receptor, specifically stretching the EL2 over the opening of the TM cleft and thus defining the path of access to the binding site.
Receptors coupled to heterotrimeric GTP-binding proteins (G proteins) are integral membrane proteins involved in the transmission of signals from the extracellular environment to the cytoplasm. The family of G protein-coupled receptors (GPCRs),1 which forms one of the largest protein families found in nature, exhibits a common structural motif consisting of seven helical transmembrane domains (TMs) (1, 2).
The mechanisms of signal transduction for GPCRs coupled to different second messengers involve common intracellular domains of the receptor. The binding of extracellular ligands to GPCRs causes yet unclear conformational changes in the receptor protein which promote its association with a distinct subset of G protein heterotrimers (1–3). On the intracellular side of the membrane this association causes the exchange of GDP for GTP bound to the G protein α subunit and the dissociation of the βγ heterodimers (1–3). In turn, both GTP-bound α subunits and βγ complexes of the G protein may initiate intracellular signaling responses by acting on effector molecules, such as adenylate cyclase or phospholipases, or directly regulating ion channel or kinase functions (4, 5).
To better understand the physiological actions of a given GPCR, it is important to identify the molecular mechanisms of recognition between ligands and receptor. Mutational mapping of hypothetical binding sites indicates that large peptide ligands, such as glycoprotein hormones, interact primarily with amino acid residues located among the extracellular loops (ELs) and the outermost segments of the transmembrane helices. In contrast, presumed interaction points for nonpeptide ligands are predominantly found in a deep ligand-binding cleft among the TMs (see refs 6–8 for reviews).
The possible involvement of the ELs in the molecular mechanism of recognition of nonpeptide ligands has not yet been described in detail. A study of chimeric A1/A3 adenosine receptors indicated that the C-terminal portion of the EL2 is essential for the high-affinity binding of xanthine antagonists (9). Site-directed mutagenesis of the human A2A receptor has revealed the involvement of specific glutamic acid residues located within EL2 in the binding of both agonists and antagonists (10). For the α1-adrenergic receptor three amino acids at the C-terminal end of EL2 have been identified to be responsible for subtype-specific antagonist binding (11). For the thyrotropin-releasing hormone receptor (THR-R) a tyrosine located two residues beyond the conserved cysteine in the EL2 has been found to be crucial for ligand binding, and in addition to this residue an asparagine in EL3 was found to be important (12).
Recently, we reported that conformational constraints of the ELs are essential for the activation of the human P2Y1 receptor (Figure 1) (13). This receptor is a GPCR stimulated by extracellular ADP and ATP (14, 15). We have investigated the role in P2Y1 receptor activation of all charged amino acids (D, E, K, and R) and cysteines in the ELs by alanine scanning mutagenesis (13). We found that two disulfide bridges are crucial for the receptor activation and receptor membrane trafficking: the first between Cys124 (EL1) and Cys202 (EL2) and the second between Cys42 (N-terminal segment) and Cys296 (EL3). Moreover, we also found that Glu209 (EL2) and Arg287 (EL3) are most likely involved in the ligand recognition process (13). Upon replacement of these two amino acids, the activity of the receptor was drastically reduced (13). Also, Asp204 (EL2) seems to be involved in the activation of the P2Y1 receptor (13).
Figure 1.

Topology of the hP2Y1 receptor showing residues proposed to be involved in multisite recognition of the agonist, ATP. Circles with bold underline indicate the amino acids involved in the meta-binding site I. Solid highlighted circles indicate the amino acids involved in the meta-binding site II. Solid highlighted circles with thick outline indicate the amino acids involved in both meta-binding site II and the principal TM binding site. Circles with thick outline indicate the amino acids involved in the principal TM binding site. Residues beyond the truncation points indicated at the cytosolic interface and N-terminal domain were not included in the model.
Through a computational approach, in the present study we provide evidence that stable conformational constraints within the extracellular region of the human P2Y1 receptor are crucial for the correct assembly of the receptor architecture and for ligand binding.
EXPERIMENTAL PROCEDURES
Computational Methodologies
All calculations were performed on a Silicon Graphics Indigo2 R8000 workstation.
The molecular simulations were carried out using the Amber (16) all-atom force field implemented in Macromodel 6.0 (17). A fixed dielectric constant of 4.0 was used throughout these calculations with a nonbonded cutoff distance of 8.0 Å. 3D receptor structure representations and docking procedures were performed using Sybyl 6.4.2 (18).
The initial coordinates of the TM region of human P2Y1 were taken from a model recently developed in our group to rationalize the binding of the ATP ligand (19). The TM structure was further modified here by adding residues 37–51 (N-terminal segment), 110–123 (EL1), 192–215 (EL2), and 284–304 (EL3). Secondary structure predictions of these regions were performed using the Chou–Fasman method (20) as implemented in Biology Work Bench 1.5 (21). To facilitate building, we have divided EL2 and EL3 into domains separated by a putative disulfide linkage (N-EL2, residues 192–202; C-EL2, residues 203–215; and N-EL3, residues 284–296; C-EL3, residues 297–303). Each loop segment was built in the predicted conformation and minimized until the rms value of the conjugate gradient (CG) was < 0.1 kcal/mol−1 Å−1. The minimized structure was then subjected to 50 ps of molecular dynamics performed at a constant temperature of 300 K using a time step of 0.001 ps. At this step, each loop segment was connected to the TM bundle. The disulfide bridges between Cys124 (EL1) and Cys202 (EL2) and between Cys42 (N-terminal segment) and Cys296 (EL3), which have been shown to be essential to maintain a high-affinity form of the P2Y1 receptor, were included throughout all calculations. The model with frozen helices was energy minimized until the rms value of the conjugate gradient was <0.1 kcal/mol−1 Å−1. The minimized structure was further refined using the following simulated annealing protocol: the structure was heated to 1500 K in 30 ps followed by 10 ps of constant temperature molecular dynamic simulation at 1500 K. Ten structures were extracted from the trajectory at 1500 K by sampling every 1 ps. Each structure was cooled to 300 K in 50 ps followed by 100 ps of constant temperature simulation at 300 K. The lengths of the bonds involving hydrogen atoms were constrained according to the SHAKE algorithm (22). The receptor structure averaged over the last 50 ps of the equilibrated time period of the molecular dynamic simulation was fully minimized until the rms value of the conjugate gradient was <0.1 kcal/mol−1 Å−1. Only one conformational family was obtained after pairwise root-mean-square deviation analysis implemented in Sybyl. One of these structures was equilibrated at 300 K for 100 ps, and the receptor structure averaged over the last 50 ps was fully minimized until the rms value of the conjugate gradient was <0.05 kcal/mol−1 Å−1.
A model of adenosine 5′-triphosphate (ATP) was constructed using the “Sketch Molecule” of Sybyl. The optimized ATP structure was obtained through semiempirical molecular orbital calculations using the AM1 Hamiltonian (23) as implemented in MOPAC 6.0 (24) (keywords: PREC, GNORM = 0.1, EF). Partial atomic charges for the ligand were imported from the MOPAC output file. ATP meta-binding sites were identified by scanning the entire surface of the ELs and the TM cavity using a rigid docking coupled to continuous energy monitoring (Dock module of Sybyl). When each final position was reached, consistent with a new local energy minimum, the receptor structure averaged over the last 50 ps of molecular dynamic simulation at 300 K was fully minimized until the rms value of the conjugate gradient was < 0.05 kcal/mol−1 Å−1. Curvature and electrostatic potential surface mapping using GRASP software (25) have been also performed to better describe the steric and electrostatic properties of all possible binding sites. The new hypothetical binding sites were selected on the basis of the lowest ligand/receptor interaction energy and electrostatic matching considerations. The interaction energy values were calculated as follows: ΔEcomplex = Ecomplex − (EL + Ereceptor).
Materials
The preparation of the human P2Y1 receptor (pCDP2Y1) and its mutant receptor constructs was previously published (13). The agonists ATP and 3′NH2-ATP were from Sigma (St. Louis, MO), and 2-MeSADP was from RBI (Natick, MA). The agonist HT-AMP was synthesized as described (26). [3H]-myo-Inositol (15 Ci/mmol) was obtained from American Radiolabeled Chemicals (St. Louis, MO). Fetal bovine serum (FBS) was from Gibco (Gaithersburg, MD). O-Phenylenediamine dihydrochloride was purchased from Sigma Chemical Co. (St. Louis, MO). DEAE-dextran was obtained from Pharmacia-LKB (Piscataway, NJ).
Plasmid Construction and Site-Directed Mutagenesis
All mutations were introduced into pCDP2Y1 (13) using standard PCR mutagenesis techniques (27). The accuracy of all PCR-derived sequences was confirmed by dideoxy sequencing of the mutant plasmids (28).
Transient Expression of Mutant Receptors in COS-7 Cells
COS-7 cells (4 × 106) were seeded into 150 mm culture dishes containing 25 mL of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 100 units/ mL penicillin, 100 μg/mL streptomycin, and 2 μmol/mL glutamine. Cells were transfected approximately 24 h later with plasmid DNA (10 μg of DNA/dish) using the DEAE-dextran method (29) for 40 min, followed by treatment with 100 μM chloroquine for 2.5 h, and grown for an additional 24 h at 37 °C and 5% CO2.
Inositol Phosphate Determination
Assays were carried out according to the general approach of Harden et al. (30). About 24 h after transfection, the cells were split into six-well plates (Costar, ~0.75 × 106 cells/well) in DMEM culture medium supplemented with 3 μCi/mL [3H]-myo-inositol. After a 24 h labeling period, cells were preincubated with 10 mM LiCl for 20 min at room temperature. The mixtures were swirled gently to ensure uniformity. Following the addition of agonists, the cells were incubated for 30 min at 37 °C in 5% CO2. The supernatant was removed by aspiration, and 750 μL of cold 20 mM formic acid was added to each well. After a 30 min incubation at 4 °C, cell extracts were neutralized with 250 μL of 60 mM NH4OH. The inositol monophosphate fraction was then isolated by anion-exchange chromatography (31). The contents of each well were applied to a small anion-exchange column (Bio-Rad AG-1-X8) that had been pretreated with 15 mL of 0.1 M formic acid/3 M ammonium formate, followed by 15 mL of water. The columns were then washed with 10 mL water, followed by 15 mL of a solution containing 5 mM sodium borate and 60 mM sodium formate. [3H]Inositol phosphates were eluted with 4.5 mL of 0.1 M formic acid/0.2 M ammonium formate and quantitated by liquid scintillation spectrometry (LKB Wallace 1215 Rackbeta scintillation counter). Pharmacological parameters were analyzed using the KaleidaGraph program (Abelbeck Software, version 3.01). Statistical analysis was performed using the Alternate t-test (InStat version 2.04, GraphPad, San Diego, CA).
RESULTS
Although numerous studies have modeled ligand binding to TM regions of GPCRs (19, 32–37), very few studies have focused on modeling of the ELs and, in particular, on the possible role that these loops might have in the ligand recognition process (12, 38–39).
We extended our previously proposed model of the ATP–P2Y1 complex (19) to include the three ELs and a segment of the N-terminal domain. Due to the lack of structural constraints, the first 36 residues (residues 1–36) in the N-terminal region were excluded from the new model. This omission should not prevent us from using the model in identifying new hypothetical ligand binding sites, because there is no indication that this region plays a significant role in ligand binding (40). A schematic representation of the human P2Y1 receptor is given in Figure 1. Application of this method resulted in an extended backbone motif for each segment as reported in Table 1. We used Chou–Fasman analysis only to obtain preliminary information about the secondary structure of the ELs. In fact, a serious problem in the structure prediction of ELs is their high flexibility. Moreover, specific solvation, local pH, role of glycosylation, ionic–protein, and protein–protein interactions are only some of the factors that could modify the organization of the extracellular loops. None of these perturbations are easily included in the molecular modeling simulations. In the present paper, we used molecular dynamics simulation as a useful tool to select representative structures among the many thermodynamically possible. In particular, we have conducted simulated annealing and molecular dynamics simulations of the human P2Y1 receptor model, and we used this average-minimized structure to improve our previously reported ATP–P2Y1 complex model as described in Experimental Procedures.
Table 1.
Secondary Structure Predictions of the Extracellular and N-Terminal Regions of the Human P2Y1 Receptora
| region (residue nos.) | sequence | prediction |
|---|---|---|
| N-terminal (37–51) | SSFKCALTKTGFQF | extended |
| EL1 (110–123) | YYFNKTDWIFGDAM | extended + turn |
| N-EL2 (192–202) | TGVRKNKTITC | extended |
| C-EL2 (203–215) | YDTTSDEYLRSYF | extended |
| N-EL3 (284–296) | LRARLDFQTPAMC | extended + turn |
| C-EL3 (296–303) | AFNDRVY | extended |
Secondary structure prediction obtained using the method of Chou and Fasman (20).
Topology and Steric Constraints in the Extracellular Loops of the Human P2Y1 Receptor
After simulated annealing and rmsd analysis only one family of P2Y1 conformers was found. The overall architecture of the average-minimized structure of the complex is shown in Figure 2. The first extracellular loop (EL1) forms a span between the ends of TM2 and TM3, with a disulfide bond present at Cys124, which is located at the end of this loop close to the extracellular end of TM3. The longer EL2 is arranged near the exofacial surface of the helical bundle. This arrangement is particularly striking, since it is located at the surface of the receptor and in proximity to TMs 3, 5, 6, and 7. We have already identified a putative binding pocket for both P2Y1 agonists and antagonists, which is located among TMs 3, 5, 6, and 7 (19). Viewed from the top, this particular configuration of EL2 seems to define an entry channel for the ATP molecule (see Discussion below). Cys202, the disulfide bond counterpart of Cys124, is located approximately in the middle of EL2. The region of EL2 beyond Cys202 spans the exofacial path of access to the TM binding cleft. The third extracellular loop forms another flat span between the end of TM6 and TM7, with a second disulfide bridge forming at a Cys residue located in the middle of EL3 (13). The N-terminal segment (residues 37–51) is located parallel to EL3 to allow for the formation of the second disulfide bond between Cys42 (N-terminal) and Cys296 (EL3). Our data seem to suggest that the presence of two disulfides bridges reduced the conformational complexity of the EL domains compared with GPCRs in which only one disulfide bond, usually between TM3 and EL2, is present. Interestingly, the folding patterns of the ELs found in our studies are close to those recently published by Colson et al. (41).
Figure 2.

hP2Y1 receptor model (TM helical bundle, extracellular and N-segment regions) viewed along the helical axes from the extracellular end (see Experimental Procedures for details). Side chains of cysteines participating in disulfide bridge formation are highlighted.
Ligand Recognition in the Human P2Y1 Receptor
Site-directed mutagenesis has been utilized for localizing agonist and antagonist recognition elements in the transmembrane cleft of the human P2Y1 receptor (19, 42). Amino acid residues in TM3, TM5, TM6, and TM7 were found to be involved in nucleotide binding to P2Y1 receptors. In particular, it is possible to distinguish three different parts of the transmembrane domain responsible for ATP binding: TM6 and TM7 are close to the adenine ring, TM3 and TM6 are close to the ribose moiety, and TM3, TM6, and TM7 are near the triphosphate chain (details are summarized in Table 2) (19, 42).
Table 2.
Residues of Meta-Binding Site I, Meta-Binding Site II, and the Principal TM Binding Site of the Human P2Y1 Receptor in Proximity (<5 Å) to Docked ATP
| adenine | ribose | triphosphate | |
|---|---|---|---|
| meta-binding site I | Thr47 (N-terminal) {ND}a | Glu209 (EL2) {>1000 fs}b | Arg287 (EL3) {>1000 fs} |
| meta-binding site II | Gln307 (TM7) {>1000 fs} | Lys280 (TM6) {>1000 fs} | Ser213 (EL2) {ND} |
| Asp204 (EL2) {32 fs} | |||
| Arg128 (TM3) {>1000 fs} | |||
| Thr222 (TM5) {10 fs} | |||
| principal TM binding | Gln307 (TM7) {360 fs} | Arg310 (TM7) {>1000 fs} | Arg128 (TM3) {>1000 fs} |
| Ser314 (TM7) {>1000 fs} | Thr222 (TM5) {10 fs} | ||
| Lys280 (TM6) {>1000 fs} | |||
| Arg310 (TM7) {>1000 fs} |
We have demonstrated using site-directed mutagenesis analysis that both Glu209 (EL2) and Arg287 (EL3) are crucial for the normal transduction of the extracellular signals (13). The E209A mutant receptor (EL2) exhibited a >1000-fold right shift in EC50 of all agonist ligands, such as 2-MeSATP, 2-MeSADP, and HT-AMP, compared with the wild-type receptor, while it responded like the wild-type receptor if Glu209 were substituted with amino acids capable of hydrogen bonding, such as Asp, Gln, or Arg. Arg287 in EL3 was impaired similarly to Glu209 when substituted by Ala, i.e., concentration–response curves where right shifted by >1000-fold of all agonist ligands, such as 2-MeSATP, 2-MeSADP, and HT-AMP, and the shape was identical. Substitution of Arg287 by Lys, another positively charged residue, increased the potency of the agonist but failed to restore wild-type activity. Moreover, the substitution of Arg287 with the negatively charged amino acid Glu drastically reduced the potency of the agonist. Taking into account all the experimental evidence, we have argued that Glu209 and Arg287 could be in proximity (ca. 3.5 Å) to form a strong and stabilizing ionic bridge in the resting form of the wild-type receptor. Analyzing the electrostatic potential surface of the P2Y1 receptor, we find support for this hypothesis. A close contact between the negative potential region corresponding to the formal negative charge of Glu209 and the positive potential region corresponding to the formal positive charge of Arg287 is present. Figure 3 shows the molecular surface of the P2Y1 receptor model colored according to electrostatic potential as calculated using GRASP.
Figure 3.

(Left) Side view of the human P2Y1 receptor model. The side chains of the important residues (Glu209 and Arg287) involved in the ionic bridge are highlighted and labeled. (Right) Surface potential of the P2Y1 receptor model, displayed with GRASP. The molecular surface is color coded by electrostatic potential. Potentials less than −20 kT are in red, those greater than 20 kT are in blue, and neutral potentials (0 kT) are in white. The ionic interaction between Glu209 and Arg287 is clearly distinguishable as a region of intense negative and positive potentials, respectively.
This model is not able to rationalize all the site-directed mutagenesis results already discussed above. For example, it was still unclear why E209D, E209Q, and E209R were fully activated by all agonist ligands, such as 2-MeSATP, 2-MeSADP, and HT-AMP, and responded in a manner indistinguishable from wild-type receptors and also why Arg287 could not be replaced with Lys or Glu (13). In fact, we can speculate about the presence of an ionic interaction between Glu209 and Arg287 in the resting form of the receptor, but we can also hypothesize that these two amino acids could be involved in the recognition of the ligand outside of the transmembrane region.
Meta-Binding Sites Involving the Extracellular Loops of the Human P2Y1 Receptor
Recently, Perlman et al. also reported evidence about the role of the ELs of the thyrotro-pin-releasing hormone receptor in ligand binding (12). Following this hypothesis, we have explored the possibility that for the P2Y1 receptor, as well as for other GPCRs in general, a multistep mechanism for the binding of small ligands may operate. Starting from our updated model of the ATP/P2Y1 receptor (19), we have explored this possibility, using a docking energy-monitor procedure. We have also mapped the curvature and electrostatic potential surface using GRASP software to better describe the steric and the electrostatic properties of all possible binding sites. In fact, we have recently reported that the electrostatic interactions between the negatively charged phosphate groups and the positively charged amino acids of the receptor are crucial for the recognition process of the ATP (19). We found that ATP can be situated in the P2Y1 receptor, in an energetically favorable conformation, within two other regions of the receptor. We have termed these additional binding sites, which are defined by the ELs, meta-binding sites. Meta-binding sites would chronologically precede association at the principal TM site. The first meta-binding site, which is located outside of the TM bundle between EL2 and EL3, represents a possible distal binding site on the P2Y1 receptor. The second meta-binding site is positioned immediately underneath the EL2. A snapshot of this multistep process is summarized in Figure 4. According to our models, we can recognize different sets of amino acids involved in the formation of different meta-binding sites (I and II) and of the principal TM binding site (see Table 2). In particular in meta-binding site I (Figure 5) there appear to be two favorable interactions between an ATP molecule and ELs 2 and 3. The side chain of Glu209 (EL2) is within hydrogen-bonding distance of O3′ of the ribose moiety at 2.8 Å, and Arg287 (EL3) appears to be involved in the coordination of α- and β-phosphates of the triphosphate chain (2.0 Å, O2α, and 1.9 Å, O3β; Figure 5). To consolidate our hypothesis, we have designed an activation experiment in which ATP was replaced with 3′-amino-3′-deoxyadenosine 5′-triphosphate (3′NH2-ATP). Under physiological conditions the 3′-NH2 group of this nucleotide is largely protonated. In this case, if there is an interaction between the substituent at the 3′-position of the ribose moiety and Glu209 (EL2), it might be possible to see a dramatic difference in the potency of the positively charged 3′NH2-ATP between the wild-type receptor and the E209R mutant. In fact, a strong electrostatic repulsion between the protonated 3′-amino group and the positively charged Arg209 side chain may occur. Accordingly, the E209R mutant receptor was activated by 3′NH2-ATP with very low potency compared with the wild-type receptor (see Figure 6).
Figure 4.

Side view of the hP2Y1–ATP complex models describing meta-binding site I, meta-binding site II, and the principal TM binding site. The side chains of the important residues in proximity to the docked ATP (Table 2) are highlighted.
Figure 5.
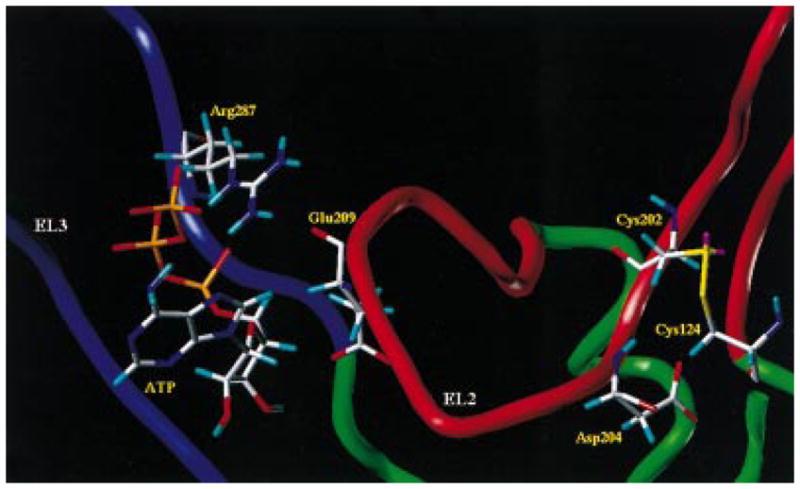
Detailed view of the meta-binding site I model. The side chains of the important residues in proximity (≤5 Å) to the docked ATP molecule are highlighted and labeled.
Figure 6.
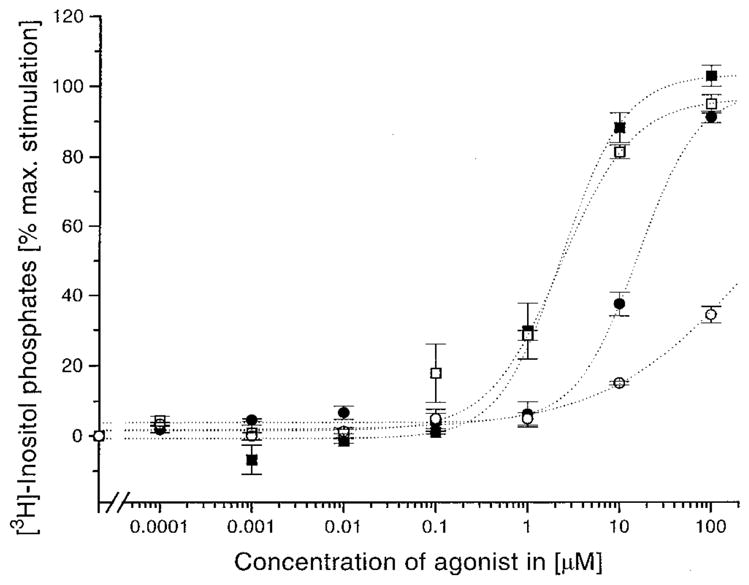
Concentration–response curves of receptors having a mutation in EL2 (E209). The wild-type human P2Y1 receptor (filled symbols) or the mutant receptor (E209R, open symbols) was transiently expressed in COS-7 cells. [3H]Inositol phosphate accumulation was measured in response to ATP (squares) or 3′NH2-ATP (circles) stimulated phospholipase C activation (see Experimental Procedures for details). Maximal response ranged from a 2.5− to 4-fold increase in [3H]inositol phosphate accumulation. Dose–response curves represent the mean of two to four replicate experiments.
There is another amino acid located in EL2, Asp204, that seems to have a role in the physiological activation of the P2Y1 receptor (13). A possibly stabilizing ionic interaction with the positively charged amino acid Lys125, located on the top part of TM3 and close to Asp204, has been excluded by site-directed mutagenesis (13). The K125A mutant receptor exhibited an apparent affinity for 2-MeSATP that was similar to that observed with the wild-type receptor. Asp204 is also in the vicinity of Arg128, which we have already found to be critical for the binding of ATP, and also close to the triphosphate side chain of ATP in meta-binding site II.
Some of the amino acids may interact with the ligand in more than one binding site; for example, Arg128 (TM3), Lys280 (TM6), and Gln307 (TM7) are integral in both meta-binding site II and the principal TM binding site. According to our receptor model, Arg195 (EL2) and Asp204 (EL2), which are probably involved in a relatively less stable meta-binding site, have only a modulatory effect on the activity of the P2Y1 receptor. Vice versa, Glu209 (EL2), Arg287 (EL3), Arg128 (TM3), and Gln307 (TM7) are essential for the receptor functionality because they are involved in the definition of the thermodynamically stable intermediates of the ATP multistep binding process.
DISCUSSION
An increasing number of GPCR subfamilies show diverse modes of ligand binding, signal generation, signal transduction through TMs, and signal transfer to the cytoplasmic environment (1–5). In particular, different ligand recognition pathways have been proposed (6–8). For large ligands such as glycoprotein hormones, the high-affinity binding interactions are with residues in the extracellular domains; for small ligands, such as biogenic amine neurotransmitters or nucleosides and nucleotides, the high-affinity binding pocket is within the TM domains. For peptides, such as angiotensin, there is evidence that both extracellular and TM domains contribute to the binding pocket. The general underlying mechanisms appear to involve changes in interactions and conformation during and after the binding process, particularly rearrangements of loops and TMs (19, 39, 43). As recently reported by Perlman et al., no direct evidence of a multistep mechanism for binding of small ligands to GPCRs has been described previously (12). Turner et al. suggested that TM residues near the extracellular surface of the parathyroid hormone (PTH) receptor may filter access of ligands to the TM bundle, but this proposal, however, has not been accompanied by supporting kinetic or computational analyses (44).
In the present paper we have rationalized, through a computational approach, the experimental evidence that stable conformational constraints within the extracellular region of human P2Y1 receptor are crucial for the correct assembly of the receptor architecture and in ligand binding. In particular, we have shown that a multistep mechanism for binding of ATP could operate. We found that ATP can be situated in the P2Y1 receptor, in an energetically favorable conformation, within two unanticipated regions of the receptor. We have termed these additional binding sites, which are defined by the ELs, meta-binding sites. Two amino acids are particularly crucial for activation of the P2Y1 receptor, Glu209 (EL2) and Arg287 (EL3). As shown in Figures 3 and 5, both of these amino acids seem to be involved in the conformational properties of ELs in the resting form of the wild-type receptor and also in the recognition of extracellular ATP (meta-binding site I).
As previously proposed by Samama et al. (45), we can hypothesize that the P2Y1 receptor, as well as other GPCRs, can exist in equilibrium between two interconvertible states, R (inactive) and R* (active). Moreover, new evidence for a multistate model of GPCR activation has recently been reported (46, 47). Considering the simple two-states model, in the absence of ligand, the inactive state R predominates, possibly because a structural constraint prevents signal generation and/or signal transduction through the TMs. Consistent with our model, this structural constraint could be represented by the ionic interaction between Glu209 and Arg287 (see Figure 3). The release of such a constraint may be related to the conversion of the receptor into the active state R*. The position of the equilibrium between R and R* can be altered by agonist binding or mutations and may be responsible for basal (agonist-independent) activity of a GPCR. We speculate that, in the active state R*, the negative charged carboxylate group of Glu209 can interact with the 3′-hydroxy group of the ribose moiety. Consistent with this model, the E209A mutant receptor is activated at >1000-fold higher agonist concentration than for the wild-type receptor. Moreover, E209D, E209Q, and E209R mutant receptors are fully activated in a manner indistinguishable from the wild-type receptor, indicating that this carboxylate group is more likely to be involved in a hydrogen-bonding interaction than in an ionic interaction. Considering the positively charged Arg287, our model suggests that this amino acid is a likely candidate for the counterion function. As shown in Figure 5, Arg287 coordinates the α- and β-phosphates of the triphosphate side chain. Consistent with this model, the R287A mutant receptor is activated at >1000-fold higher agonist concentration than the wild type. Substitution of Arg287 by Lys, another positively charged residue but with different electron density distribution, only partially restored wild-type activity (13). However, the concentration–response curve shift was sensitive to the nature of the negatively charged phosphate side chain of the agonist (the 2-methylthio 5′-triphosphate = 17-fold, the 2-methylthio 5′-diphosphate = 35-fold, and the 2-hexylthio 5′-monophosphate = 3-fold; see Figure 7). In contrast, a markedly reduced response is observed with R287Q and R287E mutant receptors (13). Hence, Arg287 appears to participate in a direct ionic interaction with the phosphate side chain of the agonist. Furthermore, the presence of the disulfide bridge between Cys124 (EL1) and Cys202 (EL2) constrains EL2 over the extracellular opening of the TM cleft, preventing the direct contact between Arg287 and ATP in the principal binding site (see Figure 4).
Figure 7.

Concentration–response curves of receptors with the mutated residue in the EL3 (R287). The wild-type human P2Y1 receptor (filled symbols) or the mutant receptor in which Arg287 was converted to lysine (R287K, open symbols) was transiently expressed in COS-7 cells. [3H]Inositol phosphate accumulation was measured in response to 2-MeSADP (squares) or HT-AMP (circles) stimulated phospholipase C activation (see Experimental Procedures for details). Maximal response ranged from a 2.5− to 4-fold increase in [3H]inositol phosphate accumulation. Dose–response curves represent the mean of two to six replicate experiments (13).
These meta-binding site I interactions are followed by secondary interactions with the remainder of the receptor (meta-binding site II), allowing an ATP molecule to move from the extracellular environment to the TM cleft (principal TM binding site). From the present modeling study, Asp204, located in EL2, seems to have a role in the definition of meta-binding site II. As shown in Figure 4, Asp204 is close to the ATP triphosphate side chain in meta-binding site II (ca. 5 Å). As demonstrated using site-directed mutagenesis, the D204A mutant receptor displayed a right-shifted concentration–response curve to stimulation using 2-MeSADP, by ca. 30-fold (13). This shift is similar for all agonists and independent of the length of the 5′-phosphate chain. This fact most likely excludes an involvement of Asp204 in a possible Mg2+ coordination. However, water molecules may be involved to bridge the two groups. Interestingly, this amino acid is located at the same position as a critical tyrosine found in the EL2 of the THR-receptor, i.e., two residues beyond the conserved cysteine (12).
In the multistep binding process presented in this paper, the binding energies calculated for ATP at both meta-binding sites are lower with respect to that calculated for the regular TM binding site. Furthermore, meta-binding site I is closer in energy to the principal TM binding site than is meta-binding site II. A schematic energy diagram is presented in Figure 8. It is important to underline that the calculated energies do not correspond to the real energetic value in a rigorous thermodynamic way. They can only be compared to each other in terms of more or less favorable states. Consequently, these molecular interaction energy values cannot be used to calculate exact values of affinities, since changes in entropy and solvation effects are not taken into account. However, meta-binding site I and the principal TM binding site, having deep minima in the energy profile diagram, may be involved in the definition of the ATP/P2Y1 macroscopic equilibrium constant (Keq) as shown in Figure 8. Since conventional pharmacological assays are performed under equilibrium conditions, we have to suppose that only the effects of those amino acids which contribute to define thermodynamically stable binding sites can be detected using this kind of assay. Moreover, the effects of all the amino acids that stabilize the receptor in its high-affinity configuration, even if they are not in direct contact with the ligand, can nevertheless be measured.
Figure 8.

Potential energy profile for the multistep binding process proposed for ATP at the human P2Y1 receptor. The interaction energy values, in kcal/mol, were calculated as follows: ΔEcomplex = Ecomplex – (EL + Ereceptor). The macroscopic equilibrium constant Keq has been defined considering the two deep minima of the energy profile diagram corresponding to the meta-binding site I and the TM binding site.
We have also shown that rigid conformational constraints in the ELs, such as disulfide bridges, appear to be required for the proper functioning of the P2Y1 receptor (13). In our model these linkages seem to be important to allow the receptor to attain a correct conformation during the multistep binding process and, in particular, in the definition of meta-binding sites. However, the role of the disulfide bridges as conformational determinants of the ELs and on the general functionality of GPCRs is still unresolved. In any case, with rare exception, for example the Mas oncogene and the cannabinoid receptors (48), which lack a disulfide bridge, all of the members of GPCRs contain a cysteine in the putative first extracellular loop (EL1) near the top of the third transmembrane domain (TM3) and another cysteine in the second extracellular loop (EL2) (49). It has been found that this pair of Cys residues forms a disulfide bond in many GPCRs (49). This linkage has been proposed to be important for overall receptor conformation and/or for correct protein trafficking to the membrane surface. We have demonstrated using alanine scanning site-directed mutagenesis that, in the ELs of the human P2Y1 receptor, two essential disulfide bridges are found. The first bridge is between Cys124 of EL1 and Cys202 of EL2, and the second is between Cys42 (N-terminal) and Cys296 of EL3, which probably constrain the receptor in a high-affinity conformation (13). In particular, the presence of a second, less common disulfide bridge between the N-terminal domain and EL3 can drastically reduce the possible movement of TM1 relative to TM7. Similar findings were made for the angiotensin (AT1) receptor (50) and for the interleukin-8 (IL-8 type A) receptor (51), and it was concluded that this second disulfide bridge would help to properly position extracellular amino acids involved in the ligand binding and signal transduction. Moreover, as recently proposed by different authors, rotations and translations of the TM domains might be crucial factors in the ligand recognition process in different GPCRs (19, 39, 43).
In conclusion, our findings are consistent with the idea that ATP binds to at least two distinct domains of the human P2Y1 receptor: the first domain located outside of the TM bundle, between EL2 and EL3, and the second domain located within the TM core. We also discuss the major role of disulfide bonds in the structure and stability of the receptor. The two disulfide bridges present in the human P2Y1 receptor are expected to constrain the loops within the receptor, specifically stretching the EL2 over the opening of the TM cleft and thus defining the path of access to the binding site.
Acknowledgments
The authors are grateful for helpful comments and encouragement provided by Dr. Robert Pearlstein of the Center for Molecular Modeling (CIT), NIH. We thank Dr. Ivar von Kügelgen of the Molecular Recognition Section, NIDDK/NIH, for helpful discussion. We also thank Prof. T. K. Harden and Prof. R. A. Nicholas of the University of North Carolina at Chapel Hill for the P2Y1 receptor plasmid.
Footnotes
Abbreviations: DMEM, Dulbecco’s modified Eagle’s medium; EL, extracellular loop; ELISA, enzyme-linked immunosorbent assay; FBS, fetal bovine serum; GPCR, G protein-coupled receptor; GDP, guanosine 5′-diphosphate; GTP, guanosine 5′-triphosphate; HA, hemagglutinin; HT-AMP, 2-(hexylthio)adenosine 5′-monophosphate; 2-MeSADP, 2-(methylthio)adenosine 5′-diphosphate; 2-MeSATP, 2-(methylthio)adenosine 5′-triphosphate; 3′NH2-ATP, 3′-amino-3′-deoxyadenosine 5′-triphosphate; PBS, phosphate-buffered saline; PCR, polymerase chain reaction; TM, (helical) transmembrane domain.
References
- 1.Wess J. FASEB J. 1997;11:346–354. [PubMed] [Google Scholar]
- 2.Ji TH, Grossman M, Ji I. J Biol Chem. 1998;273:17299–17302. doi: 10.1074/jbc.273.28.17299. [DOI] [PubMed] [Google Scholar]
- 3.Bourne HR. Curr Opin Cell Biol. 1997;9:134–142. doi: 10.1016/s0955-0674(97)80054-3. [DOI] [PubMed] [Google Scholar]
- 4.Conklin BR, Bourne HR. Cell. 1993;73:631–641. doi: 10.1016/0092-8674(93)90245-l. [DOI] [PubMed] [Google Scholar]
- 5.Claphman DE, Neer EJ. Annu Rev Pharmcol Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- 6.Baldwin JM. EMBO J. 1993;12:1693–1703. doi: 10.1002/j.1460-2075.1993.tb05814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz TW. Curr Opin Biotechnol. 1994;5:434–444. doi: 10.1016/0958-1669(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 8.Strader CD, Fong TM, Graziano MP, Tota MR. FASEB J. 1995;9:745–754. [PubMed] [Google Scholar]
- 9.Olah ME, Jacobson KA, Stiles GL. J Biol Chem. 1994;269:24692–24698. [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Jiang Q, Glashofer M, Yehle S, Wess J, Jacobson K. J Mol Pharmacol. 1996;49:683–691. [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao MM, Hwa J, Perez DM. Mol Pharmacol. 1996;50:1118–1126. [PubMed] [Google Scholar]
- 12.Perlman JH, Colson AO, Jain R, Czyzewski B, Cohen L, Osman R, Gershengorn MC. Biochemistry. 1997;36:15670–15676. doi: 10.1021/bi9713310. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann C, Moro S, Nicholas RA, Harden TK, Jacobson KA. J Biol Chem. 1999 doi: 10.1074/jbc.274.21.14639. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abbracchio MP, Burnstock G. Pharmacol Ther. 1994;64:445–475. doi: 10.1016/0163-7258(94)00048-4. [DOI] [PubMed] [Google Scholar]
- 15.Burnstock G, King BF. Drug Dev Res. 1996;38:67–71. [Google Scholar]
- 16.Weiner SJ, Kollman PA, Nguyen D, Case DA. J Comput Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 17.Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. J Comput Chem. 1990;11:440–450. [Google Scholar]
- 18.The program SYBYL 6.4. TRIPOS Associates; St. Louis, MO: 1998. [Google Scholar]
- 19.Moro S, Guo D, Camaioni E, Boyer JL, Harden KT, Jacobson KA. J Med Chem. 1998;41:1456–1466. doi: 10.1021/jm970684u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou PY, Fasman GD. Adv Enzymol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- 21.The application is available through the National Center for Supercomputing Applications at the University of Illinois at Urbana-Champaign (http://biology.ncsa.uiuc.edu).
- 22.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–333. [Google Scholar]
- 23.Dewar MJSE, Zoebisch G, Healy EF. J Am Chem Soc. 1985;107:3902–3909. [Google Scholar]
- 24.MOPAC 6.0 is available from the Quantum Chemistry Program Exchange.
- 25.Nicholls A, Sharp K, Honing B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 26.Boyer JL, Siddiqi S, Fischer B, Romera-Avila T, Jacobson KA, Harden TK. Br J Pharmacol. 1996;118:1959–1964. doi: 10.1111/j.1476-5381.1996.tb15630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higuchi R. Using PCR to Engineer DNA. In: Ehrlich HA, editor. PCR Technology. Stockton Press; New York: 1989. pp. 61–70. [Google Scholar]
- 28.Sanger R, Nicklen S, Coulson AR. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cullen BR. Methods Enzymol. 1987;152:684–704. doi: 10.1016/0076-6879(87)52074-2. [DOI] [PubMed] [Google Scholar]
- 30.Harden TK, Hawkins PT, Stephens L, Boyer JL, Downes P. Biochem J. 1988;252:583–593. doi: 10.1042/bj2520583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berridge MJ, Dawson RM, Downes CP, Heslop JP, Irvine RF. Biochem J. 1983;212:473–482. doi: 10.1042/bj2120473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Galen PJM, van Vlijmen HWT, IJzerman AP, Soudijn W. J Med Chem. 1990;33:1708–1713. doi: 10.1021/jm00168a027. [DOI] [PubMed] [Google Scholar]
- 33.Hibert MF, Trumpp-Kallmeyer S, Bruinvels A. Mol Pharmacol. 1991;40:8–15. [PubMed] [Google Scholar]
- 34.Maloneyhuss K, Lybrand TP. J Mol Biol. 1992;225:879–871. doi: 10.1016/0022-2836(92)90406-a. [DOI] [PubMed] [Google Scholar]
- 35.Teeter MM, Froimowitz M, Stec B, DuRand DJ. J Med Chem. 1994;37:2874–2888. doi: 10.1021/jm00044a008. [DOI] [PubMed] [Google Scholar]
- 36.van Rhee AM, Fischer B, van Galen PJM, Jacobson KA. Drug Des Disc. 1995;13:133–154. [PMC free article] [PubMed] [Google Scholar]
- 37.Metzger TM, Paterlini MG, Portoghese PS, Ferguson DM. Neurochem Res. 1996;21:1287–1294. doi: 10.1007/BF02532369. [DOI] [PubMed] [Google Scholar]
- 38.Scheer A, Fanelli F, Costa T, De Benedetti PG, Cotecchia S. EMBO J. 1996;15:3566–3578. [PMC free article] [PubMed] [Google Scholar]
- 39.Paterlini G, Portoghese PS, Ferguson DM. J Med Chem. 1997;40:3254–3262. doi: 10.1021/jm970252j. [DOI] [PubMed] [Google Scholar]
- 40.Hjorth SA, Schambye HT, Greenlee WJ, Schwartz TW. J Biol Chem. 1994;269:30953–30959. [PubMed] [Google Scholar]
- 41.Colson AO, Pearlman JH, Smolyar A, Gershengorn MC, Osman R. Biophys J. 1998;74:1087–1100. doi: 10.1016/S0006-3495(98)77827-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang Q, Guo D, Lee BX, van Rhee AM, Kim YC, Nicholas RA, Schachter JB, Harden TK, Jacobson A. Mol Pharmacol. 1997;52:499–507. doi: 10.1124/mol.52.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gouldson PR, Snell CR, Reynolds J Med Chem. 1997;40:3871–3886. doi: 10.1021/jm960647n. [DOI] [PubMed] [Google Scholar]
- 44.Turner PR, Bambino T, Nissenson RA. J Biol Chem. 1996;271:9205–9208. doi: 10.1074/jbc.271.16.9205. [DOI] [PubMed] [Google Scholar]
- 45.Samama P, Cotecchia S, Costa T, Lefkowitz RJ. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- 46.Chidiac P. Trends Pharmacol Sci. 1995;16:83–85. doi: 10.1016/s0165-6147(00)88986-5. [DOI] [PubMed] [Google Scholar]
- 47.Surya A, Stadel M, Knox BE. Trends Pharmacol Sci. 1998;19:243–247. doi: 10.1016/s0165-6147(98)01217-6. [DOI] [PubMed] [Google Scholar]
- 48.Probst WC, Snyder LA, Schuster DI, Brosius J, Sealfon SC. DNA Cell Biol. 1992;11:1–20. doi: 10.1089/dna.1992.11.1. [DOI] [PubMed] [Google Scholar]
- 49.Strader CD, Fong TM, Tota MR, Underwood D, Dixon RAF. Annu Rev Biochem. 1994;63:101–132. doi: 10.1146/annurev.bi.63.070194.000533. [DOI] [PubMed] [Google Scholar]
- 50.Yamano Y, Ohyama K, Chaki S, Guo DF, Inagami T. Biochem Biophys Res Commun. 1992;187:1426–1431. doi: 10.1016/0006-291x(92)90461-s. [DOI] [PubMed] [Google Scholar]
- 51.Leong SR, Kabakoff RC, Hebert CA. J Biol Chem. 1994;269:19343–19348. [PubMed] [Google Scholar]