

Abstract
Mantle cell lymphoma (MCL) is characterized by an early, widespread dissemination and residual disease after conventional treatment, but the mechanisms responsible for lymphoma cell motility and drug resistance are largely unknown. There is growing evidence suggesting that chemokine receptors and adhesion molecules are critical for malignant B-cell trafficking and homing to supportive tissue microenvironments, where they receive survival and drug resistance signals. Therefore, we examined chemokine receptor and adhesion molecule expression and function in MCL cells and their importance for migration and adhesion to marrow stromal cells (MSCs). We found that MCL cells display high levels of functional CXCR4 and CXCR5 chemokine receptors and VLA-4 adhesion molecules. We also report that MCL cells adhere and spontaneously migrate beneath MSCs in a CXCR4- and VLA-4–dependent fashion (pseudoemperipolesis). Moreover, we demonstrate that MSCs confer drug resistance to MCL cells, particularly to MCL cells that migrate beneath MSC. To target MCL-MSC interactions, we tested Plerixafor, a CXCR4 antagonist, and natalizumab, a VLA-4 antibody. Both agents blocked functional responses to the respective ligands and inhibited adhesive interactions between MCL cells and MSCs. These findings provide a rationale to further investigate the therapeutic potential of these drugs in MCL.
Introduction
Mantle cell lymphoma (MCL) is an aggressive B-cell lymphoma that accounts for approximately 6% to 8% of non-Hodgkin lymphomas and has distinct clinical and pathologic features.1,2 The majority of patients present with advanced stage disease (Ann Arbor stages III/IV) at diagnosis, and more than 90% of patients have extranodal manifestations with a high prevalence of circulating MCL cells, bone marrow, and gastrointestinal involvement. MCL cells display a distinct immunophenotype with expression of CD5, CD19, CD20, CD22, but absence of CD23. The hallmark translocation t(11;14)(q13;q32) is another characteristic in MCL, bringing the cyclin D1 (BCL1/CCND1) gene under the control of the immunoglobulin heavy chain enhancer, and resulting in an overexpression of cyclin D1.3,4 Generally, MCL is considered an incurable disease, with a continuous decline in survival, resulting in a median overall survival of only 30 to 43 months, and less than 15% long-term survivors.5,6 New therapeutic modalities, in particular combinations of intensive polychemotherapy with monoclonal antibodies (rituximab plus fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone, alternating with methotrexate/high-dose cytarabine [HCVAD], or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone [CHOP]) with or without autologous stem cell support improved remission rates, and are currently used in frontline treatment for younger MCL patients,7,8 but their impact on long-term patient outcome remains controversial. Despite the high partial remission and complete remission rates with these regimens, minimal residual disease (MRD) is common, leading to relapses in the majority of patients.
There is growing evidence suggesting that interactions between the neoplastic B cells and accessory cells in tissue microenvironments, such as the lymphatic tissues, are critical for disease progression in various B-cell malignancies.9,10 Chemokine receptors and adhesion molecules are essential for normal and malignant B-cell trafficking and homing to such tissue microenvironments, but their expression and function in MCL are largely unknown. Because drugs targeting these molecules are currently in preclinical and clinical development,11 we examined expression and function of chemokine receptors and adhesion molecules, and tested novel drugs targeting these molecules in primary MCL cells and MCL cell lines.
Normal B-cell trafficking and function largely depend on interactions between B cells and accessory cells collectively referred to as stromal cells.12,13 For example, stromal cells in secondary lymphatic tissues constitutively express chemokines, such as CXCL12 and CXCL13, which provide guidance for B-cell positioning within distinct lymph node compartments.12,14,15 According to the multistep paradigm, initially proposed by Springer,16 lymphocyte trafficking and homing require the cooperation between chemokine receptors and adhesion molecules, such as integrins, CD44, and L-selectins. Lymphocytes actively enter and home into tissue microenvironments, such as the secondary lymphatic tissues, where stromal cell networks provide guidance by secreting chemokines, establishing chemokine gradients, and expressing ligands for lymphocyte adhesion molecules. Coordinated lymphocyte entry, migration, and territoriality are essential during immune surveillance and induction of specific immune responses.14,17–19 In B-cell lymphomas/leukemias, the neoplastic B cells largely retain the capacity of their normal counterparts for trafficking and homing, as demonstrated in chronic lymphocytic leukemia (CLL), B-cell acute lymphoblastic leukemia, and follicular lymphoma cells,20 both in vitro21,22 and in vivo.23 Moreover, studies in follicular lymphoma demonstrated that accessory cells in the lymphoma microenvironment that are not part of the malignant clone, such as CD68+ macrophages or T cells, have a major impact on the clinical outcome,24–26 indicating that crosstalk between malignant B cells and stromal cells plays a key role in disease progression and/or resistance to conventional treatments. Despite the pattern of early dissemination in MCL, only a few studies today have investigated the expression and function of chemokine receptors20,27,28 and adhesion molecules29 in MCL, and the importance of these molecules for MCL-stromal cell interactions has not yet been explored. Therefore, we investigated the expression and function of important B-cell chemokine receptors and adhesion molecules in primary MCL cells and MCL cell lines, and their importance in MCL adhesion and migration beneath stromal cells. We show that MCL cells strongly adhere to and spontaneously migrate beneath mesenchymal stromal cells (MSCs), and demonstrate the importance of CXCR4 and very late antigen (VLA-4) for MCL adhesion and migration beneath MSCs, using specific inhibitors. Because stromal cell-mediated drug resistance, also termed cell adhesion-mediated drug resistance (CAM-DR), is a common feature in chronic B-cell malignancies, such as multiple myeloma (MM)30 or CLL,31 we also investigated the role of CAM-DR in MCL.
Methods
Cell purification, cell lines
Patient samples were obtained under protocols approved by the institutional review board at the M. D. Anderson Cancer Center from MCL patients fulfilling cytogenetic (t(11;14) verification) and immunophenotypic criteria for MCL. Informed consent was obtained in accordance with the Declaration of Helsinki for samples used in the study. Peripheral blood mononuclear cells were isolated by density gradient centrifugation over Ficoll Paque (GE Healthcare, Little Chalfont, United Kingdom). Cells were used fresh or viably frozen in fetal bovine serum (FBS; SAFC Biosciences, Lenexa, KS) plus 10% dimethyl sulfoxide (Sigma-Aldrich, St Louis, MO) for storage in liquid nitrogen. All MCL samples examined contained more than 90% MCL B cells, as determined by fluorescence-activated cell sorter (FACS) analysis with anti-CD5 and anti-CD19 monoclonal antibodies (mAbs). Control peripheral blood samples were obtained from healthy volunteers. The murine stromal cell line M2-10B4 was purchased from ATCC (Manassas, VA). Cells were maintained in RPMI 1640 medium supplemented with 2.05 mM l-glutamine (HyClone Laboratories, Logan, UT) and containing 10% FBS as well as penicillin-streptomycin (Cellgro, Hemdon, VA). The MCL cell lines SP-53, Mino, JeKo-1, and Granta 519 were cultured as previously described.22
Chemokines, antibodies, inhibitors, and flow cytometry
Synthetic human CXCL12 (stromal cell derived factor-1) was purchased from Upstate Biotechnology (Lake Placid, NY). CXCL13 (BCA-1) was purchased from R&D Systems (Minneapolis, MN). The following mAbs specific for human surface antigens were used: anti–CXCR5-phycoerythrin (PE) from R&D Systems, anti–CD5-fluorescein isothiocyanate (FITC), anti–CD44-FITC, anti–CD49d-PE, anti–CD62L-FITC, anti–CD5-FITC, anti–CD19-allophycocyanine, anti–CXCR4-PE (12G5), anti–CXCR3-PE, and appropriate isotype controls from BD Biosciences (San Jose, CA). For inhibition experiments, the CXCR4 chemokine receptor antagonist AMD3100 (Plerixafor, Mozobil) was purchased from Sigma-Aldrich and provided by Genzyme (Cambridge, MA); pertussis toxin was used to inhibit G-protein signaling via chemokine receptors. Anti–VLA-4 (CD49d) mAb (natalizumab, Tysabri) was provided by Biogen (Cambridge, MA). A small cyclic CS-1 fibronectin peptide containing the minimal CS-1-VLA-4 binding motif “LDV” (H-CWLDVC-NH2) and a control peptide in which the sequence was scrambled (KCDLWCK) were synthesized and purchased from Sigma-Genosys (The Woodlands, TX) and used as described previously.32
For flow cytometry, cells were adjusted to a concentration 5 × 106 cells/mL in RPMI 1640 with 0.5% bovine serum albumin (FACS buffer). A total of 5 × 105 cells were stained with saturating antibody concentration for 30 minutes at 4°C, washed 2 times, and then analyzed on a FACSCalibur (BD Biosciences). MCL cells were gated based on their CD19 expression. CD5+ B cells were gated based on the simultaneous expression of CD5 and CD19; at least 5000 CD5+ B cells were acquired per stain. Mean fluorescence intensity ratios (MFIRs) were calculated by dividing the mean fluorescence intensity of analyzed marker by the mean fluorescence intensity of respective isotype control. Flow cytometry data were analyzed using FlowJo software (TreeStar, Ashland, OR).
Chemotaxis assay
MCL cells were suspended in FACS buffer, and 5 × 105 cells were added to the top chambers of 6.5-mm diameter Transwell inserts (Corning Life Sciences, Acton, MA) with a pore size of 5 μm. Filters then were transferred to wells containing medium with or without CXCL12 or CXCL13. For inhibition experiments, cells were preincubated for 1 hour with 100 μg/mL Plerixafor or 1 μg/mL pertussis toxin before they were added to the inserts. In an additional set of experiments to differentiate between chemotaxis and chemokinesis, CXCL12 or CXCL13 was added at the same concentrations to both the top and lower chambers. The chambers were incubated for 3 hours at 37°C in 5% CO2. After this incubation, the cells in the lower chamber were suspended and divided into aliquots for counting with FACSCalibur, as described.21
Actin polymerization
MCL cells were suspended in FACS buffer in concentration 1.25 × 106 cells/mL and incubated with 200 ng/mL CXCL12 or CXCL13 at 37°C for various times, as described.21 At the indicated time points, 400 μL of the cell suspension was added to 100 μL of a staining solution containing 4 × 10−7 M FITC-labeled phalloidin, 0.5 mg/mL l-alpha-lysophosphatidylcholine, and 37 wt% formaldehyde in water (all from Sigma-Aldrich). The fixed cells were analyzed by flow cytometry, and all time points were plotted relative to the mean relative fluorescence of the sample before addition of the chemokines.
In vitro migration assay of MCL cells beneath marrow stromal cells (pseudoemperipolesis)
M2-10B4 stromal cells were seeded onto collagen-coated 12-well plates at a concentration of 1.5 × 105 cells per well in RPMI 1640 with 10% FBS. After overnight incubation, MCL cells were added onto the confluent stromal cell layers to a final concentration of 106 cells per well. For inhibition experiments, MCL cells were preincubated for 1 hour with 100 μg/mL Plerixafor or 1 μg/mL pertussis toxin, or 2, 5, or 10 μg/mL natalizumab before they were added to the wells. As a control antibody, we used MOPC-21 at the concentration 200 μg/mL. We also performed pretreatment with 10 or 100 μg/mL small cyclic CS-1 peptide inhibitor or control scrambled peptide at the same concentrations. The plates then were incubated for 6 hours at 37°C in 5% CO2, after which supernatant cells were removed by washing. Stromal cell layers were photographed, then single-cell suspensions were obtained by adding trypsin, and the migrated cell fraction was quantified by flow cytometry, as previously described.21
Cell viability testing
To test the protective effect of M2-10B4 cells, MCL cells treated with 5, 10, or 20 μM fludarabine (dephosphorylated 9-β-D-arabinofuranosyl-2-fluoroadenine, F-ara-A; Sigma-Aldrich), or 10 μM 4-hydroperoxycyclophosphamide (4-HC; kindly provided by Dr W. Plunkett, Department of Experimental Therapeutics, M. D. Anderson Cancer Center) were plated with or without M2-10B4 cells. Determination of MCL cells viability after 24, 48, and 72 hours of treatment was based on the analysis of mitochondrial transmembrane potential by 3,3′ dihexyloxacarbocyanine iodide (DiOC6, Invitrogen, Carlsbad, CA) and cell membrane permeability with propidium iodide (PI), as previously described.21 To distinguish between MCL cells and stromal cells, we used a leukocyte gate that excluded large granular stromal cells, based on their forward- and side-scatter characteristics.
Western blotting
For p44/p42 mitogen-activated protein kinase (MAPK) immunoblot assays, 107 MCL cells were serum-starved for 2 hours, stimulated with CXCL12 at 250 ng/mL and CXCL13 at 1 μg/mL for different time points, and then lysed. To investigate the role of blocking CXCR4 in signal transduction, cells were pretreated with 100 μg/mL Plerixafor for 1 hour and then stimulated with CXCL12. Protein content was determined using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA) reagent. Equal amounts of protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and were transferred onto nitrocellulose membranes (Bio-Rad). Western blot analysis was performed using the phospho-p44/42 (Thr202/Tyr204) rabbit polyclonal antibodies that specifically recognize the phosphorylated form of p44/42, the p44/42 MAP kinase total antibodies, and β-actin (13E5) rabbit mAbs (all from Cell Signaling Technology, Danvers, MA). Immunoreactive bands were visualized using peroxidase-labeled goat anti–rabbit secondary antibody (GE Healthcare) and enhanced using the enhanced chemiluminescence detection system (GE Healthcare).
Data analysis and statistics
Results are shown as mean plus or minus SEM of at least 3 experiments each. For statistical comparison between groups, the Student paired t test was used. Analyses were performed using GraphPad Prism 4 software for Macintosh (GraphPad Software, San Diego, CA). A P value less than .05 was considered statistically significant. Flow cytometric data were analyzed using FlowJo software (TreeStar).
Results
Expression of cell surface migration-related molecules on MCL B cells
We characterized the expression of adhesion molecules, such as CD44, VLA-4, and CD62L, as well as CXC chemokine receptors CXCR3, CXCR4, and CXCR5 in malignant B cells from lymphoma patients and cell lines. We found that MCL B cells from patients display high cell surface expression of CD44 and CD49d, but not CD62L. As shown in Table 1, SP-53, MINO, and Granta 519 cell lines demonstrated high levels of CD44 expression, and all of the MCL cell lines were strongly positive for CD49d. All MCL cell lines except for SP-53 were negative for CD62L. Analysis of CXC chemokine receptor expression showed that in all cases CXCR4 and CXCR5 are highly expressed, whereas CXCR3 expression is very low. According to MFIR data, patients' MCL cells demonstrate higher level of CXCR5 expression compared with MCL cell lines. Figure 1 illustrates the expression of adhesion molecules and chemokine receptors in MCL cell lines: SP-53 shows the highest expression of CXCR4 and a similar level of CXCR5, comparing MINO and JeKo-1. Granta 519 was found to be dimly positive for CXCR4 with very low CXCR5 expression. We also examined the expression of CXCR4, CXCR5, and CD49d in normal CD5+ B cells from peripheral blood (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article) and found that, in comparison with MCL B cells, normal CD5+ B cells demonstrated higher levels of CD49d and CXCR5 expression, whereas the level of CXCR4 expression was comparable (Table S1).
Table 1.
Phenotype of MCL cells derived from patients with MCL and cell lines
| MFIR | MCL patients (n = 6) | SP-53 | MINO | JeKo-1 | Granta 519 |
|---|---|---|---|---|---|
| CD44 | 42.5 ± 9.5 | 64.5 | 39.0 | 4.2 | 47.9 |
| CD49d | 14.1 ± 1.9 | 38.6 | 36.0 | 42.4 | 14.4 |
| CD62L | 1.3 ± 0.2 | 7.9 | 1.3 | 1.2 | 1.2 |
| CXCR4 | 53.5 ± 27.8 | 121.0 | 56.4 | 42.4 | 6.3 |
| CXCR5 | 35.4 ± 5.9 | 14.0 | 10.7 | 6.0 | 1.0 |
| CXCR3 | 2.0 ± 0.5 | 1.1 | 1.3 | 1.1 | 1.8 |
Phenotypic characteristics of MCL cells presented as mean fluorescence intensity ratio (MFIR) for major migration-related molecules. Gating on CD19-expressing cells identified MCL cells. Patient data shown are mean ± SEM values of 6 experiments with MCL cells from different patients.
Figure 1.
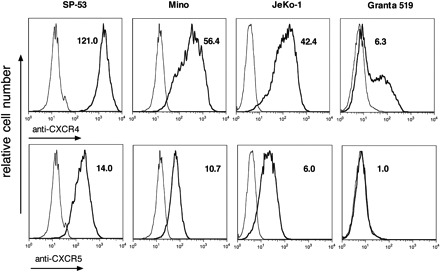
MCL cell lines demonstrate a high level of CXCR4 and CXCR5 expression. Overlay histogram plots depict the relative CXCR4 and CXCR5 fluorescence intensity (bold line) of CD19+ MCL cells in comparison with isotype control stain (thin line). The MFIRs are displayed next to each histogram. The MCL lines SP-53, MINO, and JeKo-1 demonstrate high levels of CXCR4 and CXCR5 expression, whereas in the EBV+ cell line Granta 519 expression of these chemokine receptors is low.
CXCL12 and CXCL13 induce actin polymerization in MCL cells
Reorganization of the actin cytoskeleton is an early event in the migratory response to chemokines. To evaluate the ability of CXCL12 and CXCL13 chemokines to induce changes in filamentous actin (F-actin), MCL cells were treated with 200 ng/mL CXCL12 and CXCL13. As shown in Figure 2A, stimulation with these chemokines induces a transient increase in F-actin that peaks 15 seconds after exposure to CXCL12 and CXCL13. Actin polymerization after CXCL12 stimulation was completely inhibited by preincubation of MCL cells for 1 hour with 100 μg/mL of the CXCR4 antagonist Plerixafor (Figure 2A,B) or 1 μg/mL of pertussis toxin, indicating that this response can be blocked by these inhibitors and is mediated through pertussis toxin–sensitive Gi proteins.
Figure 2.
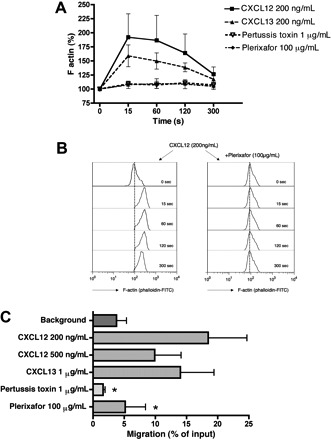
Chemotaxis and actin polymerization in primary MCL cells in response to CXCL12 and CXCL13. (A) Actin polymerization of MCL cells in response to CXCL12 and CXCL13. Intracellular F-actin content in MCL cells was determined at the time points indicated on the horizontal axis after the addition of 200 ng/mL CXCL12 or CXCL13. Inhibition of CXCL12-induced actin polymerization was detected after preincubation with Plerixafor or pertussis toxin. The relative F-actin content compared with samples before chemokine stimulation (100%) is displayed on the vertical axis and is the mean plus or minus SEM of 4 MCL samples from different patients. (B) Effect of Plerixafor on actin polymerization in response to CXCL12 stimulation. Remarkable inhibition of actin changes mediated by CXCL12 was demonstrated after pretreatment with Plerixafor. The histograms show representative results from one of 4 experiments with MCL B cells from different patients. (C) Displayed is the mean relative chemotaxis of primary MCL cells toward the chemokines CXCL12 or CXCL13 under the conditions displayed on the vertical axis. MCL cells display relatively high levels of spontaneous migration toward wells without chemokine (control). CXCL12 and CXCL13 both induced chemotaxis of MCL cells, and CXCL12-induced chemotaxis is inhibited by pretreatment with pertussis toxin or Plerixafor. Results are percentages of migrated cells relative to input and are mean ± SEM of 4 experiments. *Significant difference compared with control sample (P < .05).
Chemotaxis of MCL cells in response to CXCL12 and CXCL13
Figure 2C depicts the chemotactic response of MCL cells from patients to the chemokines CXCL12 and CXCL13. The maximum migration of MCL B cells was elicited using 200 ng/mL CXCL12; at this concentration, 18.5% (± 6.2%) of input cells (mean ± SEM, n = 4) migrated through the micropore filters. Stimulation with 500 ng/mL CXCL12 resulted in migration of 9.9% (± 4.2%) of input MCL cells (mean ± SEM, n = 4), and 1 μg/mL CXCL13 induced the migration of 14.0% (± 5.4%) of input MCL cells. Chemotaxis of primary MCL cells was significantly inhibited by preincubation of the MCL cells with the CXCR4 antagonist Plerixafor (100 μg/mL) or pertussis toxin (1 μg/mL) to levels that were 5.2% (± 3.3%) for Plerixafor or 1.6% (± 0.3%) of input cells for pertussis toxin.
All the tested MCL cell lines except for Granta 519 demonstrated a major chemotactic response to the CXCL12. SP-53 cells displayed the highest level of chemotaxis with 35.7% (± 5.7%) of input cells that migrated toward 200 ng/mL CXCL12, followed by 25.8% (± 2.8%) of input cells for MINO and 6.7% (± 0.9%) of input cells for JeKo-1 (mean ± SEM, n = 3 for each experiment). Increasing the concentration of CXCL12 to 500 ng/mL resulted in reduced levels of chemotaxis that were 29.1% (± 3.7%) for SP-53, 16.2% (± 0.4%) for MINO, and 3.7% (± 0.5%) for JeKo-1, consistent with the bell-shaped dose-response curve that is typical for chemokines. Stimulation with 1 μg/mL CXCL13 also induced major chemotactic response in SP-53 cells (43.4% ± 9.8%), in MINO cells (16.7% ± 2.8%), and JeKo-1 (4.3% ± 0.5%, mean ± SEM, n = 3 for each experiment). In chemokinesis experiments, in which CXCL12 was added at 200 ng/mL to both chambers, only 0.55% (± 0.19%) of input SP-53 cells or 1.85% (± 0.05%) of input MINO cells migrated to the lower chambers (mean ± SEM, n = 3 for each experiment). Similar results were obtained with CXCL13 added at 1 μg/mL to both chambers, which resulted in migration of 0.30% (± 0.08%) of input SP-53 cells or 4.2% (± 0.6%) of input MINO cells, demonstrating that these chemokines induce MCL chemotaxis, but not random motility/chemokinesis. These results are summarized in Figure S2.
Specific inhibition of CXCR4 with 32 μg/mL of Plerixafor partially abrogated the migration of MCL cells derived from cell lines (data not shown); we therefore used the concentration of 100 μg/mL in subsequent experiments. Preincubation of MCL cells with 100 μg/mL led to significant blocking of CXCL12-induced migration with the quantity of migrated cells reduced to 0.3% (± 0.1%) of input cells for SP-53, 0.5% (± 0.3%) for MINO, and 0.3% (± 0.0%) for JeKo-1 (mean ± SEM, n = 3 for each experiment). Blocking of Gi proteins with 1 μg/mL pertussis toxin also resulted in a significant reduction of chemotaxis toward CXCL12 to the levels that were 0.5% plus or minus 0.5% of input cells in SP-53, 4.8% plus or minus 1.7% in MINO, and 3.0% plus or minus 0.9% in JeKo-1, delineating that this process is dependent on activation of Gi proteins. Pretreatment of the MCL cells with 5 μg/mL natalizumab did not affect CXCL12-induced chemotaxis and actin polymerization (data not shown).
Plerixafor, natalizumab, pertussis toxin, and CS-1 peptide inhibit spontaneous migration of MCL cell lines beneath stromal cells (pseudoemperipolesis)
In coculture of MCL cells with M2-10B4 bone MSCs (Figure 3A), we noticed high levels of spontaneous migration of MCL beneath the stromal cells (Figure 3B). This striking phenomenon, called pseudoemperipolesis, was previously reported for CLL cells21 and other leukemia cells,33 and is characterized by the dark appearance of the cells that have migrated into the same focal plane as the stromal cells, whereas the more superficial, nonmigrated, cells remain refractive. In previous studies, we have established that B-cell pseudoemperipolesis is dependent on CXCR4 activation of the B cells by CXCL12 secreted by stromal cells, as well as VLA-4 binding to respective stromal cell ligands.21 Consistent with the lack of active CXCR4 (as established in chemotaxis and actin polymerization assays), we found that Granta 519 did not migrate beneath the MSCs. For blocking experiments, we chose the lines that demonstrated the highest level of pseudoemperipolesis, SP-53 and MINO. In quantification experiments, we noticed that 55.8% plus or minus 3.2% of input SP-53 cells and 20.2% plus or minus 3.0% of input MINO cells spontaneously migrated beneath the MSCs (mean ± SEM, n ≥ 3).
Figure 3.
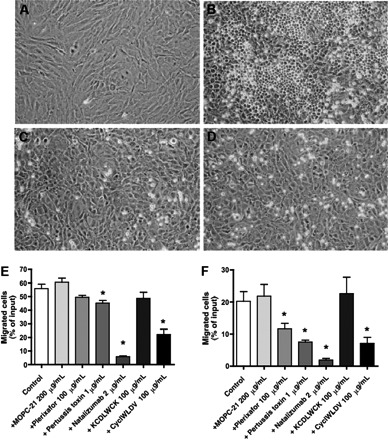
Effects of natalizumab, Plerixafor, pertussis toxin, and CS-1 peptide on the migration of MCL cells beneath stromal cells (pseudoemperipolesis). To demonstrate the effect of blocking VLA-4 integrins on pseudoemperipolesis, a confluent layer of M2-10B4 marrow stromal cells was established (A), and untreated (B) or pretreated (with natalizumab in panel C, with CS-1 peptide in panel D) MCL cells were seeded onto the stromal cell layer. After 6 hours, nonmigrated cells were removed by vigorous washing. Migrated cells are characterized by a dark appearance, whereas nonmigrated cells remain bright. The stromal cell layer containing the migrated MCL cells was photographed (100× magnification). These photomicrographs illustrate that pseudoemperipolesis of MCL cells after treatment with natalizumab (C) and CS-1 peptide inhibitor (D) was greatly reduced compared with untreated controls (B). Cells were imaged using a phase contrast microscope (Model ELWD 0.3; Nikon, Garden City, NY) with a 10×/0.25 NA objective lens. Images were captured with a Nikon D40 digital camera (Nikon, Tokyo, Japan) using Camera Control Pro software (Nikon) and processed with Adobe Photoshop 9.0 software (Adobe Systems, San Jose, CA). The MCL cell lines SP-53 (E) and MINO (F) were left untreated (controls) or pretreated with the agents displayed on the horizontal axis and then incubated on confluent MSC layers. After incubation, the nonmigrated cells were vigorously washed off, and the MCL cells that had migrated into the MSC layer were quantified by FACS. The mean ± SEM relative pseudoemperipolesis is shown for 3 independent experiments for each cell line. *Significant inhibition of pseudoemperipolesis compared with control sample (P < .05).
To investigate the importance of CXCR4 and VLA-4 in MCL migration beneath MSCs, we used specific (Plerixafor, natalizumab, CS-1 peptide) and nonspecific (pertussis toxin) inhibitors, or controls, and then quantified pseudoemperipolesis. As shown in Figure 3E, we noticed decreased migration of SP-53 cells beneath MSCs after pretreatment of the cells with Plerixafor, which did not reach significance (P < .07). However, significant inhibition after incubation with pertussis toxin (P < .01) and a significant blocking of migration were observed after pretreatment with the CS-1 peptide in both concentrations: 10 and 100 μg/mL (P < .01 and P < .02, respectively). In the MINO cells (Figure 3F), both Plerixafor and pertussis toxin pretreatment resulted in a significant reduction of MCL cell pseudoemperipolesis (P < .04 and P < .01, respectively). Pretreatment with small CS-1 peptide inhibitor (10 and 100 μg/mL) also resulted in significant inhibition of pseudoemperipolesis of MINO cells (P < .005 and P < .015, respectively). A control antibody (MOPC-21) or control scrambled peptides did not significantly affect pseudoemperipolesis in both MCL lines.
Interestingly, and in contrast to the moderate effect of Plerixafor, preincubation with natalizumab resulted in a robust, significant inhibition of pseudoemperipolesis of MCL cells, even at very low concentrations. For example, 2 μg/mL of natalizumab reduced MCL pseudoemperipolesis to 5.8% plus or minus 0.5% of input cells for SP-53 cells (P < .02) and to 1.9% plus or minus 0.5% for MINO cell (mean ± SEM, n = 3, P < .01). Higher concentrations of natalizumab (5 and 10 μg/mL) did not further inhibit the pseudoemperipolesis. Figure 3C and D illustrates the lack of migration of MINO cells beneath MSCs after pretreatment with 5 μg/mL natalizumab or 100 μg/mL CS-1 peptide.
Stromal cells protect MCL cells from cytotoxic effect of fludarabine and 4-hydroperoxycyclophosphamide
We have previously established that MSCs protect CLL B-cells from fludarabine-induced apoptosis and cell death in a contact-dependent fashion.31 To determine whether MCL cells also show CAM-DR in cocultures with MSCs, we tested the protective effect of SP-53 and MINO cell lines treated with different concentrations of fludarabine. In titration experiments with 5, 10, and 20 μM F-ara-A, we found that the effect of treatment in both cell lines was directly dose-dependent and M2-10B4 protected MCL cells from apoptosis in all drug concentrations tested (data not shown). Figure 4A depicts the protective effect of M2-10B4 cells on the survival of SP-53 MCL cells treated with 10 μM F-ara-A for 24, 48, and 72 hours with or without stromal cells. The mean viabilities for SP-53 cells treated in the presence or absence of stromal cells were 91.7% plus or minus 2.4% versus 92.2% plus or minus 1.2% at 24 hours, 88.2% plus or minus 1.6% versus 68.9% plus or minus 4.3% at 48 hours, and 87.8% plus or minus 3.8% versus 44.4% plus or minus 3.8% at 72 hours. Results are presented as the mean plus or minus SEM relative to untreated controls. In MINO cells, this effect was more pronounced, as illustrated in Figure 4B. The mean viabilities for of MINO cells treated in the presence or absence of stromal cells were 92.6% plus or minus 5.3% versus 46.9% plus or minus 7.3%, 89.8% plus or minus 4.0% versus 9.0% plus or minus 2.8%, and 80.2% plus or minus 6.7% versus 7.5% plus or minus 1.0%, respectively.
Figure 4.
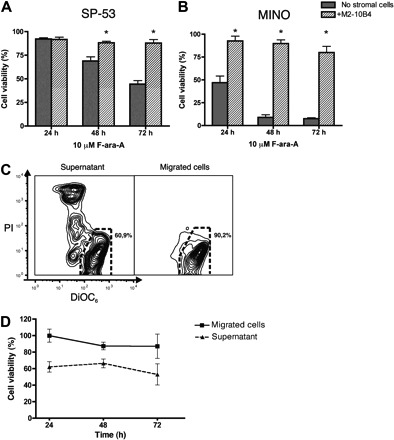
Marrow stromal cells protect MCL cells from F-ara-A-induced apoptosis. (A,B) Mean viability of SP-53 cells (A) or MINO cells (B) treated with F-ara-A at the time points displayed on the horizontal axis. MCL viability was higher when MCL cells were cocultured with M2-10B4 MSC cells (▨) compared with MCL without stromal cell support ([graphic024]). Results are presented as mean relative viability compared with untreated controls (100%) and are the mean ± SEM values of triplicates. *Significant protection of MCL cells from F-ara-A cytotoxicity compared with control sample (P < .05). (C) Cell viability of MCL cells was determined by staining with DiOC6 and PI. Contour plot represents supernatant and migrated fractions of pretreated with 10 μM F-ara-A SP-53 cells after 72 hours of cultivation on M2-10B4. The predominance of vital cells (DiOC6bright, PIexclusion) was detected for migrated fraction (right panel) compared with supernatant fraction (left panel). The percentage of vital cells is displayed in each contour map. (D) Viability of migrated and supernatant fractions of MCL cells pretreated with 10 μM F-ara-A after 24, 48, and 72 hours of cultivation on stromal cells. Results are represented relative to untreated controls and are mean ± SEM values of 3 different experiments.
To determine the effect of pseudoemperipolesis on CAM-DR in MCL, we assessed MCL cell viability separately on nonmigrated “supernatant” MCL cells, the cell fraction in MSC cocultures that did not migrate beneath the stromal cells versus the “migrated” MCL cells that had spontaneously migrated beneath the MSCs at 3 time points after treatment with 10 μM fludarabine. Migrated SP-53 cells survived the treatment with fludarabine significantly better compared with supernatant cells (Figure 4C); and, as depicted in Figure 4D, this difference was significant at each time point. The mean viabilities for SP-53 cells were 100.0% plus or minus 4.0% in the migrated versus 62.1% plus or minus 3.2% in the nonmigrated fraction after 24 hours, 87.4% plus or minus 2.3% versus 66.4% plus or minus 2.6% after 48 hours, and 87.2% plus or minus 7.3% versus 53% plus or minus 6.4% after 72 hours, respectively. Results are presented as the mean plus or minus SEM relative to untreated controls.
We also tested the cytotoxicity of 4-HC, the bioactive form of cyclophosphamide, in MCL cocultures with M2-10B4. As illustrated in Figure 5A, the mean viabilities for SP-53 cells treated with 10 μM 4-HC in the presence or absence of stromal cells were 62.3% plus or minus 2.4% versus 46.2% plus or minus 0.4% at 24 hours and 28.2% plus or minus 0.8% versus 24.0% plus or minus 2.5% at 48 hours. Results are presented as the mean plus or minus SEM relative to the viability of untreated controls. In MINO cells, the protective effect of stromal cells on 4-HC–induced MCL cell death was remarkable (and significant at all time points). As shown in Figure 5B, the mean viabilities for MINO cells treated with 10 μM 4-HC in the presence or absence of stromal cells were 98.2% plus or minus 1.9% versus 28.8% plus or minus 0.6% after 24 hours and 72.4% plus or minus 2.0% versus 1.9% plus or minus 0.1% after 48 hours. Results are presented as the mean plus or minus SEM relative to untreated controls.
Figure 5.
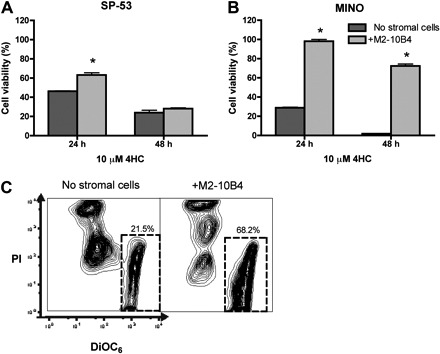
Marrow stromal cells protect MCL cells from 4-HC-induced cytotoxicity. The mean viability of SP-53 cells (A) and MINO cells (B) treated with 4-HC is presented at the different time points displayed on the horizontal axis. MCL cell viability was increased when MCL cells were cocultured with M2-10B4 ( ), compared with MCL in suspension and without stromal cell support (). Results are presented as mean relative viability compared with untreated controls (100%) and are the mean ± SEM of triplicates. *Protection of MCL cells from 4-HC-induced cytotoxicity with significantly higher viabilities compared with controls (P < .05). (C) Contour plots that depict the viability of MINO MCL cells, as detected by staining with DiOC6 and PI after 24 hours of culture with 10 μM 4-HC, in the presence or absence of M2-10B4 stromal cells, as indicated above each of the plots. The proportion of viable cells is indicated above each of the gates that define viable cells by bright DiOC6 staining and PI exclusion.
), compared with MCL in suspension and without stromal cell support (). Results are presented as mean relative viability compared with untreated controls (100%) and are the mean ± SEM of triplicates. *Protection of MCL cells from 4-HC-induced cytotoxicity with significantly higher viabilities compared with controls (P < .05). (C) Contour plots that depict the viability of MINO MCL cells, as detected by staining with DiOC6 and PI after 24 hours of culture with 10 μM 4-HC, in the presence or absence of M2-10B4 stromal cells, as indicated above each of the plots. The proportion of viable cells is indicated above each of the gates that define viable cells by bright DiOC6 staining and PI exclusion.
CXCL12 and CXCL13 stimulate p44/p42 MAP-kinase activation in MCL cells
Transient activation of p44/p42 mitogen-activated protein (MAP)–kinase (ERK 1/2) is an inducible signaling pathway related to cell survival and growth. We stimulated SP-53 MCL cells with CXCL12 or CXCL13 at the indicated time points. As shown in Figure 6, CXCL12 induced a rapid, transient phosphorylation/activation of p44/p42 MAP-kinase within 2 minutes (we found the same effect after 1 minute of stimulation; data not shown). Pretreatment of cells with Plerixafor resulted in significant abrogation of CXCL12 mediated activation of MAPK.
Figure 6.
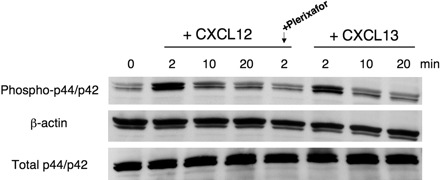
Effects of CXCL12 and CXCL13 stimulation on p44/p42 MAPK activation. SP-53 cells were stimulated with 250 ng/mL CXCL12 or 1 μg/mL CXCL13 for different times. To block CXCR4-derived signaling, cells were preincubated for 1 hour with 100 μg/mL Plerixafor and then stimulated with CXCL12. MCL cell lysates were probed with antiphospho p44/p42, antiβ-actin mAbs, and (on separate gels) with anti-p44/p42 mAbs. As indicated, maximum p44/p42 activation occurs within 2 minutes of stimulation with a subsequent decline in signal intensity. Pretreatment with Plerixafor effectively inhibits CXCL12-mediated p44/p42 MAPK activation. Results shown are representative of 3 experiments.
Discussion
In this study, we characterized the expression and function of the B-cell chemokine receptors CXCR4 and CXCR5 and the VLA-4 adhesion molecules (CD49d) on primary MCL B cells and MCL cell lines. We found that the phenotype of primary MCL cells is characterized by high-level expression of CXCR4, CXCR5, CD44, and CD49d, whereas the expression of CXCR3 and CD62L was largely negative. In CLL, the phenotypic signature of CXCR4high+CD49d+CD62L− is known to be associated with an adverse prognosis.34,35 High expression of CXCR4 and CXCR5 suggests that MCL cells use these receptors for dissemination of the disease that characterizes MCL progression because interactions of CXCR4 and CXCR5 with their respective ligands (CXCL12 and CXCL13) are critical for normal B cells trafficking and homing to secondary lymphatic tissues.13–15,36 In cooperation with chemokine receptors, VLA-4/CD49d integrins play an important role in B-cell trafficking and homing within tissue microenvironments, such as the marrow and the lymphatic tissues.37,38
Comparing chemokine receptor profiles among MCL lines, we noticed that, in contrast to the other MCL lines or primary MCL cells, Granta 519 displayed only low levels of CXCR4 and was CXCR5 negative, and did not migrate toward CXCL12 or CXCL13. This could be the result of Epstein-Barr virus (EBV) transformation of this particular cell line39 because EBV infection previously was shown to cause down-regulation of CXCR4 and known to reduce responsiveness to CXCL12.40
Functional testing of CXCR4 and CXCR5 in chemotaxis and actin polymerization assays demonstrated high levels of chemotaxis and actin polymerization in MCL cells in response to the respective chemokines that were comparable with those reported in CLL cells.21 We also demonstrated that CXCL12-induced chemotaxis and actin polymerization, in both MCL patient samples and MCL cell lines, can be completely abrogated by pretreatment with Plerixafor, a selective CXCR4 inhibitor, or pertussis toxin, indicating that CXCL12-induced migration and signaling in MCL are largely dependent on activation of CXCR4 and CXCR4-coupled Gi proteins.
Next, we asked the question of how MCL would interact with MSCs that constitutively secret CXCL12 and display ligands for VLA-4, such as fibronectin. We found that MCL cells spontaneously migrate beneath MSCs, and the in vitro phenomenon “pseudoemperipolesis”21,33 was abundant in MCL (Figure 3). These data support the concept that MCL cells use chemokine receptors and adhesion molecules for trafficking and homing to tissue microenvironments, where stromal cells constitutively express respective ligands and provide supportive stromal niches for MCL cells.
Once MCLs adhere to MSCs, they are largely protected from the cytotoxic effect of F-ara-A and 4-HC (Figures 4,5); in particular, the MCL cell fraction that migrated beneath the MSCs was almost completely protected, suggesting that stromal cell–mediated drug resistance is an important drug resistance mechanism in MCL. Collectively, these experiments support a model in which MCL cells reside in tissue microenvironments, such as the marrow and the secondary lymphatic tissues, where they are protected and nourished by MSCs. Hence, disruption of the heterotypic adhesion may liberate MCLs from MSC niches and make them more accessible to conventional drugs. To test this hypothesis, we evaluated compounds that target both CXCR4 chemokine receptors (Plerixafor, pertussis toxin) and VLA-4 integrins (natalizumab, cyclic CS-1 peptides). We found that both Plerixafor and pertussis toxin can effectively inhibit activation of MCL cells in response to CXCL12 in chemotaxis and actin polymerization assays (Figure 2). Moreover, we noticed that pretreatment with natalizumab and cyclic CS-1 peptide was highly effective in inhibiting MCL cell migration beneath MSCs (Figure 3). Interestingly, we noticed that, in the pseudoemperipolesis assay, blocking of CXCR4 with Plerixafor or pertussis toxin was less effective compared with natalizumab and cyclic CS-1 peptides. One possible explanation for these differences could be based on the multistep model of cell migration,16 which describes that adhesion molecules, such as VLA-4, are critical for the early phase of adhesion to MSCs.33,41 Once this initial adhesion is established, chemokine receptor activation is responsible for later phases of adhesion and subsequent migration into tissues16 or beneath MSCs.9 As such, targeting VLA-4 might be more effective in blocking pseudoemperipolesis because it targets the upstream early phase of heterotypic adhesion, whereas blocking of CXCR4 modulates later downstream events. Moreover, MCL cells display high levels of spontaneous migration, which is independent from chemokine receptor activation and accounts for the high levels of spontaneous migration of the MCL cells (∼ 5% of input cells) toward chemotaxis chambers without chemokine (Figure 2C). Once adhesion to MSCs is established via VLA-4, spontaneous, chemokine-independent MCL cell migration may be sufficient to drive migration of a significant proportion of the MCL cells beneath the MSCs in the pseudoemperipolesis assay. However, it is questionable whether these in vitro effects would translate into differences in clinical application of these new agents. Currently, Plerixafor is used clinically for autologous stem cell mobilization in patients with lymphomas and MM, and early phase clinical trials of this drug in patients with leukemias (AML, CLL) are ongoing. In the later trials, the idea is to mobilize the leukemia cells from protective tissue microenvironments.42 Both Plerixafor43,44 and natalizumab45,46 have been demonstrated to be effective in mobilizing various hematopoietic cells from tissues to the blood. Based on these clinical data with Plerixafor, it would be anticipated that targeting of CXCR4 in MCL patients could also mobilize MCL cells from protective tissue niches and thereby make them better accessible for cytotoxic therapy. A similar, or even more pronounced, effect would be anticipated for natalizumab, based on the data presented in this study.
Stroma-mediated drug resistance, also called CAM-DR,30 is a well-defined phenomenon demonstrated for CLL,31,47 acute leukemias,48,49 and MM.30,50 In this study, we demonstrated that marrow stromal cells are highly effective in protecting MCL cells from chemotherapy-induced cell death (fludarabine, cyclophosphamide). MCL cells treated with fludarabine or cyclophosphamide displayed significantly higher viability than MCL cells without stromal cell support (Figures 4,5). Interestingly, MCL cells that had migrated beneath the MSCs were much more protected from F-ara-A than MCL cells that were in the nonmigrated supernatant fraction, a notion that has not previously been reported and that highlights the importance of close physical proximity between the neoplastic B cells and the MSCs for mediating drug resistance. Even though the relevance of stromal cell niches for MCL cells has not yet been established in vivo, it is tempting to speculate that stromal cells in the marrow and/or secondary lymphatic tissues establish such niches in which MCL cells could survive chemotherapies and then provide a reservoir for MRD and relapses. Currently, the architecture of distinct niches for hematopoietic and tissue stem cells and the mechanisms that govern stem cell migration, homing, and homeostasis are emerging.51 Hematopoietic stem cells localize to CXCL12+ stromal cells near the marrow vasculature (vascular niche) or the endosteum (osteoblast niche).52 Brain cancer stem cells also reside in vascular niches,53 indicating that normal stem cells and cancer cells have fundamentally similar niche requirements51 and suggesting that also lymphoma cells might preferentially localize to hematopoietic stem cell niches. In MCL, the marrow is a common site of MRD and relapses, and contamination of autologous stem cell products by MCL cells is considered a major reason for recurrence of the disease after autologous stem cell transplantation.54 Conceivably, although treatment eliminates the bulk of MCL cells, residual MCL cells can survive in protective niches where they receive signals from accessory cells that promote survival and drug resistance and pave the way to relapse.
Constitutive secretion of CXCL12 and CXCL13 by stromal cells induces not only signal for migration and cell adhesion, but also activates downstream signaling pathways, such as the MAPK pathway. The Ras-Raf-MAP (extracellular signal-regulated kinase/ERK) kinase pathway is an evolutionary, conserved signaling pathway that is involved in the control of many fundamental cellular processes, including cell proliferation, survival, differentiation, cell death, motility, and metabolism.55 Here, we demonstrated the activation of p44/42 MAPK in MCL cells after stimulation with chemokines, CXCL12 and CXCL13, and the inhibition of CXCL12-induced p44/42 MAP kinase activation by Plerixafor (Figure 6). These findings suggest that CXCL12 and CXCL13 activate MCL cells not only for migration to stromal cells secreting these chemokines but subsequently also for initiating signaling events that could contribute to MCL cell survival and/or proliferation. The inhibition of p44/42 MAPK activation in response to CXCL12 by Plerixafor exemplifies the efficacy of this antagonist in blocking CXCR4 and provides further rationale to explore this agent in treatment of MCL.
In conclusion, this study demonstrates that MCL cells display high levels of chemokine receptors and adhesion molecules that allow adhesive interactions between MCL cells and MSCs and potentially other types of tissue stromal cells. We also report that MCL cells adhere and spontaneously migrate beneath MSCs in a CXCR4- and VLA-4–dependent fashion (pseudoemperipolesis). Moreover, we demonstrate that MSCs confer drug resistance (CAM-DR) to MCL cells, a primary drug resistance mechanism that could be responsible for MRD and therefore of high clinical relevance. In particular, MCL cells that had migrated beneath MSCs were largely protected from the cytotoxic effect of fludarabine. These observations support a therapeutic concept that attempts to mobilize the MCL cells hiding in stromal niches to overcome CAM-DR and to improve our current treatments of MCL patients. We addressed this question using 2 agents that currently are in clinical use for inhibition of CXCR4 and VLA-4, Plerixafor and natalizumab. Both drugs were highly effective in blocking functional responses to the respective ligands in MCL cells and inhibited adhesion of MCL cells to MSCs, indicating that these drugs are potential candidates for clinical trials that target the MCL microenvironment. Such studies could lead us toward new therapeutic avenues for MCL patients.
Acknowledgments
The authors thank Dr D. Sampath and Dr W. Plunkett for valuable suggestions regarding the 4-HC experiments.
This work was supported by an ASCO Career Development Award (Alexandria, VA; J.A.B.), a CLL Global Research Foundation grant (Houston, TX; J.A.B.), and a Kimmel Scholar Award by the Sidney Kimmel Foundation for Cancer Research (Baltimore, MD; J.A.B.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.V.K. performed the experiments, designed the figures, and wrote the paper; A.T.T. provided MCL samples and lines and analyzed data; R.J.F. analyzed data and reviewed the manuscript; and J.A.B. designed the research, supervised the study, analyzed the data, and wrote and revised the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jan A. Burger, Department of Leukemia, Unit 428, University of Texas M. D. Anderson Cancer Center, PO Box 301402, Houston, TX 77230-1402; e-mail: jaburger@mdanderson.org.
References
- 1.Fisher RI, Dahlberg S, Nathwani BN, Banks PM, Miller TP, Grogan TM. A clinical analysis of two indolent lymphoma entities: mantle cell lymphoma and marginal zone lymphoma (including the mucosa-associated lymphoid tissue and monocytoid B-cell subcategories). A Southwest Oncology Group study. Blood. 1995;85:1075–1082. [PubMed] [Google Scholar]
- 2.O'Connor OA. Mantle cell lymphoma: identifying novel molecular targets in growth and survival pathways. Hematology Am Soc Hematol Educ Program. 2007:270–276. doi: 10.1182/asheducation-2007.1.270. [DOI] [PubMed] [Google Scholar]
- 3.Raffeld M, Jaffe ES. bcl-1, t(11;14), and mantle cell-derived lymphomas. Blood. 1991;78:259–263. [PubMed] [Google Scholar]
- 4.Bosch F, Jares P, Campo E, et al. PRAD-1/cyclin D1 gene overexpression in chronic lymphoproliferative disorders: a highly specific marker of mantle cell lymphoma. Blood. 1994;84:2726–2732. [PubMed] [Google Scholar]
- 5.Argatoff LH, Connors JM, Klasa RJ, Horsman DE, Gascoyne RD. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood. 1997;89:2067–2078. [PubMed] [Google Scholar]
- 6.Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood. 2008;111:558–565. doi: 10.1182/blood-2007-06-095331. [DOI] [PubMed] [Google Scholar]
- 7.Fayad L, Thomas D, Romaguera J. Update of the M. D. Anderson Cancer Center experience with hyper-CVAD and rituximab for the treatment of mantle cell and Burkitt-type lymphomas. Clin Lymphoma Myeloma. 2007;8(suppl 2):S57–S62. doi: 10.3816/clm.2007.s.034. [DOI] [PubMed] [Google Scholar]
- 8.Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol. 2005;23:1984–1992. doi: 10.1200/JCO.2005.08.133. [DOI] [PubMed] [Google Scholar]
- 9.Burger JA, Kipps TJ. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk Lymphoma. 2002;43:461–466. doi: 10.1080/10428190290011921. [DOI] [PubMed] [Google Scholar]
- 10.Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23:43–52. doi: 10.1038/leu.2008.299. [DOI] [PubMed] [Google Scholar]
- 12.Okada T, Ngo VN, Ekland EH, et al. Chemokine requirements for B cell entry to lymph nodes and Peyer's patches. J Exp Med. 2002;196:65–75. doi: 10.1084/jem.20020201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bajenoff M, Egen JG, Koo LY, et al. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reif K, Ekland EH, Ohl L, et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature. 2002;416:94–99. doi: 10.1038/416094a. [DOI] [PubMed] [Google Scholar]
- 15.Allen CD, Ansel KM, Low C, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- 16.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 17.Ansel KM, Ngo VN, Hyman PL, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 18.Campbell DJ, Kim CH, Butcher EC. Chemokines in the systemic organization of immunity. Immunol Rev. 2003;195:58–71. doi: 10.1034/j.1600-065x.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- 19.Schwickert TA, Lindquist RL, Shakhar G, et al. In vivo imaging of germinal centres reveals a dynamic open structure. Nature. 2007;446:83–87. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]
- 20.Trentin L, Cabrelle A, Facco M, et al. Homeostatic chemokines drive migration of malignant B cells in patients with non-Hodgkin lymphomas. Blood. 2004;104:502–508. doi: 10.1182/blood-2003-09-3103. [DOI] [PubMed] [Google Scholar]
- 21.Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999;94:3658–3667. [PubMed] [Google Scholar]
- 22.Burkle A, Niedermeier M, Schmitt-Graff A, Wierda WG, Keating MJ, Burger JA. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood. 2007;110:3316–3325. doi: 10.1182/blood-2007-05-089409. [DOI] [PubMed] [Google Scholar]
- 23.Sipkins DA, Wei X, Wu JW, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dave SS, Wright G, Tan B, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351:2159–2169. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- 25.Farinha P, Masoudi H, Skinnider BF, et al. Analysis of multiple biomarkers shows that lymphoma-associated macrophage (LAM) content is an independent predictor of survival in follicular lymphoma (FL). Blood. 2005;106:2169–2174. doi: 10.1182/blood-2005-04-1565. [DOI] [PubMed] [Google Scholar]
- 26.Byers RJ, Sakhinia E, Joseph P, et al. Clinical quantitation of immune signature in follicular lymphoma by RT-PCR-based gene expression profiling. Blood. 2008;111:4764–4770. doi: 10.1182/blood-2007-10-115915. [DOI] [PubMed] [Google Scholar]
- 27.Durig J, Schmucker U, Duhrsen U. Differential expression of chemokine receptors in B cell malignancies. Leukemia. 2001;15:752–756. doi: 10.1038/sj.leu.2402107. [DOI] [PubMed] [Google Scholar]
- 28.Corcione A, Arduino N, Ferretti E, et al. CCL19 and CXCL12 trigger in vitro chemotaxis of human mantle cell lymphoma B cells. Clin Cancer Res. 2004;10:964–971. doi: 10.1158/1078-0432.ccr-1182-3. [DOI] [PubMed] [Google Scholar]
- 29.Terol MJ, Lopez-Guillermo A, Bosch F, et al. Expression of beta-integrin adhesion molecules in non-Hodgkin's lymphoma: correlation with clinical and evolutive features. J Clin Oncol. 1999;17:1869–1875. doi: 10.1200/JCO.1999.17.6.1869. [DOI] [PubMed] [Google Scholar]
- 30.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–1667. [PMC free article] [PubMed] [Google Scholar]
- 31.Burger M, Hartmann T, Krome M, et al. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood. 2005;106:1824–1830. doi: 10.1182/blood-2004-12-4918. [DOI] [PubMed] [Google Scholar]
- 32.Burger JA, Zvaifler NJ, Tsukada N, Firestein GS, Kipps TJ. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism. J Clin Invest. 2001;107:305–315. doi: 10.1172/JCI11092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyake K, Hasunuma Y, Yagita H, Kimoto M. Requirement for VLA-4 and VLA-5 integrins in lymphoma cells binding to and migration beneath stromal cells in culture. J Cell Biol. 1992;119:653–662. doi: 10.1083/jcb.119.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shanafelt TD, Geyer SM, Bone ND, et al. CD49d expression is an independent predictor of overall survival in patients with chronic lymphocytic leukaemia: a prognostic parameter with therapeutic potential. Br J Haematol. 2008;140:537–546. doi: 10.1111/j.1365-2141.2007.06965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zucchetto A, Bomben R, Dal Bo M, et al. A scoring system based on the expression of six surface molecules allows the identification of three prognostic risk groups in B-cell chronic lymphocytic leukemia. J Cell Physiol. 2006;207:354–363. doi: 10.1002/jcp.20570. [DOI] [PubMed] [Google Scholar]
- 36.Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 37.Miyake K, Weissman IL, Greenberger JS, Kincade PW. Evidence for a role of the integrin VLA-4 in lympho-hemopoiesis. J Exp Med. 1991;173:599–607. doi: 10.1084/jem.173.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo CG, Lu TT, Cyster JG. Integrin-dependence of lymphocyte entry into the splenic white pulp. J Exp Med. 2003;197:353–361. doi: 10.1084/jem.20021569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakayama T, Fujisawa R, Izawa D, Hieshima K, Takada K, Yoshie O. Human B cells immortalized with Epstein-Barr virus upregulate CCR6 and CCR10 and downregulate CXCR4 and CXCR5. J Virol. 2002;76:3072–3077. doi: 10.1128/JVI.76.6.3072-3077.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ehlin-Henriksson B, Mowafi F, Klein G, Nilsson A. Epstein-Barr virus infection negatively impacts the CXCR4-dependent migration of tonsillar B cells. Immunology. 2006;117:379–385. doi: 10.1111/j.1365-2567.2005.02311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patrick CW, Jr, Juneja HS, Lee S, Schmalstieg FC, McIntire LV. Heterotypic adherence between human B-lymphoblastic and pre-B-lymphoblastic cells and marrow stromal cells is a biphasic event: integrin very late antigen-4 alpha mediates only the early phase of the heterotypic adhesion. Blood. 1995;85:168–178. [PubMed] [Google Scholar]
- 42.Uy GL, Rettig MP, Ramirez P, Nervi B, Abboud CN, DiPersio JF. Kinetics of human and murine mobilization of acute myelogenous leukemia in response to AMD3100. Blood. 2007;110:265a. [Google Scholar]
- 43.Broxmeyer HE, Orschell CM, Clapp DW, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Devine SM, Vij R, Rettig M, et al. Rapid mobilization of functional donor hematopoietic cells without G-CSF using AMD3100, an antagonist of the CXCR4/SDF-1 interaction. Blood. 2008;112:990–998. doi: 10.1182/blood-2007-12-130179. [DOI] [PubMed] [Google Scholar]
- 45.Bonig H, Wundes A, Chang KH, Lucas S, Papayannopoulou T. Increased numbers of circulating hematopoietic stem/progenitor cells are chronically maintained in patients treated with the CD49d blocking antibody natalizumab. Blood. 2008;111:3439–3441. doi: 10.1182/blood-2007-09-112052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zohren F, Toutzaris D, Klarner V, Hartung HP, Kieseier B, Haas R. The monoclonal anti–VLA-4 antibody natalizumab mobilizes CD34+ hematopoietic progenitor cells in humans. Blood. 2008;111:3893–3895. doi: 10.1182/blood-2007-10-120329. [DOI] [PubMed] [Google Scholar]
- 47.Panayiotidis P, Jones D, Ganeshaguru K, Foroni L, Hoffbrand AV. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br J Haematol. 1996;92:97–103. doi: 10.1046/j.1365-2141.1996.00305.x. [DOI] [PubMed] [Google Scholar]
- 48.Hall BM, Gibson LF. Regulation of lymphoid and myeloid leukemic cell survival: role of stromal cell adhesion molecules. Leuk Lymphoma. 2004;45:35–48. doi: 10.1080/1042819031000139620. [DOI] [PubMed] [Google Scholar]
- 49.Zeng Z, Samudio IJ, Munsell M, et al. Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol Cancer Ther. 2006;5:3113–3121. doi: 10.1158/1535-7163.MCT-06-0228. [DOI] [PubMed] [Google Scholar]
- 50.Chatterjee M, Honemann D, Lentzsch S, et al. In the presence of bone marrow stromal cells human multiple myeloma cells become independent of the IL-6/gp130/STAT3 pathway. Blood. 2002;100:3311–3318. doi: 10.1182/blood-2002-01-0102. [DOI] [PubMed] [Google Scholar]
- 51.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 53.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 54.Pott C, Schrader C, Gesk S, et al. Quantitative assessment of molecular remission after high-dose therapy with autologous stem cell transplantation predicts long-term remission in mantle cell lymphoma. Blood. 2006;107:2271–2278. doi: 10.1182/blood-2005-07-2845. [DOI] [PubMed] [Google Scholar]
- 55.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]