

Abstract
Despite major therapeutic advances, most mature B-cell malignancies remain incurable. Compelling evidence suggests that crosstalk with accessory stromal cells in specialized tissue microenvironments, such as the bone marrow and secondary lymphoid organs, favors disease progression by promoting malignant B-cell growth and drug resistance. Therefore, disrupting the crosstalk between malignant B cells and their milieu is an attractive novel strategy for treating selected mature B-cell malignancies. Here we summarize the current knowledge about the cellular and molecular interactions between neoplastic B lymphocytes and accessory cells that shape a supportive microenvironment, and the potential therapeutic targets that are emerging, together with the new problems they raise. We discuss clinically relevant aspects and provide an outlook into future biologically oriented therapeutic strategies. We anticipate a paradigm shift in the treatment of selected B-cell malignancies, moving from targeting primarily the malignant cells toward combining cytotoxic drugs with agents that interfere with the microenvironment's proactive role. Such approaches hopefully will help eliminating residual disease, thereby improving our current therapeutic efforts.
Introduction
The microenvironment is the compilation of accessory cells that within individual organs work as a team through cell-cell contacts and active molecular crosstalk to provide a functional scaffolding to parenchymal cells. In solid tumors, a microenvironment instrumental to the survival and propagation of malignant cells is built up by the concurrence of inflammatory cells that produce growth factors, new vessel formation that provides nutrients, and immune tolerance that avoids immune-mediated elimination. Conceivably, this conducive microenvironment may be an appropriate soil to host cancer stem cells (CSCs). Signals from the tumor microenvironment are a major hurdle to cancer cell eradication, and its neoangiogenetic component has become an attractive target for treatment strategies that aim at perturbing the nurturing capacity of the tumor cell milieu.1
Blood cancers develop in specialized tissue microenvironments, such as bone marrow (BM) and secondary lymphoid organs. These microenvironments are characterized by different populations of accessory stromal and T cells that interact with malignant cells and promote tumor growth and drug resistance.2,3 Malignant blood cells apparently have highly variable degrees of dependency on signals from the microenvironment. To make matters more complex, the affinity and dependency on accessory stromal cells for tumor growth and progression can change over time, with the evolution of stroma-dependent or stroma-independent subclones.
The issue of microenvironment-targeted treatment has recently gained increasing attention in hemato-oncology, and the enthusiasm in this field is justified by a number of new drugs that are targeted toward the microenvironment, such as thalidomide and lenalidomide, plerixafor, and natalizumab.4 At this point, the main questions are which blood malignancies may mostly benefit from microenvironment-targeted approaches, what are the potential therapeutic targets, and what are the problems they raise.
Microenvironment and blood cancers
The relationships between neoplastic blood cells and microenvironment appear to follow 3 major patterns whose better definition may provide clues for future microenvironment-targeted treatments.
The first pattern may be referred to as loss of interconnection with the microenvironmental network, which occurs when a genetic abnormality provides transformed cells with a sustained proliferation advantage that is largely autonomous and independent of microenvironmental signals. A typical example is Burkitt lymphoma, where virtually all malignant B cells are determined to proliferate because of the chromosomal translocation that leads to perennial c-myc gene activation. Accordingly, in Burkitt lymphoma, the microenvironment appears to have a limited role in planning new treatment strategies.
The second pattern is a dysfunctional environment, where the neoplastic cells engage in deregulated interactions with the supportive stroma that provide the malignant cells with growth- and drug-resistance signals. Examples are the acute leukemias and myelodysplastic syndromes, where leukemia stem cells (LSCs) escape the tightly regulated cell growth- and proliferation-control within the hematopoietic niches, and instead parasitize in the supportive hematopoieic microenvironment. Here, the dependency of the neoplastic cells on the stromal counterparts for growth and survival is partially retained and may lead to the survival of clones endowed with a higher affinity for the microenvironment.4,5 Accordingly, in these diseases, targeting the microenvironment is justified as an attempt to target stromal cell–dependent subclones, which otherwise survive conventional treatments and pave the way for relapses.
The third pattern, a friendly, regulated coexistence of the malignant cells and the microenvironment, is apparent in subsets of B-cell tumors, such as chronic lymphocytic leukemia (CLL), follicular lymphoma (FL), mucosa-associated lymphoid tissue (MALT) lymphomas, and multiple myeloma (MM). Here, the interactions between the malignant cells and the microenvironment largely resemble the pattern that the normal counterpart B cells engage in with their respective microenvironments. Consequently, the proliferative drive for the malignant cells is, at least initially, largely dependent on external signals from the microenvironment, such as antigens, cytokines, and cell-cell interactions, and the neoplastic cells undergo apoptosis unless their survival is reinforced by these external stimuli. These interactions are not targeted by our current “conventional” treatments, which may explain why, despite major therapeutic advances, some B-cell malignancies still remain incurable. Based on this concept, these tumors are expected to be particularly responsive to microenvironment-directed treatment approaches.
Bone marrow and secondary lymphoid organs have different microenvironments
Blood-forming BM and secondary lymphoid organs have entirely different microenvironments, each finely tuned to support different aspects of lymphocyte maturation and differentiation. The BM harbors hematopoietic stem cells (HSCs) and hosts the development of mature B cells from committed progenitors. The latter process is primarily concerned with the events that lead to the production of cells endowed with a functional antigen (Ag) receptor (B-cell receptor [BCR]). Mature B cells migrate to secondary lymphoid organs where they are exposed to Ag within germinal centers (GCs) of secondary lymphoid follicles. The microenvironment of GC allows maturing B cells to interact with CD4+ T cells for the necessary help on Ag recognition and with specialized stromal cells, follicular dendritic cells (FDCs), for the required quality control after affinity maturation.6,7 The Ag encounter triggers the proliferation, maturation, and final differentiation into effector plasma cells (PCs) and memory B cells.8
In the BM, accessory “feeder” cells maintain HSCs within specialized “niches,” which are close to the marrow vasculature (vascular niche) or to the endosteum (osteoblast niche).9 The concept of niches appears to apply to CSCs as well. As an example, LSCs not only share phenotypic and functional characteristics with normal HSCs10 but also preferentially localize within HSC niches11 (Figure 1A). The BM development of B cells from early progenitors requires their intimate contact with a heterogeneous population of adherent cells collectively referred to as “stroma.”12 The importance of stromal cells for hematopoiesis was initially demonstrated in long-term BM cultures13 and was used by Whitlock and Witte to develop a culture system to study the early stages of B-cell maturation.14
Figure 1.
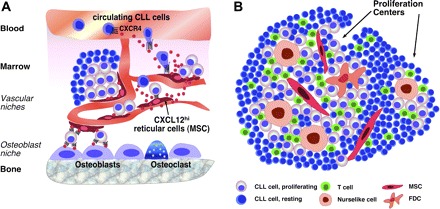
Cellular interactions. (A) Marrow and (B) lymphatic tissue microenvironments in mature B-cell tumors. (A) MSCs (arrow), also called reticular cells, are scattered throughout the marrow cavity and constitutively secrete high levels of the chemokine CXCL12 (SDF-1). MSCs colocalize with the vasculature, forming so-called “vascular niches.” CXCL12 secretion by MSC induces CXCL12 gradients that can attract circulating neoplastic B cells via CXCR4 receptors expressed on CLLs, MM, and other malignant B cells. Circulating lymphoma cells may become attracted by CXCL12 gradients to home to the marrow where contact with MSCs provides them with growth and survival signals. Mesenchymal-derived osteoblasts are specialized fibroblasts critical for bone formation and able to secrete CXCL12. Therefore, interaction with osteoblasts is an alternative, additional niche where lymphoma cells can home. These cellular interactions also confer drug resistance to leukemia/lymphoma cells and may therefore account for MRD. (B) In secondary lymphoid tissues, CLL cells and other lymphoma cells can interact with a variety of accessory cells, such as MSCs, monocyte-derived NLCs, which are similar to LAMs, and T cells. The presence of FDCs in lymphoid tissues in CLL is controversial. Formation of proliferation centers is a hallmark of CLL histopathology. Interactions between CLL and accessory cells within proliferation centers are critical for providing growth and survival signals to CLL B cells, inducing their proliferation and resembling interactions between normal, antigen-stimulated B cells and accessory cells (antigen-presenting cells, T cells) during GC reaction. CLL cells outside the proliferation centers are resting and considered the nonproliferative compartment.
Implications for mature B-cell malignancies
The microenvironment of mature B-cell tumors mirrors the microenvironment of the tissue where the tumor develops. It shares the general properties of cancer microenvironment with the presence of abundant different populations of stromal cells that favor the survival of malignant cells, various subsets of T cells that may support the disease progression, overcoming the antitumor effect exerted by still other specific T-cell subsets, and endothelial cells that organize neoangiogenetic microvessels. The clinical relevance of microenvironment is exemplified by the observation that the length of survival among patients with FL correlates with the molecular features of nonmalignant immune cells present in the tumor at diagnosis,15 and those differences in immune cells, fibrosis, and angiogenesis in tumor environment influence the survival after treatment of diffuse large B-cell lymphoma (DLBCL).16 LSCs have not (yet) been identified in B-cell tumors, and it remains unresolved whether these tumors are nurtured by LSCs that find their optimal soil in specific microenvironments or whether they are rather uniform mature B-cell clones that become exposed to microenvironmental growth signals in specific tissues. Furthermore, and because of the specific features of B cells, other players, such as Ag stimulation and the molecules involved in lymphocyte traffic, are more specifically operating in the microenvironments of B-cell tumors. Angiogenesis also is a major player in the microenvironment of B-cell lymphomas, although its role in B-cell tumors is currently less well defined compared with solid tumors,17 where antiangiogenic strategies have become an important therapeutic modality in metastatic disease. B-cell lymphoma growth is enhanced by at least 2 angiogenic mechanisms: autocrine stimulation of tumor cells via expression of vascular endothelial growth factor and vascular endothelial growth factor receptors by lymphoma cells, such as CLL,18 DLBCL, and mantle cell lymphoma cells, as well as paracrine effects on local neovascularization and recruitment of circulating endothelial progenitors. The efficacy of antiangiogenic treatment approaches in mature B-cell malignancies is currently explored in clinical trials.17
Antigen stimulation in mature B-cell malignancies
The very raison d'être of mature B cells is their Ag receptor. It follows that in specific B-cell malignancies the concept of microenvironment as a regulator of malignant B-cell growth is tightly linked to the possible role of Ag stimulation.19 Chronic BCR stimulation by latent microbial or auto-Ag can trigger the development and expansion of malignant B cells. Classic examples are the development of gastric MALT lymphomas in the context of chronic Helicobacter pylori infection20,21 and of lymphomas within salivary glands in Sjogren syndrome or the thyroid in Hashimoto thyroiditis. The list of B-cell tumors where microorganisms provide antigenic lymphomagenic stimuli is growing and includes hepatitis C virus in splenic marginal zone lymphoma,22 Borrelia in cutaneous marginal zone lymphoma,22,23 Chlamydia psittaci in ocular adnexal marginal zone lymphoma,22,24 and possibly also Campylobacter jejuni in immunoproliferations of the small intestine.22
The existing data also indicate that molecular structures normally involved in eliminating cellular debris, scavenging apoptotic cells, and providing a first line of defense against pathogenic bacteria may trigger and/or facilitate the onset and evolution also of CLL.25–27
Most patients with FL have somatically mutated immunoglobulin variable (IGVH) genes with intraclonal variation, consistent with their origin from GC. Surprisingly, these IGVH genes almost always carry introduced motifs available for N-glycosylation that are uncommon in normal B cells, very rare in CLL, undetectable in MM, although evident in a proportion of DLBCLs, suggesting a site-specific role.28 Most novel glycosylation sites are located in the complementary-determining regions sites, possibly precluding conventional Ag binding and contain oligomannose glycans, which are expressed by tumor cell surface IgM. As binding studies indicate that the oligomannose glycans occupying the variable regions are accessible to mannose-binding lectin, the possibility is raised, that within malignant GC, the Ag stimulation operating in other B-cell tumors might be mimicked by the interaction of the tumor cell BCR with mannose-binding molecules of innate immunity.29 A possible BCR reactivity has also been shown in DLBCL where it appears to carry a prognostic significance.30 The majority of examined primary DLBCLs were found to exhibit both tonic- and ligand-induced BCR signaling, which could be identified by transcriptional profiles and was abrogated by an inhibitor of spleen tyrosine kinase (Syk).30
In MM both the IgVH mutational status31 and the actual paraprotein reactivity identified in some instances32 document that Ag stimulation is somehow instrumental in triggering the onset of the diseases. Whether a persistent Ag stimulation may fuel the disease by providing persistent waves of Ag-specific plasmablasts is a matter of speculation.
The issue becomes how we might use all this information for designing therapeutic strategies. One obvious possibility is to identify the stimulating Ag, especially if it is of microbial origin and interferes with its activity, an approach successfully applied to H pylori– and C psittaci–related lymphomas. Another option is to learn how to manipulate the B-cell response triggered by the BCR stimulation. The possibility of targeting the signal transduction pathways activated in malignant B cells by microenvironmental interactions exists considering the impressively rapid progress in the field of (kinase-) signaling inhibitors. Unfortunately, signal transduction pathways are generally redundant: if one is switched off, others remain intact and can bypass the inhibitors. Master signaling pathways tend to be used in various cell types, and targeting these pathways might lead to unwanted severe side effects.
Malignant B-cell trafficking, chemokine receptors, and adhesion molecules
Evidence is growing that malignant B cells exploit the physiologic mechanisms of tissue-specific lymphocyte migration and homing to access supportive microenvironmental niches.
The exquisite homing to and within the BM of malignant B cells, such as MM33 and CLL cells,34 is mediated by the chemokine receptor CXCR4.35 This allows malignant B cells to interact with CXCL12-expressing mesenchymal stromal cells (MSCs),5 which in turn provide ligands for adhesion molecules, such as VLA-4 (CD49d) or CD44.36 VLA-4–mediated adhesion to fibronectin confers a survival advantage and inhibits drug-induced apoptosis (cell adhesion–mediated drug resistance) in MM37 and other hematopoietic malignancies. Activation of CXCR4 and CXCR5 induces signals related to cell growth, such as p44/42 MAP kinase-(ERK1/2) and STAT3,38 and prolongs the survival of CLL cells,39,40 indicating that chemokine receptor activation not only attracts neoplastic B cells but also directly regulates their fate. MM PCs express on the membrane adhesion molecules, such as H-CAM (CD44), ICAM-I (CD54), N-CAM (CD56), LFA-3 (CD58), the proteoglycan syndecan, a receptor for hyaluronan-mediated motility, and frequently also CD11/CD18.36,41,42 The interaction of MM surface adhesion structures with their homologous ligands within the BM microenvironment allows malignant PCs to be entrapped within the BM stromal cell web. There, they encounter locally produced cytokines enriched on the extracellular matrix protein layers.
FL43,44 and mantle cell lymphoma cells45 also express surface chemokine receptors, particularly CXCR4 and CXCR5,44,45 and adhesion molecules that allow for the homing and retention within supportive stromal niches. Anti-CD49d monoclonal antibodies or peptides that compete with CD49d ligands inhibit malignant B-cell adhesion46,47 to stromal cells and enhance the activity of cytotoxic drugs. Accordingly, drugs targeting CD49d, other adhesion molecules, or their stromal ligands, should also be evaluated in selected B-cell malignancies.
Lessons from the analysis of different B-cell malignancies
Individual B-cell malignancies have unique microenvironment properties whose detailed analysis raises a number of issues important for planning new therapeutic strategies.
Microenvironment in CLL
CLL cells relentlessly accumulate in vivo but rapidly undergo spontaneous apoptosis in vitro, implying that their apoptosis resistance, rather than being an intrinsic feature of leukemic cell, depends on external signals.2,3,19 This is not surprising considering that also the maturation stages of normal B cells are highly dependent on microenvironment signals.48 Starting from the original studies, which used unselected BM stromal cells,39,49 growing evidence suggests that different types of stromal cells, such as monocyte-derived nurselike cells (NLCs),39 MSCs,39,49 or even FDCs50 protect CLL cells in coculture and are an integral part of the CLL microenvironment.
NLCs owe their name to the similarities with thymic nurse cells that nurture developing thymocytes.39 In vitro, NLCs differentiate from blood monocytes cocultured with CLL cells.39 In vivo, NLCs can be found in the spleen and lymphoid tissues of CLL patients.51 They attract CLL cells by secreting CXCL1239 and CXCL1352 and protect CLL cells from spontaneous or drug-induced apoptosis through CXCL12,39,40 B cell–activating factor of the tumor necrosis factor family (BAFF), a proliferation-inducing ligand,40 CD31, and plexin-B153 (Figure 2).
Figure 2.
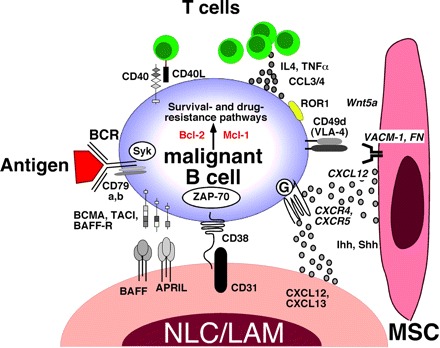
Molecular crosstalk between malignant B cells, exemplified for CLL B cells, and the microenvironment. This figure displays the molecules involved in crosstalk between CLL cells and accessory cells in the marrow and/or lymphoid tissue microenvironments. Contact between CLL cells (and various other mature B-cell lymphomas, as detailed in the text) and NLCs/LAMs or MSCs is established and maintained by chemokine receptors and adhesion molecules. NLCs express the chemokines CXCL12 and CXCL13, whereas MSCs predominantly express CXCL12. NLCs and MSCs attract CLL cells via the G protein–coupled chemokine receptors CXCR4 and CXCR5, which are expressed at high levels on CLL cells. Integrins, particularly VLA-4 integrins (CD49d), expressed on the surface of CLL cells cooperate with chemokine receptors in establishing cell-cell adhesion through respective ligands on the stromal cells (VCAM-1 and fibronectin/FN). NLCs also express the TNF family members BAFF and a proliferation-inducing ligand, providing survival signals to CLL cells via corresponding receptors (BCMA, TACI, BAFF-R). CD38 expression allows CLL cells to interact with CD31, the ligand for CD38, expressed by stromal and NLCs. Ligation of CD38 activates ZAP-70 and downstream survival pathways. Self and/or environmental antigens (Ag) are considered a key factor in stimulation and expansion of the CLL clone. The nature and source of Ag and its mode of presentation in CLLs are unknown and currently the focus of intensive research. Stimulation of the BCR complex (BCR and CD79a,b) induces downstream signaling by recruitment and activation of Syk and ZAP-70. BCR stimulation and coculture with NLCs also induce CLL cells to secrete high levels of the chemokines CCL3 and CCL4, which are potent T cell–attracting chemokines. Through this mechanism, CLL cells can actively recruit T cells for cognate T-cell interactions with CLL cells. CD40L+ T cells are preferentially found in CLL proliferation centers and can interact with CLL cells via CD40. Cytokines secreted by T cells or CLL cells, such as IL-4, or TNF-α are considered important regulators of CLL cell survival. Hedgehog (Hh) proteins, such as the indian (Ihh) and sonic (Shh) hedgehog proteins, are stromal cell–derived factors that can regulate survival of CLL and other mature B-cell malignancies.54 ROR1, an oncofetal antigen with CLL-restricted expression, may function as receptor for Wnt5a,55 expressed by stromal cells. Collectively, this crosstalk between CLL cells and accessory cells results in activation of survival and drug resistance pathways, such as those provided by Bcl-2 and Mcl-1.
MSCs are known to profoundly influence the development and progression of various cancers.56 In CLL38 and other B-cell malignancies, such as MM57 as well as in a murine mature B-cell lymphoma model,54 MSC-derived survival and drug resistance signals are largely adhesion-dependent. Coculture with MSC results in the rapid, spontaneous migration of a fraction of CLL cells beneath and underneath the MSCs (pseudoemperipolesis), inducing a cobblestone-like appearance.34 This migration depends on CXCR4 and VLA-4 expression by leukemia cells34 and exemplifies the migratory potential and the need for adhesive interactions between CLL cells and the microenvironment. Adhesion molecules, particularly VLA-4 integrins, along with CD44 and gelatinase B/matrix metalloproteinase-9,58 cooperate with chemokine receptors in CLL cell adhesion to stromal cells.46 Interestingly, VLA-4 expression on CLL cells has prognostic impact,59 indicating the relevance of these interactions in vivo.
In CLL tissues, a unique histopathology hallmark is apparent: the presence of focal aggregates of proliferating cells that cluster in pseudofollicular structures, named proliferation centers or pseudofollicles (Figure 1B).3 The architecture and cellular composition of proliferation centers include different Ag-presenting cells and numerous CD4+ T cells25,60 (Figure 1B), suggesting that some of the mechanisms required for the expansion of Ag-specific normal B cells within GC may also be operating in CLL. Still the processes accounting for the development and maintenance of pseudofollicles are unknown. CXCL13, a chemokine critical in establishing the architecture of normal follicles, probably plays a role. CXCL13 is constitutively secreted by FDCs and NLCs and is a potent chemoattractant for CLL cells.52 Recent data suggest that CLL cells are not mere passive seeds that need to home into the appropriate soil but rather active players that create a supportive microenvironment. CLL cells, after BCR stimulation or exposure to NLCs, secrete chemokines, such as CCL22,60 CCL3, and CCL4,61,62 which can attract other supportive cells, such as T cells. This indicates the possibility that the CLL microenvironment is created and maintained through a dynamic, interactive coevolution between leukemic and normal bystander cells.
The role of T cells in CLL can be seen from different, nonmutually exclusive angles. One possibility is that T-cell subsets may help favor disease progression, overcoming the antitumor effect exerted by other specific T-cell subsets. In this context, the role and functional significance of regulatory T cells (Treg) and γδ T cells in the CLL microenvironment are imperfectly defined.63 Another possibility is that T-cell abnormalities may provide the biologic basis of the significant immune deficiency that is typical of CLL. The recently identified impaired formation of immunologic synapse might represent an important element of immune dysfunction,64 especially because this abnormality can also be induced in healthy allogeneic T cells after contact with CLL cells. Further, the possibility of reversing this defect with the immunomodulatory drug lenalidomide is of therapeutic interest.64,65
The overall number of circulating T cells, oligoclonal in both the CD4 and the CD8 compartment,66 is increased in untreated CLL patients. It is still unresolved whether these expanded T cells display immune reactivity to the malignant clone or rather target microbial antigens that are more prevalent in CLL patients because of their chronic immunosuppression. Functionally, they can stimulate CLL cell growth and survival by secreting cytokines, such as interleukin-4 (IL-4) or tumor necrosis factor-α (TNF-α)19 (Figure 2). Within proliferation centers, a significant proportion of T cells display CD40L (CD154),25 a member of the TNF superfamily that may bind to CD40+ CLL cells, rescuing them from apoptosis.67 Conversely, CD40 crosslinking on CLL cells induces up-regulation of CD80 and CD54 and turns nonimmunogenic CLL cells into effective T-cell stimulators.68 CLL cells engineered to express CD154 by adenoviral gene transfer can crosslink CD40 on bystander CLL cells and induce the same sequence of activation and immune recognition. When this concept was moved into the clinic as a novel form of immunotherapy, CLL patients were infused with their cells transduced in vitro with an adenovirus encoding CD40L (Ad-CD154).69 Recently, 3 patients treated with Ad-CD154–transduced CLL cells were shown to make antibodies to ROR1,55 an oncofetal antigen with surface expression restricted to CLL B cells. Collectively, these data suggest that CD40 activation of CLL cells can result in different outcomes that are not mutually contradictory: activation of prosurvival and proliferation pathways, if triggered by CD154+ T cells in the context of proliferation centers, or immune recognition and induction of a specific immune response, if triggered in the context of Ad-CD154–transduced CLL cells.
Microenvironment in follicular lymphoma
A proactive involvement of the microenvironment appears to be operating in some indolent lymphomas, which tend to retain the architecture and the cellular microenvironment of normal lymph nodes. This is particularly true in FL, which faithfully recapitulates the general architecture of normal follicles, including the presence of T cells and of a meshwork of FDCs, even when the infiltrate localizes in the BM and in nonlymphoid organs. FL cells, like normal GC cells, are notoriously difficult to grow in vitro where they rapidly undergo apoptosis unless stimulated through CD40,70 a main signaling pathway of T- and B-cell interactions. It is also apparent in marginal zone B-cell lymphomas of MALT-type, where malignant cells maintain an association with epithelial cells. These observations indicate that the pioneer colonizing lymphoma cells that land in the appropriate soil may contribute to render it much more welcoming and suggest the coevolution of malignant cells and microenvironment.
Given that the composition of the microenvironment appears to be a key prognostic factor in FL,15 the relevant question becomes how such a conducive microenvironment is assembled. It has to be understood whether the non-B cellular elements in an FL infiltrate outside a lymph node, such as T cells and FDCs, are imported from outside (eg, from the original lymph node lesion) together with malignant B cells or whether they are differentiating in situ from local cells recruited from the different microenvironment elements of the different organs involved. If the latter hypothesis is correct, it remains to be established which T-cell subsets are implicated and through which mechanisms and especially which local stromal cells are recruited and how the stromal cells differentiate into FDCs. In this context, a relevant role may be played by MSCs, which can be part of the normal BM and lymphoid tissue microenvironment and, as such, might represent the soil where the neoplastic B cells seed. Alternatively, they may coevolve in the tumor microenvironment, as it has been shown in some solid tumors where carcinoma-associated fibroblasts provide growth signals by secreting CXCL12.71
Lymphoma-associated macrophages (LAMs) confirm the critical role of myelomonocytic cells in shaping the microenvironment of some B-cell malignancies. Microarray and immunohistochemistry studies15,72,73 suggested a central role of LAMs in the pathogenesis and prognosis of FL, indicating the responsiveness of FLs to microenvironmental signals, especially delivered by these cells. Because NLCs and LAMs share lineage and high CD68 expression, they may represent a common master cellular regulator in the microenvironment of different B-cell tumors. Considering the general role of macrophages in cancer progression,74 it is not surprising to find them as central players in the FL microenvironment, although one would have also expected FDCs to be identified, given the critical role of Ag stimulation in B-cell malignancies and the role of mesenchymal FDCs6 as prototype Ag-presenting cells in normal lymphoid follicles. Still, Ag presentation by myelomonocytic cells was long known as an alternative form of Ag delivery to B cells and has recently gained new attention.75,76 Rapid in vivo delivery of immune complexes to macrophages in the lymph node subcapsular sinus and their capture by follicular B cells were demonstrated using multiphoton intravital microscopy of lymph nodes.75 It is tempting to speculate that similar events might operate between malignant B cells and NLCs or LAMs in the microenvironment of CLL and FL.
Neoplastic follicles contain numerous T cells usually found in normal GC, including follicular T cells. The role of T cells in shaping FL microenvironment is unclear, some probably favoring tumor progression by production of cytokines and/or direct contact.77 Of interest, FOXP3+CD4+CD25+ regulatory T cells are enriched in FL nodes.78 Tregs play a critical role in the inhibition of self-reactive immune responses and, as such, have been implicated in the suppression of tumor-reactive effector T cells in FL, thereby fostering an immune-privileged microenvironment.79 It is therefore a relevant observation that FL B cells may be able to induce Foxp3 expression and regulatory function in conventional T cells within the tumor microenvironment,80,81 thereby allowing FL cells to evade the immune system detection. However, the biologic and prognostic role of various non-neoplastic T-cell populations in FL infiltrates is poorly understood, and results from a number of immunohistochemical studies investigating the number of CD3+, CD4+, and CD8+ T-cell populations are highly contradictory.72,73,82–86 de Jong et al recently reviewed 9 studies in FL, often reporting contradictory findings of FL infiltration by accessory T cells and other stromal cells (macrophages, FDCs), and their impact on patients' prognosis.85 Lessons from these studies are that the prognostic role of certain T-cell populations might be highly dependent on specific treatment protocols and that the spatial distribution of T-cell subsets (eg, intrafollicular or extrafollicular) might be of biologic importance.
Microenvironment in MM
MM has long been the paradigmatic model for investigating the role of microenvironment in blood cancers. The MM PC precursor is either an activated B memory cell or a plasmablast31,87 generated in peripheral lymphoid organs during secondary T cell–dependent antibody response, programmed to home to the BM and committed to differentiate in close association with the BM microenvironment.36 Malignant PCs capitalize on the same interactions and stimuli provided by BM stromal cells that allow for the development, survival, and differentiation of normal lympho-hemopoietic progenitors. MM malignant PCs produce a number of cytokines that include interleukin-1β, TNF-β, and monocyte-macrophage colony stimulating factor,36 which activate the microenvironment. In turn, the cells of BM microenvironment produce several cytokines relevant to the growth and survival of MM PCs epitomized by IL-6 and insulin-like growth factor-I (reviewed by Hideshima et al36). The physical contact between MM PC and BM stromal cells increases IL-6 production.88 BM microenvironmental stromal cells both in mice and in human PC tumors are well equipped with a large series of adhesion and extracellular matrix proteins that mediate homotypic and heterotypic interactions and provide anchorage sites to cells selectively exposed to locally released growth factors.36
The MM microenvironment has 2 cardinal features. First, neoangiogenesis is especially prominent. The density of newly formed microvessels is proportional to the PC labeling index; it increases with stage and has a strong prognostic value.89 MM cells produce angiogenic cytokines90; in turn, endothelial cells stimulate MM cell growth and protect them from drugs.91 The role of neoangiogenesis, which is also evident in other blood cancers spanning from CLL92 to myelofibrosis,93 as a therapeutic target in MM is still unclear. As an example, a number of drugs, such as thalidomide, lenalidomide, and bortezomib, appear able to counteract the nurturing effect of the BM milieu in MM, but their precise relationship with a direct antiangiogenetic effect has not been proven. More recently, the possibility has emerged that dasatinib may target tumor vessels.94
The second important property of the MM microenvironment is that several of the cytokines that function as growth and survival factors for monoclonal PC (such as IL-6 and insulin-like growth factor) are also produced in the context of bone remodeling, leading to the concept of the vicious cycle between bone resorption and tumor growth. This notion is at the basis of the microenvironment-targeted treatment centered on blocking osteoclast-mediated bone resorption, indirectly suppressing the sequela of osteoclast activity.
These features still fail to disclose why MM PCs localize uniquely within the BM where bone resorption and tumor expansion stimulate and excite each other, creating a vicious cycle. The problems that have to be solved include which properties of BM microenvironment allow such a unique situation, whether specific populations of stromal cells are involved, how osteoimmunology may provide much needed clues, and how the modifications of microenvironment that occur in MM relate to the microenvironment features of monoclonal gammopathy of undetermined significance.
Potential therapeutic targets: the issue of malignant B-cell niches
Overall, we are only starting to learn which pathways deliver critical survival and drug-resistance signals in the interactions that occur between B-cell malignancies and their microenvironments. The definition of the architecture of distinct niches for hematopoietic and tissue SC and their relationships with CSC niches, together with the mechanisms that govern SC migration, homing, and homeostasis,95 may translate into studies of B-cell malignancies. Particularly in the early stages, the pathways used by largely stroma-dependent clones are similar to those used during early B-cell development in the marrow and Ag-triggered B-cell expansion in follicles.
In this context, a tissue of crucial importance is the BM. Besides MM, which is by definition a BM disease, the BM is a common site of minimal residual disease and the source of relapses in patients with B-cell tumors, such as CLL. One possible explanation is that, although conventional treatment eliminates the bulk of clonal elements, residual malignant cells lurk in protective niches where they receive signals from accessory cells that promote survival and drug resistance (Figure 1A). It is reasonable to suggest that these niches may have similarities with the niches that protect HSCs, including the presence of stromal cells, T cells, and endothelial cells. This situation would be optimal to pave the way to relapse. A similar organization might occur in MM, where it is plausible to consider that transformed migrating plasmablasts reaching the BM may organize themselves in niches that would provide the environment necessary for their differentiation into nonmigratory PCs that will progressively and successfully occupy the marrow.
Considering that different types of stromal cells are being implicated in the growth of mature B-cell tumors, a precise definition of which cells and through which pathways are operating in vivo in different malignancies becomes essential. The compelling evidence that interactions with different types of stromal cells, such as MSCs or NLCs, confer survival- and drug-resistance signals to malignant B cells offers attractive therapeutic avenues. At the same time, it cannot be overlooked that stromal cells are essential for the tightly regulated tissue homeostasis, providing niches to normal SCs, such as HSCs. This may result in a double-edged sword as exemplified when considering CXCR4 as a molecular therapeutic target. Given the importance of CXCR4 for malignant B-cell adhesion to MSCs,34 and more generally, its significance for CSC homing to protective niches,96 CXCR4 antagonists (plerixafor/AMD3100, or T140 analogs) could be useful for mobilizing cancer cells for a more effective exposure to anticancer drugs.4 However, this approach also mobilizes normal HSCs (and potentially other SCs) and exposes them to cytotoxic drugs outside their protective niches, which might result in increased toxicity. Combinations of CXCR4 antagonists with drugs that target predominantly the malignant cells, such as monoclonal antibodies (ie, rituximab, alemtuzumab), could help avoid this potential hazard.
Targeting the microenvironment: clinical and preclinical studies
Preliminary data from an ongoing clinical trial in patients with relapsed leukemia, which combines plerixafor with chemotherapy, suggest safety and efficacy of the concept to mobilize (using a CXCR4 antagonist) and then target tumor cells outside their protective niches.97 Related clinical studies include a CLL trial that combines plerixafor with rituximab, and a trial in MM with the T140 analog BKT140, but preliminary data are not yet available.
Another therapeutic approach that recently entered the clinical stage is related to the Syk. Syk is a key component of BCR signaling and can be blocked by R406, a small-molecule Syk inhibitor. R406 is active in preclinical models in CLL62,98 and DLBCL30 and displayed impressive activity in CLL patients in a first clinical trial.99 Other therapeutic targets are adhesion molecules, such as VLA-445 or CD44 as an alternative to CXCR4 antagonists for mobilization of neoplastic cells from their protective environment. In a first clinical trial in myeloma patients, the anti–VLA-4 antibody natalizumab (Biogen IDEC), approved for the treatment of multiple sclerosis, is currently being investigated.
Surface molecules or signaling pathways that are involved in crosstalk between malignant B cells and their microenvironment (Figure 2) also represent attractive therapeutic targets. The phosphoinositide 3-kinases are a key signaling pathway in cancer, including B-cell malignancies.100 Small molecule phosphoinositide 3-kinase inhibitors effectively antagonize stromal cell–derived migration, survival, and drug resistance signals in CLL.100 These drugs are expected to become available for targeting stroma-derived survival signals in B-cell malignancies in the near future. Immunomodulatory drugs, such as thalidomide and its successor, lenalidomide, are clinically active in mature B-cell malignancies, including MM and CLL, and their activity has been related to modulation of the microenvironment, rather than the malignant B cells. Here, the precise mechanism(s) remain unknown and are currently the subject of intensive research. Finally, discovery and targeting of the causative agents that induce B-cell expansion in the various B-cell malignancies could be a quintessential goal of our efforts in this field, and the discoveries related to H pylori and its role in development of MALT lymphoma, as discussed before, are a paradigm for this concept. Whether or not the concept of CSC applies to B-cell malignancies remains to be explored, but the microenvironment dependency and the concept of niches merge HSC, CSC, and several B-cell malignancies. Likely, a more proper understanding of these aspects will allow movement toward additional targeted treatment options.
Acknowledgments
This work was supported by the Chronic Lymphocytic Leukemia Global Research Foundation (J.A.B., P.G., A.R., and F.C.-C.), a Kimmel Scholar Award by the Sidney Kimmel Foundation for Cancer Research (J.A.B.), an American Society of Clinical Oncology Career Development Award (J.A.B.), and the Associazione Italiana per la Ricerca sul Cancro, Milano (P.G., F.C.-C.).
Authorship
Contribution: J.A.B. designed the figures and wrote the paper; and P.G., A.R., and F.C.-C. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jan A. Burger, Department of Leukemia, Unit 428, University of Texas M. D. Anderson Cancer Center, PO Box 301402, Houston, TX 77230-1402; e-mail: jaburger@mdanderson.org; or Federico Caligaris-Cappio, Universita Vita-Salute San Raffaele, via Olgettina 60, 20132 Milano, Italy; e-mail: caligaris.federico@hsr.it.
References
- 1.Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7(2):139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 2.Burger JA, Kipps TJ. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk Lymphoma. 2002;43(3):461–466. doi: 10.1080/10428190290011921. [DOI] [PubMed] [Google Scholar]
- 3.Caligaris-Cappio F. Role of the microenvironment in chronic lymphocytic leukaemia. Br J Haematol. 2003;123(3):380–388. doi: 10.1046/j.1365-2141.2003.04679.x. [DOI] [PubMed] [Google Scholar]
- 4.Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23(1):43–52. doi: 10.1038/leu.2008.299. [DOI] [PubMed] [Google Scholar]
- 5.Burger JA, Burkle A. The CXCR4 chemokine receptor in acute and chronic leukaemia: a marrow homing receptor and potential therapeutic target. Br J Haematol. 2007;137(4):288–296. doi: 10.1111/j.1365-2141.2007.06590.x. [DOI] [PubMed] [Google Scholar]
- 6.Cyster JG, Ansel KM, Reif K, et al. Follicular stromal cells and lymphocyte homing to follicles. Immunol Rev. 2000;176:181–193. doi: 10.1034/j.1600-065x.2000.00618.x. [DOI] [PubMed] [Google Scholar]
- 7.Harwood NE, Batista FD. New insights into the early molecular events underlying B cell activation. Immunity. 2008;28(5):609–619. doi: 10.1016/j.immuni.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 8.LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570–1580. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 10.Rossi DJ, Jamieson CH, Weissman IL. Stems cells and the pathways to aging and cancer. Cell. 2008;132(4):681–696. doi: 10.1016/j.cell.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 11.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322(5909):1861–1865. doi: 10.1126/science.1164390. [DOI] [PubMed] [Google Scholar]
- 12.Nagasawa T. Microenvironmental niches in the bone marrow required for B-cell development. Nat Rev Immunol. 2006;6(2):107–116. doi: 10.1038/nri1780. [DOI] [PubMed] [Google Scholar]
- 13.Dexter TM, Allen TD, Lajtha LG. Conditions controlling the proliferation of haemopoietic stem cells in vitro. J Cell Physiol. 1977;91(3):335–344. doi: 10.1002/jcp.1040910303. [DOI] [PubMed] [Google Scholar]
- 14.Whitlock CA, Witte ON. Long-term culture of B lymphocytes and their precursors from murine bone marrow. Proc Natl Acad Sci U S A. 1982;79(11):3608–3612. doi: 10.1073/pnas.79.11.3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dave SS, Wright G, Tan B, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351(21):2159–2169. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- 16.Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359(22):2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruan J, Hajjar K, Rafii S, Leonard JP. Angiogenesis and antiangiogenic therapy in non-Hodgkin's lymphoma. Ann Oncol. 2009;20(3):413–424. doi: 10.1093/annonc/mdn666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh AK, Shanafelt TD, Cimmino A, et al. Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in CLL B cells. Blood. 2009;113(22):5568–5574. doi: 10.1182/blood-2008-10-185686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 20.Zucca E, Bertoni F, Roggero E, et al. Molecular analysis of the progression from Helicobacter pylori-associated chronic gastritis to mucosa-associated lymphoid-tissue lymphoma of the stomach. N Engl J Med. 1998;338(12):804–810. doi: 10.1056/NEJM199803193381205. [DOI] [PubMed] [Google Scholar]
- 21.Farinha P, Gascoyne RD. Molecular pathogenesis of mucosa-associated lymphoid tissue lymphoma. J Clin Oncol. 2005;23(26):6370–6378. doi: 10.1200/JCO.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 22.Suarez F, Lortholary O, Hermine O, Lecuit M. Infection-associated lymphomas derived from marginal zone B cells: a model of antigen-driven lymphoproliferation. Blood. 2006;107(8):3034–3044. doi: 10.1182/blood-2005-09-3679. [DOI] [PubMed] [Google Scholar]
- 23.Kutting B, Bonsmann G, Metze D, Luger TA, Cerroni L. Borrelia burgdorferi-associated primary cutaneous B cell lymphoma: complete clearing of skin lesions after antibiotic pulse therapy or intralesional injection of interferon alfa-2a. J Am Acad Dermatol. 1997;36(2):311–314. doi: 10.1016/s0190-9622(97)80405-7. [DOI] [PubMed] [Google Scholar]
- 24.Ferreri AJ, Guidoboni M, Ponzoni M, et al. Evidence for an association between Chlamydia psittaci and ocular adnexal lymphomas. J Natl Cancer Inst. 2004;96(8):586–594. doi: 10.1093/jnci/djh102. [DOI] [PubMed] [Google Scholar]
- 25.Stevenson FK, Caligaris-Cappio F. Chronic lymphocytic leukemia: revelations from the B-cell receptor. Blood. 2004;103(12):4389–4395. doi: 10.1182/blood-2003-12-4312. [DOI] [PubMed] [Google Scholar]
- 26.Chu CC, Catera R, Hatzi K, et al. Chronic lymphocytic leukemia antibodies with a common stereotypic rearrangement recognize nonmuscle myosin heavy chain IIA. Blood. 2008;112(13):5122–5129. doi: 10.1182/blood-2008-06-162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanemo Myhrinder A, Hellqvist E, Sidorova E, et al. A new perspective: molecular motifs on oxidized LDL, apoptotic cells, and bacteria are targets for chronic lymphocytic leukemia antibodies. Blood. 2008;111(7):3838–3848. doi: 10.1182/blood-2007-11-125450. [DOI] [PubMed] [Google Scholar]
- 28.Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, Stevenson FK. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood. 2002;99(7):2562–2568. doi: 10.1182/blood.v99.7.2562. [DOI] [PubMed] [Google Scholar]
- 29.Radcliffe CM, Arnold JN, Suter DM, et al. Human follicular lymphoma cells contain oligomannose glycans in the antigen-binding site of the B-cell receptor. J Biol Chem. 2007;282(10):7405–7415. doi: 10.1074/jbc.M602690200. [DOI] [PubMed] [Google Scholar]
- 30.Chen L, Monti S, Juszczynski P, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood. 2008;111(4):2230–2237. doi: 10.1182/blood-2007-07-100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor BJ, Kriangkum J, Pittman JA, et al. Analysis of clonotypic switch junctions reveals multiple myeloma originates from a single class switch event with ongoing mutation in the isotype-switched progeny. Blood. 2008;112(5):1894–1903. doi: 10.1182/blood-2008-01-129221. [DOI] [PubMed] [Google Scholar]
- 32.Sompuram SR, Bastas G, Vani K, Bogen SA. Accurate identification of paraprotein antigen targets by epitope reconstruction. Blood. 2008;111(1):302–308. doi: 10.1182/blood-2007-05-090654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hargreaves DC, Hyman PL, Lu TT, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. 2001;194(1):45–56. doi: 10.1084/jem.194.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999;94(11):3658–3667. [PubMed] [Google Scholar]
- 35.Sipkins DA, Wei X, Wu JW, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435(7044):969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7(8):585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 37.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93(5):1658–1667. [PMC free article] [PubMed] [Google Scholar]
- 38.Burger M, Hartmann T, Krome M, et al. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood. 2005;106(5):1824–1830. doi: 10.1182/blood-2004-12-4918. [DOI] [PubMed] [Google Scholar]
- 39.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell'Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–2663. [PubMed] [Google Scholar]
- 40.Nishio M, Endo T, Tsukada N, et al. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood. 2005;106(3):1012–1020. doi: 10.1182/blood-2004-03-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robledo MM, Sanz-Rodriguez F, Hidalgo A, Teixido J. Differential use of very late antigen-4 and -5 integrins by hematopoietic precursors and myeloma cells to adhere to transforming growth factor-beta1-treated bone marrow stroma. J Biol Chem. 1998;273(20):12056–12060. doi: 10.1074/jbc.273.20.12056. [DOI] [PubMed] [Google Scholar]
- 42.Sahara N, Takeshita A. Prognostic significance of surface markers expressed in multiple myeloma: CD56 and other antigens. Leuk Lymphoma. 2004;45(1):61–65. doi: 10.1080/1042819031000149377. [DOI] [PubMed] [Google Scholar]
- 43.Durig J, Schmucker U, Duhrsen U. Differential expression of chemokine receptors in B cell malignancies. Leukemia. 2001;15(5):752–756. doi: 10.1038/sj.leu.2402107. [DOI] [PubMed] [Google Scholar]
- 44.Trentin L, Cabrelle A, Facco M, et al. Homeostatic chemokines drive migration of malignant B cells in patients with non-Hodgkin lymphomas. Blood. 2004;104(2):502–508. doi: 10.1182/blood-2003-09-3103. [DOI] [PubMed] [Google Scholar]
- 45.Kurtova AV, Tamayo AT, Ford RJ, Burger JA. Mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d): importance for interactions with the stromal microenvironment and specific targeting. Blood. 2009;113(19):4604–4613. doi: 10.1182/blood-2008-10-185827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burger JA, Zvaifler NJ, Tsukada N, Firestein GS, Kipps TJ. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism. J Clin Invest. 2001;107(3):305–315. doi: 10.1172/JCI11092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mori Y, Shimizu N, Dallas M, et al. Anti-alpha4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood. 2004;104(7):2149–2154. doi: 10.1182/blood-2004-01-0236. [DOI] [PubMed] [Google Scholar]
- 48.Monroe JG, Dorshkind K. Fate decisions regulating bone marrow and peripheral B lymphocyte development. Adv Immunol. 2007;95:1–50. doi: 10.1016/S0065-2776(07)95001-4. [DOI] [PubMed] [Google Scholar]
- 49.Lagneaux L, Delforge A, Bron D, De Bruyn C, Stryckmans P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood. 1998;91(7):2387–2396. [PubMed] [Google Scholar]
- 50.Pedersen IM, Kitada S, Leoni LM, et al. Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood. 2002;100(5):1795–1801. [PubMed] [Google Scholar]
- 51.Tsukada N, Burger JA, Zvaifler NJ, Kipps TJ. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood. 2002;99(3):1030–1037. doi: 10.1182/blood.v99.3.1030. [DOI] [PubMed] [Google Scholar]
- 52.Burkle A, Niedermeier M, Schmitt-Graff A, Wierda WG, Keating MJ, Burger JA. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood. 2007;110(9):3316–3325. doi: 10.1182/blood-2007-05-089409. [DOI] [PubMed] [Google Scholar]
- 53.Deaglio S, Vaisitti T, Bergui L, et al. CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood. 2005;105(8):3042–3050. doi: 10.1182/blood-2004-10-3873. [DOI] [PubMed] [Google Scholar]
- 54.Dierks C, Grbic J, Zirlik K, et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007;13(8):944–951. doi: 10.1038/nm1614. [DOI] [PubMed] [Google Scholar]
- 55.Fukuda T, Chen L, Endo T, et al. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc Natl Acad Sci U S A. 2008;105(8):3047–3052. doi: 10.1073/pnas.0712148105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 57.Azab AK, Runnels JM, Pitsillides C, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–4351. doi: 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Redondo-Munoz J, Ugarte-Berzal E, Garcia-Marco JA, et al. Alpha4beta1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood. 2008;112(1):169–178. doi: 10.1182/blood-2007-08-109249. [DOI] [PubMed] [Google Scholar]
- 59.Gattei V, Bulian P, Del Principe MI, et al. Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood. 2008;111(2):865–873. doi: 10.1182/blood-2007-05-092486. [DOI] [PubMed] [Google Scholar]
- 60.Ghia P, Strola G, Granziero L, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol. 2002;32(5):1403–1413. doi: 10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 61.Burger JA, Quiroga MP, Hartmann E, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood. 2009;113(13):3050–3058. doi: 10.1182/blood-2008-07-170415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Quiroga MP, Balakrishnan K, Kurtova AV, et al. B-cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: specific targeting with a novel Syk inhibitor, R406. Blood. 2009;114:1029–1037. doi: 10.1182/blood-2009-03-212837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beyer M, Kochanek M, Darabi K, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005;106(6):2018–2025. doi: 10.1182/blood-2005-02-0642. [DOI] [PubMed] [Google Scholar]
- 64.Ramsay AG, Johnson AJ, Lee AM, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest. 2008;118(7):2427–2437. doi: 10.1172/JCI35017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gorgun G, Ramsay AG, Holderried TA, et al. E(mu)-TCL1 mice represent a model for immunotherapeutic reversal of chronic lymphocytic leukemia-induced T-cell dysfunction. Proc Natl Acad Sci U S A. 2009;106(15):6250–6255. doi: 10.1073/pnas.0901166106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rezvany MR, Jeddi-Tehrani M, Wigzell H, Osterborg A, Mellstedt H. Leukemia-associated monoclonal and oligoclonal TCR-BV use in patients with B-cell chronic lymphocytic leukemia. Blood. 2003;101(3):1063–1070. doi: 10.1182/blood-2002-03-0746. [DOI] [PubMed] [Google Scholar]
- 67.Kitada S, Zapata JM, Andreeff M, Reed JC. Bryostatin and CD40-ligand enhance apoptosis resistance and induce expression of cell survival genes in B-cell chronic lymphocytic leukaemia. Br J Haematol. 1999;106(4):995–1004. doi: 10.1046/j.1365-2141.1999.01642.x. [DOI] [PubMed] [Google Scholar]
- 68.Ranheim EA, Kipps TJ. Activated T cells induce expression of B7/BB1 on normal or leukemic B cells through a CD40-dependent signal. J Exp Med. 1993;177(4):925–935. doi: 10.1084/jem.177.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wierda WG, Cantwell MJ, Woods SJ, Rassenti LZ, Prussak CE, Kipps TJ. CD40-ligand (CD154) gene therapy for chronic lymphocytic leukemia. Blood. 2000;96(9):2917–2924. [PubMed] [Google Scholar]
- 70.Ghia P, Boussiotis VA, Schultze JL, et al. Unbalanced expression of bcl-2 family proteins in follicular lymphoma: contribution of CD40 signaling in promoting survival. Blood. 1998;91(1):244–251. [PubMed] [Google Scholar]
- 71.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 72.Glas AM, Knoops L, Delahaye L, et al. Gene-expression and immunohistochemical study of specific T-cell subsets and accessory cell types in the transformation and prognosis of follicular lymphoma. J Clin Oncol. 2007;25(4):390–398. doi: 10.1200/JCO.2006.06.1648. [DOI] [PubMed] [Google Scholar]
- 73.Farinha P, Masoudi H, Skinnider BF, et al. Analysis of multiple biomarkers shows that lymphoma-associated macrophage (LAM) content is an independent predictor of survival in follicular lymphoma (FL). Blood. 2005;106(6):2169–2174. doi: 10.1182/blood-2005-04-1565. [DOI] [PubMed] [Google Scholar]
- 74.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 75.Junt T, Moseman EA, Iannacone M, et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature. 2007;450(7166):110–114. doi: 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 76.Carrasco YR, Batista FD. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity. 2007;27(1):160–171. doi: 10.1016/j.immuni.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 77.Ghia P, Caligaris-Cappio F. The indispensable role of microenvironment in the natural history of low-grade B-cell neoplasms. Adv Cancer Res. 2000;79:157–173. doi: 10.1016/s0065-230x(00)79005-1. [DOI] [PubMed] [Google Scholar]
- 78.Carreras J, Lopez-Guillermo A, Fox BC, et al. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood. 2006;108(9):2957–2964. doi: 10.1182/blood-2006-04-018218. [DOI] [PubMed] [Google Scholar]
- 79.Hilchey SP, De A, Rimsza LM, Bankert RB, Bernstein SH. Follicular lymphoma intratumoral CD4+CD25+GITR+ regulatory T cells potently suppress CD3/CD28-costimulated autologous and allogeneic CD8+CD25- and CD4+CD25- T cells. J Immunol. 2007;178(7):4051–4061. doi: 10.4049/jimmunol.178.7.4051. [DOI] [PubMed] [Google Scholar]
- 80.Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. CD70+ non-Hodgkin lymphoma B cells induce Foxp3 expression and regulatory function in intratumoral CD4+CD25 T cells. Blood. 2007;110(7):2537–2544. doi: 10.1182/blood-2007-03-082578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mittal S, Marshall NA, Duncan L, Culligan DJ, Barker RN, Vickers MA. Local and systemic induction of CD4+CD25+ regulatory T-cell population by non-Hodgkin lymphoma. Blood. 2008;111(11):5359–5370. doi: 10.1182/blood-2007-08-105395. [DOI] [PubMed] [Google Scholar]
- 82.Lee AM, Clear AJ, Calaminici M, et al. Number of CD4+ cells and location of forkhead box protein P3-positive cells in diagnostic follicular lymphoma tissue microarrays correlates with outcome. J Clin Oncol. 2006;24(31):5052–5059. doi: 10.1200/JCO.2006.06.4642. [DOI] [PubMed] [Google Scholar]
- 83.Alvaro T, Lejeune M, Salvado MT, et al. Immunohistochemical patterns of reactive microenvironment are associated with clinicobiologic behavior in follicular lymphoma patients. J Clin Oncol. 2006;24(34):5350–5357. doi: 10.1200/JCO.2006.06.4766. [DOI] [PubMed] [Google Scholar]
- 84.Klapper W, Hoster E, Rolver L, et al. Tumor sclerosis but not cell proliferation or malignancy grade is a prognostic marker in advanced-stage follicular lymphoma: the German Low Grade Lymphoma Study Group. J Clin Oncol. 2007;25(22):3330–3336. doi: 10.1200/JCO.2006.10.5833. [DOI] [PubMed] [Google Scholar]
- 85.de Jong D, Koster A, Hagenbeek A, et al. Impact of the tumor microenvironment on prognosis in follicular lymphoma is dependent on specific treatment protocols. Haematologica. 2009;94(1):70–77. doi: 10.3324/haematol.13574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taskinen M, Karjalainen-Lindsberg ML, Nyman H, Eerola LM, Leppa S. A high tumor-associated macrophage content predicts favorable outcome in follicular lymphoma patients treated with rituximab and cyclophosphamide-doxorubicin-vincristine-prednisone. Clin Cancer Res. 2007;13(19):5784–5789. doi: 10.1158/1078-0432.CCR-07-0778. [DOI] [PubMed] [Google Scholar]
- 87.Ralph QM, Brisco MJ, Joshua DE, Brown R, Gibson J, Morley AA. Advancement of multiple myeloma from diagnosis through plateau phase to progression does not involve a new B-cell clone: evidence from the Ig heavy chain gene. Blood. 1993;82(1):202–206. [PubMed] [Google Scholar]
- 88.Gunn WG, Conley A, Deininger L, Olson SD, Prockop DJ, Gregory CA. A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: a potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells. 2006;24(4):986–991. doi: 10.1634/stemcells.2005-0220. [DOI] [PubMed] [Google Scholar]
- 89.Vacca A, Ribatti D. Bone marrow angiogenesis in multiple myeloma. Leukemia. 2006;20(2):193–199. doi: 10.1038/sj.leu.2404067. [DOI] [PubMed] [Google Scholar]
- 90.Hose D, Moreaux J, Meissner T, et al. Induction of angiogenesis by normal and malignant plasma cells. Blood. 2009;114:128–143. doi: 10.1182/blood-2008-10-184226. [DOI] [PubMed] [Google Scholar]
- 91.Kumar S, Witzig TE, Timm M, et al. Expression of VEGF and its receptors by myeloma cells. Leukemia. 2003;17(10):2025–2031. doi: 10.1038/sj.leu.2403084. [DOI] [PubMed] [Google Scholar]
- 92.Kay NE. Angiogenesis revisited in CLL. Leuk Res. 2007;31(11):1459–1460. doi: 10.1016/j.leukres.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 93.Mesa RA, Hanson CA, Rajkumar SV, Schroeder G, Tefferi A. Evaluation and clinical correlations of bone marrow angiogenesis in myelofibrosis with myeloid metaplasia. Blood. 2000;96(10):3374–3380. [PubMed] [Google Scholar]
- 94.Coluccia AM, Cirulli T, Neri P, et al. Validation of PDGFRbeta and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood. 2008;112(4):1346–1356. doi: 10.1182/blood-2007-10-116590. [DOI] [PubMed] [Google Scholar]
- 95.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132(4):598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Laird DJ, von Andrian UH, Wagers AJ. Stem cell trafficking in tissue development, growth, and disease. Cell. 2008;132(4):612–630. doi: 10.1016/j.cell.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 97.Uy GL, Rettig MP, McFarland KM, et al. Mobilization and chemosensitization of AML with the CXCR4 antagonist Plerixafor (AMD3100): a phase I/II study of AMD3100+MEC in patients with relapsed or refractory disease. Blood. 2008;112(11):678a. [Google Scholar]
- 98.Gobessi S, Laurenti L, Longo PG, et al. Inhibition of constitutive and BCR-induced Syk activation downregulates Mcl-1 and induces apoptosis in chronic lymphocytic leukemia B cells. Leukemia. 2009;23(4):686–697. doi: 10.1038/leu.2008.346. [DOI] [PubMed] [Google Scholar]
- 99.Friedberg J, Sharman J, Schaefer-Cutillo J, et al. Fostamatinib disodium (FosD), an oral inhibitor of Syk, is well-tolerated and has significant clinical activity in diffuse large B cell lymphoma (DLBCL) and chronic lymphocytic leukemia (SLL/CLL). Blood. 2008;112(11):3a. [Google Scholar]
- 100.Niedermeier M, Hennessy BT, Knight ZA, et al. Isoform-selective phosphoinositide 3′-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: a novel therapeutic approach. Blood. 2009;113(22):5549–5557. doi: 10.1182/blood-2008-06-165068. [DOI] [PMC free article] [PubMed] [Google Scholar]