

Abstract
The asymmetric alkylation of acyclic ketones is a longstanding challenge in organic synthesis. We report diastereoselective and enantioselective allylic substitutions with acyclic α-alkoxy ketones catalyzed by a metallacyclic iridium complex to form products with contiguous stereogenic centers derived from the nucleophile and electrophile. These reactions occur between allyl methyl carbonates and unstabilized copper(I) enolates generated in situ from acyclic α-alkoxy ketones. The resulting products can be readily converted to enantioenriched tertiary alcohols and tetrahydrofuran derivatives without erosion of enantiomeric purity.
Keywords: alkylation, asymmetric catalysis, ketones, iridium, diastereoselectivity
Graphical Abstract

The asymmetric alkylation of acyclic ketones is a longstanding synthetic challenge. We report diastereoselective and enantioselective allylic substitutions with acyclic α-alkoxy ketones catalyzed by a metallacyclic iridium complex to form products containing two contiguous stereogenic centers, one derived from the nucleophile and one from the electrophile. These reactions occur between allyl methyl carbonates and unstabilized copper(I) enolates generated in situ from acyclic α-alkoxy ketones. The resulting products can be readily converted to enantioenriched tertiary alcohols and tetrahydrofuran derivatives without erosion of ee.
Transition metal-catalyzed asymmetric allylation of enolates serves as an efficient and reliable method to construct carbon-carbon bonds with high levels of asymmetric induction.[1] The majority of these reactions form products containing a single stereocenter from a prochiral enolate as the nucleophile or a prochiral allylic compound as the electrophile. If both nucleophile and electrophile are prochiral, synthetically valuable dyads containing contiguous stereocenters could be assembled in a catalytic and stereoselective fashion.[2] However, this transformation is challenging because a new bond needs to be formed between two sterically hindered prochiral carbons with control of both absolute and relative configurations.
Metallacyclic iridium complexes catalyze allylic substitutions with a variety of carbon and heteroatom nucleophiles regio- and enantioselectively.[1b, 3] Although reactions have been reported between prochiral enolates and prochiral electrophiles to afford products containing vicinal tetra-substituted and tertiary stereocenters with excellent diastereo- and enantioselectivity, reactions with unstabilized, acyclic, prochiral ketones have not been reported.[4] The main challenge facing this transformation results from the lack of control of the geometry of the unstabilized enolate of an α-branched acyclic ketone. In contrast to cyclic enolates, the backbone of the nucleophile does not dictate the geometry. Also, because α-branched, acyclic ketones do not readily form enamines, the use of amine auxiliaries has not been effective to control the geometry.[5]
In the presence of suitable metal cations, acyclic carbonyl compounds bearing α-heteroatoms form enolates with a defined geometry created by chelation. This structure has been exploited for the allylation of glycine derivatives.[6] However, these reactions occurred with low diastereoselectivity when forming products containing adjacent tetra-substituted and tertiary stereocenters.[6a] With the same strategy, Evans and co-workers achieved diastereoselective allylations of α-hydroxy, as well as α-alkoxy or α-siloxy acetophenone catalyzed by an achiral rhodium catalyst.[7] However, only products containing adjacent tertiary and tri-substituted stereocenters bearing oxygen were formed, and no enantioselective transformation was reported. We envisioned that this strategy could be followed to achieve the enantioselective allylation of unstabilized ketones with cyclometallic iridium catalysts we developed.[8]
Herein, we report diastereo- and enantioselective allylic alkylations with unstabilized enolates of acyclic α-alkoxy ketones catalyzed by iridium complex 3 (Scheme 1). The geometry of the enolates is controlled through chelation in the presence of a copper(I) cation. These reactions form, with high diastereo- and enantioselectivity, products containing vicinal oxygen-bearing tetra-substituted and tertiary stereocenters. Products containing an O-MOM (methoxymethyl) group on the tertiary alcohol were formed in good yield with high dr and ee, and these products can be readily converted to tertiary alcohols and tetrahydrofuran (THF) derivatives without erosion of enantiomeric purity.
Scheme 1.

Iridium-catalyzed diastereo- and enantioselective allylations with unstabilized copper(I) enolates of acyclic α-alkoxy ketones.
To assess the potential of developing an iridium-catalyzed allylation of an acyclic ketone enolate, we conducted the reactions between O-methyl benzoin (1a) and methyl cinnamyl carbonate (2a) (Table 1). Treatment of 1a and 2a with Ir complex 3 in the presence of LHMDS at 5 °C for 12 h furnished the branched product 4aa in 95% yield (combined yield of two diastereoisomers), but with a low dr of 2.0:1 (entry 1). The reaction conducted after addition of LiCl[4o] to the lithium enolate gave the product with a lower dr of 1:1.1, slightly favoring the formation of the other diastereoisomer (entry 2). The reaction with added ZnCl2[6] afforded 4aa with excellent diastereoselectivity (16:1 dr, entry 3), albeit in a lower yield of 72%. In contrast, the reaction conducted with added CuI occurred with a higher diastereomeric ratio to 5.7:1 while maintaining excellent conversion to yield 4aa in 97% yield (entry 4). Similarly, Evans and coworkers observed higher diastereoselectivity with the copper(I) enolate of α-hydroxy acetophenone derivatives than with the corresponding lithium enolate.[7]
Table 1.
Evaluation of reaction conditions for the Ir-catalyzed allylation.[a]

| ||||
|---|---|---|---|---|
| Entry | Additive | Yield [%][b] | dr[c] | ee [%][d] |
| 1 | - | 95 | 2.0:1 | n.d. |
| 2 | LiCl | >99 | 1:1.1 | n.d. |
| 3 | ZnCl2 | 72 | 16:1 | n.d. |
| 4 | CuI | 97 | 5.7:1 | n.d. |
| 5 | CuCl | 58 | 10:1 | n.d. |
| 6 | CuBr | >99 (>99) | 14:1 | 92 |
| 7 | CuCN | 34 | 2.5:1 | n.d. |
| 8 | CuOAc | 52 | 1.5:1 | n.d. |
| 9 | CuSCN | 94 | 2.3:1 | n.d. |
| 10[e] | CuBr | 42 | 2.7:1 | n.d. |
| 11[f] | CuBr | >99 | 14:1 | 93 |
| 12[g] | CuBr | >99 | 12:1 | 95 |
| 13 | CuBr2 | 0 | - | n.d. |
The molar ratio of 1a/2a/3/LHMDS/additive = 2/1/0.02/2/2. The absolute configuration of 4aa was assigned by analogy.
Combined yield of two diastereoisomers. Determined by 1H NMR analysis with mesitylene as internal standard. The yield in parentheses is an isolated yield of two diastereoisomers.
Determined by 1H NMR analysis of crude reaction mixtures.
Determined by chiral SFC analysis of the major isomer.
KHMDS was used as the base instead of LHMDS.
1 equiv of CuBr was used.
0.5 equiv of CuBr was used.
n.d. = not determined.
Because the anion of the copper(I) salt could influence the transmetalation, we further evaluated a series of copper(I) salts. Reactions conducted with added CuBr occurred with a higher diastereomeric ratio of 14:1 with an excellent ee of 92% (entry 6). The major diastereoisomer was isolated in 93% yield (see SI for details). Reactions with other copper(I) additives, such as CuCl, CuCN, CuOAc or CuSCN, occurred in significantly lower yield (58%, entry 5) or with lower dr (1.5–2.5:1, entries 7–9).[9]
The identity of the cation of the anionic base was crucial to obtaining high yields and diastereoselectivities. Reactions conducted with KHMDS instead of LHMDS afforded only 42% yield of 4aa with a significantly lower dr of 2.7:1 (entry 10). Reactions conducted with one equivalent of copper in place of two equivalents led to excellent reactivity and afforded the product in identical dr and ee (entry 11), but reactions with 0.5 equiv occurred with lower dr (although slightly higher ee) as shown in entry 12.[10] In all cases, the branched product was obtained exclusively. Reactions run with CuBr2 as additive gave no product, indicating the critical role of the copper(I) cation in this reaction, rather than a copper(II) cation that might be formed by disproportionation or oxidation of copper(I) salt (entry 13).
The scope of the allylic electrophiles that underwent the Ir-catalyzed allylic substitution with acyclic α-alkoxy ketones is summarized in Table 2. Various para-substituted cinnamyl carbonates are suitable electrophiles. Electron-neutral (4ab), electron-donating (4ac) and electron-withdrawing (4ah, 4ai) functional groups on the cinnamyl aryl ring were all tolerated in this reaction, and the corresponding products were formed in excellent yield (≥94%) with high dr (≥10:1) and ee (≥90%). Cinnamyl carbonates bearing halogens at the para- or meta-position reacted cleanly, furnishing 4ad–ag in ≥92% yield, ≥12:1 dr and ≥91% ee.[11] The absolute stereochemistry of 4ag was established by single crystal X-ray diffraction.
Table 2.
Ir-catalyzed allylations of acyclic α-alkoxy ketone enolates: scope of allylic carbonates.[a]

|
The molar ratio of 1a/2/3/LHMDS/CuBr = 2/1/0.02/2/2. The absolute configurations were assigned by analogy. The structure of 4ag was determined by X-ray analysis. The yields were reported as the combined yields of two diastereoisomers. The diastereomeric ratios were determined by 1H NMR analysis of crude reaction mixtures. The enantiomeric excesses were determined by chiral SFC analysis of major isomers.
0.5 equiv. of CuBr was used.
The reaction also occurred with allylic carbonates containing heteroaryl, alkenyl and alkyl substituents. The reaction of thienyl carbonate 2k afforded 4ak in high yield with excellent diastereo- and enantioselectivity (>99%, >20:1 dr, 92% ee). Methyl sorbyl carbonate (2l) reacted to form product 4al in 75% yield with >20:1 dr and 94% ee. Even the simple crotyl carbonate (2m) reacted to form product 4am in good yield, although the dr and ee (71%, 8:1 dr and 88% ee) were slightly lower than those with aryl-substituted allylic carbonates.
The scope of the acyclic α-alkoxy ketones that underwent the Ir-catalyzed allylation is summarized in Table 3.[12–13] MOM (1b), MEM (methoxyethoxymethyl, 1c), and PMB (para-methoxybenzyl, 1d) protected benzoins underwent allylation in high yield with excellent diastereo- and enantioselectivity. The reaction between O-MOM benzoin 1b and cinnamyl carbonate 2a required a higher catalyst loading of 4 mol% to reach full conversion within 12 hours. Several acyclic O-Me benzoin derivatives bearing identical substituents at both aryl rings, such as 1e and 1f, as well as their O-MOM analogues 1i and 1j were suitable for this transformation (4ea, 4fa, 4ic and 4jk, ≥84%, ≥7:1 dr, ≥94% ee).
Table 3.
Ir-catalyzed allylation of acyclic α-alkoxy ketone enolates: scope of ketones.[a]

|
The molar ratio of 1/2/3/LHMDS/CuBr = 2/1/0.02/2/2. The absolute configurations were assigned by analogy. The yields were reported as the combined yields of two diastereoisomers. The diastereomeric ratios were determined by 1H NMR analysis of crude reaction mixtures. The enantiomeric excesses were determined by chiral SFC analysis of major isomers.
4 mol% of 3 was used.
1 equiv of CuBr was used.
0.5 equiv of CuBr was used.
cinnamyl acetate was used instead of cinnamyl carbonate, and the reaction time was elongated to 36 h. The yield was reported as the isolated yield of the major diastereomer.
The reactions with nucleophiles derived from non-symmetrical benzoins were also examined. Benzoin 1g, bearing a thienyl group, underwent allylation in quantitative yield with excellent dr of 15:1 and 96% ee (4ga). Benzoin analogue 1h, containing an i-butyl group, reacted with methyl cinnamyl carbonate 2a in low yield of 38% and low branched/linear selectivity of 4:1 (5:1 dr for the branched product).[14] However, the identical reaction with the less reactive cinnamyl acetate as the electrophile afforded branched product 4ha exclusively in high yield (74%, isolated yield of the major diastereomer) with acceptable diastereoselectivity and excellent enantioselectivity (6:1 dr, >99% ee).
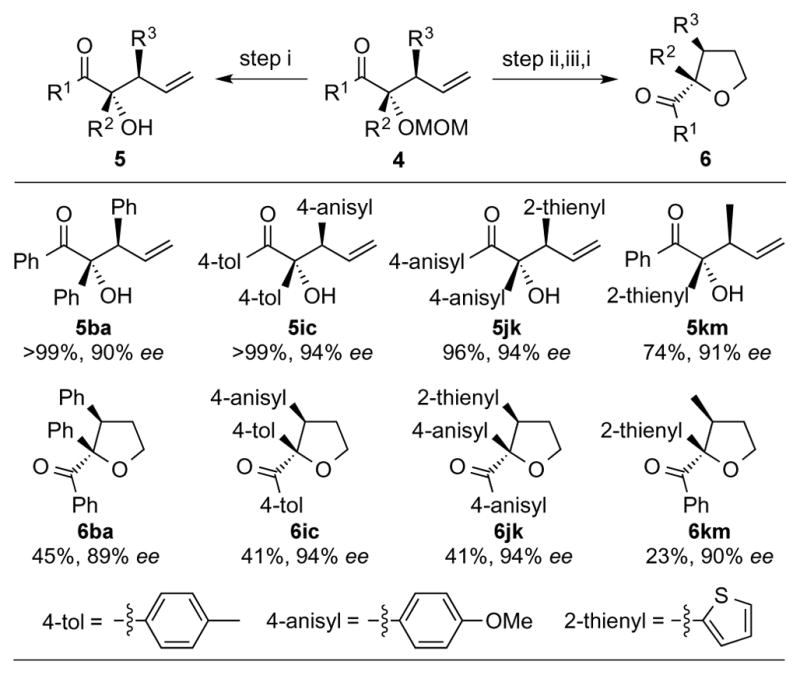
Allylation products containing an O-MOM group on the tertiary alcohol were readily transformed to enantioenriched tertiary alcohols 5 containing adjacent tertiary stereogenic centers (Table 2). Deprotection of 4ba by reaction with acidic Dowex-50W-X8 resin[15] (step i) afforded the corresponding alcohols 5ba in quantitative yield without any erosion of enantiomeric purity. The synthetic value of these allylated benzoin derivatives was further demonstrated by their transformation into highly substituted THF derivatives. Hydroboration of 4ba with 9-BBN, followed by oxidation (step ii), yielded the terminal alcohol, which was subsequently converted to the corresponding tosylate (step iii). Removal of the MOM protecting group (step i) afforded the free tertiary alcohol, which underwent a 5-exo-tet cyclization in situ to furnish THF derivative 6ba in 45% yield over 3 steps.
These synthetic sequences were also applied to allylation products bearing different substituents R1, R2 and R3. In all cases, substrates 4ic (R1 = R2 = 4-tol, R3 = 4-anisyl), 4jk (R1 = R2 = 4-anisy, R3 = 2-thienyl) and 4km (R1 = Ph, R2 = 2-thienyl and R3 = Me) afforded the corresponding tertiary alcohols 5ic, 5jk and 5km in high yield without erosion of enantiomeric purity. Similarly, the corresponding THF derivatives 6ic, 6jk and 6km were obtained in high enantiomeric purity.
The olefin moiety is a useful precursor to many functional groups. For example, ozonolysis, hydrogenation and the combination of hydroboration and oxidation of the products of allylation 4aa afforded aldehyde 7aa, ketone 8aa, and primary alcohol 9aa, respectively, in high yields without erosion of enantiomeric purity (Scheme 2).
Scheme 2.

Derivatizations of 4aa.
In summary, we have developed Ir-catalyzed diastereo- and enantioselective allylic substitutions with unstabilized copper(I) enolates of acyclic α-alkoxy ketones. Employing metallacyclic complex 3 as the catalyst, LHMDS as the base and CuBr as the additive, allylation reactions gave the products containing vicinal tetra-substituted and tertiary stereocenters in high yield with excellent dr and ee. The geometry of the enolates is controlled by chelation in the presence of a copper(I) cation. The synthetic utility of this method was demonstrated by the synthesis of enantioenriched THF derivatives and tertiary alcohols containing adjacent tertiary stereogenic centers. Studies to gain insight into the origin of diastereoselectivity in this reaction are ongoing in our laboratories.
Supplementary Material
Table 4.
Synthesis of enantioenriched tertiary alcohols 5 and tetrahydrofuran derivatives 6.[a]
|
The yield for 6 is reported as the overall yield of 3 steps. The absolute structures were assigned by analogy. Steps: i) Dowex-50W-X8 (H+ form), MeOH/H2O, 65 °C. ii) 9-BBN then NaBO3•4H2O. iii) TsCl, TEA.
Footnotes
Financial support provided by the Director, Office of Science, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231 and the NIH-NIGMS (GM-55382).
References
- 1.a) Ding CH, Hou XL. In: Comprehensive Organic Synthesis II. 2. Knochel P, editor. Elsevier; Amsterdam: 2014. p. 648. [Google Scholar]; b) Hartwig JF, Stanley LM. Acc Chem Res. 2010;43:1461. doi: 10.1021/ar100047x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu Y, Han SJ, Liu WB, Stoltz BM. Acc Chem Res. 2015;48:740. doi: 10.1021/ar5004658. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lu Z, Ma S. Angew Chem Int Ed. 2008;47:258. doi: 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]; e) Oliver S, Evans PA. Synthesis. 2013;45:3179. [Google Scholar]; f) Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- 2.a) Christoffers J, Baro A. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Wiley-VCH; Weinheim: 2005. [Google Scholar]; b) Douglas CJ, Overman LE. Proc Natl Acad Sci USA. 2004;101:5363. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Quasdorf KW, Overman LE. Nature. 2014;516:181. doi: 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; e) Trost BM, Jiang C. Synthesis. 2006:369. [Google Scholar]; f) Yus M, González-Gómez JC, Foubelo F. Chem Rev. 2013;113:5595. doi: 10.1021/cr400008h. [DOI] [PubMed] [Google Scholar]
- 3.a) Hartwig JF, Pouy MJ. Top Organomet Chem. 2011;34:169. [Google Scholar]; b) Helmchen G. Iridium Complexes in Organic Synthesis. Wiley-VCH; Weinheim: 2009. p. 211. [Google Scholar]; c) Helmchen G, Dahnz A, Dubon P, Schelwies M, Weihofen R. Chem Commun. 2007:675. doi: 10.1039/b614169b. [DOI] [PubMed] [Google Scholar]; d) Liu W-B, Xia J-B, You S-L. Top Organomet Chem. 2012;38:155. [Google Scholar]; e) Teichert JF, Feringa BL. Angew Chem Int Ed. 2010;49:2486. doi: 10.1002/anie.200904948. [DOI] [PubMed] [Google Scholar]; f) Tosatti P, Nelson A, Marsden SP. Org Biomol Chem. 2012;10:3147. doi: 10.1039/c2ob07086c. [DOI] [PubMed] [Google Scholar]
- 4.Chen W, Chen M, Hartwig JF. J Am Chem Soc. 2014;136:15825. doi: 10.1021/ja506500u.Chen W, Hartwig JF. J Am Chem Soc. 2013;136:377. doi: 10.1021/ja410650e.Chen W, Hartwig JF. J Am Chem Soc. 2013;135:2068. doi: 10.1021/ja311363a.Liu WB, Reeves CM, Virgil SC, Stoltz BM. J Am Chem Soc. 2013;135:10626. doi: 10.1021/ja4052075.. For reactions of α-branched aldehydes, see: Krautwald S, Sarlah D, Schafroth MA, Carreira EM. Science. 2013;340:1065. doi: 10.1126/science.1237068.Krautwald S, Schafroth MA, Sarlah D, Carreira EM. J Am Chem Soc. 2014;136:3020. doi: 10.1021/ja5003247.Sandmeier T, Krautwald S, Zipfel HF, Carreira EM. Angew Chem Int Ed. 2015;54:14363. doi: 10.1002/anie.201506933.. For reactions of stabilized enolates of β-ketoesters, see: Liu WB, Reeves CM, Stoltz BM. J Am Chem Soc. 2013;135:17298. doi: 10.1021/ja4097829.. For related work with Mo and Pd, see: Braun M, Laicher F, Meier T. Angew Chem Int Ed. 2000;39:3494. doi: 10.1002/1521-3773(20001002)39:19<3494::aid-anie3494>3.0.co;2-4.Chen JP, Ding CH, Liu W, Hou XL, Dai LX. J Am Chem Soc. 2010;132:15493. doi: 10.1021/ja106703y.Trost BM, Dogra K, Franzini M. J Am Chem Soc. 2004;126:1944. doi: 10.1021/ja031539a.Trost BM, Miller JR, Hoffman CM. J Am Chem Soc. 2011;133:8165. doi: 10.1021/ja2029602.Trost BM, Zhang Y. J Am Chem Soc. 2006;128:4590. doi: 10.1021/ja060560j.Trost BM, Zhang Y. J Am Chem Soc. 2007;129:14548. doi: 10.1021/ja0755717.Zheng WH, Zheng BH, Zhang Y, Hou XL. J Am Chem Soc. 2007;129:7718. doi: 10.1021/ja071098l.
- 5.Mahrwald R. Modern Aldol Reactions. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 6.a) Kanayama T, Yoshida K, Miyabe H, Kimachi T, Takemoto Y. J Org Chem. 2003;68:6197. doi: 10.1021/jo034638f. [DOI] [PubMed] [Google Scholar]; b) Kazmaier U, Zumpe FL. Angew Chem Int Ed. 1999;38:1468. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1468::AID-ANIE1468>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]; c) Huwig K, Schultz K, Kazmaier U. Angew Chem Int Ed. 2015;54:9120. doi: 10.1002/anie.201502975. [DOI] [PubMed] [Google Scholar]
- 7.Evans PA, Lawler MJ. J Am Chem Soc. 2004;126:8642. doi: 10.1021/ja049080n.. For related work with α-hydroxy esters, see: Kiefer A, Gawas G, Kazmaier U. Eur J Org Chem. 2015:5810.
- 8.a) Ohmura T, Hartwig JF. J Am Chem Soc. 2002;124:15164. doi: 10.1021/ja028614m. [DOI] [PubMed] [Google Scholar]; b) Kiener CA, Shu C, Incarvito C, Hartwig JF. J Am Chem Soc. 2003;125:14272. doi: 10.1021/ja038319h. [DOI] [PubMed] [Google Scholar]
- 9.We also investigated the reactions with metallacyclic catalyst generated in situ, with the pre-formed catalysts bearing different ligands, and at different temperatures. The reaction catalyzed by the pre-formed complex 3 at 5 °C or 0 °C afforded the product in the highest yield and dr. See Table S1 in SI for details.
- 10.The solution of copper enolate was heterogeneous and was transferred into the vial containing allyl carbonate 2 and catalyst 3. The loss of CuBr was inevitable and unable to be measured. Considering the low price of CuBr and that inadequate amount of CuBr would result in lower diastereoselectivity in this reaction (Table 1, entry 11 versus entry 12), we decided to use excessive CuBr (2 equiv) as the additive for further study.
- 11.As observed for the test substrate 2a (see Table 1, entry 6 vs. entry 12), the reaction of 3,4-dichlorocinnamyl carbonate (2g) in the presence of 2 equiv of CuBr gave the product 4ag in a similar 91% yield, higher 15:1 dr, but slightly lower 89% ee than the reaction with 1 equiv of CuBr (see SI for details), whereas the same reaction with 0.5 equiv of CuBr resulted in similar yield (92%), slightly lower dr (12:1), but excellent ee (96%).
- 12.The reaction of 2-methoxyacetophenone with cinnamyl methyl carbonate afforded the product quantitatively with dr of 1.9:1. It is possible that the initially formed product underwent epimerization under the reaction conditions to cause the dr of the final product to be low.
- 13.In several cases (4ca, 4ea–4ga, 4ic, 4km), viscous solutions of copper enolates were observed if 2 equiv of CuBr were added, and the consumption of limiting allylic carbonates was not completed within 12 hours. However, if less CuBr (1 or 0.5 equiv, see SI for details) was added, full conversion of the allylic carbonates was obtained within 12 hours.
- 14.Approximately 30% of allylic carbonate remained, and about 30% of the material was a mixture of uncharacterized products.
- 15.Seto H, Mander LN. Synth Commun. 1992;22:2823. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.