

Abstract
The process is commonly known as epileptogenesis refers to the cascade of molecular and cellular changes that transform the brain to make it hyperexcitable and capable of generate recurrent spontaneous seizures. Unfortunately, our understanding of the molecular changes that affect the brain during epileptogenesis remains incomplete. Recent evidence suggests that dysfunction of cation-chloride transporters (CCCs) might be one of the factors that contribute to the deficits in inhibitory neurotransmission observed during epileptogenesis. This study analyzed the cell surface expression of CCCs during epileptogenesis and during chronic epilepsy to evaluate if a loss of CCCs from the plasma membrane might contribute to hyperexcitability. Alterations in the plasma membrane expression of CCCs were mostly detected during the early phase of the epileptogenic period, suggesting that dysfunction of CCCs might contribute to the alterations in the chloride gradient previously detected. Together, the findings presented here suggest that aberrant regulation of the plasma membrane levels of CCCs might contribute to the impartment of GABAergic neurotransmission and that CCCs dysfunction might be relevant for the initial appearance of spontaneous seizures.
Keywords: KCC2, NKCC1, epileptogenesis, transporter trafficking
INTRODUCTION
In humans, occurrence of a severe head injury, prolonged febrile seizures, stroke or status epilepticus (SE) may precede the appearance of overt spontaneous seizures by months or years [19]. In animal models, an experimental brain injury is followed by a latent period with no overt seizure activity yet a number of pathophysiological and structural alterations have been described [8, 25, 26]. The myriad of cellular and molecular events leading to formation of hyperexcitable networks and the manifestation of overt spontaneous seizures is, as a whole, known as epileptogenesis [8, 11, 25, 26]. In rodents, experimental SE promotes a transient decrease in GABAergic drive that coincides with the appearance of spontaneous seizures [10]. The deficit in GABAergic neurotransmission and the increase in neuronal excitability detected during the early phase of epileptogenesis appear to result, at least in part, from a lack of functional GABAA receptors (GABAAR) [9, 11, 18].
The generation of “classical” postsynaptic inhibition through GABAAR activation requires the maintenance of low intracellular chloride concentrations that is mostly set by cation-chloride cotransporters (CCCs) [2, 17]. In mature neurons, the potassium-chloride cotransporter 2 (KCC2) is the main chloride extruder maintaining intracellular chloride concentrations below electrochemical equilibrium while the opposite effect, intracellular chloride accumulation, is facilitated by the sodium-potassium-chloride cotransporter 1 (NKCC1) [3, 17]. In central neurons from adult hippocampus and cortex KCC2 activity governs the chloride gradient and loss of KCC2 from the plasma membrane promotes a reversal in the chloride gradient and reversal of GABAAR potential (EGABA) [17]. However, it is also possible that subtle changes in NKCC1 plasma membrane expression might exert a dominant effect on the chloride gradient and change EGABA [20]. Some evidence that alterations in CCCs expression might heavily contribute to abnormal synchronization and hyperexcitability during epileptogenesis has been observed in adult rats following pilocarpine-induced SE [1, 23]. Unfortunately, the full extent of the molecular remodeling affecting GABAergic neurotransmission and its contribution to epileptogenesis is not fully understood.
In previous independent studies, we analyzed the cell surface expression of GABAAR during the “latent” and “chronic” period of epilepsy and discovered deficits on the cell surface expression of GABAAR subunits [12, 13]. Here, the cell surface expression of KCC2 and NKCC1 was analyzed in tissue obtained during the epileptogenic period and in tissue obtained from chronically epileptic animals enduring both frequent and infrequent seizures. The results obtained show a prominent shift in the cell surface expression of KCC2 and NKCC1 during the early phase of epileptogenesis, with little to no change observed during the chronic stage. The results presented here suggest that hyperexcitability during epileptogenesis may be a cumulative effect of multiple deficits impacting GABAergic neurotransmission.
MATERIALS AND METHODS
The methods described here are similar to those described elsewhere, for further details please refer to our previous reports [12, 13]. Samples were isolated from animals described in the above-mentioned studies and repurposed for the analysis of KCC2 and NKCC1 immunoreactivity.
Induction of Status Epilepticus
Adult male Sprague Dawley rats (Charles River, Wilmington, MA) were housed in a temperature-controlled vivarium with food and water ad libitum. Rats were injected intraperitoneally with scopolamine methyl nitrate (1 mg/kg) 30 minutes before administration of pilocarpine hydrochloride (385 mg/kg), as previously described [4, 12, 13, 27]. Diazepam (6 mg/kg; Hospira, Lake Forest, IL) was administered to stop seizure progression. Control rats were handled similarly but treated with a subconvulsive dose of pilocarpine (1/10 of the full dose, 38.5 mg/kg) and a reduced dose of diazepam (1/10 of the full dose, 0.6 mg/kg). Animal procedures were performed in accordance with Institutional Animal Care and Use Committee regulations and approved protocols by the University of Colorado Anschutz Medical Campus.
EEG acquisition and analysis
Rats were implanted with EEG electrodes as previously described [13]. Following recovery, rats were placed in a recording chamber equipped with flexible cables attached to a commutator (i.e., electric swivel) and recorded 24 hours/day using an automatic Pinnacle digital video-EEG system. Tissue samples were collected either ≤3 hours from the last seizure or ≥24 hours after the last seizure. Thus, tissue was collected only if seizures were observed during the previous 3 hours (frequent/recent seizures group), or if no seizures were detected in the last 24 hours (infrequent seizures group) [13]
Biotinylation Procedure
The protocol used was a modification of methods previously reported [14, 15]. Briefly, to label plasma membrane proteins, hippocampal slices were bathed in aCSF containing 1 mg/ml sulfo-NH-SS-biotin (Thermo Scientific, Rockford, IL). Slices were rinsed in aCSF containing 100 mM glycine (quenching buffer) to eliminate unreacted biotin. Hippocampal regions of interest were isolated by microdissection. Tissue was lysed in RIPA buffer containing protease and phosphatase inhibitors. An aliquot of cleared lysate was mixed with a half volume of Laemmli buffer and labeled as “lysate fraction”. Biotinylated proteins were batch extracted using Ultralink avidin-conjugated beads (Thermo Scientific, Rockford, IL). Proteins in the biotinylated fraction were diluted to the same extent than proteins in the total lysate, so that immunoreactivity in the lysate and biotinylated fractions is proportional when equal volumes of lysate and biotinylated fraction are analyzed.
Western Blot
Protein samples were separated in SDS-polyacrylamide gels, transferred to nitrocellulose membranes and incubated with primary and secondary antibodies. Polyclonal rabbit antibodies for KCC2 were obtained from Millipore (Billerica, MA). A monoclonal mouse antibody to detect NKCC (T4) was obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Department of Biology, Iowa City, IA). This monoclonal antibody (T4) was generated against a fusion protein that encompassed the 310 C-terminal residues of human NKCC [21, 22]. Since the C-terminus of NKCC1 and NKCC2 share more than a 90% of identity, the T4 antibody recognizes both polypeptides. Because NKCC2 is not expressed in the vertebrate brain, the immunoreactivity detected by T4 antibody in brain samples corresponds to NKCC1 [22]. To estimate potential variability in protein content and loading, blots were also probed with an anti-actin antibody from Sigma (St. Louis, MO). Anti-rabbit or anti-mouse secondary antibodies conjugated to horseradish peroxidase were from GE Health Care (Piscataway, NJ) or Jackson Immunoresearch laboratories (West Grove, PA), respectively. Immunoreactive bands were visualized using Super Signal West Dura chemiluminiscent substrate (Pierce, Rockford, IL, USA) and film. After scanning the films, immunoreactive bands of the appropriate size were quantified using Image J (NIH, Bethesda, MD, USA). Immunoreactivity for the bands of interest was normalized to actin immunoreactivity and compared to control values. Data is presented as the mean ± SEM.
Statistical analysis
Statistical analyses were performed using GraphPad InStat (GraphPad Software, Inc., San Diego, CA, USA). Differences between groups were determined by one-way analysis de variance (ANOVA) followed by Bonferroni post hoc test. For these analyses, p values < 0.05 were considered significant.
RESULTS
To characterize the effects of SE on CCCs, the cell surface expression of KCC2 and NKCC1 was analyzed in hippocampal tissue collected at different time points after induction of SE. Western blot analysis of samples from the CA1 region of hippocampus obtained 1-day post-SE showed reduced KCC2 immunoreactivity in the biotinylated (cell surface) fraction (Fig. 1). KCC2 loss was also evident in the lysate and intracellular (non-biotinylated) fractions (Fig 1), suggesting that after internalization there is a net loss of KCC2 expression. Blots were also probed with antibodies directed against NKCC transporters (T4 mAb) and a strong immunoreactive band was easily detected in tissue CA1 lysates. The signal detected corresponded to either a single broad band or a doublet of ~150-170 kDa (Fig 1), a band pattern previously reported in samples from rat and human neocortex [22]. Unexpectedly, NKCC1 immunoreactivity was mostly absent from the biotinylated fraction (cell surface) while the majority of NKCC1 signal was detected in the non-biotinylated fraction (intracellular). Longer exposure allowed detection of a faint but quantifiable band in the biotinylated fraction (Fig 1). Under these conditions, no change in NKCC1 was detected in the biotinylated fraction (cell surface) at 1-day post-SE (Fig 1E). Analysis of lysates and biotinylated samples from DG did not show significant changes in KCC2 immunoreactivity (Fig. 2). A significant change was, however, detected in the intracellular fraction. Finally, analysis of NKCC1 in DG samples did not show any apparent changes (Fig. 2).
Figure 1. Cell surface expression of CCCs in CA1 region of hippocampus.
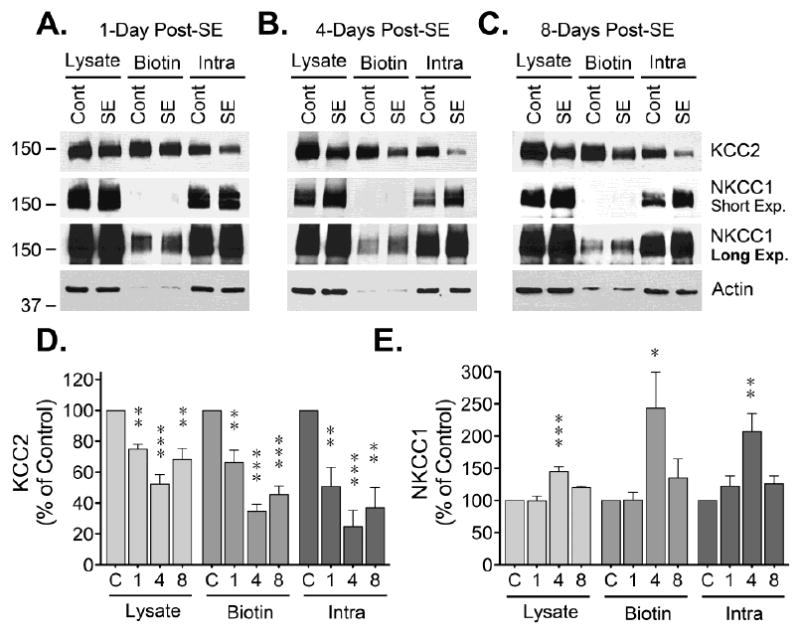
Hippocampal slices from control (Cont) or SE animals (at 1, 4 or 8 days post-SE) were labelled with a cell impermeable biotinylation reagent. After labeling, hippocampal slices were microdissected to isolated CA1 and DG. Biotinylated proteins (“cell surface proteins”) were batch extracted using avidin-conjugated beads. Identical volumes of lysate, biotinylated and non-biotinylated fractions were analyzed by Western blot. (A, B, C) Representative KCC2, NKCC and actin blots for samples obtained at 1, 4 or 8 days post-SE. (D) Densitometry analysis of KCC2 immunoreactivity normalized to actin is presented as the mean ± SEM of four to five independent experiments. (E) Densitometry analysis of NKCC1 immunoreactivity normalized to actin presented as the mean ± SEM of four to five independent experiments. The signal detected at different time points after SE was compared to control by ANOVA followed by Bonferroni post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001).
Figure 2. Cell surface expression of CCCs in DG of hippocampus.
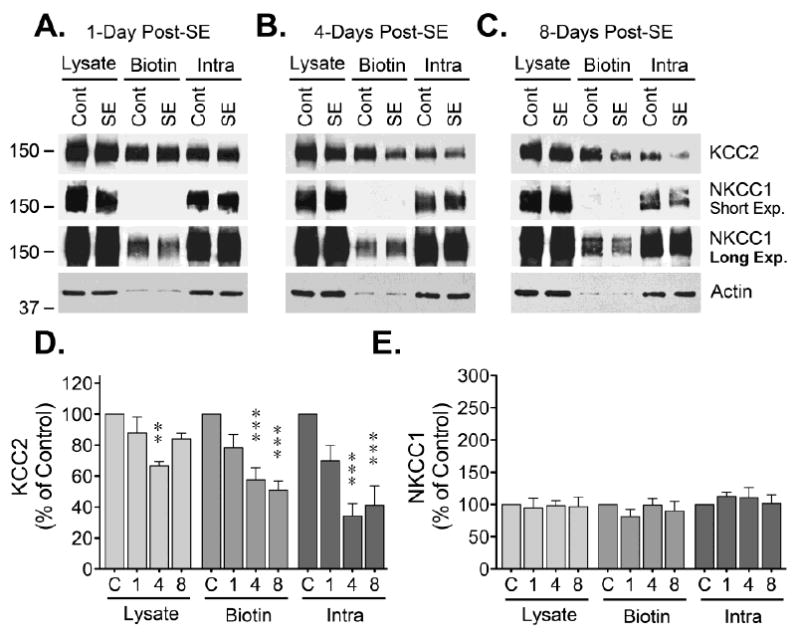
After labeling and separation of the biotinylated proteins, lysate, biotinylated and non-biotinylated samples from DG were analyzed by Western blot. (A, B, C) Representative blots showing KCC2, NKCC and actin blots at 1, 4 or 8 days post-SE. (D) Densitometry analysis of KCC2 immunoreactivity normalized to actin is presented as the mean ± SEM of four to five independent experiments. (E) Densitometry analysis of NKCC1 immunoreactivity normalized to actin represented as the mean ± SEM of four to five independent experiments. The signal detected at the different time points after SE was compared to controls by ANOVA followed by Bonferroni post hoc test (**p < 0.01, ***p < 0.001).
Samples from the CA1 region obtained 4-days post-SE evidenced a pronounced decrease in KCC2 immunoreactivity in the lysate, biotinylated and non-biotinylated fractions (Fig 1). Interestingly, in these same samples, there is a significant increase on NKCC1 immunoreactivity in both the lysate and non-biotinylated (intracellular) fraction. Longer exposure times allowed the quantization of NKCC1 in the biotinylated fraction and showed that in addition to an increase in total expression, there is a prominent (243 ± 56% of control) and significant (*p < 0.05) increase in NKCC1 levels at the plasma membrane (Fig 1E). At this time point, samples from DG also showed a significant decrease in KCC2 immunoreactivity in all fractions analyzed, but no significant change was detected for NKCC1 (Fig. 2).
Evaluation of CA1 samples collected 8 days post-SE revealed that KCC2 immunoreactivity remained significantly reduced, but with an upward trend towards control levels. In the case of NKCC1, a significant increase in lysate immunoreactivity was still evident, but no significant change was detected on the biotinylated or intracellular fractions, indicating that NKCC1 levels also trend towards control levels (Fig. 1). Samples from DG show that at 8 days post-SE the decrease in KCC2 immunoreactivity remained significant in all fractions and, in contrast, no significant change was detected on NKCC1 immunoreactivity (Fig 2).
In a previous study, our group found that the occurrence of spontaneous seizures is associated with alterations in GABAARs [13]. For those studies, we analyzed two groups of animals: one with frequent seizures that had on average 26.9 ± 16.5 seizures/day and a second one with infrequent seizures that had on average 3.5 ± 4.6 seizures/day (for additional details see [13]). Our findings suggested that epileptic animals experiencing frequent/recent seizures (within 3 hours of tissue collection) have similar levels of GABAAR at the plasma membrane but show abnormalities in phasic and tonic inhibition. In contrast, animals presenting infrequent seizures (no seizures in the 24 hours prior to tissue collection) have increased levels of GABAAR at the plasma membrane and functional responses similar to controls [13]. To further investigate possible molecular alterations that occur during chronic epilepsy, tissue obtained from animals experiencing frequent/recent or infrequent seizures was analyzed to assess the cell surface levels of KCC2 and NKCC1.
Tissue from the CA1 region showed a significant reduction in KCC2 cell surface expression in animals with recent/frequent seizures but no significant change was detected in animals with infrequent seizures (Fig. 3). KCC2 levels were also decreased in the intracellular fraction of both groups of epileptic animals but no change was detected in the total lysates. Despite an upward trend, no significant change was detected on NKCC1 in the lysate, biotinylated or intracellular fractions of the CA1 (Fig. 3). Finally, DG samples from chronically epileptic animals showed no significant changes in KCC2 or NKCC1 immunoreactivity (Fig. 3). NKCC1 expression showed an upward trend by the effect was not consistently detected in all samples analyzed, rendering non-significant results. Thus, the only significant change evident in tissue obtained from chronically epileptic animals was a reduction on KCC2 observed in the CA1 region of rats with frequent/recent seizures.
Figure 3. Cell surface expression of CCCs in CA1 and DG of chronically epileptic animals.
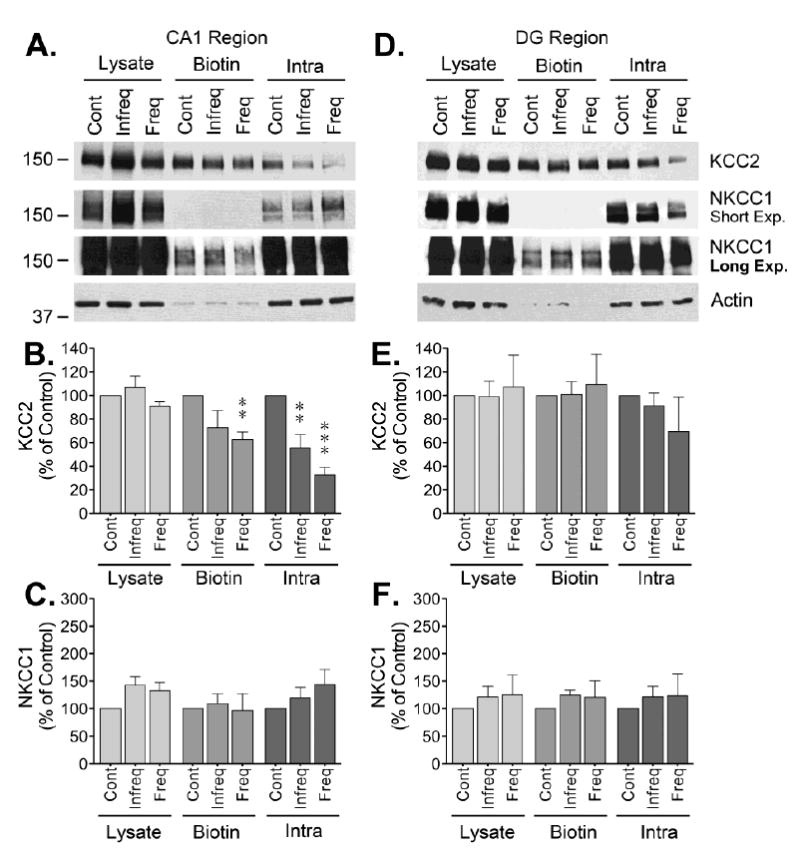
Following labeling, microdissection and batch extraction of biotinylated proteins, samples from the CA1 and DG of hippocampus obtained from chronically epileptic animals with either recent/frequent or infrequent seizures were analyzed by Western blot. (A and D) Representative blots for KCC2, NKCC1 and actin. Densitometry analysis of (B) KCC2 or (C) NKCC1, immunoreactivity in CA1 samples from epileptic animals normalized to actin is presented as the mean ± SEM of five independent experiments. Densitometry analysis of (E) KCC2 or (F) NKCC1 immunoreactivity in DG of chronically epileptic animals normalized to actin presented as the mean ± SEM of five independent experiments. The signal detected at the epileptic animals was compared to controls by ANOVA followed by Bonferroni post hoc test (**p < 0.01, ***p < 0.001).
DISCUSSION
At steady state, KCC2 molecules are distributed between the plasma membrane and an intracellular pool, but neuronal stimulation might alter the relative distribution of KCC2 molecules between these two compartments [5, 24]. Thus, the pool of functionally active KCC2 represents a fraction of the total protein that can be actively controlled by insertion and removal of transporters [16, 17, 20]. In tissue obtained from control animals, the rough distribution of KCC2 corresponds to ~65% being present at the cell surface while ~35% located within an intracellular pool. Under control conditions, the yield of recovery from the extraction protocol for KCC2 was 107 ± 6% (n=19), suggesting optimal recovery. Additional validation was provided by the observation that only ~8% of the intracellular protein actin was detected in the biotinylated fraction (cell surface) while the bulk of signal (~80%) was detected in the non-biotinylated (intracellular) fraction with a yield close to 90%.
In total lysates, the antibody used to detect NKCC1 revealed a strong immunoreactive band of the appropriate molecular size (~150-170 KDa) [22]. Unexpectedly, after fractionation, the vast majority of NKCC1 was detected in the intracellular pool with little expression at the plasma membrane and a yield of only 55 ± 4% (n=19, control animals). It is unclear why NKCC1 yield is low, since extraction of other proteins, including GABAAR [12, 13], is much more efficient. The poor yield of NKCC1 recovery could be due to properties intrinsic to this transporter that might include: the inaccessibility during the biotinylation procedure, insolubility of the protein during the extraction process or less efficiency during immunoblotting due to the skewed distribution between intracellular and cell surface fractions. Despite this caveat, the results presented here suggest that although KCC2 protein is more evenly distributed among plasma membrane and intracellular pools, the bulk of NKCC1 is detected on the intracellular pool.
A recent report indicates that SE induction triggers a rapid loss of KCC2 from the plasma membrane due to enhanced internalization [28], suggesting that expression of transporters at the plasma membrane regulates the onset and severity of SE. Analysis of KCC2 cell surface expression showed a reduction in KCC2 levels within ~30 minutes of SE [28]. The evidence presented here suggests that in the CA1 region of hippocampus there is a maximal reduction in KCC2 cell surface expression and a prominent (243 ± 56 of control levels, n=5) and significant (*p < 0.05) increase in NKCC1 cell surface expression at 4-days post-SE. Interestingly, application of the NKCC1 inhibitor bumetanide produces a significantly shift in EGABA unmasking and increase in NKCC1 activity in tissue obtained 1 week after SE [1]. Together, these observations suggest a transient shift (reversal) in the plasma membrane levels of KCC2 and NKCC1 that can be detected during the early phase of epileptogenesis and that this reversal might alter the balance between excitation/inhibition and contribute to the appearance of spontaneous seizures.
A previous report demonstrated that in the pilocarpine model of epilepsy, KCC2 expression is down regulated after SE but it returns to control levels in chronically epileptic animals [23]. In the current study, KCC2 loss was no evident in tissue lysates from chronically epileptic animals but a significant reduction in KCC2 cell surface levels was detected in the CA1 region of animals with frequent/recent seizures. In addition, the levels of NKCC1 at the plasma membrane were not increased. Staining of autopsy or biopsy specimens using the same antibody employed here (T4) showed that NKCC1 immunoreactivity can be detected in the soma and proximal dendritic processes of granule cells from DG and pryramidal cells from the CA1 region [22]. In non-sclerotic hippocampus, colocalization of NKCC1 and KCC2 was detected in the majority of neurons but in sclerotic CA1 the degree of colocalization decreased and ~20% of NKCC1-immunoreactive neurons do not express KCC2. Interestingly, only a small fraction of pyramidal cells discharge during interictal events while the majority of cells are strongly inhibited and do not participate in the epileptic network [6]. The difference between discharging and inhibited pyramidal cells apparently results from differential EGABA, raising the possibility that a small number of pacemaker cells with increased NKCC1 and reduced KCC2 are involved in the generation of epileptic bursts [7]. The lack of evidence for increased levels of NKCC1 at plasma membrane in epileptic tissue presented here favors the idea that a massive increase in NKCC1 might not be necessary to facilitate hyperexcitability.
In summary, alterations in the plasma membrane expression of KCC2 and NKCC1 were mostly detected during the early phase of epileptogenesis. These observations are in line with previous reports demonstrating alterations in the chloride gradient and the reversal of EGABA during the same time frame and suggest that aberrant function of CCCs might be more relevant for the initial appearance of spontaneous seizures.
Highlights.
Differential alterations in the plasma membrane expression of cation-chloride transporters were observed during epileptogenesis.
A shift in the cell surface expression of KCC2 and NKCC1 was detected during the early phase of epileptogenesis
No change in the cell surface expression of KCC2 or NKCC1 was detected at the chronic stage.
Hyperexcitability during epileptogenesis may be a cumulative effect of multiple deficits impacting different aspects of GABAergic neurotransmission
Acknowledgments
The author gratefully acknowledges Heidi L. Grabenstatter, Yasmin Cruz Del Angel and Jessica Carlsen for their contributions during the studies aimed to analyze the impact of spontaneous seizures on GABAAR. This work was supported by grants K01-NS069583 and R01-NS089698 from the National Institutes of Health to MIG.
Abbreviations used
- CCCs
cation-chloride transporters
- GABAAR
GABAA receptors
- SE
status epilepticus
- CA1
Cornus Ammonis 1
- DG
Dentate Gyrus
- aCSF
artificial cerebrospinal fluid
- sulfo-NH-SS-biotin
sulfosuccinimidyl-2-[biotinamido]ethyl-1,3-dithiopropionate
- PBS
phosphate buffered saline
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The author declares no conflict of interest.
References
- 1.Barmashenko G, Hefft S, Aertsen A, Kirschstein T, Kohling R. Positive shifts of the GABAA receptor reversal potential due to altered chloride homeostasis is widespread after status epilepticus. Epilepsia. 2011;52:1570–1578. doi: 10.1111/j.1528-1167.2011.03247.x. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Ari Y, Khalilov I, Kahle KT, Cherubini E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist. 2012;18:467–486. doi: 10.1177/1073858412438697. [DOI] [PubMed] [Google Scholar]
- 3.Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- 5.Chamma I, Heubl M, Chevy Q, Renner M, Moutkine I, Eugene E, Poncer JC, Levi S. Activity-dependent regulation of the K/Cl transporter KCC2 membrane diffusion, clustering, and function in hippocampal neurons. J Neurosci. 2013;33:15488–15503. doi: 10.1523/JNEUROSCI.5889-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- 7.Cohen I, Navarro V, Le Duigou C, Miles R. Mesial temporal lobe epilepsy: a pathological replay of developmental mechanisms? Biol Cell. 2003;95:329–333. doi: 10.1016/s0248-4900(03)00081-9. [DOI] [PubMed] [Google Scholar]
- 8.Curia G, Longo D, Biagini G, Jones RS, Avoli M. The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods. 2008;172:143–157. doi: 10.1016/j.jneumeth.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Hassar L, Esclapez M, Bernard C. Hyperexcitability of the CA1 hippocampal region during epileptogenesis. Epilepsia. 2007;48(Suppl 5):131–139. doi: 10.1111/j.1528-1167.2007.01301.x. [DOI] [PubMed] [Google Scholar]
- 10.El-Hassar L, Milh M, Wendling F, Ferrand N, Esclapez M, Bernard C. Cell domain-dependent changes in the glutamatergic and GABAergic drives during epileptogenesis in the rat CA1 region. J Physiol. 2007;578:193–211. doi: 10.1113/jphysiol.2006.119297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fritschy JM. Epilepsy, E/I Balance and GABA(A) Receptor Plasticity. Front Mol Neurosci. 2008;1:5. doi: 10.3389/neuro.02.005.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.González MI, Cruz Del Angel Y, Brooks-Kayal A. Down-regulation of gephyrin and GABAA receptor subunits during epileptogenesis in the CA1 region of hippocampus. Epilepsia. 2013;54:616–624. doi: 10.1111/epi.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.González MI, Grabenstatter HL, Cea-Del Rio CA, Cruz Del Angel Y, Carlsen J, Laoprasert RP, White AM, Huntsman MM, Brooks-Kayal A. Seizure-related regulation of GABAA receptors in spontaneously epileptic rats. Neurobiol Dis. 2015;77:246–256. doi: 10.1016/j.nbd.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.González MI, Krizman-Genda E, Robinson MB. Caveolin-1 regulates the delivery and endocytosis of the glutamate transporter, excitatory amino acid carrier 1. J Biol Chem. 2007;282:29855–29865. doi: 10.1074/jbc.M704738200. [DOI] [PubMed] [Google Scholar]
- 15.Holman D, Henley JM. A novel method for monitoring the cell surface expression of heteromeric protein complexes in dispersed neurons and acute hippocampal slices. J Neurosci Methods. 2007;160:302–308. doi: 10.1016/j.jneumeth.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, Moss SJ. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013;36:726–737. doi: 10.1016/j.tins.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci. 2014;15:637–654. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leroy C, Poisbeau P, Keller AF, Nehlig A. Pharmacological plasticity of GABA(A) receptors at dentate gyrus synapses in a rat model of temporal lobe epilepsy. J Physiol. 2004;557:473–487. doi: 10.1113/jphysiol.2003.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loscher W, Brandt C. Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol Rev. 2010;62:668–700. doi: 10.1124/pr.110.003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2013;69:62–74. doi: 10.1016/j.neuropharm.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 21.Lytle C, Xu JC, Biemesderfer D, Forbush B., 3rd Distribution and diversity of Na-K-Cl cotransport proteins: a study with monoclonal antibodies. Am J Physiol. 1995;269:C1496–1505. doi: 10.1152/ajpcell.1995.269.6.C1496. [DOI] [PubMed] [Google Scholar]
- 22.Muñoz A, Mendez P, DeFelipe J, Alvarez-Leefmans FJ. Cation-chloride cotransporters and GABA-ergic innervation in the human epileptic hippocampus. Epilepsia. 2007;48:663–673. doi: 10.1111/j.1528-1167.2007.00986.x. [DOI] [PubMed] [Google Scholar]
- 23.Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puskarjov M, Ahmad F, Kaila K, Blaesse P. Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J Neurosci. 2012;32:11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scorza FA, Arida RM, Naffah-Mazzacoratti Mda G, Scerni DA, Calderazzo L, Cavalheiro EA. The pilocarpine model of epilepsy: what have we learned? An Acad Bras Cienc. 2009;81:345–365. doi: 10.1590/s0001-37652009000300003. [DOI] [PubMed] [Google Scholar]
- 26.Sharma AK, Reams RY, Jordan WH, Miller MA, Thacker HL, Snyder PW. Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol Pathol. 2007;35:984–999. doi: 10.1080/01926230701748305. [DOI] [PubMed] [Google Scholar]
- 27.Shumate MD, Lin DD, Gibbs JW, 3rd, Holloway KL, Coulter DA. GABA(A) receptor function in epileptic human dentate granule cells: comparison to epileptic and control rat. Epilepsy Res. 1998;32:114–128. doi: 10.1016/s0920-1211(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 28.Silayeva L, Deeb TZ, Hines RM, Kelley MR, Munoz MB, Lee HH, Brandon NJ, Dunlop J, Maguire J, Davies PA, Moss SJ. KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc Natl Acad Sci U S A. 2015;112:3523–3528. doi: 10.1073/pnas.1415126112. [DOI] [PMC free article] [PubMed] [Google Scholar]