

Abstract
The sulfonamide class of antibiotics has been in continuous use for over 70 years. They are thought to act by directly inhibiting dihydropteroate synthase (DHPS), and also acting as prodrugs that sequester pterin pools by forming dead end pterin-sulfonamide conjugates. In this study, eight pterin-sulfonamide conjugates were synthesized using a novel synthetic strategy and their biochemical and microbiological properties were investigated. The conjugates were shown to competitively inhibit DHPS, and inhibition was enhanced by the presence of pyrophosphate that is crucial to catalysis and is known to promote an ordering of the DHPS active site. The co-crystal structure of Yersinia pestis DHPS bound to one of the more potent conjugates revealed a mode of binding that is similar to that of the enzymatic product analog pteroic acid. The antimicrobial activities of the pterin-sulfonamide conjugates were measured against Escherichia coli in the presence and absence of folate precursors and dependent metabolites. These results show that the conjugates have appreciable antibacterial activity and act by an on target, anti-folate pathway mechanism rather than as simple dead end products.
Graphical Abstract

Sulfonamides were the some of the first successful antimicrobial agents and have been used continuously since the 1940s to treat a wide variety of Gram-positive, Gram-negative and protozoal infections. 1–3 Sulfonamides are usually prescribed in combination with the dihydrofolate reductase inhibitor trimethoprim to maximize therapeutic response and reduce the development of resistance.4, 5 Today, this drug combination is relied upon as a valuable therapeutic option in two important areas: (1) as an inexpensive oral treatment for community acquired bacterial infections resistant to frontline agents such as penicillins and fluoroquinolones; and (2) as a prophylaxis treatment for the prevention of Pneumocystis jirovecii pneumonia in immune-compromised cancer and HIV patients.6, 7 Over time, sulfonamide use has been compromised by the emergence of resistance and the poor tolerance of patients to long-term treatment. 6, 8
Sulfonamides target the enzyme dihydropteroate synthase (DHPS) and mimic the substrate p-aminobenzoic acid (pABA).1, 9 We previously reported detailed structural, biochemical and computational studies on the catalytic mechanism of DHPS.10 DHPS catalyzes the conjugation of 6-hydroxymethyl-7,8-dihydropterin pyrophosphate (DHPPP) with pABA to form 7,8-dihydropteroate, an intermediate in the folate biosynthesis pathway (Figure 1A).10 In addition to inhibiting DHPS, sulfonamides can also act as alternative substrates by forming dead end pterin-sulfonamide products, which may also inhibit enzymes later in the pathway (Figure 1B).10–12 In this role, the contribution to the overall antimicrobial potency of this class of antibiotics is poorly defined. For the purpose of continuing our studies on the inhibition of DHPS and its therapeutic potential, we sought to chemically synthesize a panel of pterin-sulfonamide conjugates.
Figure 1.

1A: Reaction catalyzed by dihydropteroate synthase; 1B: General structure of pterinsulfonamide conjugates
Repeating the previously published alkylation based route13 to these conjugates, we obtained only incorrect isomeric products, challenging the previously ascribed pharmacological properties.14, 15 Consequently, a new synthetic route to rapidly and accurately generate pterin-sulfonamide conjugates was developed. The new conjugates were tested for inhibition of DHPS and co-crystallized with the enzyme to characterize their mode of binding. Microbiological profiling using E. coli indicated that pterin-sulfonamides are not only good inhibitors of DHPS but also have substantial antimicrobial activity that can be antagonized by folate precursors.
Our initial strategy to synthesize the conjugates was based on the method detailed by Piper and Montgomery.13 Briefly, commercially available (2,4-diaminopteridin-6-yl)methanol 1 was treated with triphenyl phosphine and bromine to afford the 6-bromomethyl pterin analog 2,13 which was expected to couple with the appropriate sulfonamide, followed by 4-deamination, to give target compounds in three steps (Scheme 1). However, when compound 2 reacted with sulfamethoxazole 3 in the presence of N,N-diisopropylethylamine (DIEA), only isomeric sulfonamide linked compound 4 was isolated as the main product, instead of the desired aniline linked pterin-sulfamethoxazole 15. Attempts to modify the synthesis by protection of the sulfonamide or alter the alkylating conditions failed.16–18
Scheme 1.

Initial synthetic strategy.
An alternative synthetic route that provided the necessary flexibility to conjugate a large variety of sulfonamides to pterin was developed around the key acyl protected formyl-pterin intermediate 13 shown in Scheme 2. Treatment of commercially available 2-amino-5-(chloromethyl)-3-cyanopyrazine 1-oxide 5 with phosphorus trichloride at room temperature in tetrahydrofuran solution resulted in smooth deoxygenation to give 6,19 which was treated with pyridine with stirring overnight at room temperature to afford pyridinium salt 7.20 Salt 7 reacted with N,N-dimethyl-4-nitrosoaniline in the presence of potassium carbonate to give nitrone 8, which was then treated with 6N HCl to give aldehyde 9. Quantitative conversion of 9 to its dimethyl acetal in the presence of DOWEX 50WEX8-100 ion-exchange resin, followed by condensation with guanidine carbonate in the presence of sodium methoxide, afforded 2,4-diamino pterin 10. Treatment of 10 with hot 5% sodium hydroxide gave 6-formylpterindine dimethyl acetal 11, which was treated with acetic anhydride at 100 °C to give 2-acetyl protected 12. The 6-formylpterine dimethyl acetal 12 was hydrolyzed with the treatment of formic acid to afford 6-formylpterin 13.21 Coupling of 6- formylpterin 13 with different sulfonamides or dapsone (4-[(4-aminobenzene)sulfonyl]aniline) was carried out smoothly in acetic acid to give the corresponding Schiff base compound, which was subsequently reduced with borane tert-butylamine complex to give 14 a–h.22 Deprotection of 14 ah with 0.1 N NaOH afforded target compounds 15-22.23
Scheme 2.

Synthetic method for production of pterin-sulfonamide conjugates.
Compounds 15–22 were all tested for inhibition of B. anthracis DHPS using a fluorescence polarization (FP) based competition binding assay, performed in the absence or presence of 2 mM sodium pyrophosphate (Table 1 and Supplementary Material). 25 Pyrophosphate (PPi) is the biochemical byproduct of DHPS catalysis, and its inclusion in the FP assay allowed us to confirm the site-specific binding of pterin-sulfonamide conjugates. More specifically, we have previously observed that PPi stabilizes two flexible loops in the DHPS active site that form the pABA-sulfonamide binding site.10
Table 1.
Inhibitory activities of pterin-sulfa conjugates.
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | R |
BaDHPS in vitro FP Binding Inhibition IC50 (µM) |
Minimum Inhibitory Concentration E. coli K12, in µMb,c |
|||
| (−) PPia | (+) 2 mM PPia |
M9 media | M9 plus 5 µg/mL PABA |
M9 plus 20 µg/mL Methionine |
||
| 15 | 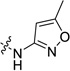 |
20.6±3.5 | 3.4±0.1 | 10.9 (0.8)c | 467 (263)c | 29 (1.5)c |
| 16 | 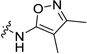 |
22.5±1.0 | 8.1± 0.7 | 28.3 (1.5) | >412 (249) | 47 (5.9) |
| 17 | 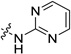 |
19.3±0.6 | 9.4± 0.7 | 17.1 (1.6) | >470 (666) | 78 (6.2) |
| 18 | 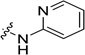 |
>62.5 | 2.5± 0.2 | 29.5 (2.7) | >471 (669) | 98 (6.3) |
| 19 | 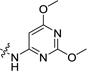 |
26.8±3.0 | 1.9± 0.1 | 17.2 (1.6) | >412 (537) | 86 (3.4) |
| 20 | 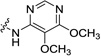 |
15.2±0.6 | 11.5±0.8 | 34.3 (3.8) | >412 (>644) | 172 (10.1) |
| 21 | 17.8±0.8 | 11.3±1.1 | 197 (25) | 236 (>934) | 236 (50) | |
| 22 | 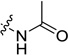 |
18.6±0.8 | 17.1±1.2 | 136 (22) | >514 (>934) | >200 (58) |
| Ampicillin | ND | ND | 1.58 | 2 | 2.1 | |
| Chloramphenicol | ND | ND | 8.5 | 9.7 | 9.7 | |
| Trimethroprim | ND | ND | 1.7 | 1.4 | 0.7 | |
Present as sodium pyrophosphate. Average of 3 experiments, ±SEM.
Performed according to 24.
Corresponding values for parent sulfonamides shown in parentheses.
As a group, the pterin-sulfonamide conjugated compounds are more efficient than SMX (FP IC50 >62.5 µM) at displacing the probe from the active site in the FP competition assay. These compounds show an even more efficient displacement of probe when PPi is added to the assay. Among all analogs, pterin-sulfadimethoxine (pterin-SDM) conjugate 19 and pterin-sulfamethoxazole (pterin-SMX) conjugate 15 have the highest potency against BaDHPS, with IC50 values of 1.9 µM and 3.4 µM, respectively, in the presence of PPi. Conjugation of pterin with other sulfa analogs resulted in compounds with good inhibitory activity with the weakest inhibition observed for the sulfanilamide conjugate 22.
To establish the mode of binding of the pterin-sulfonamide conjugates in the DHPS active site, we soaked compound 16 into apo crystals of Yersinia pestis DHPS (YpDHPS) and determined the structure of the complex to 2.1 Å resolution (PDB ID: 5JQ9). As anticipated, 16 occupies both the pterin and pABA binding pocket of DHPS (Figure 2A). The mode of binding is very similar to those of pteroic acid26 and the sulfathiazole-pterin adduct that was generated catalytically in crystallo 10 (Figures 2B and 2C). Residues D96, N115, D185, K221 and R255 interact with the pterin moiety, and F190, K221 and S222 interact with the sulfonamide/pABA groups (YpDHPS residue numbering). We have also determined the structure of the DHPS near transition state structure,10 and its superposition onto the 16 complex shows how closely the compound mimics the binding modes of pterin and pABA. Note that weak electron density adjacent to 16 (Figure 2A) corresponds to the DHPS anion-binding pocket, which is occupied by sulfate and pyrophosphate in the DHP-STZ and transition state structures, respectively (Figures 2C and 2D). Occupancy of the anion-binding pocket by pyrophosphate and the resulting stabilization of loop2 (Figure 2D) would explain our observation that pyrophosphate augments the binding of the conjugate compounds.
Figure 2.

A. Yersinia pestis DHPS (YpDHPS) with compound 16 at the active site. Also shown is the omit map (mFo-DFc) showing the electron density of 16 contoured at 2σ (PDB ID: 5JQ9). B. YpDHPS with the product analog pteroic acid bound at the active site (PDB ID: 3TYU). C. Bacillus anthracis DHPS (BaDHPS) with the pterin-sulfathiazole (DHP-STZ) catalytic product bound at the active site (PDB ID: 3TYE). D. Superimposition of the YpDHPS 16 complex and the YpDHPS near transition state complex (PDB ID: 3TYZ). In all figures, the DHPS cartoon and carbon atoms are grey, the oxygen, nitrogen and sulfur atoms are red, blue and yellow respectively, and the compound carbons are cyan. In (D), the transition state structure is shown as pink cartoon and carbon atoms, the ordered loop2 is shown in green, and the essential Mg2+ ion is shown.
The antibacterial activities of these conjugate compounds and their corresponding parent sulfonamides were tested against Escherichia coli K12 (Table 1) in M9 minimal media 27 suitable for testing sulfonamide activity. Under these conditions, most conjugates showed appreciable antibacterial activity, with the exception of the sulfanilamide and dapsone hybrids (21, 22) mirroring the weaker direct activity of the corresponding parent sulfonamides.24 The sulfamethoxazole hybrid (15) showed the most potency with a MIC of 10.9 µM. Overall the minimal inhibitory concentration activities for the conjugates were usually around 10 times weaker than for the corresponding parent sulfonamides (compound 15 10.9 µM, vs sulfamethoxazole 0.8 µM).
We next explored whether the observed antimicrobial activities are a result of on target biochemical inhibition. MIC activities were examined in the presence of the added folate precursor pABA or the dependent metabolite methionine (Table 1). Addition of pABA completely ablated MIC activity for all conjugates28, and a similar but less dramatic effect was seen when growth media was supplemented with methionine. These data mirrored the MIC shifts we observed with the corresponding sulfonamides, supporting the hypothesis that the pterin conjugates function as direct antibacterial compounds through inhibition of the folate pathway,
In conclusion, we have demonstrated that pterin-sulfonamide conjugates have antibacterial action through biochemical inhibition of DHPS and antagonism of the folate pathway. It is important to note that the conjugates synthesized in this study are in the oxidized form of pterin and not the reduced form dihydropterin that is formed by the natural biochemical action of DHPS on DHPPP and sulfonamides, which likely accounts for some of the reduction in pharmacological activity. However, our structures reveal that the oxidation state of the pterin ring has little effect on how it binds into the pterin pocket (compare Figures 2A and 2B with 2C and 2D).
The lower MIC activity of the conjugates compared to their corresponding sulfonamides may be attributable to differences in penetration and efflux of the pterin sulfonamide conjugates vs sulfonamides. Further studies are ongoing to study the susceptibility to efflux, capacity to inhibit enzymes later in the pathway and in cell metabolism of the conjugates. The pharmacological advantages that pterin-sulfonamide conjugates may have over sulfonamides are also worthy of further study. The conjugates may have a lower propensity for sulfonamide mediated anaphylaxis, as the oxidatively reactive aniline group in the sulfonamides is hindered by the pterin conjugation.29 Another important observation from this study is the enhanced binding to DHPS by these conjugates that presumably results from the introduction of a pterin anchor. The pterin anchor also has implications for the development of resistance to these conjugates. As previously noted,30 the pterin pocket is highly conserved and unlikely to tolerate mutations that might decrease binding affinity. This is not true of the isolated sulfonamides because the pABA-binding site can tolerate such mutations.10 Thus, the activity of the conjugates against defined sulfonamide resistant strains should be determined.
Supplementary Material
Acknowledgments
This research was supported by funding from the National Institutes of Health (grant R01AI070721 to R.E.L. and S.W.W.) and the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Woods DD. The Relation of p-aminobenzoic Acid to the Mechanism of the Action of Sulphanilamide. British Journal of Experimental Pathology. 1940;21:74–90. [Google Scholar]
- 2.Miller AK. Folic Acid and Biotin Synthesis by Sulfonamide-sensitive and Sulfonamide-resistant Strains of Escherichia coli. Experimental Biology and Medicine. 1944;57:151–153. [Google Scholar]
- 3.Strauss E, Lowell FC, Finland M. OBSERVATIONS ON THE INHIBITION OF SULFONAMIDE ACTION BY PARA-AMINOBENZOIC ACID. J Clin Invest. 1941;20:189–197. doi: 10.1172/JCI101211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warner P, Maniar AC. Combined action of sulfadiazine and trimethoprim. Appl Microbiol. 1966;14:299–300. doi: 10.1128/am.14.2.299-300.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bushby SR, Hitchings GH. Trimethoprim, a sulphonamide potentiator. British Journal of Pharmacology and Chemotherapy. 1968;33:72–90. doi: 10.1111/j.1476-5381.1968.tb00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiholm BE, Emanuelsson S. Drug-related blood dyscrasias in a Swedish reporting system, 1985–1994. Eur J Haematol Suppl. 1996;60:42–46. doi: 10.1111/j.1600-0609.1996.tb01644.x. [DOI] [PubMed] [Google Scholar]
- 7.Skold O. Sulfonamide resistance: mechanisms and trends. Drug Resist Updat. 2000;3:155–160. doi: 10.1054/drup.2000.0146. [DOI] [PubMed] [Google Scholar]
- 8.Ho JM, Juurlink DN. Considerations when prescribing trimethoprim-sulfamethoxazole. CMAJ. 2011;183:1851–1858. doi: 10.1503/cmaj.111152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bermingham A, Derrick JP. The folic acid biosynthesis pathway in bacteria: evaluation of potential for antibacterial drug discovery. Bioessays. 2002;24:637–648. doi: 10.1002/bies.10114. [DOI] [PubMed] [Google Scholar]
- 10.Yun MK, Wu Y, Li Z, Zhao Y, Waddell MB, Ferreira AM, Lee RE, Bashford D, White SW. Catalysis and sulfa drug resistance in dihydropteroate synthase. Science. 2012;335:1110–1114. doi: 10.1126/science.1214641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roland S, Ferone R, Harvey RJ, Styles VL, Morrison RW. The characteristics and significance of sulfonamides as substrates for Escherichia coli dihydropteroate synthase. J Biol Chem. 1979;254:10337–10345. [PubMed] [Google Scholar]
- 12.Chakraborty S, Gruber T, Barry CE, 3rd, Boshoff HI, Rhee KY. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science. 2013;339:88–91. doi: 10.1126/science.1228980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piper JR, Montgomery JA. Preparation of 6-(bromomethyl)-2,4-pteridinediamine hydrobromide and its use in improved syntheses of methotrexate and related compounds. J Org Chem. 1977;42:208–211. doi: 10.1021/jo00422a005. [DOI] [PubMed] [Google Scholar]
- 14.Fröhlich LG, Kotsonis P, Traub H, Taghavi-Moghadam S, Al-Masoudi N, Hofmann H, Strobel H, Matter H, Pfleiderer W, Schmidt HHHW. Inhibition of Neuronal Nitric Oxide Synthase by 4-Amino Pteridine Derivatives: Structure–Activity Relationship of Antagonists of (6R)-5,6,7,8-Tetrahydrobiopterin Cofactor. Journal of Medicinal Chemistry. 1999;42:4108–4121. doi: 10.1021/jm981129a. [DOI] [PubMed] [Google Scholar]
- 15.Cavazzuti A, Paglietti G, Hunter WN, Gamarro F, Piras S, Loriga M, Allecca S, Corona P, McLuskey K, Tulloch L, Gibellini F, Ferrari S, Costi MP. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proceedings of the National Academy of Sciences. 2008;105:1448–1453. doi: 10.1073/pnas.0704384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel O, Satchell J, Baell J, Fernley R, Coloe P, Macreadie I. Inhibition studies of sulfonamide-containing folate analogs in yeast. Microb Drug Resist. 2003;9:139–146. doi: 10.1089/107662903765826723. [DOI] [PubMed] [Google Scholar]
- 17.Hill B, Liu Y, Taylor SD. Synthesis of alpha-fluorosulfonamides by electrophilic fluorination. Org Lett. 2004;6:4285–4288. doi: 10.1021/ol048249z. [DOI] [PubMed] [Google Scholar]
- 18.Marques SM, Enyedy EA, Supuran CT, Krupenko NI, Krupenko SA, Santos MA. Pteridine-sulfonamide conjugates as dual inhibitors of carbonic anhydrases and dihydrofolate reductase with potential antitumor activity. Bioorg Med Chem. 2010;18:5081–5089. doi: 10.1016/j.bmc.2010.05.072. [DOI] [PubMed] [Google Scholar]
- 19.Taylor EC, Kobayashi T. Pteridines. XXXII. 2-Amino-3-cyano-5-chloromethylpyrazine 1-oxide and its conversion to 6-alkenyl-substituted pteridines. The Journal of Organic Chemistry. 1973;38:2817–2821. [Google Scholar]
- 20.Taylor EC, Henrie RN, Portnoy RC. Pteridines. 44. A convenient synthesis of 6-formylpterin. The Journal of Organic Chemistry. 1978;43:736–737. [Google Scholar]
- 21.Sletzinger M, Reinhold D, Grier J, Beachem M, Tishler M. The Synthesis of Pteroylglutamic Acid. Journal of the American Chemical Society. 1955;77:6365–6367. [Google Scholar]
- 22.Freisleben A, Schieberle P, Rychlik M. Syntheses of Labeled Vitamers of Folic Acid to Be Used as Internal Standards in Stable Isotope Dilution Assays. Journal of Agricultural and Food Chemistry. 2002;50:4760–4768. doi: 10.1021/jf025571k. [DOI] [PubMed] [Google Scholar]
- 23.General procedure for the synthesis of target compounds: Eg. 4-((2-Amino-4-oxo-3,4-dihydropteridin-6-yl)methylamino)-N-(5-methylisoxazol-3-yl)benzenesulfonamide (15): A mixture of N-(6-((4-(N-(5-methylisoxazol-3-yl)sulfamoyl)phenylamino)methyl)-4-oxo-3,4-dihydropteridin-2-yl)acetamide (0.12 g, 0.255 mmol) in 0.1 M NaOH (24 mL) was stirred at room temperature overnight. The reaction was quenched by adjusting the pH to 8 with diluted HCl. The reaction solution was concentrated to about 5mL, then the reaction solution was adjusted to pH 5~6 with diluted HCl and filtered, washed with water and MeOH, then dried to give a yellow solid (0.106 g, 97%); 1H NMR (400 MHz, DMSO-d6): δ 2.25 (s, 3 H), 4.46 (d, 2 H, J = 5.6 Hz), 6.03 (s, 1 H), 6.68, 7.50 (dd, 4 H, J = 8.8 Hz), 7.07 (br.s, 2 H, D2O exch), 7.33 (br.t, 1 H, J = 5.6 Hz, D2O exch), 8.64 (s, 1 H), 11.09 (br.s, 1 H, D2O exch), 11.57 (br.s, 1 H, D2O exch); m/z 429.2 (ES+H+).
- 24.Zlitni S, Ferruccio LF, Brown ED. Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat Chem Biol. 2013;9:796–804. doi: 10.1038/nchembio.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Hammoudeh D, Lin W, Das S, Yun MK, Li Z, Griffith E, Chen T, White SW, Lee RE. Development of a pterin-based fluorescent probe for screening dihydropteroate synthase. Bioconjug Chem. 2011;22:2110–2117. doi: 10.1021/bc200346e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Babaoglu K, Qi J, Lee RE, White SW. Crystal structure of 7,8-dihydropteroate synthase from Bacillus anthracis: mechanism and novel inhibitor design. Structure. 2004;12:1705–1717. doi: 10.1016/j.str.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 27.ATCC Medium 2511. The carbon source in this media is glucose.
- 28.Bock L, Miller GH, Schaper KJ, Seydel JK. Sulfonamide structure-activity relationships in a cell-free system. 2. Proof for the formation of a sulfonamide-containing folate analog. J Med Chem. 1974;17:23–28. doi: 10.1021/jm00247a006. [DOI] [PubMed] [Google Scholar]
- 29.Smith DA, Jones RM. The sulfonamide group as a structural alert: A distorted story? Curr Opin Drug Discov Devel. 2008;11:72–79. [PubMed] [Google Scholar]
- 30.Hammoudeh DI, Zhao Y, White SW, Lee RE. Replacing sulfa drugs with novel DHPS inhibitors. Future Med Chem. 2013;5:1331–1340. doi: 10.4155/fmc.13.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.