

Abstract
Proteins often possess highly specific biological activities that make them potential therapeutics, but their physical and chemical instabilities during formulation, storage, and delivery have limited their medical use. Therefore, engineering of nano-sized vehicles to stabilize protein therapeutics and to allow for targeted treatment of complex diseases, such as cancer, is of considerable interest. A micelle-like nanoparticle (NP) was designed for both, tumor targeting and stimulus-triggered release of the apoptotic protein cytochrome c (Cyt c). This system is composed of a Cyt c NP stabilized by a folate-receptor targeting amphiphilic copolymer (FA-PEG-PLGA) attached to Cyt c through a redox-sensitive bond. FA-PEG-PLGA-S-S-Cyt c NPs exhibited excellent stability under extracellular physiological conditions, whereas once in the intracellular reducing environment, Cyt c was released from the conjugate. Under the same conditions, the folate-decorated NP reduced folate receptor positive HeLa cell viability to 20% while the same complex without FA only reduced it to 80%. Confocal microscopy showed that the FA-PEG-PLGA-S-S-Cyt c NPs were internalized by HeLa cells and were capable of endosomal escape. The specificity of the folate receptor-mediated internalization was confirmed by the lack of uptake by two folate receptor deficient cell lines: A549 and NIH-3T3. Finally, the potential as anti-tumor therapy of our folate-decorated Cyt c-based NPs was confirmed with an in vivo brain tumor model. In conclusion, we were able to create a stable, selective, and smart nanosized Cyt c delivery system.
Keywords: active targeting, biodegradable polymer, drug delivery, nanoparticle, protein drug, triggered release
Graphical abstract
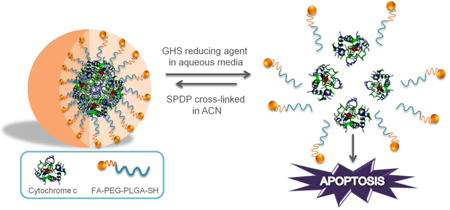
1. Introduction
The inactivation of the upstream components of the signaling pathways (such as the p53 pathway) that activate the release of cytochrome c (Cyt c) from the mitochondria to the cyto-plasm in response to DNA damage, disable apoptosis in many cancer cells.1 This explains the limited efficiency of many classic chemotherapeutic agents (e.g., Taxol®) that cause DNA damage leading to p53-dependent apoptosis. Such limitations and a generally low therapeutic index have spurred efforts to identify new and more effective chemotherapeutic agents that can be tolerable in high doses and act independently of the p53 pathway. For example, some previous studies have demonstrated that the delivery of an apoptotic protein, such as Cyt c, directly to the cytoplasm of various cancer cell lines induced apoptosis in them.2-5 However, due to several protein delivery and stability issues,6 the use of proteins as therapeutics is limited. A major challenge in the development of delivery systems that seek to improve protein drugs is to achieve high drug loading of the device and an efficient release of the active protein at the target site.7 Recently, we overcame payload limitations commonly seen in delivery systems with therapeutic proteins6,7 by designing protein-based nanoparticles (NPs) coated with a biodegradable polymer PLGA.8 The designed Cyt c drug delivery system incorporates a triggered release mechanism mediated by the reducing environment inside of the cells and it showed improved cytotoxicity and biocompatibility when compared to previous reported Cyt c formulations.2,5
For the in vivo application of this kind of NP delivery system additional considerations are necessary. Although the NP size takes advantage of passive targeting, i.e., through the enhanced permeation and retention (EPR) effect characteristic of tumors,9-12 it is not the sole determinant of tumor specificity in in vivo studies. The surface of the NP should be modified (e.g., by attaching PEG molecules) to increase the blood circulation half-life of the system. The attachment of targeting moieties (i.e., active targeting) also improves the internalization and intra-cellular delivery of NPs. Only two FDA approved nano-formulations for cancer therapy (i.e., Abraxane and Rexin-G) have combined passive targeting with active targeting. At present there are few NP formulations of drug delivery systems which combine active targeting and triggered release to treat cancer. In this work we improve our previous smart-release nano-carrier by transforming it into an active delivery system, using a folate-poly(ethylene glycol)-poly(lactic-co-glycolic acid)-thiol (FA-PEG-PLGA-SH) conjugate to coat Cyt c-based NPs. This new vehicle for therapeutic proteins possesses the stability advantages of a micellar-like NP system: the hydrophobic polymer shell region (PLGA) stabilizes the protein core, whereas the hydrophilic polymer shell region (FA-PEG) facilitates the suspension in aqueous media, making the particle an appropriate candidate for intravenous administration. Typically, polymeric micelles consisting of amphiphilic polymers that are able to spontaneously self-assemble into core-shell nanostructures are used for the loading of hydrophobic drugs.13,14 In our case, a polymer/protein micelle-like NP is formed in an organic medium mediated by a cross-linking reaction between the amphiphilic polymer and the amine groups on the hydrophilic protein NP surface (Figure 1).
Figure 1.
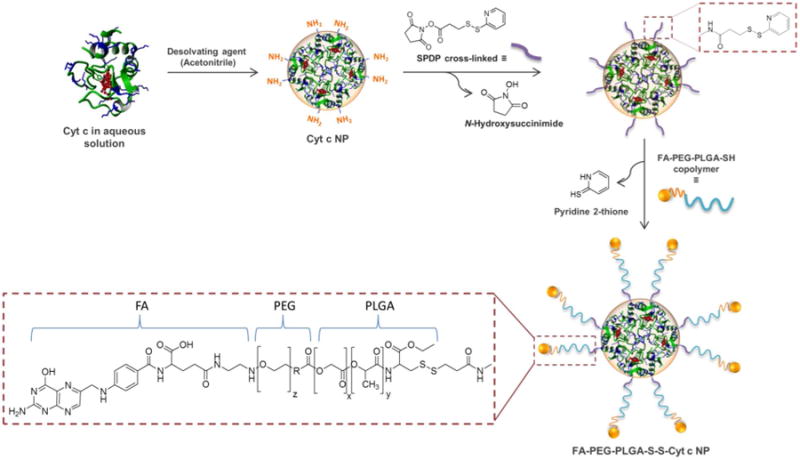
Synthesis of polymer/protein Cyt c-based NPs. Cyt c NPs are made insoluble in aqueous media by a coat consisting of FA-PEG-PLGA-SH copolymer.
We chose folic acid (FA) as the ligand displayed on the surface of Cyt c NPs because the folate receptor (FR) is a well-known tumor marker that binds folate-drug conjugates with high affinity and carries them into the cells via receptor-mediated endocytosis.11,15 FA is important in the formation of new cells because it is required in one carbon metabolic reactions and consequently is essential for the synthesis of nucleotide bases.10,15 The folate receptor is overexpressed by tumor cells in several tumors but in normal tissue it is low in concentration.16 In addition, folate receptors present in normal cells (i.e., lung, brain, and kidneys cells) are inaccessible to the NP delivered via intravenous routes.15,16 Our new drug delivery vehicle combines passive and active targeting capabilities with a stabilized nanoparticle protein formulation to facilitate an intracellular reduction-triggered mechanism for the release of Cyt c.
2. Experimental Section
2.1. Chemicals and Reagents
Cytochrome c from equine heart, reduced glutathione ethyl ester, 3-nitro-2-pyridinesulfenyl chloride (Npys-Cl), O-methyl-O′-succinylpolyethylene glycol (PEG-COOH, 2,000 Da), fluorescein isothiocyanate isomer I (FITC), protease inhibitor cocktail, N,N-diisopropylethylamine (DIEA), and tributhylphosphine (PBu3) were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile (HPLC grade) and dimethylformamide (DMF) were purchased from Fisher (Waltham, MA). Succinimidyl-3-(2-pyridyldithio)propionate (SPDP) was purchased from Proteochem (Denver, CO). Poly(lactide-co-glycolide) thiol end-capped (PLGA-SH, 30,000 Da, copolymer ratio 1:1) and folate-poly(ethylene glycol)-carboxylic acid (Folate-PEG-COOH, 3,000 Da) were purchased from Akina, Inc (West Lafayette, IN). 4′, 6-Diamidino-2-phenylindole (DAPI), propidium iodide (PI), and FM-4-64 membrane stain were purchased from Invitrogen (Grand Island, NY). All reagents were used without further purification. All other chemicals were from various commercial suppliers and were at least of analytical grade. HeLa, A549, and NIH-3T3 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and grown according to ATCC instructions.
2.2. Synthesis of FA-PEG-PLGA-SH
To protect the thiol group of PLGA-SH with Npys,17 PLGA-SH (500 mg, 0.016 mmol) was dissolved in 25 ml of anhydrous DMF and reacted with Npys-Cl (3.3 mg, 0.024 mmol) overnight at RT (Figure 2). The resultant product PLGA-Npys was dialyzed against distilled water overnight using 10,000 g/mol MW cut off dialysis tubing. The product precipitated in the tubing. The pellet was collected by centrifugation (10,000 rpm for 15 min at 4°C in a Hermle Z323K centrifuge) and then dried by lyophilization. PLGA was attached to folate-conjugated poly(ethylene glycol) (FA-PEG-COOH) to synthesize a FA-PEG-PLGA copolymer. The free alcohol group on PLGA-Npys was linked to the terminal carboxyl group on the FA-PEG-COOH. Briefly, 400 mg of PLGA-Npys (0.0133 mmol) and 120 mg of FA-PEG-COOH (0.04 mmol) were dissolved in 20 mL of anhydrous DMF. An excess of N,N-diisopropylethylamine (DIEA) was added dropwise to the resulting polymer solution and the reaction mixture was stirred overnight. The resulting mixture was dialyzed against distilled water overnight using 10,000 g/mol MW cut off dialysis tubing. The precipitated product FA-PEG-PLGA-Npys was pelleted by centrifugation and the pellet was dried by lyophilization. The thiol group in PLGA was then deprotected with tributylphosphine (PBu3). Briefly, 200 mg of FA-PEG-PLGA-Npys (0.006 mmol) was dissolved in 10 mL of anhydrous DMF and was treated with an excess of PBu3 overnight at RT. The resulting mixture was dialyzed against water over-night, the precipitated product FA-PEG-PLGA-SH was pelleted by centrifugation and the pellet was dried by lyophilization. All reactions were carried out using flame-dried round bottom flasks under a N2 atmosphere. The formation of folate-conjugated copolymer, FA-PEG-PLGA-SH, was monitored and confirmed by 1H NMR analysis. NMR spectra were recorded on a Bruker AV-500 MHz spectrometer using dimethyl sulfoxide-d6 as solvent (DMSO-d6). Chemical shifts (δ) were calibrated relative to the DMSO-d6 signal at 2.50 ppm 1H NMR (500 MHz, DMSO-d6) δ: 8.53 (s, FA, 1H), 7.97 (s, FA, 2H), 6.68 (s, FA, 2H), 5.22 (m, PLGA), 4.93 (m, PLGA), 4.7 (s, FA, 2H), 3.34 (s, PEG/PLGA), 1.50 (m, PLGA) (Figure 2). A control polymer PEG-PLGA-SH (DMSO-d6) was synthesized following the same procedure using a modified PEG not carrying the terminal folate modification. 1 H NMR (500 MHz, DMSO-d6) δ: 5.22 (m, PLGA), 4.93 (m, PLGA), 3.34 (s, PEG/PLGA), 1.50 (m, PLGA).
Figure 2.
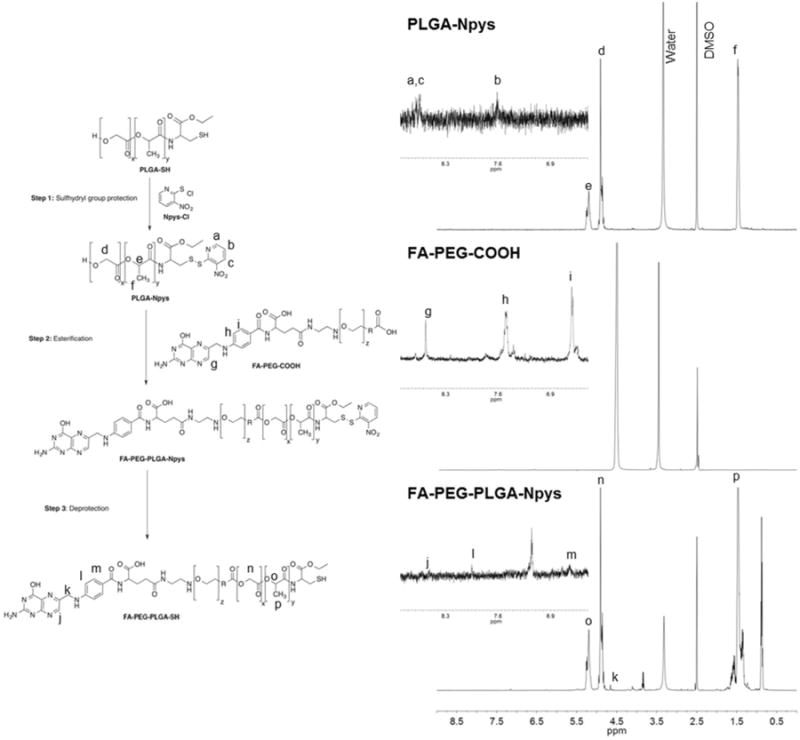
Synthesis of the FA-PEG-PLGA-SH conjugate. The starting reagents (FA-PEG-COOH and the Npys protected PLGA) and product were characterized by 1H NMR. The 1H NMR spectrum of the FA-PEG-PLGA-SH conjugate demonstrates the presence of the folate group.
2.3. Synthesis of FA-PEG-PLGA-S-S-Cyt c NP
Cyt c nanoparticles were obtained using a method developed by our laboratory (Figure 1).8 Briefly, 10 mg/ml of Cyt c in the presence of the excipient methyl-β-cyclodextrin (1:8 w/w) in nanopure water was solvent-precipitated by adding acetonitrile at a 1:4 volume ratio. Following the Cyt c nanoprecipitation, the SPDP linker (0.8 mg, 1:3 Cyt c-to-linker molar ratio) was added directly to the resulting suspension to modify the nanoparticle surface with the linker. This releases N-hydroxysuccinimide. After 30 min, 30 mg of FA-PEG-PLGA-SH (0.0009 mmol) was dissolved in 10 ml of acetonitrile and added to the mixture to form a thiol bond with the Cyt c attached linker releasing pyridine 2-thione. The mixture was allowed to react at RT for 18 h. To stop the reaction and to remove unreacted reagents, the nanoparticles were washed by multiple cycles of resuspension/centrifugation (8,000 rpm, 15 min). The supernatant was used to determine the level of covalent modification of the nanoparticles with the copolymer by measuring the concentration of pyridine-2-thione at 365 nm using the extinction coefficient 8.08×103 M-1cm-1.
2.4. In vitro release of Cyt c
The release of Cyt c from FA-PEG-PLGA-S-S-Cyt c NP was measured as described.8 In brief, 0.5 mg of FA-PEG-PLGA-S-S-Cyt c NP powder was suspended by sonication in 1 ml of 50 mM PBS with 1 mM EDTA at pH 7.4 and glutathione (GHS) concentrations of 0, 0.001, and 10 mM simulating extra- and intracellular conditions.5 Incubation was performed for various times at 37 °C, and the NPs were pelleted by centrifugation at 14,000 rpm for 10 min. The supernatant was removed and used to determine the concentration of released Cyt c. The protein concentration was determined by spectrophotometric analysis from the heme absorbance at 408 nm. The pellet was resuspended in GHS-PBS buffer. The amount of released protein was used to construct cumulative release profiles. The experiments were performed in triplicate, the results averaged, and the standard deviations calculated.
2.5. Zeta potential measurements
Zeta potential values of the different formulations of Cyt c NPs were determined using a Zetasizer Nanoseries from Malvern. The samples were dispersed in distilled water and subjected to ultrasonication at 240 W for 60 sec prior to the measurements. Zeta potential measurements were repeated three times for each sample, and the data are reported as averages with the standard deviations.
2.6. Dynamic light scattering (DLS) and scanning electron microscopy (SEM)
Particle size of FA-PEG-PLGA-S-S-Cyt c NP were determined by dynamic light scattering using a DynaPro Titan. The samples were dispersed in DMF and subjected to ultrasonication at 240 W for 30 sec prior to the measurements. SEM of the different formulations of Cyt c NPs was performed using a JEOL 5800LV scanning electron microscope at 20 kV. The samples were coated with gold for 10 sec to a thickness of 10 nm using a Denton Vacuum DV-502A.
2.7. Cell-free caspase-3 assay
The cell lysate was obtained as described by us.5 Briefly, FA-PEG-PLGA-Cyt c NP was reacted with 10 mM GHS, centrifuged at 8,000 rpm for 15 min and the supernatant was removed to determine the concentration of released Cyt c. The cell-free reaction was initiated by adding 100 μg/mL of Cyt c to freshly purified cytosol (3 mg/ml) in a total reaction volume of 50 μL. NPs of the non-apoptotic protein α-lactalbumin at 100 μg/mL were employed as negative control and Cyt c from the commercial source as positive control. The reaction was incubated at 37°C for 150 min. Next, the caspase-3 and caspase-9 assays were performed following the manufacturer's protocol (CaspACE™ assay; Promega, Madison, WI). The plate was incubated overnight at room temperature and the absorbance at 405 nm was measured in each well using a Thermo Scientific Multiskan FC. All measurements were performed in triplicate.
2.8. Cell culture
Human cervical carcinoma (HeLa), human lung carcinoma (A549), and mouse embryonic fibroblast (NIH-3T3) cells were maintained in accordance with the ATCC protocol. The GL261 glioma cell line derived from C57BL/6 mice was obtained from the NCI (Frederick, MD). The HeLa and A549 cells were cultured in minimum essential medium (MEM) and NIH-3T3 and GL261 cells in Dulbecco's modified Eagle's medium (DMEM). Both media contained 1% L-glutamine, 10% fetal bovine serum (FBS), and 1% penicillin in a humidified incubator with 5% CO2 and 95% air at 37°C. All experiments were conducted before cells reached 25 passages.
2.9. MTS Assay
Mitochondrial function was measured using the CellTiter 96 aqueous non-radioactive cell proliferation assay from Promega Corporation. HeLa cells (5,000 cells/well) were seeded in 96-well plates and incubated with serial dilutions (12.5, 25 and 50 μg/mL of Cyt c) of FA-PEG-PLGA-S-S-Cyt c NP and PEG-PLGA-S-S-Cyt c NP for 6 h. Controls, such as, 65 μg/mL FA-PEG-PLGA-SH and PEG-PLGA-SH were also tested. T-test analysis was used for comparison of two independent groups for cell viability. A difference between folate-targeted and folate-free Cyt c NP was considered statistically significant at a p-value of <0.05.
2.10. Investigation of Apoptosis Induction in Hela Cells by Confocal Laser Scanning Microscopy (CLSM)
HeLa cells (25,000 cells/well) were seeded in chambered cover-glass (4-wells). The cells were incubated with FA-PEG-PLGA-S-S-Cyt c NP at a Cyt c concentration of 12.5 μg/mL at 37°C for 6 h. For detection of apoptosis-dependent nuclear fragmentation, the cells were washed with PBS (1X) and incubated with DAPI (300 nM) and thereafter with PI (75 μM) for 5 min each. HeLa cells were then fixed using 3.7% formaldehyde. The coverslips were examined under a Zeiss laser-scanning microscope 510 using a 40× oil immersion objective. Co-localization of DAPI and PI upon internalization into HeLa cells was determined, which is representative of highly condensed and fragmented chromatin in apoptotic cells.2,5,18 DAPI was excited at 405 nm and its emission was detected at 420-480 nm. PI was excited at 561 nm and was detected above 575 nm.
2.11. In vitro Cellular Uptake and Endosomal Escape of Folate-targeted Cyt c-based NP
The internalization of FA-PEG-PLGA-S-S-Cyt c NPs and the ability to escape endosomal entrapment was determined by CLSM. 25,000 HeLa and A549 cells and 50,000 NIH-3T3 cells were seeded in chambered cover-glass (4-wells). To execute these experiments, NPs were modified with fluorescein isothiocyanate (FITC) via the amine group.19 HeLa cells were seeded and incubated at 37°C for 6 h with both: FITC-labeled Cyt c-based NPs (12.5 μg/mL of Cyt c) and an endosome marker (FM-4-64; 10 μg/mL). In an independent experiment, HeLa, A549 and NIH-3T3 cells were incubated only with FITC-labeled Cyt c-based NPs for 0.5 h, 2 h, and 6 h. Afterwards, the medium was removed, and the cells were washed with PBS three times followed by fixation of the cells with 3.7% formaldehyde. For endosomal escape analysis, the coverslips were examined under a Zeiss laser-scanning microscope 510 using a 100× objective and excitation at 488 nm. FITC-label Cyt c NPs and endosome marker fluorescence were detected at wavelengths between 513 and 588 nm and between 599 and 738 nm, respectively. For cellular uptake analysis, the coverslips were examined using a 40× objective. FITC-label Cyt c NP fluorescence was excited at 488 and detected above 505 nm. Mann-Whitney analysis was used for comparison of two independent groups for cellular uptake fluorescent intensity measurements. A difference between folate-positive (HeLa) and folate-negative (A549 or NIH-3T3) cell line was considered statistically significant at a p-value of <0.05.
2.12. Intracranial Implantation of Glioma Cells
All experiments with live animals were performed in compliance with the relevant laws and institutional guidelines of the National Institutes of Health, and institutional committees have approved the experiments. All surgery was performed under isoflurane anesthesia, and all efforts were made to minimize suffering. To test the effectiveness of FA-PEG-PLGA-S-S-Cyt c NPs in an animal model, tumor-bearing C57BL/6 mice (12-16 week old) were prepared by delivering 1 μl of GL261 glioma cells suspension (2×104 cells/μl in PBS) into the brain through a small opening in the cranial window (depth: 3 mm, duration: 2 min).20
2.13. Ex vivo Cellular Uptake of Folate-targeted Cyt c-based NP
14 d after implantation of GL261 cells, mice were decapitated and the brain removed from the skull in ice-cold (2-4°C) artificial cerebrospinal fluid (ACSF) composed of: 126 mM NaCl, 2.5 mM KCl, 1.2 mM NaH2PO4, 1.0 mM MgCl2, 2.0 mM CaCl2, 25 mM glucose, 25 mM NaHCO3, saturated with 95% O2 and 5% CO2. The slices (250 μm) were cut from the tumor containing tissue using a vibroslicer (Leica VT 1000S) and placed in an incubation chamber containing ACSF at 35°C. FITC-label Cyt c NPs was added to the incubation chamber for the final concentration 100 μg/ml. After four hours, slices were washed in fresh ACSF, placed on microscope slides and covered with glass coverslips. Slices were immediately visualized using an Olympus Fluoview FV1000 confocal microscope (Olympus, Japan) with 4×-10× objectives or 40-60× oil immersion objectives.
2.14. Local Administration of Folate-targeted Cyt c-based NP to glioma tumor
Tumors were allowed to grow for five days after implantation and then mini-osmotic pumps (Alzet®, DURECT, model 2004) were installed for local continous administration of drugs (Calbiochem, Billerica, MA, USA) to the tumor. Animals were anesthetized and a 3 mm brain infusion cannula connected to a pump was set up at the previous tumor implantation site using a brain infusion kit (Alzet®, DURECT). The pumps were placed subcutaneously on the mouse back. The drug was infused at 1 μl/h, 100 mg/ml of FA-PEG-PLGA-S-S-Cyt c NPs over 3 days. Pumps with normal saline solution were used as a control.
2.15. In vivo tumor size evaluation
Seven days following FA-PEG-PLGA-S-S-Cyt c NPs administration, animals were anesthetized with pentobarbital (50 mg/kg) and transcardially perfused with PBS followed by 4% paraformaldehyde (PFA). Brains were removed and postfixed in 4% PFA/PBS for 24 h at 4°C, followed by 0.15 M, 0.5 M, and 0.8 M sucrose at 4°C until fully dehydrated. Brains were then frozen-embedded in Cryo-M-Bed embedding compound (Bright Instrument, Huntingdon, England) and cut using a Vibratome UltraPro 5000 cryostat (American Instrument, Haverhill, MA). The sliced tumor tissues (10 μm) were stained by Hematoxylin and Eosin (H&E) and observed by an Olympus BX51WI microscope. Tumor size was calculated as the sum of tumor area × section thickness for each section containing a tumor tissue. Results were expressed as mean ± standard deviation (SD). Statistical probability was calculated using GraphPad software. Unpaired t-tests were used to determine significance between groups. A difference in tumor size between tumor-bearing mice treated with Cyt c-based NP and PBS was considered statistically significant at a p-value of <0.05.
2.16. In vivo apoptosis assay
Immunocytochemical detection of signs of apoptosis was performed next day following administration of FA-PEG-PLGA-S-S-Cyt c NPs to the tumors. 10 μm coronal frozen brain slices encompassing the entire tumor were proseeded with TUNEL In Situ Cell Detection Kit, Roche, according to the manufacturer protocol and further evaluated with an Olympus BX51WI microscope and Q-capture Pro software (Olympus).
3. Results
In this study we coupled a folic acid-functionalized PEG molecule to a selected PLGA polymer in order to include an active tumor-targeting component in our drug delivery system and to protect it from the reticular endotelial system (RES). The complete synthesis of the FA-PEG-PLGA-SH conjugate is shown in Figure 2. The first step involved protection of the sulfhydryl group with Npys-Cl affording the disulfide PLGA-Npys polymer. Then, esterification of PLGA-Npys polymer with FA-PEG-COOH mediated by DIEA afforded the FA-PEG-PLGA-Npys ester. Finally, the deprotection of the Npys with PBu3 resulted in the desired conjugated FA-PEG-PLGA-SH thiol. The product was characterized by 1H NMR spectroscopy (Figure 2). A control polymer (PEG-PLGA-SH) was synthesized following the same procedure using a modified PEG not carrying the terminal folate modification. This polymer was used as a control for folate receptor-positive cell line selectivity experiments.
The resulting FA-PEG-PLGA-SH conjugate was chemically linked to surface lysine residues of Cyt c NPs. To assure the NP integrity during the coating process and to avoid NP aggregation, Cyt c NPs were not subjected to centrifugation after their SPDP modification. The SPDP and polymer conjugated coating procedures were performed consecutively. The characterization of the covalent modification of the FA-PEG-PLGA-SH conjugate onto the NPs was determined by quantitative ultraviolet-visible (UV-Vis) spectrophotometric analysis. The sulfhydryl-reactive portion of the SPDP reagents is the 2-pyridyldithio group, which reacts with sulfhydryls resulting in the displacement of a pyridine-2-thione group. Figure 3A shows the characteristic increase in the absorbance at 280 nm and 365 nm resulting from a reaction between SPDP with the reducing agent dithiothreitol (DTT). The same trend was observed for the released pyridine-2-thione group when the SPDP at the NP surface reacted with FA-PEG-PLGA-SH. A similar increase in absorbance occurred when the same FA-PEG-PLGA coated NP was constructed using the non-apoptotic protein α-lactalbumin (LA). This was not observed when SPDP reacted with the lysine at the surface of the Cyt c NPs or when the polymer was not attached to the SPDP-Cyt c NPs. The molar ratio of Cyt c-to-FA-PEG-PLGA after the coating reaction was calculated from the released pyridine-2-thione absorbance and was found to be 2:1.
Figure 3.
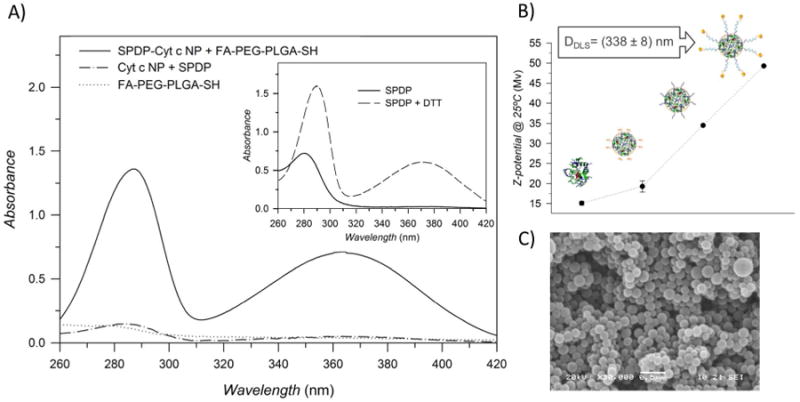
Characterization of the modification of protein-based NPs with FA-PEG-PLGA-SH. A) UV-VIS spectra of the supernatant recovery after the coating process. The release of the pyridine-2-thione group produced an absorbance increase at around 280 nm and 365 nm. B) Z-potential of Cyt c in water before and after nanoprecipitation and after each coating step. The particle diameter of the resulted nanoparticle was measured with DLS. C) SEM image of the resulting Cyt c-based NPs coated with FA-PEG-PLGA.
Zeta potential values were used to qualitatively confirm the NP surface modification. An increment in Zeta potential positive value after both the protein nanoprecipitation and the NP coating step was observed (Figure 3B). Here it is important to clarify that Cyt c NP was not encapsulated by the copolymer but decorated at the surface with the copolymer. After the NP modification, the total amount of Cyt c (a positively charged protein) in the resulting system is higher than the negatively charged copolymer. Therefore, the resulting Cyt c-based NPs coated with FA-PEG-PLGA remained positively charged and the increment in charge is mainly due to the characteristic stability of micellar-like NP system in aqueous medium. After coating with the FA-PEG-PLGA copolymer the magnitude of the zeta potential of Cyt c-based NPs was greater than +30 mV, which typically is associated with a high degree of colloidal stability. Suspensions with a low negative or positive zeta potential value (values of only 20 mV or less) will eventually undergo detrimental aggregation due to Van Der Waals inter-particle attractions.21 Since opsonization and uptake by macrophages of the RES of negatively charged species result in reduced circulation time in blood, the positive charge of our micelle-like NPs may result in increased circulation time.9 The particle diameter of the resulting Cyt c-based NPs coated with FA-PEG-PLGA suspended in aqueous solution was 338 ± 8 nm and therefore it should be able to accumulate in the tumor through passive targeting. However, we acknowledge that molecules with positive charge rapidly bind to vascular endothelial cells, which could reduce tumor drug accumulation by means of the EPR effect. A scanning electron microscopy (SEM) image was obtained to determine the NP shape after the coating process. SEM images of lyophilized FA-PEG-PLGA-S-S-Cyt c NP show a spherical regular shape and confirm the nanometer range particle size determined by DSL (Figure 3C).
We measured the in vitro redox-response kinetics and release, as well as the caspase activation capability of coated Cyt c NPs after stimulus-induced disassembly. The FA-PEG-PLGA coated Cyt c NPs exhibited excellent release properties at an intracellular glutathione concentration (10 mM) but not at an extracellular glutathione concentration (1 μM) due to the presence of the S-S bond in the linker (Figure 4A). We found that only a small amount of Cyt c was released using no or 1 μM of glutathione. In contrast, when exposing the FA-PEG-PLGA-S-S-Cyt c NPs to 10 mM glutathione, Cyt c was completely released from the micelle-like system in less than 24 h. In contrast, the system initially developed (i.e., PLGA-S-S-Cyt c NP) showed an unwanted release of almost 20% under extracellular conditions and only around 80% of the protein was released under reducing conditions.8 The stability of the improved Cyt c-based NPs in an aqueous non-reducing media may be attributed to their significantly higher surface charges or zeta potential resulting from the micelle-like structure (see Figure 3B).
Figure 4.
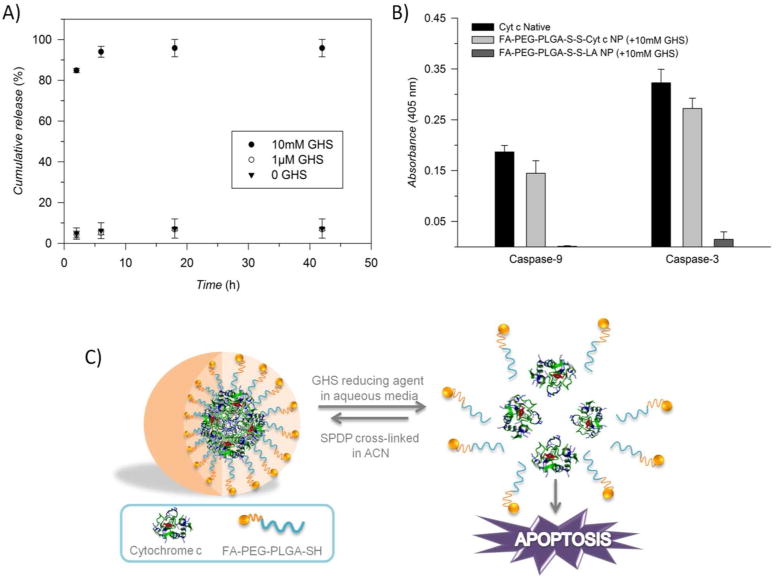
A) Cumulative release of Cyt c from FA-PEG-PLGA-coated NPs after exposure to various conditions of GHS. B) The capability of Cyt c-based NPs coated with FA-PEG-PLGA-SH to activate caspase-9 and -3, after exposure to the reducing agent glutathione (GHS). This capability is contrasted with native Cyt c and FA-PEG-PLGA-S-S-LA NP (LA= α-lactalbumin). C) Scheme of Cyt c release in the intracellular reducing environment to induce apoptosis.
Once in the cytosol, cytochrome c interacts with the apoptotic protease activating factor 1 (Apaf-1), resulting in the processing and activation of pro-caspase-9 in the presence of dATP. Mature caspase-9 activates, in turn, procaspase-3, leading to the activation of a caspase cascade which plays a fundamental role in the initiation and execution of apoptosis.22,23 Since Cyt c is a cell membrane impermeable protein,24 the capability of Cyt c to still interact with Apaf-1 and induce apoptosis after NP formulation was tested in a cell-free system. The integrity of the soluble protein after the nanoprecipitation procedure was compared with native Cyt c and with a non-apoptotic protein [(LA)-based NPs]. As expected, no caspase activation occurred upon addition of LA to fresh cytosol, while reduced Cyt c-based NPs were able to activate both caspase 9 and caspase 3 almost as well as native Cyt c did (Figure 4B). Figure 4C shows a schematic representation of how Cyt c is inducing apoptosis when released from the micellar-like NP in the reducing environment inside of the cells.
Once we confirmed that the Cyt c-based NPs coated with FA-PEG-PLGA-SH had the capability to interact with Apaf-1 and induce apoptosis upon rehydration when exposed to a reducing medium, confocal laser-scanning microscopy (CLSM) was conducted to investigate the situation after cellular uptake. Cells undergoing apoptosis display characteristic nuclear segmentation and chromatin condensation in the cell nuclei. HeLa cells, a folate receptor overexpressing carcinoma cell line,25 were incubated with the corresponding samples for 6 h and the cells were stained with PI and DAPI. Co-localization of DAPI and PI occurred when cells were incubated with folate-decorated Cyt c-based NPs (Figure 5A), which is indicative of ongoing apoptosis. In contrast, few cells show co-localization of DAPI and PI and those cells show less intense PI staining when they were incubated with folate-free Cyt c-based NPs. Cells without treatment or treated with the FA-PEG-PLGA-SH block polymer or FA-PEG-PLGA-S-S-LA NPs showed no indication for dye co-localization and thus apoptosis. For the detection of adherent apoptotic cells we used a Cyt c concentration of 12.5 μg/mL whereas for our previous system (PLGA coated Cyt c NPs) we used 25 μg/mL. When using a Cyt c concentration of 25 μg/mL for FA-PEG-PLGA-S-S-Cyt c NPs no adherent cells were present in our experiments suggesting an enhanced cytotoxic effect of FA-PEG-PLGA-coated Cyt c-based NPs.
Figure 5.
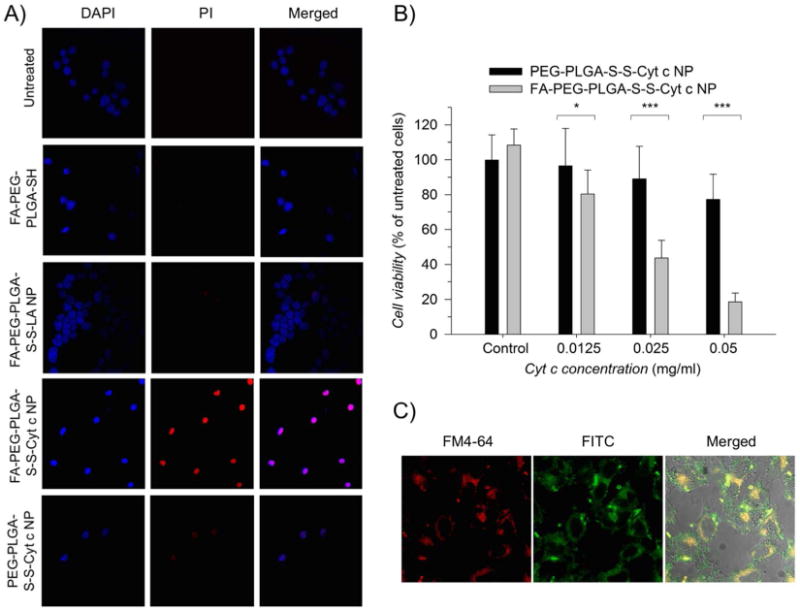
A) Study of DAPI and propidium iodine (PI) co-localization for the detection of apoptotic cells. Selective induction of apoptosis observed in HeLa cells incubated with the FA-PEG-PLGA-S-S-Cyt c NPs. No cellular or minimal apoptosis observed in untreated HeLa cells or when incubated with controls: FA-PEG-PLGA-SH conjugate, FA-PEG-PLGA-S-S-LA NPs, and PEG-PLGA-Cyt c NPs. B) Comparison of folate decorated and folate-free Cyt c-based NP cyto-toxicity in HeLa cells (all groups were statistical significance even at 0.125 mg/ml where the error bars overlap). HeLa cell incubated with polymers alone (control) had no effect on the cell viability. Asterisks (***) and (*) indicate statistical significance at p<0.0001 and p<0.05, respectively. The error bars are the calculated SD. C) Confocal fluorescence images of HeLa cells after incubation for 6h with FM4-64 (labeling endosomes), FITC labeled Cyt c-based NPs, and the merged view of A and B. Yellow color indicates localization of the NPs in the endosomes.
To investigate if the FA targeting moiety enhances the cytotoxic effect of Cyt c-based NP, HeLa cells were also incubated with FA-PEG-PLGA-S-S-Cyt c NPs and PEG-PLGA-S-S-Cyt c NPs at different Cyt c concentrations for 6h. Cyt c NPs coated with the FA-PEG-PLGA-SH copolymer induced a significant reduction in cell viability to 20% after 6h of incubation, in particular at the 50 μg/ml Cyt c concentration (Figure 5B). In contrast, Cyt c NPs coated with the PEG-PLGA-SH copolymer only reduced cell viability to 80% under the same conditions. Similar results have been observed before with other folate-functionalized systems and their folate-free counterparts.26,27 Indeed, while PEGylation reduces the rate of RES uptake and increases the circulation half-life of various types of NPs, PEG-modified molecules sometimes display slower uptake into tumor cells.28 Combining the passive EPR-mediated targeting significant with an additional ligand-mediating targeting not only amplifies the specificity of these therapeutic NPs but also facilitates their cellular uptake and aids in overcoming the PEG dilemma.9,10,29 As controls, FA-PEG-PLGA-SH and PEG-PLGA-SH conjugates were added to HeLa cells at the same concentration as in the corresponding experiments at the highest FA-PEG-PLGA-S-S-Cyt c NP or PEG-PLGA-S-S-Cyt c NP concentrations, respectively. No significant cytotoxicity was observed after 6 h with either compound.
The ability to avoid endosomal entrapment of our folate receptor targeted Cyt c NPs was also interrogated. To execute these experiments, Cyt c-based NPs labeled with FITC and the endosome marker FM-4-64 were used. Green fluorescence observed in the micrographs is due to the internalization of the folate-decorated Cyt c-based NPs, and red fluorescence shows labeled endosomes. Yellow fluorescence is due to the co-localization of the two dyes and shows Cyt c NP entrapment in endosomes.30 As depicted in Figure 5C, after 6 h of incubation a significant amount of Cyt c is located in the cytoplasm while yellow fluorescence indicates NPs being present in endosomes. These observations are compatible with the expected uptake by endocytosis.
The effect of our folate-decorated NPs on folate-deficient cell lines was also tested as an additional control. The folate-positive human cervical carcinoma HeLa cell line was compared with two well studied folate-negative cell lines: human lung carcinoma cells (A549) and mouse embryo fibroblasts (NIH-3T3).25,31 Confocal fluorescence microscopy showed an enhanced fluorescence in the cytoplasm of HeLa cells after 6h of incubation with FA-PEG-PLGA-S-S-Cyt c NPs whereas no significant or minimal internalization was observed in A549 and NIH-3T3 cells (Figure 6A). This experiment confirms a preferential intracellular uptake by folate-receptor-positive cancer cells over folate-receptor-negative cells, indicating that the cellular uptake occurred via a folate-receptor-mediated endocytosis mechanism. 32,33
Figure 6.
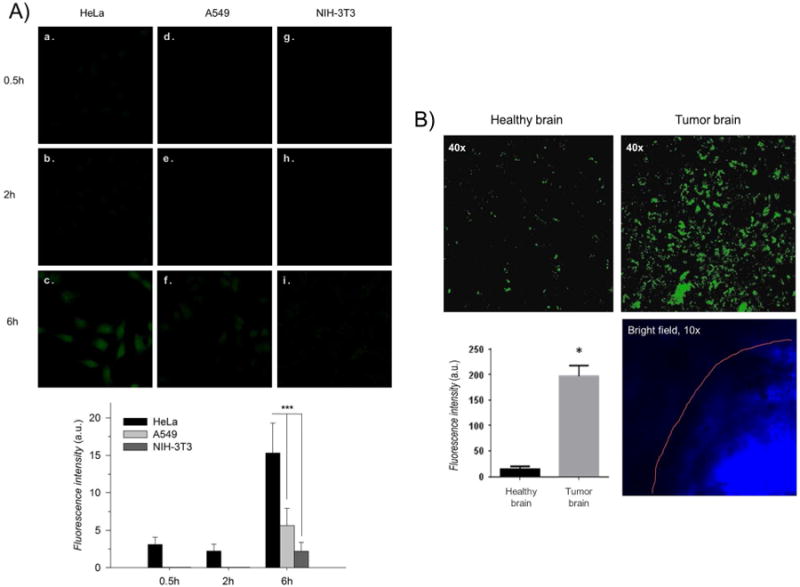
In vitro and ex vivo internalization of the folate-decorated Cyt c-based NPs observed by confocal microscopy after different incubation times. A) A folate-positive carcinoma HeLa cell line (a-c) showed better uptake behavior in comparison with folate-deficient carcinoma A549 (d-f) and a normal NIH-3T3 (g-i) cell lines. B) Brain slices from a tumor-bearing mouse showed more intense fluorescence signals of FITC labeled Cyt c-based NPs in the tumor cells in comparison with the healthy brain tissue. Asterisks (***) and (*) indicate statistical significance at p<0.0001 and p<0.05, respectively. The error bars are the calculated SD.
Indeed, many are the factors determinant of drug delivery and tumor specificity in in vivo studies. For example, the blood–brain barrier (BBB) and poor tissue penetration represents the biggest challenge for brain tumor targeted therapy.34 In this study we also tested the potential of a local delivery of our folate-decorated NPs to treat malignant glioma, the most frequent primary tumor within the central nervous system (CNS). Studies have confirmed the high expression levels of folate receptor in gliomas.35 Thus, a study in ex vivo brain tissue from GL261 glioma-bearing mice was conducted to investigate the glioma preferential uptake of our folate-decorated NPs. The results confirmed the tumor-targeted delivery of folate-decorated Cyt c-based NPs (Figure 6B). After 2 h tissue exposure to Cyt c-based NPs labeled with FITC, the fluorescence signal was mostly detected in the area of tumor. The healthy brain tissue has also demonstrated some non-specific accumulation of fluorescence, but the intensity of the fluorescence in healthy tissue showed 20-times less accumulation of Cyt c-based NPs compared to the tumor tissue area.
We evaluated the in vivo therapeutic potential by administrating Cyt c-based NPs intra-tumorally to C57BL/6 mice bearing GL261 glioma tumors. The delivery of NPs was through the mini-osmotic pump in order to avoid the possible insufficiency of the drug into the brain tissue, related to the blood brain barrier. Evaluation of tumor size seven days after the treatment termination revealed, that animals that received Cyt c-based NP treatment developed tumors 40% smaller in size compared to the control mice (PBS treatment) (Figure 7A). In addition, in vivo TUNEL investigation of tumors, performed on the next day after the treatment termination, revealed strong signs of apoptosis in tumor area but not in healthy tissue (Figure 7B) in all animals that received Cyt c-based NP treatment. The treatment provided did not cause any adverse effect on animals, including neurological effects (decreased activity/responsiveness, weight loss, or abnormal posture), indicating the lack of healthy brain tissue damage. All of these data together confirm the strong cytotoxic abilities of our folate-decorated Cyt c-based NPs, specific for glioma tumors, but not for the healthy brain tissue.
Figure 7.
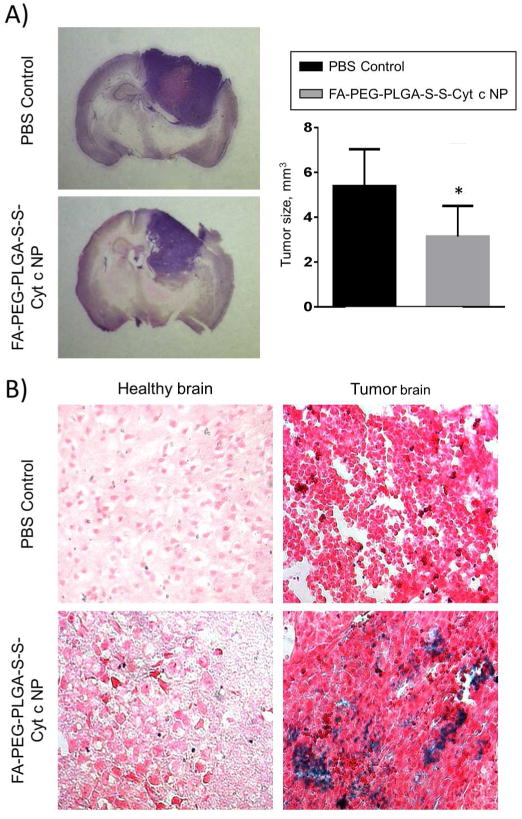
Evaluation of tumor size and signs of apoptosis in C57BL/6 mice with implanted GL261 glioma tumor after administration of folate-decorated Cyt c-based NPs; control animals received administration of PBS. A) Images and calculations of tumor size in mice with implanted glioma tumor 7 after the continuous 3 day administration of the drug. Calculations of tumor size were compared by t-test statistic analysis; mean ± SD and significant difference from control (*) is shown (p<0.05). B) TUNEL detection of apoptosis in healthy and tumor tissue areas of brain in mice the day after the continuous 3 d drug administration.
4. Conclusions
Increasing the drug accumulation in the tumor region and reducing toxicity to normal cells is a promising approach to tumor treatment. Nano-sized drug delivery systems have been extensively used to passively target tumors with anticancer agents and accumulate specifically in tumor cells. However, the lack of success in improving overall survival makes it imperative to further develop and refine drug delivery systems as well as to identify new and more effective chemotherapeutic agents. Indeed, for the EPR effect to work, not only the size but also the surface properties of drug delivery nanoparticles must be controlled to avoid uptake by the RES and to facilitate the selective cellular uptake by tumor cells. We tested if the incorporation of a folate-modified PEG molecule would improve our previous Cyt c-based NPs stabilized by PLGA. The resulting Cyt c-based NPs surrounded with FA-PEG-PLGA chains (i.e., FA-PEG-PLGA-S-S-Cyt c NP) are similar to micellar nanocarriers based on amphiphilic block copolymers. Nevertheless, polymer micelle nanocarriers are typically used with hydrophobic drugs, whereas in our case the amphiphilic polymer was employed to stabilize hydrophilic protein NPs. In addition, our micellar-like protein-based NPs were designed for both, the tumor targeting and stimuli release of Cyt c. A redox-responsive linker between the amphiphilic polymer and the protein allowed for the smart release of Cyt c inside the cell triggered by reducing intracellular conditions. Further-more, the folate-decorated system showed selective targeting for folate-receptor overexpressing cells and a potent cytotoxic effect after being internalized by the cells by receptor-mediated endocytosis. In a tumor-bearing mice model, folate-decorated Cyt c-based NPs were tumor tissue selective, caused massive apoptosis in tumor cells, and by this, effectively suppressed the tumor growth.
Our results suggest that such tumor-targeting protein-based NPs are potentially powerful drug delivery systems avoiding problems associated with many common cytotoxic agents. This system is made of biocompatible and biodegradable materials, thus it should reduce side effect by systemic toxicity associated with other materials. The previous limitations of delivery devices for therapeutic proteins, such as low protein loading and poor protein stability were also overcome with this system. The nano-size should be able to increase drug concentration in tumors through passive targeting. In addition, this system combines active targeting and triggered release strategies which improve the drug internalization and allow the maximum release of the drug at the targeted site. These redox-responsive polymer-coated protein-based NPs serve as a platform for the creation of drug delivery systems utilizing different apoptotic inducing proteins or pharmaceutical proteins for other therapeutics applications.
Supplementary Material
Acknowledgments
This work was supported in part by the Institute for Functional Nanomaterials (NSF Cooperative Agreement 1002410). M.M., A.C., C.M.F., T.G., and A.M. were supported by fellowships from the NIH Research Initiative for Scientific Enhancement (RISE) Program (2R25GM061151-12). M.M. was also supported by fellowship from National Science Foundation (NSF) GK-12 Program (Grant No. 0841338), C.M.F. by the Institute for Functional Nanomaterials (IFN) at the University of Puerto Rico (Grant No. EPS-1002410) and A.C. by the National Institutes of Health (NIH) through the Support of Competitive Research (SCoRE) Program (Grant No. 5SC1GM084826-04). A.D.T. and S.A.L-R. were supported by a NIH SC1 grant (Grant No. 5SC1CA19050402) provided by the NIGMS and NCI. A.D.T. is also supported by funding from the Puerto Rico Science, Technology, and Research Trust (Agreement No. 2013-000019), the University of Puerto Rico Score Stabilization Grant, and the Department of Chemistry at UPR RP. L.K. and J.D. were supported by a NIH grant (Grant No. 1SC2GM102040-01A1, 8G12MD007583-27, R25GM110513). The authors are very grateful to Mr. Bismark Madera, M.T., for his outstanding work and dedication to the confocal imaging experiments. The confocal microscopy experiments were performed in the NeuroImaging Facilities supported by NIH Grant P20GM103642.
Footnotes
Supporting Information: Figure S1. Near-UV Circular dichroism (CD) spectra show that no major changes in Cyt c tertiary structure occurred following the precipitation procedure using ACN as organic solvent.
Notes: The authors declare no competing financial interests and no conflicts of interest in this work.
References
- 1.Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13(3):332–337. doi: 10.1016/s0955-0674(00)00216-7. [DOI] [PubMed] [Google Scholar]
- 2.Santra S, Kaittanis C, Perez JM. Cytochrome C encapsulating theranostic nanoparticles: a novel bifunctional system for targeted delivery of therapeutic membrane-impermeable proteins to tumors and imaging of cancer therapy. Mol Pharm. 2010;7:1209–1222. doi: 10.1021/mp100043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim SK, Foote MB, Huang L. The targeted intracellular delivery of cytochrome C protein to tumors using lipid-apolipoprotein nanoparticles. Biomaterials. 2012;33:3959–3966. doi: 10.1016/j.biomaterials.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morales-Cruz M, Flores-Fernandez GM, Orellano EA, Rodriguez-Martinez JA, Ruiz M, Griebenow K. Two-step nanoprecipitation for the production of protein-loaded PLGA nano-spheres. Results Pharma Sci. 2012;2:79–85. doi: 10.1016/j.rinphs.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendez J, Morales-Cruz M, Delgado Y, Figueroa CM, Orellano EA, Morales M, Monteagudo A, Griebenow K. Delivery of chemically glycosylated cytochrome c immobilized in mesoporous silica nanoparticles induces apoptosis in HeLa cancer cells. Mol Pharm. 2014;11:102–111. doi: 10.1021/mp400400j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown LR. Commercial challenges of protein drug delivery. Expert Opin Drug Delivery. 2005;2:29–42. doi: 10.1517/17425247.2.1.29. [DOI] [PubMed] [Google Scholar]
- 7.Ye Q, Asherman J, Stevenson M, Brownson E, Katre NV. DepoFoam technology: a vehicle for controlled delivery of protein and peptide drugs. J Controlled Release. 2000;64:155–166. doi: 10.1016/s0168-3659(99)00146-7. [DOI] [PubMed] [Google Scholar]
- 8.Morales-Cruz M, Figueroa CM, Gonzalez-Robles T, Delgado Y, Molina A, Mendez J, Morales M, Griebenow K. Activation of caspase-dependent apoptosis by intracellular delivery of Cytochrome c-based nanoparticles. J Nanobiotechnol. 2014;12:33. doi: 10.1186/s12951-014-0033-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang J, Nakamura H, Maeda H. The EPR effect. Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–151. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Controlled Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 11.Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 12.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharmacol. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomasina J, Lheureux S, Gauduchon P, Rault S, Malzert-Freon A. Nanocarriers for the targeted treatment of ovarian cancers. Biomaterials. 2013;34:1073–1101. doi: 10.1016/j.biomaterials.2012.10.055. [DOI] [PubMed] [Google Scholar]
- 14.Kim D, Lee ES, Park K, Kwon IC, Bae YH. Doxorubicin loaded pH-sensitive micelle: antitumoral efficacy against ovarian A2780/DOXR tumor. Pharm Res. 2008;25:2074–2082. doi: 10.1007/s11095-008-9603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaracz S, Chen J, Kuznetsova LV, Ojima L. Recent advances in tumor-targeting anti-cancer drug conjugates. Bioorg Med Chem. 2005;13:5043–5054. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 16.Paulos CM, Reddy JA, Leamon CP, Turk MJ, Low PS. Ligand binding and kinetics of folate receptor recycling in vivo: Impact on receptor-mediated drug delivery. Mol Pharmacol. 2004;66:1406–1414. doi: 10.1124/mol.104.003723. [DOI] [PubMed] [Google Scholar]
- 17.Zuberi S, Glen A, Hider RC, Bansal SS. Synthesis of asymmetric systines. Tetrahedron Letters. 1998;39:7567–7570. [Google Scholar]
- 18.Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9 apoptosome. Journal of Cell Science. 2010;123:3209–3214. doi: 10.1242/jcs.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamprecht A, Schafer U, Lehr CM. Structural analysis of microparticles by confocal laser scanning microscopy. AAPS PharmSciTech. 2000;1:E17. doi: 10.1208/pt010317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rolón-Reyes K, Kucheryavykh YV, Cubano LA, Inyushin M, Skatchkov SN, Eaton MJ, Harrison JK, Kucheryavykh LY. Microglia Activate Migration of Glioma Cells through a Pyk2 Intracellular Pathway. PLoS One. 2015;10:1–18. doi: 10.1371/journal.pone.0131059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Honary S, Zahir F. Effect of Zeta Potential on the Properties of Nano-Drug Delivery Systems - A Review (Part 2) Trop J Pharm Res. 2013;12:265–273. [Google Scholar]
- 22.Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES. Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation. J Biol Chem. 1999;274:17941–17945. doi: 10.1074/jbc.274.25.17941. [DOI] [PubMed] [Google Scholar]
- 23.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slowing II, Trewyn BG, Lin VSY. Mesoporous silica nanoparticles for intracellular delivery of membrane-impermeable proteins. J Am Chem Soc. 2007;129:8845–8849. doi: 10.1021/ja0719780. [DOI] [PubMed] [Google Scholar]
- 25.Ding N, Lu Y, Lee RJ, Yang C, Huang L, Liu J, Xiang G. Folate receptor-targeted fluorescent paramagnetic bimodal liposomes for tumor imaging. Int J Nanomed. 2011;6:2513–2520. doi: 10.2147/IJN.S23934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo WJ, Lee RJ. Receptor-targeted gene delivery via folate-conjugated polyethyl-enimine. AAPS Pharmsci. 1999;1 doi: 10.1208/ps010419. article 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang XQ, Deng WJ, Fu LW, Blanco E, Gao JM, Quan DP, Shuai XT. Folate-functionalized polymeric micelles for tumor targeted delivery of a potent multidrug-resistance modulator FG020326. J Biomed Mater Res A. 2008;86:48–60. doi: 10.1002/jbm.a.31537. [DOI] [PubMed] [Google Scholar]
- 28.Morachis JM, Mahmoud EA, Almutairi A. Physical and Chemical Strategies for Therapeutic Delivery by Using Polymeric Nanoparticles. Pharmacol Rev. 2012;64:505–519. doi: 10.1124/pr.111.005363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroeder A, Heller DA, Winslow MM, Dahlman JE, Pratt GW, Langer R, Jacks T, Anderson DG. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2012;12:39–50. doi: 10.1038/nrc3180. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh P, Yang XC, Arvizo R, Zhu ZJ, Agasti SS, Mo ZH, Rotello VM. Intra-cellular Delivery of a Membrane-Impermeable Enzyme in Active Form Using Functionalized Gold Nanoparticles. J Am Chem Soc. 2010;132:2642–2645. doi: 10.1021/ja907887z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng D, Song YC, Shi W, Li XH, Ma HM. Distinguishing Folate-Receptor-Positive Cells from Folate-Receptor-Negative Cells Using a Fluorescence Off-On Nanoprobe. Anal Chem. 2013;85:6530–6535. doi: 10.1021/ac401377n. [DOI] [PubMed] [Google Scholar]
- 32.Yoo HS, Park TG. Folate-receptor-targeted delivery of doxorubicin nano-aggregates stabilized by doxorubicin-PEG-folate conjugate. J Controlled Release. 2004;100:247–256. doi: 10.1016/j.jconrel.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 33.Song EQ, Zhang ZL, Luo QY, Lu W, Shi YB, Pang DW. Tumor Cell Targeting Using Folate-Conjugated Fluorescent Quantum Dots and Receptor-Mediated Endocytosis. Clin Chem. 2009;55:955–963. doi: 10.1373/clinchem.2008.113423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nance E, Zhang C, Shih TY, Xu Q, Schuster BS, Hanes J. Brain-penetrating nano-particles improve paclitaxel efficacy in malignant glioma following local administration. ACS Nano. 2014;8:10655–10664. doi: 10.1021/nn504210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu H, Zhan Y, Qu Y, Qi X, Li J, Yu C. Changes of folate receptor-α protein expression in human gliomas and its clinical relevance. Zhonghua Wai Ke Za Zhi. 2014;52:202–207. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.