

Abstract
Adenylosuccinate lyase (ADSL) deficiency is a rare autosomal recessive neurometabolic disorder that presents with a broad-spectrum of neurological and physiological symptoms. The ADSL gene produces an enzyme with binary molecular roles in de novo purine synthesis and purine nucleotide recycling. The biochemical phenotype of ADSL deficiency, accumulation of SAICAr and succinyladenosine (S-Ado) in biofluids of affected individuals, serves as the traditional target for diagnosis with targeted quantitative urine purine analysis employed as the predominate method of detection. In this study, we report the diagnosis of ADSL deficiency using an alternative method, untargeted metabolomic profiling, an analytical scheme capable of generating semi-quantitative z-score values for over 1000 unique compounds in a single analysis of a specimen. Using this method to analyze plasma, we diagnosed ADSL deficiency in four patients and confirmed these findings with targeted quantitative biochemical analysis and molecular genetic testing. ADSL deficiency is part of a large a group of neurometabolic disorders, with a wide range of severity and sharing a broad differential diagnosis. This phenotypic similarity among these many inborn errors of metabolism (IEMs) has classically stood as a hurdle in their initial diagnosis and subsequent treatment. The findings presented here demonstrate the clinical utility of metabolomic profiling in the diagnosis of ADSL deficiency and highlights the potential of this technology in the diagnostic evaluation of individuals with neurologic phenotypes.
Abbreviations: AICAR, aminoimidazole carboxamide ribotide; ADSL, adenylosuccinate lyase; IEM, inborn errors of metabolism; Global MAPSSM, Global Metabolomic Assisted Pathway Screen; SAICAR, succinylaminoimidazole carboxamide ribotide
Keywords: Metabolomic profiling, ADSL deficiency, Intellectual disability, Succinyladenosine
1. Introduction
Undifferentiated (non-specific) phenotypes such as non-syndromic intellectual disability and seizures, pose a challenge to clinicians as the differential diagnosis is very broad and comprehensive biochemical testing for these disorders requires many different tests done on various fluid types. Standard biochemical and molecular studies are often not revealing in these cases, often leading to diagnostic odysseys for the patients and families. Adenylosuccinate lyase (ADSL) deficiency (OMIM: 103050) is a rare, autosomal recessive inborn error of metabolism (IEM) that may present with a wide range of neurological symptoms. The clinical presentation typically includes intellectual disability, seizures, microcephaly, autism spectrum disorder, and respiratory failure. Three different forms have been recognized defined by the age of onset and the progression of symptoms: the fatal neonatal form, the severe form or type I presenting within the first months of life characterized by a severe clinical course, and type II, which is the mild/moderate form presenting within the first years of life with hypotonia and developmental delay [1]. Approximately 80 individuals have been reported in the literature, with 5–10% of cases being the fatal neonatal form, 70–80% of cases being the severe form or type I and 15–20% of type II form. The overall incidence is unclear [1].
ADSL (EC 4.3.2.2) exists as a homotetramer and catalyzes two pathways of purine nucleotide metabolism: de novo purine synthesis and the purine nucleotide cycle [1] (Fig. 1). In the de novo pathway, ADSL catalyzes the conversion of succinylaminoimidazole carboxamide ribotide (SAICAR) into aminoimidazole carboxamide ribotide (AICAR) (Fig. 1). In the purine nucleotide cycle, ADSL catalyzes the formation of adenylate (AMP) from adenylosuccinate (S-AMP) during the conversion of inosine monophosphate (IMP) into adenine nucleotides (Fig. 1). Biochemically, ADSL deficiency can be identified by the presence of SAICAr and succinyladenosine (S-Ado) in biologic fluids [2], which are otherwise not detected or not elevated [3].
Fig. 1.

Adenylosuccinate lyase and purine nucleotide metabolism. Adenylosuccinate lyase catalyzes two pathways of purine nucleotide metabolism: de novo purine synthesis and the purine nucleotide cycle. Deficiency of ADSL results in blocks in these pathways, causing an increase in SAICAr, as well as an increase in S-Ado, as indicated by the red arrows. Abbreviations are as follows: PRPP, phosphoribosylpyrophosphate; SAICAR, succinylaminoimidazole carboxamide ribotide; SAICAr, succinylaminoimidazole carboxamide riboside; AICAR, aminoimidazole carbozamide ribotide; FAICAR, formyloaminoimidazole carboxamide ribotide; IMP, inosine monophosphate; S-AMP, adenylosuccinate; S-Ado, succinyladenosine; AMP, adenine monophosphate; XMP, xanthine monophosphate; GMP, guanine monophosphate; ADSL, adenylosuccinate lyase; AICAR TF, aminoimidazole carboxamide riboside transformylase; IMP CH, inosine monophosphate cyclohydrolase. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Here, we report four patients with ADSL deficiency of which three were identified through untargeted metabolomic profiling of plasma and confirmed by targeted quantitative urine purine analysis and targeted molecular testing. Global metabolomic profiling is a semi-quantitative tandem mass spectrometry-based technique utilized in clinical screening for inborn errors of metabolism [4].
2. Methods
2.1. Untargeted metabolomic profiling
Metabolomic profiling (Global MAPS®) was performed by Baylor Miraca Genetics Laboratories (Houston, TX) and Metabolon, Inc. (Durham, NC), as described previously [4], [5], [6] with few modifications [7]. Small molecules were extracted in an 80% methanol solution and subjected to four analyses: two LC-MS/MS analyses in positive mode and two LC-MS/MS analyses in negative mode. All chromatographic separations were completed using an ACQUITY UPLC (Waters) equipped with either a Waters BEH C18 column or a Waters BEH Amide column, depending on the method, followed by analysis with an Q-Exactive high resolution mass spectrometer (Thermo-Finnigan) [5], [7]. Metabolites were identified with known chemical structure by matching the ion chromatographic retention index, nominal mass, and mass spectral fragmentation signatures with reference library entries created from authentic standard metabolites under the identical analytical procedure as the experimental samples [6]. Currently, the reference library contains entries for ~ 2500 unique human metabolites. Semi- quantitative analysis was achieved by comparing patient samples to a set of invariant anchor specimen included in each batch. Raw spectral intensity values were normalized to the anchor samples, log transformed, and compared to a normal reference population to generate z-score values.
2.2. Urine purine analysis
The enzymatic synthesis of succinyladenosine and determination of purine metabolites by LC-MS/MS were performed as previously described [8]. Briefly, the assay separation was performed on an Acquity UPLC BEH C18 column (1.7 μM IVD 2.1 * 500, Waters Corporation, Milford, USA). The gradient elution was performed with 0.1% formic acid/2 mM ammonium acetate (buffer A) and 0.1% formic acid/2 mM ammonium acetate in methanol (buffer B). The gradient profile began with 100% buffer A, followed by a linear increase to 40% buffer B over 1.5 min and an increase to 100% buffer B at 1.8 min. The column was then regenerated with 100% buffer A for another 2.5 min. The flow rate was 0.5 mL/min. A mixture of 15N2-adenine (Sigma #644331), 13C5-adenosine (CIL #CLM3678–0.05), 13C10, 15N5-guanosine (CIL #CLM-3808-LAS-5), 1,3-15N2–xanthine (CIL #NLM-1698) and U-15N5-deoxyadenosine (CIL #NLM-3895) were used as internal standards. The detection of the analytes was carried out using an Acquity TQ tandem MS (Waters) in the multiple reaction monitoring mode.
2.3. Molecular analysis
2.3.1. ADSL sequence analysis
Clinical targeted ADSL gene sequencing was undertaken for patient F1 by Baylor Genetics Laboratory and the details of the method is as follows. The coding regions of the ADSL (NM_000026.2) gene were PCR amplified and then sequenced in the forward and reverse directions using automated fluorescent dideoxy sequencing methods. Nucleotide 1 corresponds to the A of the start codon ATG. Variants detected in exons and in introns within up to 20 bp of the exon/intron boundaries were reported. This analysis does not detect large heterozygous deletions or duplications, inversions, or mutations within the promoter or deep intronic regions.
2.3.2. Whole exome sequencing
Clinical whole-exome sequencing was undertaken by Baylor Genetics Laboratory, and the details of their clinical sequencing platform have been reported previously [9]. In brief, after exon capture and sequencing, variants are called from reads with a minimum of 20 × depth coverage [10]. Variants of suspected clinical significance were confirmed by dideoxy (Sanger) sequencing.
3. Results
3.1. Clinical phenotype
Over a six month time period, we identified four patients with ADSL deficiency from three different families, by metabolomic profiling. The first two families (F1and F2) include single probands, while the third family has three siblings, all with variable levels of developmental delay (F3-1, F3-2 and F3-3); however, only two of the siblings, F3-2 and F3-3, were found to have ADSL deficiency, as shown in pedigree (Fig. S1). Table 1, summarizes the clinical and molecular data for the four patients studied. The age of onset for all four patients was within 2–4 months of birth, but the age of diagnosis varied between 7 months to 3 years.
Table 1.
Phenotype and genotype of patients. Summary of the clinical and molecular data of the four patients from three families.
| F1 | F2 | F3-1 | F3-2 | F3-3 | |
|---|---|---|---|---|---|
| Sex | F | M | M | M | M |
| Age of onset | 4 mo | 4 mo | NA | 2 mo | 3 mo |
| Age of diagnosis | 2 yrs | 7 mo | Unaffected sibling | 3 yrs | 18 mo |
| Developmental delay | Yes | Yes | Speech delay | Yes | Yes |
| Tone | Mildly decreased | Mildly increased | Normal | Mildly increased | Increased |
| Seizures | No | Yes | No | Yes | No |
| Head circumference | Normal | Macrocephaly | Normal | Normal | Microcephaly |
| Eye phenotype | Strabismus | Eye deviation | Normal | Ocular motor apraxia | Exotropia |
| MRI | Normal | Abnormal | Normal | Normal | Normal |
| Genotype | c.736A > G c.947T > C | c.1277 G > A | Normal | c.1277G > A | c.1277G > A |
| Amino acid change | p.Lys246Glu p.Met316Tyr | p.Arg426His | NA | p.Arg426His | p.Arg426His |
| Inheritance | Compound heterozygous | Homozygous | NA | Homozygous | Homozygous |
| Consanguinity | No | Yes | No | No | No |
Abbreviations are as follows: MRI, magnetic resonance imaging; F, female; M, male; NA, not available; mo, months; yrs, years.
Each of the four patients had varying degrees of developmental delay, which prompted referrals for specialist evaluation and testing. Seizures were observed in only 2 of our patients, F2 and F3-2. On physical examination, F-2 exhibited macrocephaly, F1 and F3-2 had normal head circumference, and F3-3 had relative microcephaly with a prominent forehead and bitemporal narrowing. All four individuals showed some form of an ocular phenotype, mainly strabismus, and in addition, F3-2 had ocular motor apraxia, while F3-3 had subnormal vision and myopic astigmatism. Their neurologic exam revealed increased muscle tone.
Brain MRIs on F1, F3-2 and F3-3 were interpreted as normal. F2 had an MRI at 4 months of age that was notable for prominence of the Sylvian fissure and extra-axial spaces. Repeat MRI at 7 months showed symmetric restricted diffusion in the bilateral dentate nuclei, dorsal brainstem, thalami, subthalamic regions, and globus pallidus with corresponding T2/FLAIR signal abnormalities, as well as further widening of the Sylvian fissures and extra-axial spaces.
3.2. Metabolomic profiling
All of the patients had extensive testing including plasma amino acid analysis, urine organic acid analysis, plasma acylcarnitine analysis, blood lactate & pyruvate, CSF amino acid analysis, and blood ammonia, which were non-diagnostic. Metabolomic profiling performed on plasma of patients F1, F2 and F3-2 revealed a marked elevation in succinyladenosine (Z-score of 7.7, 9.5 and 7.3, respectively) (Fig. 2A and B), which is one of the two known biomarkers for ADSL deficiency. Molecules identified in this assay with Z-scores above or below ± 2 are listed in Supplemental Data. Patient F2 also had a marked elevation in succinyladenosine (Z-score of 11.1) (Table S1) on CSF metabolomic profiling. SAICAr is not detected by the metabolomics platform and is not reported. No other metabolite in the purine pathway is perturbed.
Fig. 2.
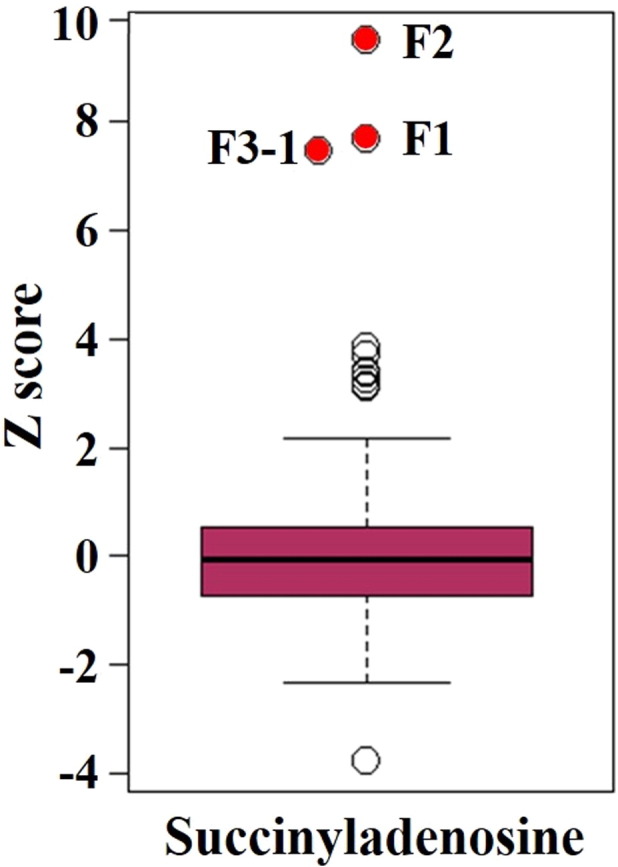
Elevated succinyladenosine levels support a diagnosis of ADSL deficiency. Succinyladenosine is a byproduct of the purine nucleotide cycle, involving conversion of adenylosuccinate (S-AMP) to adenylate (AMP). Succinyladenosine levels in the patient plasmas are the highest when compared to 625 unaffected samples. Elevation of succinyladenosine is indicative of ADSL deficiency. Red dots indicate the patients and open circles indicate outliers. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Biochemical confirmation of ADSL deficiency
Patient F3-3 did not have a metabolomic profiling. Since patient F3-3 had a similar clinical phenotype to his sibling F3-2, a targeted urine purine panel was ordered to test for ADSL deficiency. Subsequently urine purine panel analysis confirmed the significantly elevated levels of succinyladenosine in patients F1, F3-2 and F3-3 (281.9, 96.9, 123.8 millimoles/mol creatinine, respectively; normal range is 0–30.2 millimoles/mol creatinine) (Table 2). Sibling F3-1, with mild speech delay, had normal urine succinyladenosine levels. In case of patient F2, a urine purine panel analysis was not performed, but CSF analysis confirmed the elevation in succinyladenosine (673.50 μmol/L, normal ranges 0.74–4.92 μmol/L).
Table 2.
Urine purine analysis. Targeted quantitative analysis of purines revealed significant elevation of succinyladenosine in the urine of F1, F3-2 and F3-3, confirming the semi-quantitative metabolomic findings in plasma and consistent with a diagnosis of ADSL deficiency.
| Metabolite | Normal rangea (0–2 yrs) |
Normal rangea (3–99 yrs) |
F1 valuea | F3-1 valuea | F32 valuea | F3-3 valuea |
|---|---|---|---|---|---|---|
| Xanthine | 0–59 | 0–47.3 | 13.7 | 0.2 | 7.2 | 4.6 |
| Inosine | 0–4.8 | 0–4.8 | 3.8 | 0.4 | 4.1 | 2.1 |
| Guanosine | 0–7.3 | 0–5.3 | 0.6 | 0.2 | 1.7 | 1.8 |
| Adenine | 0–2.9 | 0–1.3 | 0.0 | 0.0 | 0.0 | 2.0 |
| Adenosine | 0–3.8 | 0–3.4 | 6.5 | 0.3 | 7.2 | 6.3 |
| Deoxyadenosine | 0–4.1 | 0–3.2 | 5.4 | 0.4 | 2.0 | 7.2 |
| Hypoxanthine | 0–30.9 | 0–38.6 | 12.2 | 4.1 | 33.7 | 17.2 |
| Succinyladenosine | 0–30.2 | 0–34.6 | 281.9 | 1.0 | 123.8 | 96.9 |
Millimoles/mol creatinine.
3.4. Molecular findings further support ADSL deficiency diagnosis
Sanger sequencing of the ADSL gene on patient F1 revealed compound heterozygosity for two missense variants. The first missense variant (ADSL NM_000026.3 c.736 A > G) resulted in substitution of glutamate for lysine at amino acid position 246 (p. Lys246Glu) (Table 1). This variant was classified by the performing laboratory as a pathogenic variant, as it was previously reported in association with ADSL deficiency [11]. However, another missense variant (ADSL NM_000026.3 c.947T > C) resulting in substitution of tyrosine for methionine at amino acid position 316 (p.Met316Tyr) was classified as a variant of unknown significance as it had not been reported previously in public population databases. SIFT and Polyphen-2 algorithms predicted this variant to be tolerated.
In the case of patient F2, an infantile epilepsy panel revealed a homozygous pathogenic mutation in ADSL (ADSL NM_000026.3 c.1277G>A, p.Arg426His) (Table 1). Whole exome sequencing (sent concomitantly with the metabolomics profiling) on patient F3-3 revealed a homozygous pathogenic mutation in ADSL (ADSL NM_000026.3 c.1277G > A, p.Arg426His) and was confirmed by Sanger sequencing on his parents and sibling F3-2 (Table 1). The unaffected sibling, F3-1 who had mild speech delay but was clearly not as delayed as his two brothers, does not have this variant and both parents were identified as carriers. Patient F2 is from consanguineous parents; however, there is no consanguinity in the other two families as per the clinical records, and the WES data showed no absence of heterozygosity (AOH) in the third family (F3).
3.5. ADSL variants, population frequency, and literature review
There are 38 ADSL variants reported in the Exome Aggregation Consortium (ExAc) database, and none of these variants are homozygous (Table S3). According to the ExAC database, c.1277G > A, p.Arg426His is the most common variant and is carried on 31 chromosomes in the heterozygous state only, with an allelic frequency of 0.000255 and a carrier frequency of ~ 1/2000. Out of the 31 individuals, 28 are from non-Finnish Europeans, and one each from African, Asian and Latino populations (Table S2).
To date fourteen patients with homozygous ADSL c.1277G > A, p.Arg426His variant, including three from the current study, are reported [11], [12], [13], [14], [15] (Table 3). The age of onset for the majority of these patients (10/12) is very early, with an average age of onset at six months and only two patients with age of onset beyond two years [12]. Most patients are diagnosed with ADSL deficiency after a significant amount of time since the onset of symptoms. Out of fourteen patients, five are female, eight are male, and one unknown, indicating there is no obvious sex bias. There is not enough information available to calculate the mortality rate. Some of the symptoms that are consistent among the patients are developmental delay (14/14), seizures (10/12), hypotonia/change in tone (7/10), eye issues (8/11) and MRI abnormalities (8/11) (Table 3). Even though all fourteen patients show developmental delay, they vary significantly in terms of severity. Significant variation is observed in terms of head circumference, six patients being microcephalic, one macrocephalic and three within the normal range. Out of fourteen patients, four individuals are reported to be products of consanguineous relationships (Table 3).
Table 3.
Clinical features of patients with ADSL deficiency and homozygous c.1277G > A, p.Arg426His variant. A review of the ADSL deficiency literature is provided.
| Patient | Sex | AO | AD | DD | Tone | Seizures | HC | EP | MRI | Genotype | Cons. | Source |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | NA | 5 yrs | Yes | Normal | No | Normal | No | NA | c.1277 G > A | No | Marie et al. (1999) |
| 2 | M | 6 mo | 22 mo | Yes | Normal | NA | Microcephaly | Strabismus | Normal | c.1277 G > A | No | Marie et al. (1999) |
| 3 | F | 9 weeks | 3 mo | Yes | Normal | Yes | NA | No | Abnormal | c.1277 G > A | No | Marie et al. (1999) |
| 4 | U | NA | NA | Yes | NA | NA | NA | NA | NA | c.1277 G > A | No | Marie et al. (1999) |
| 5 | M | 2 weeks | NA | Yes | Hypotonia | Yes | NA | NA | NA | c.1277 G > A | No | Kmoch et al. (2000) |
| 6 | F | 2 yrs | 10 yrs | Yes | Hypotonia | Yes | Microcephaly | No | Abnormal | c.1277 G > A | Yes | Edery et al. (2003) |
| 7 | M | 2 yrs | 9 yrs | Yes | NA | Yes | Microcephaly | Abnormal movements | Abnormal | c.1277 G > A | Yes | Edery et al. (2003) |
| 8 | M | 6 mo | 5 yrs | Yes | NA | Yes | Microcephaly | Abnormal movements | Abnormal | c.1277 G > A | Yes | Edery et al. (2003) |
| 9 | F | 6 mo | 1.5 yrs | Yes | NA | Yes | Normal | Strabismus | Abnormal | c.1277 G > A | No | Jurecka et al. (2008) |
| 10 | F | 1 mo | NA | Yes | Hypotonia | Yes | Microcephaly | Strabismus | Abnormal | c.1277 G > A | No | Jurecka et al. (2008) |
| 11 | M | 2 mo | 4 yrs | Yes | Hypotonia | Yes | NA | NA | Abnormal | c.1277 G > A | No | Henneke et al. (2010) |
| 12 | M | 4 mo | 7 mo | Yes | Increased | Yes | Macrocephaly | Eye deviation | Abnormal | c.1277 G > A | Yes | Current study |
| 13 | M | 2 mo | 3 yrs | Yes | Increased | Yes | Normal | Oculomotor apraxia | Normal | c.1277 G > A | No | Current study |
| 14 | M | 3 mo | 18 mo | Yes | Increased | No | Microcephaly | Exotropia | Normal | c.1277 G > A | No | Current study |
Abbreviations are as follows: AO, age of onset; AD, age of diagnosis; DD, developmental delay; HC, head circumference; EP, eye phenotype; MRI, magnetic resonance imaging; AA change, amino acid change; Cons., consanguinity; F, female; M, male; U, unknown; NA, not available; mo, months; yrs., years.
4. Discussion
ADSL deficiency is an autosomal recessive disorder due to mutations in the ADSL gene. Recent evidence demonstrates that proteins involved in purine biosynthesis, including ADSL, form a multienzyme complex named the “purinosome” [16]. The purinosome could be disrupted by functional or structural defects in ADSL or any other subunit of the complex. The phenotypic severity of ADSL deficiency is mainly associated with structural stability and residual catalytic capacity of the purinosome complex [17], [18]. ADSL deficiency results in accumulation of SAICAr and S-Ado in urine and CSF and to lesser extent, in plasma (Fig. 1). Indeed, the sensitivity of S-Ado detection in plasma is much lower than that in urine and CSF [19]. Several simple screening procedures [20], [21] were developed for detection of disorders of purine metabolism; most of the methods are based on the detection of abnormal metabolites, SAICAr and S-Ado, in the urine and CSF through HPLC and tandem mass spectrometry [2], [19].
In the literature, 11 unrelated patients are reported with a homozygous change of c.1277G > A, p.Arg426His (Table 3, Table S3), the most common pathogenic variant responsible for ADSL deficiency. Even though all 14 patients (three from current study) have a common genotype (homozygous c.1277G > A, p.Arg426His), their phenotype varies significantly. There is a considerable range in terms of age of onset and the severity, as well as progression of the disease in these patients, which is suggestive of a modifier effect [12], [14], [22]. Among our three patients with the homozygous c.1277G > A, p.Arg426His variant, F2 is the youngest and has most severe form of the disease. Interfamilial variability of the disease phenotype was described previously [12] and was also observed in F3-2 and F3-3 in our study.
It is noteworthy that there is a significant gap between the age of onset and age at which diagnosis was made (Table 3). There is no known effective treatment at this time; however, there have been several unsuccessful attempts toward developing a treatment [14], [23], [24]. Considering the progressive neurodegenerative course of the disease, one would expect that if a treatment were developed, that early diagnosis would improve the outcome. Also an early diagnosis is essential for prenatal and pre-implantation diagnosis in a subsequent pregnancy.
While the presence of seizures (observed in F2 and F3-2) would support the inclusion of a purine panel in the diagnostic workup, F1 and F3-3 did not have seizures, which are typically considered a consistent feature of ADSL deficiency. The neurologic phenotype in F1and F3-3 included intellectual disability and autism spectrum disorder (ASD), which are common and non-specific symptoms of a large number of genetic and neurometabolic disorders, hence making the diagnosis challenging given the broad differential diagnosis. Identification of four patients from three families within six months in our laboratory raises the possibility of this disorder being misdiagnosed or remaining undiagnosed in the broader population. In individuals such as F1and F3-3, metabolomic profiling may be a helpful tool in screening for these rare neurometabolic disorders. The untargeted nature of metabolomic profiling allows screening for many metabolic disorders in one plasma sample [4]. In the case of patient F1, the SIFT and Polyphen-2 algorithms predicted the variant, c.947T > C, p.Met316Tyr, to be tolerated, while it was not. This highlights the utility of metabolomic profiling as a functional study in assessing the pathogenicity of variants of unknown significance. A similar untargeted metabolomics analysis using urine samples was reported last year whereas here, we show diagnosis of ADSL deficiency using plasma samples [25]. These cases of ADSL deficiency highlight the use of metabolomic profiling as a screening tool, as multiple common and rare metabolic disorders may be screened for in a single test rather than requiring multiple targeted and specific tests and allowing for the identification of individuals who may have less common presentations of disease [4], [7].
Disclosure
TRD, GC, LH, JN, PSA, MJM, ALC, VRS, MFW, QS, LTE and SHE are employees of Baylor College of Medicine. Baylor College of Medicine and Baylor Genetics Laboratory have a joint partnership and derive revenue from metabolomics testing.
Acknowledgements
We thank Dr. Adam Kennedy for his technical support. This project was supported, in part, by the T32 GM07526-37 Medical Genetics Research Fellowship Program (MJM).
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ymgmr.2016.07.007.
Appendix A. Supplementary data
Supplementary data.
References
- 1.Jurecka A., Zikanova M., Tylki-Szymanska A., Krijt J., Bogdanska A., Gradowska W., Mullerova K., Sykut-Cegielska J., Kmoch S., Pronicka E. Clinical, biochemical and molecular findings in seven Polish patients with adenylosuccinate lyase deficiency. Mol. Genet. Metab. 2008;94:435–442. doi: 10.1016/j.ymgme.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Zikanova M., Krijt J., Skopova V., Krijt M., Baresova V., Kmoch S. Screening for adenylosuccinate lyase deficiency using tandem mass spectrometry analysis of succinylpurines in neonatal dried blood spots. Clin. Biochem. 2015;48:2–7. doi: 10.1016/j.clinbiochem.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Jurecka A., Zikanova M., Kmoch S., Tylki-Szymańska A. Adenylosuccinate lyase deficiency. J. Inherit. Metab. Dis. 2015;38:231–242. doi: 10.1007/s10545-014-9755-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller M.J., Kennedy A.D., Eckhart A.D., Burrage L.C., Wulff J.E., Miller L.A.D., Milburn M.V., Ryals J.A., Beaudet A.L., Sun Q., Sutton V.R., Elsea S.H. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2015:1–11. doi: 10.1007/s10545-015-9843-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans A.M., DeHaven C.D., Barrett T., Mitchell M., Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 2009;81:6656–6667. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- 6.DeHaven C.D., Evans A.M., Dai H., Lawton K.A. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J Cheminform. 2010;2:9. doi: 10.1186/1758-2946-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atwal P.S., Donti T.R., Cardon A.L., Bacino C.A., Sun Q., Emrick L., Reid Sutton V., Elsea S.H. Aromatic l-amino acid decarboxylase deficiency diagnosed by clinical metabolomic profiling of plasma. Mol. Genet. Metab. 2015;115:91–94. doi: 10.1016/j.ymgme.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 8.Sun Q. Urine purine metabolite determination by UPLC-tandem mass spectrometry. Methods Mol. Biol. 2016;1378:227–235. doi: 10.1007/978-1-4939-3182-8_24. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y., Muzny D.M., Xia F., Niu Z., Person R., Ding Y., Ward P., Braxton A., Wang M., Buhay C., Veeraraghavan N., Hawes A., Chiang T., Leduc M., Beuten J., Zhang J., He W., Scull J., Willis A., Landsverk M., Craigen W.J., Bekheirnia M.R., Stray-Pedersen A., Liu P., Wen S., Alcaraz W., Cui H., Walkiewicz M., Reid J., Bainbridge M., Patel A., Boerwinkle E., Beaudet A.L., Lupski J.R., Plon S.E., Gibbs R.A., C.M. Eng Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y., Muzny D.M., Reid J.G., Bainbridge M.N., Willis A., Ward P.A., Braxton A., Beuten J., Xia F., Niu Z., Hardison M., Person R., Bekheirnia M.R., Leduc M.S., Kirby A., Pham P., Scull J., Wang M., Ding Y., Plon S.E., Lupski J.R., Beaudet A.L., Gibbs R.A., Eng C.M. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marie S., Cuppens H., Heuterspreute M., Jaspers M., Tola E.Z., Gu X.X., Legius E., Vincent M.F., Jaeken J., Cassiman J.J., Van den Berghe G. Mutation analysis in adenylosuccinate lyase deficiency: eight novel mutations in the re-evaluated full ADSL coding sequence. Hum. Mutat. 1999;13:197–202. doi: 10.1002/(SICI)1098-1004(1999)13:3<197::AID-HUMU3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 12.Edery P., Chabrier S., Ceballos-Picot I., Marie S., Vincent M.F., Tardieu M. Intrafamilial variability in the phenotypic expression of adenylosuccinate lyase deficiency: a report on three patients. Am. J. Med. Genet. 2003;A120a:185–190. doi: 10.1002/ajmg.a.20176. [DOI] [PubMed] [Google Scholar]
- 13.Henneke M., Dreha-Kulaczewski S., Brockmann K., van der Graaf M., Willemsen M.A., Engelke U., Dechent P., Heerschap A., Helms G., Wevers R.A., Gartner J. In vivo proton MR spectroscopy findings specific for adenylosuccinate lyase deficiency. NMR Biomed. 2010;23:441–445. doi: 10.1002/nbm.1480. [DOI] [PubMed] [Google Scholar]
- 14.Jurecka A., Tylki-Szymanska A., Zikanova M., Krijt J., Kmoch S. d-ribose therapy in four polish patients with adenylosuccinate lyase deficiency: absence of positive effect. J. Inherit. Metab. Dis. 2008;31:329–332. doi: 10.1007/s10545-008-0904-z. [DOI] [PubMed] [Google Scholar]
- 15.Kmoch S., Hartmannova H., Stiburkova B., Krijt J., Zikanova M., Sebesta I. Human adenylosuccinate lyase (ADSL), cloning and characterization of full-length cDNA and its isoform, gene structure and molecular basis for ADSL deficiency in six patients. Hum. Mol. Genet. 2000;9:1501–1513. doi: 10.1093/hmg/9.10.1501. [DOI] [PubMed] [Google Scholar]
- 16.An S., Kumar R., Sheets E.D., Benkovic S.J. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008;320:103–106. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- 17.Baresova V., Skopova V., Sikora J., Patterson D., Sovova J., Zikanova M., Kmoch S. Mutations of atic and adsl affect purinosome assembly in cultured skin fibroblasts from patients with aica-ribosiduria and adsl deficiency. Hum. Mol. Genet. 2012;21:1534–1543. doi: 10.1093/hmg/ddr591. [DOI] [PubMed] [Google Scholar]
- 18.Spiegel E.K., Colman R.F., Patterson D. Adenylosuccinate lyase deficiency. Mol. Genet. Metab. 2006;89:19–31. doi: 10.1016/j.ymgme.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 19.Marinaki A., Champion M., Kurian M., Simmonds H., Marie S., Vincent M.-F., Van den Berghe G., Duley J., Fairbanks L. Adenylosuccinate lyase deficiency—first British case. Nucleosides Nucleotides Nucleic Acids. 2004;23:1231–1233. doi: 10.1081/NCN-200027494. [DOI] [PubMed] [Google Scholar]
- 20.Laikind P.K., Seegmiller J.E., Gruber H.E. Detection of 5'-phosphoribosyl-4-(N-succinylcarboxamide)-5-aminoimidazole in urine by use of the Bratton-Marshall reaction: identification of patients deficient in adenylosuccinate lyase activity. Anal. Biochem. 1986;156:81–90. doi: 10.1016/0003-2697(86)90158-2. [DOI] [PubMed] [Google Scholar]
- 21.Maddocks J., Reed T. Urine test for adenylosuccinase deficiency in autistic children. Lancet Lond. Engl. 1989;1:158–159. doi: 10.1016/s0140-6736(89)91172-0. [DOI] [PubMed] [Google Scholar]
- 22.Race V., Marie S., Vincent M.F., Van den Berghe G. Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum. Mol. Genet. 2000;9:2159–2165. doi: 10.1093/hmg/9.14.2159. [DOI] [PubMed] [Google Scholar]
- 23.Salerno C., Crifo C., Curatolo P., Ciardo F. Effect of uridine administration to a patient with adenylosuccinate lyase deficiency. Adv. Exp. Med. Biol. 2000;486:75–78. doi: 10.1007/0-306-46843-3_14. [DOI] [PubMed] [Google Scholar]
- 24.Salerno C., D'Eufemia P., Finocchiaro R., Celli M., Spalice A., Iannetti P., Crifo C., Giardini O. Effect of d-ribose on purine synthesis and neurological symptoms in a patient with adenylosuccinase deficiency. Biochim. Biophys. Acta. 1999;1453:135–140. doi: 10.1016/s0925-4439(98)00093-3. [DOI] [PubMed] [Google Scholar]
- 25.Janeckova H., Kalivodova A., Najdekr L., Friedecky D., Hron K., Bruheim P., Adam T. Untargeted metabolomics analysis of urine samples in the diagnosis of some inherited metabolic disorders. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2015;159(4):582–585. doi: 10.5507/bp.2014.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data.