

Abstract
Combination treatment for non-small cell lung cancer (NSCLC) is becoming more popular due to the anticipation that it may be more effective than single drug treatment. In addition, there are efforts to genetically screen patients for specific mutations in light of attempting to administer specific anticancer agents that are most effective. In this study, we evaluate the anticancer and anti-angiogenic effects of low dose celecoxib-erlotinib combination in NSCLC in vitro and in vivo. In NSCLC cells harboring epidermal growth factor receptor (EGFR) mutations, combination celecoxib-erlotinib treatment led to synergistic cell death, but there was minimal efficacy in NSCLC cells with wild-type EGFR. In xenograft models, combination treatment also demonstrated greater inhibition of tumor growth compared to individual treatment. The anti-tumor effect observed was secondary to the targeting of angiogenesis, evidenced by decreased vascular endothelial growth factor A (VEGFA) levels and decreased levels of CD31 and microvessel density. Combination treatment targets angiogenesis through the modulation of of the PI3K/AKT and ERK/Raf1-1 pathway in NSCLC with EGFR exon 19 deletions. These findings may have significant clinical implications in patients with tumors harboring EGFR exon 19 deletions as they may be particularly sensitive to this regimen.
Keywords: EGFR, NSCLC, celecoxib, erlotinib, angiogenesis
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide and non-small cell lung cancer (NSCLC) comprises about 85% of all types of lung cancer [1]. NSCLC is strongly associated with epidermal growth factor receptor (EGFR) mutations. It is estimated that more than 60% of NSCLC have EGFR mutations that lead to constitutive activation of downstream pathways [2]. The over-expression of EGFR has been shown to have a direct correlation with poor prognosis, indicating that this mutation and pathologic mechanism may be a suitable target for anti-cancer agents. There have been great advancements in developing small molecule inhibitors that specifically target EGFR. Erlotinib is an EGFR-specific tyrosine kinase inhibitor (TKI) approved for the clinical treatment of advanced stages of NSCLC [3]. Erlotinib binds to EGFR and inhibits the activation of its downstream signaling pathways involved in cell proliferation, survival, angiogenesis, invasion, and migration [4].
NSCLC patients with EGFR mutations initially respond well to erlotinib treatment; however, patients develop acquired resistance to erlotinib approximately 10 months after initial therapy [5]. The two most recognized mechanisms that promote erlotinib acquired resistance are T790M point mutation in exon 21 in EGFR and MET amplification. Although several other mechanisms have been identified (insulin-like growth factor-1 and AXL tyrosine kinase receptor overexpression and ABCG2 efflux of drug transporters), 30% of mechanisms remain unknown [6]. Similarly, in the case of cisplatin, despite good initial response, patients often develop chemotherapy resistance. It is shown that cisplatin treatment results in increased EGFR phosphorylation and activation of its downstream target kinases in cisplatin-resistant cancer cells. Additionally, cisplatin can cause EGFR translocation to the nucleus where it contributes to resistance by restoring DNA repair activity [7]. Therefore, overcoming these resistant mechanisms through combination treatment is of great interest.
Several studies have shown that non-steroidal anti-inflammatory drugs (NSAID) are able to prevent the development of various human cancers, including lung cancer, even if the exact molecular mechanisms in chemoprevention of NSAID are not clearly understood [8]. There is good correlation between high levels of COX-2 and tumor cell sensitivity to NSAIDs. As a result, COX-2 has become a natural target for anti-cancer agents and selective COX-2 inhibitors, such as celecoxib and rofecoxib, have been considered for therapy. The induction of COX-2 and up-regulation of the prostaglandin cascade play a significant role in carcinogenesis by promoting cell division, induction of vascular endothelial growth factor and stimulation of an anti-apoptotic pathway. Treatment with COX-2 inhibitor, such as celecoxib, in a cancer chemoprevention trial reduced the risk of developing familial adenomatous polyposis (FAP), some cases of which would inevitably progress to fully-fledged colon cancer [9]. The pathways by which EGFR and COX-2 contribute to carcinogenesis have been separately considered and targeted; increasing evidence indicates a tight connection between these two pathways [10]. EGFR and COX-2 signaling pathways form a positive feedback loop. Activation of EGFR has been shown to induce increased COX-2 expression in various normal and tumor cell lines. Both transforming growth factor-α and epidermal growth factor, ligands of EGFR, were found to induce COX-2 expression. Expression of COX-2 is regulated at both the transcriptional and posttranscriptional level. The signaling pathway involved in COX-2 induction via EGFR varies depending on the type of cells and inducers, but the Raf signaling pathways mainly contribute to both increased transcriptional and posttranscriptional control [11]. One explanation for the linkage between EGFR/mitogen-activated protein kinase and transcriptional activation of COX-2 may be the activation of transcription factors such as cyclic AMP response element-binding protein/activating transcription factor and activator protein-1 by mitogen-activated protein kinase signaling [12]. Binding sites for these transcription activators have been identified in the COX-2 promoter region. On the other hand, COX-2 induces transactivation or increased expression of EGFR. Transactivation of EGFR by PGE2, a major prostaglandin involved in carcinogenesis, has been well documented but the process seems to be quite complex and cell type dependent. PGE2 trans-activated EGFR and triggered the activation of extracellular signal regulated kinase-2 pathways in normal gastric epithelial cells and colon cancer cells, inducing cell proliferation in vitro and in vivo [13].
Angiogenesis is a fundamental step for tissue growth and maturation, and this process is often pathologically used by tumors to grow, expand, and metastasize [14]. One of the most potent growth factors that mediate angiogenesis is vascular endothelial growth factor (VEGF) as it recruits and stimulates the growth of endothelial cells, leading to increased vascularity. Angiogenesis has been understood to be an important therapeutic target, and drugs targeting VEGF such as a bevacizumab has been developed and approved for clinical use [15]. Over the past several years, there have been studies suggesting that both erlotinib and celecoxib individually are able to target angiogenesis in various cancers [16]. However, details regarding their mechanisms of action and the effects of combining these two drugs have yet to be elucidated. In this study, we showed that low dose erlotinib-celecoxib combination treatment was able to induce synergistic cell death in EGFR-mutated HCC827 and H1650 cells in vitro and we also evaluated its effectiveness in xenograft mouse models. In addition, we examined whether combination erlotinib-celecoxib treatment is able to inhibit angiogenesis at greater degrees compared to single drug treatment alone. The effected pathway and regulatory proteins were also identified.
Materials and methods
Cell culture
Human non-small cell lung carcinoma cell lines (A549, NCI-H292, H1650 and HCC827) were purchased from American Type Culture Col-lection (Manassas, VA). The EGFR mutation status of the main cell lines of interest is reported in Table 1. Cells were maintained in RPMI 1640 media containing 10% FBS and 37°C in a humidified incubator with 5% CO2.
Table 1.
A report of the EGFR status of multiple NSCLC cell lines is shown
| NSCLC cell lines | EGFR status |
|---|---|
| A549 | Wild type |
| H292 | Wild type |
| HCC827 | Exon 19 deletion |
| H1650 | Exon 19 deletion |
Cell viability assay
Four treatment groups were devised to control, celecoxib-alone, erlotinib-alone, and celecoxib-erlotinib combination. All cell lines were treated for 3 days and cell viability assay was measured using a CCK8 kit (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instruction at different indicated time points [17]. Data were from three separate experiments with four replications each time.
RNA interference
Oligonucleotides for human EGFR siRNA kit were purchased from OriGene (Rockville, MD, USA). The kit contains three predesigned duplexes targeting a specific gene of interest, and we used a pool of three target siRNAs to ensure work efficiency [18]. Cells were transfected with EGFR siRNA or non-specific siRNA (0.15 μg/well for 96 well culture plates and 2 μg/well for 6 well culture plates) using the opti-MEM plus X-tremeGENE siRNA transfection reagent (Roche, Mannheim, Germany) according to the instruction manual. After 24 h post-transfection, the cells were further treated with celecoxib-alone, erlotinib-alone, and celecoxib-erlotinib combination for western blot analyses and proliferation assay.
Immunocytochemistry
After human umbilical vein endothelial cells (HUVECs) on glass coverslips were treated by indicated agents, they were fixed by pre-cold acetone, and then rinsed three times with PBS. The cells were permeabilized in 0.1% Triton X-100 and incubated with 1% BSA/PBS to block nonspecific binding. According to the manufacturer’s protocol for ABC detection kit (Abcam), 3,3’-diaminobenzidine (DAB) staining was performed to detect CD31 (Abcam; 1:200 overnight incubation at 4°C). Images were taken and analyzed using the ZEN 2011 imaging software on a Zeiss invert microscope (CarlZeiss, Hallbergnoos, Germany) under 400-fold magnification [19].
Scratch-induced migration assay
HUVECs cells were synchronized by in a low serum medium for 8 h, and the cell monolayers were then damaged with a micropipette to create a linear wound 2 mm in width. The indicated concentrations of indicated agents (plus 4 ng·mL-1 VEGFA for HUVECs) were added and the cells were incubated for 24 h. Images were acquired using a microscope (Zeiss, Jena, Germany) and the results are expressed as the percentage inhibition rate of migration compared with untreated cells [20].
Transwell migration assay
Transwell filter inserts (Billerica, MA, USA) were pre-coated with Matrigel for 30 min at 37°C. For HUVEC assays, the bottom chambers were filled with EGM-2 with or without VEGFA (4 ng·mL-1) and the top chambers were seeded with 4 × 104 cells. Various concentrations of indicated agents were added to the top and bottom chambers and the plates were incubated for 24 h. Migrated cells were stained with 0.1% crystal violet and imaged microscopically. Cells were enumerated and the results are expressed as the percentage inhibition rate of migration compared with untreated cells [21].
Flow cytometric analysis
Cells were seeded in 6-well cell culture plates and treated with various agents for indicated time period. For cell cycle analysis, cells were detached with trypsin and washed with cold PBS. Precipitated cells were fixed by 500 μL cold 70% ethanol overnight at -20°C. After being washed in PBS, fixed cells were then incubated with RNase at 37°C for 30 min and stained with propidium iodide (PI) for 15 min at room temperature in dark and immediately analyzed by flow cytometry (FACS Calibur, BD Biosciences, San Jose, CA). For cell apoptosis analysis, cells were detached with trypsin and washed with cold PBS. Resuspended cells in 500 μL binding buffer were double stained with FITC-conjugated Annexin V and PI. After 15 min of incubation at room temperature in dark, samples were immediately analyzed by flow cytometry [22].
Colony formation assay
HCC827 (1 × 103) and H1650 (1 × 103) cells then were plated onto a 6-well tissue culture plate in complete medium and incubated at 37°C. HCC827 and H1650 cells were pretreated with DMSO (< 0.1%), erlotinib-alone or celecoxib-erlotinib combination. Cells were allowed to grow in complete medium for 14 days. Then cells were fixed and stained with 1% crystal violet and counted under the microscope [23].
Western blot analysis
Cells were harvested and lysed in a RIPA buffer (Cell Signaling) containing protease inhibitors (Complete Mini EDTA-free; Roche Applied Science). Total protein (20 μg) was denatured, separated with 4-20% SDS-PAGE (Criterion TGX, Bio-Rad) and transferred to an Immuno-Blot™ polyvinylidene difluoride (PVDF) membrane (Bio-Rad). After blocking the membrane in 5% non-fat milk powder dissolved in phosphate-buffered saline (PBS), membranes were incubated overnight in 3% non-fat milk powder or 5% BSA at 4°C with primary antibodies. Afterwards, membranes were incubated with an HRP-conjugated secondary antibody (Dako Cytomation) diluted 1:1000. Target proteins were detected by the ECL system (Millipore, Braunschweig, Germany) and visualized with the ChemiDoc XRS system (Bio-Rad, Hercules, CA, USA).
In vivo matrigel plug angiogenesis assay
In vivo angiogenesis assay was tested through the use of the Matrigel plug assay. Matrigel (500 ul; BD Biosciences) was mixed with recombinant human VEGFA (R&D Systems, Inc, Minneapolis, MN), and then appropriate drugs were added: vehicle, erlotinib (15 mg/kg), celecoxib (20 mg/kg), or combination of erlotinib and celecoxib (each group containing four mice). Matrigel mixes were injected subcutaneously into the ventral abdomen of mice. Mice were then sacrificed, and Matrigel plugs were removed on day 21. Each plug was weighed, and following manufacturer’s protocol, Drabkin’s solutions (Sigma-Aldrich) were added and incubated for 30 minutes at room temperature. The hemoglobin contents in the Matrigel plugs were then read at 540 nm and analyzed [24].
Xenograft mice
The protocol was approved by the Institutional Animal Care and Use Committee of the China Pharmaceutical University. This study used 4- to 5-week-old female athymic nu/nu mice from Shanghai Slack laboratory animal co., LTD. They were acclimated for 1 week on arrival to our animal facility before testing was initiated. The local committee for animal care approved all animal studies. A total of 5 × 106 cells was suspended in 100 μl of serum-free RPMI 1640 and subcutaneously injected into the flank. When tumor volume reached 100 cm3, mice were randomly assigned to four treatment groups: vehicle, erlotinib (15 mg/kg), celecoxib (20 mg/kg), or combination of erlotinib and celecoxib (each group containing five to six mice). Treatments were administered every 2 days by intraperitoneal injection. The length and width of the tumor were measured using a digital caliper, and the volume of the tumor was calculated using the formula: length × (width)2/2. In addition, before each treatment, mice were weighed to monitor signs of drug toxicity. On day 25 of treatment, the mice were sacrificed and tumors were harvested for histologic and molecular analyses [25].
Immunohistochemistry
Tissue sections were stained with hematoxylin and eosin (H&E) by the UC Davis Cancer Center Biorepository Department. All harvested tumors were fixed with 4% paraformaldehyde overnight in a cold room and then embedded with paraffin. On the day of immunohistochemistry, the paraffin-embedded tumor tissues were deparaffinized and rehydrated. Sections were pretreated with heat-mediated antigen retrieval using Tris/EDTA pH 9.0 buffer. Then, sections were incubated with hydrogen peroxidase blocking agent (Abcam) for 20 minutes at room temperature and washed with phosphate-buffered saline twice for 3 minutes each time. TUNEL staining and immunohistological detection of anti-CD31, anti-Ki67 and anti-CD31 in tumor tissues were performed according to the manufacturers’ instructions [26].
Microvessel density quantification
Due to CD31’s endothelial cell specificity, it is the most common marker to detect angiogenesis and is widely used to detect the presence of microvasculature. The Zeiss Axio Vision 4 Light Microscope was used to visualize tumor sections stained with CD31. Microvessel density (MVD) was quantified using ImageJ, and the percentage area of CD31 was calculated by imaging four different 20 × high power fields through the hotspot method [27].
Statistical analysis
The data were presented as mean ± SD. Differences in the results of two groups were evaluated using either two-tailed Student’s t test or one-way ANOVA followed by post hoc Dunnett’s test. The differences with P < 0.05 were considered statistically significant.
Results
Combination treatment in EGFR wild-type and mutated NSCLC cell lines
To examine the effects of celecoxib and rofecoxib on cell viability as single agents, we performed CCK-8 assay assays by using EGFR wild-type and mutated NSCLC cell lines. Rofecoxib had limited effect on reducing cell viability in both EGFR wild-type and mutated cell lines (Figure 1A). In contrast, celecoxib was much more potent in EGFR mutated NSCLC cell lines, with an estimated IC50 in the range of 2-2.5 μM (Figure 1B). The effect of celecoxib depended, at least partially, on EGFR, because siRNA knockdown of EGFR in HCC827 and H1650 cells attenuated the reduction in cell viability in response to celecoxib (Figure 1C and 1D). These results together demonstrate that celecoxib is much more potent than rofecoxib in decreasing EGFR mutated NSCLC cell viability. Based on these findings, we focused on celecoxib to examine the effects of its combination with erlotinib on NSCLC cell viability. As shown in Figure 1E, co-treatment of HCC827 and H1650 cells with celecoxib and erlotinib led to a more pronounced decrease in cell viability compared with treatment with erlotinib alone.
Figure 1.

Celecoxib with erlotinib show a synergistic effect on reducing viability of EGFR-mutated NSCLC cells. A and B. Celecoxib, but not rofecoxib, reduces cell viability of HCC827 and H1650 cells. Cells were treated with either drug at the indicated concentration for 72 h before MTT assays were performed. Data from three independent experiments were represented as mean ± SD. C and D. EGFR is important for the ability of celecoxib to reduce cell viability. HCC827 and H1650 cells stably expressing siRNA against EGFR or scrambled control were treated with celecoxib for 3 d before MTT assays were performed. E. Cotreatment of celecoxib with erlotinib increases the sensitivity of HCC827 and H1650 cells to erlotinib. Cells were treated with various concentration of erlotinib as indicated in the absence or presence of 2 μM celecoxib for 3 d before MTT assays were performed. F. Celecoxib with erlotinib cooperatively induced apoptosis in HCC827 and H1650 cells. Cells were treated with DMSO, celecoxib, erlotinib alone, or in combination for 72 h, before used in the annexin V staining assay. Data from three independent experiments were represented as mean ± SD. **P < .001 vs control. G. HCC827 or H1650 cells were treated with DMSO, celecoxib, erlotinib alone, or in combination for 48 h. Western blot analysis showed that cleaved PARP was higher in cells after combination treatment.
We next used annexin V staining assays to further explore the mechanism underlying the synergistic effect of the combination. Although erlotinib modestly induced apoptosis in HCC827 and H1650 cells, celecoxib alone did not (Figure 1F). However, the combination of celecoxib with erlotinib greatly enhances the apoptotic activity of erlotinib (Figure 1F). Similarly, HCC827 and H1650 cells displayed higher sensitivity to combination treatment-induced apoptosis than cells treatment with each drug individually as demonstrated by PARP cleavage (Figure 1G).
The effect of combination treatment in EGFR mutated NSCLC cell lines resistance
Based on the synergistic effect of combining celecoxib and erlotinib, we hypothesized that combining celecoxib with erlotinib may also influence the emergence of acquired resistance to erlotinib in EGFR mutated NSCLC cells. We first assessed this possibility in colony formation assays in vitro. HCC827 and H1650 cells were seeded at a low density in the presence of erlotinib, either alone or together with celecoxib, and were then analyzed for the growth of resistant colonies from single cells. As shown in Figure 2A, treatment with erlotinib plus celecoxib significantly reduced the number of colonies formed compared with treatment with erlotinib alone. As a second approach to address this question, we adapted a well-established in vitro assay for the development of drug resistance, in which HCC827 and H1650 cells were chronically treated with stepwise increased concentration of erlotinib alone (ER, erlotinib-resistant) or erlotinib together with celecoxib (EMR, erlotinib and celecoxib-resistant). Cells were selected at each step until they resumed the normal growth kinetics of the untreated parental line before moving to the next step. After 7 week of treatment, cells from both treatment groups were evaluated for their sensitivities to erlotinib as evaluated by the ability of erlotinib (as a single agent) to suppress phosphorylation of EGFR and to suppress cell growth. As shown in Figure 2B, HCC827-ER and H1650-ER were less sensitive to erlotinib compared with parental cells in regard to cell growth suppression, and these cells were also less sensitive to erlotinib in regard to inhibiting EGFR and its down-regulation kinases phosphorylation (Figure 2C). Interestingly, HCC827-ECR and H1650-ECR cells, which were selected for resistance to the combination therapy, retained sensitivity to erlotinib as a single agent (Figure 2B) and were intermediate between the parent cells and erlotinib resistant cells in regard to erlotinib inhibition of EGFR activation (Figure 2C). These results indicate that co-treatment with erlotinib and celecoxib suppresses the emergence of erlotinib resistance in NSCLC cells with EGFR mutation.
Figure 2.

Cotreatment of celecoxib and erlotinib delays the emergence of erlotinib resistance in cultured NSCLC cells with EGFR mutation. A. Cotreatment of celecoxib and erlotinib delayed the formation of HCC827 and H1650 erlotinib-resistant colonies. Cells were seeded at a low density in the presence of erlotinib, either alone or together with celecoxib, and were then analyzed for the growth of resistant colonies from single cells. Cells were stained with crystal violet after 15 d. B. Cotreatment of celecoxib and erlotinib delayed the emergence of erlotinib resistance in HCC627 and H1650 cells in the in vitro resistance development assays. Cells were treated with stepwise increased concentration of erlotinib alone (ER, erlotinib-resistant) or erlotinib together with fixed 0.3 mM of celecoxib (ECR, erlotinib- and celecoxib-resistant). Cells were selected at each step until they resumed the normal growth kinetics of the untreated parental line before moving to the next step. After 7 week of treatment, cells from both cells-ER and cells-ECR groups were seeded in 96-well plates and treated with 0, 3, or 5 μM of erlotinib for 3 d before MTT assays were performed. Data from three independent experiments were represented as mean ± SD. C. Cells from both ER and ECR groups were treated with 3 μM erlotinib for 72 h. Cell lysates were used for Western blotting with indicated antibodies.
Combination treatment inhibits tumor growth in HCC827 xenograft mice
Finally, we investigated the effects of the erlotinib/celecoxib combination on EGFR-driven NSCLC tumor growth in vivo by using xenograft model. HCC827 or H1650 cells were implanted into the xenograft mice to assess their sensitivities to various treatment options. Once tumor volumes reached between 80 and 100 mm3, animals were randomly assigned to four groups that were administered vehicle, erlotinib, celecoxib, or erlotinib/celecoxib combination by oral gavage twice per day. As shown in Figure 3A, although tumors in the vehicle-treated animals progressed steadily, those tumors treated with erlotinib or celecoxib, respectively, showed slight and significant inhibition of tumor growth, but no evidence of tumor regression. In contrast, animals treated with the combination of erlotinib and celecoxib showed significant reduction in tumor size. On day 25, the combination group had significantly smaller mean tumor weight compared to control, erlotinib, and celecoxib (Figure 3C). None of the treatment groups demonstrated a weight loss of more than 10%, indicating no significant signs of toxicity (Figure 3B). Next, we determined whether this combination therapy was associated with apoptosis of tumor cells assessed through Ki-67 and TUNEL staining, respectively. Tumor tissues from mice receiving vehicle treatment showed no apoptosis in the tumor cells in either xenograft model. Treatment of mice with erlotinib in combination with celecoxib significantly increased apoptosis in tumor cells compared to single agent alone in both HCC827 and H1650 xenograft models (Figure 3D and 3E). We then investigated the impact of combination treatment on EGFR and Raf-1 signaling in vivo. As expected, in vivo treatment with the erlotinib-celecoxib resulted in EGFR, ERK, PI3K, AKT and Raf-1 significantly synergistic inactivation compared with celecoxib or erlotinib alone in two xenograft models (Figure 3F). This supports that erlotinib-celecoxib combination is not only effective in vitro but also acts as an effective anti-cancer regimen in vivo as well.
Figure 3.

Combination treatment leads to tumor regression in mouse models. A. Nude mice bearing NSCLC cells xenograft tumors were treated with vehicle, erlotinib (20 mg/kg), celecoxib (100 mg/kg), or the combination of celecoxib and erlotinib twice per day when tumor volume reached between 80 and 100 mm3. The two-way ANOVA test was performed to compare between the erlotinib group vs. the combination group. B. Tumor weight. **P < 0.01 compared to control, ##P < 0.01 compared to erlotinib. C. Body weights were shown at indicated time points (± SD, n = 6 mice each group). D and E. Representative results were shown with Ki67 and TUNEL staining in two xenografted tumor. F. Combination treatment with celecoxib and erlotinib cooperatively induced inactivation of EGFR signaling in tumors.
Combination treatment decreased angiogenesis in NSCLC xenograft models
It is noteworthy that during harvesting and gross examination of the resected tumors, there was less blood loss among the tumors that underwent combination treatment. Because erlotinib shows anti-angiogenic activity in xenografted human tumors in mice, we examined microvessel density (MVD) in NSCLC HCC827 and H1650 xenograft models after treatment. Microvessel density in the tumor xenografts was determined by immunohistochemical staining against CD31. Treatment with celecoxib alone did not decrease MVD in xenograft model. However, the combination group had the least amount of blood vessels among the treatment groups. The control group had the highest MVD, followed by celecoxib, then erlotinib, and then combination. Compared to control and erlotinib- and celecoxib-treated groups, combination treatment had significantly less MVD (Figure 4A). In vivo Matrigel plug angiogenesis assays were performed to test the effects of combination treatment on hemoglobin contents. Matrigels containing combination drugs resulted in the least hemoglobin contents compared to Matrigels containing vehicle, erlotinib, and celecoxib (Figure 4B). Western blot analysis further verified these findings. The combination group had the lowest levels of CD31, VEGF-A and this was significantly lower than control, celecoxib, and erlotinib (Figure 4C).
Figure 4.
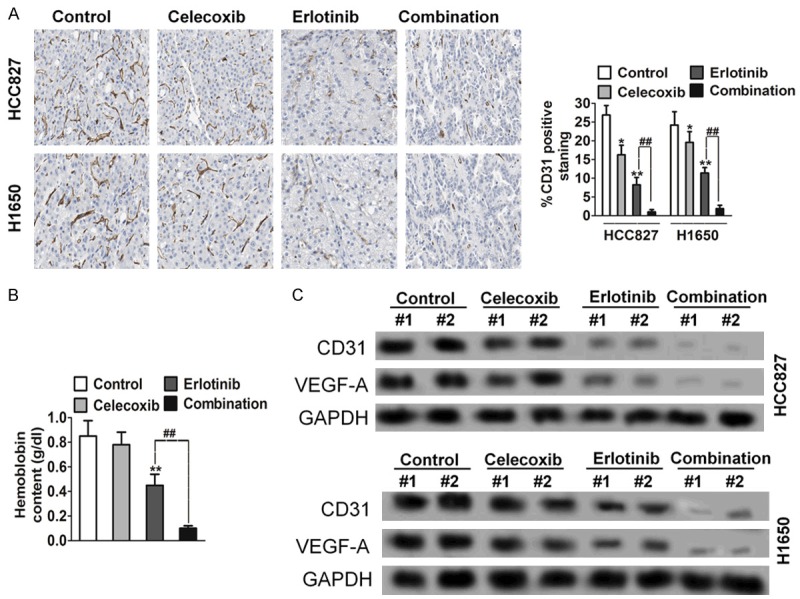
Combination treatment with celecoxib and erlotinib decreases microvessel density in NSCLC cancer xenograft models. A. The tumors were resected and processed for immunohistochemical evaluation of CD31. Columns indicate means of density of CD31-positive count. *P < 0.05, **P < 0.01 compared to control, ##P < 0.01 compared to erlotinib. B. In vivo angiogenesis was performed by Matrigel plug assay. The Matrigel plug with combination drugs had the lowest hemoglobin contents compared to Matrigel plug with control, erlotinib alone, and celecoxib alone. C. CD31 and VEGF-A levels from Matrigel plug were analyzed through Western blot analysis. Compared to plug from control, erlotinib, and celecoxib groups, significantly lower CD31 and VEGF-A levels were measured in the combination treatment group.
Combination treatment targets angiogenesis through down-regulation of VEGF-A in vitro and in vivo
VEGF is one of the most potent pro-angiogenic peptides known, and modulation of this peptide will likely have significant consequence on angiogenesis [28]. On treatment of HCC827 and H1650 cells in vitro, both erlotinib and celecoxib individually resulted in lower levels of VEGF-A compared to control. However, combination treatment had the greatest effect in lowering VEGF-A levels among four treatment groups (Figure 5A and 5B). Consistently, however, the reduction of VEGF-A with combination treatment was most profound in vivo. We next considered if celecoxib or erlotinib was capable of modulating the production of angiogenic molecules by cancer cells, which in turn might influence the tumor environment. To assess this, we analyzed the effect of celecoxib or erlotinib on the expression of angiogenic factors in cells carrying mutant EGFR. We found that, upon celecoxib or erlotinib co-treatment, multiple mediators of angiogenesis (e.g., CXCL1-2-3, IL-1β, and IL-8 and VEGF-C) were down-regulated in HCC727 and H1650 (Figure 5C). Oncogenic EGFR is a key player in the activation of the PI3K-AKT and ERK signaling cascade. We therefore reasoned that the control of VEGF-A expression by celecoxib or erlotinib might be caused by the inhibition of PI3K/AKT and ERK signaling (Figure 5D).
Figure 5.
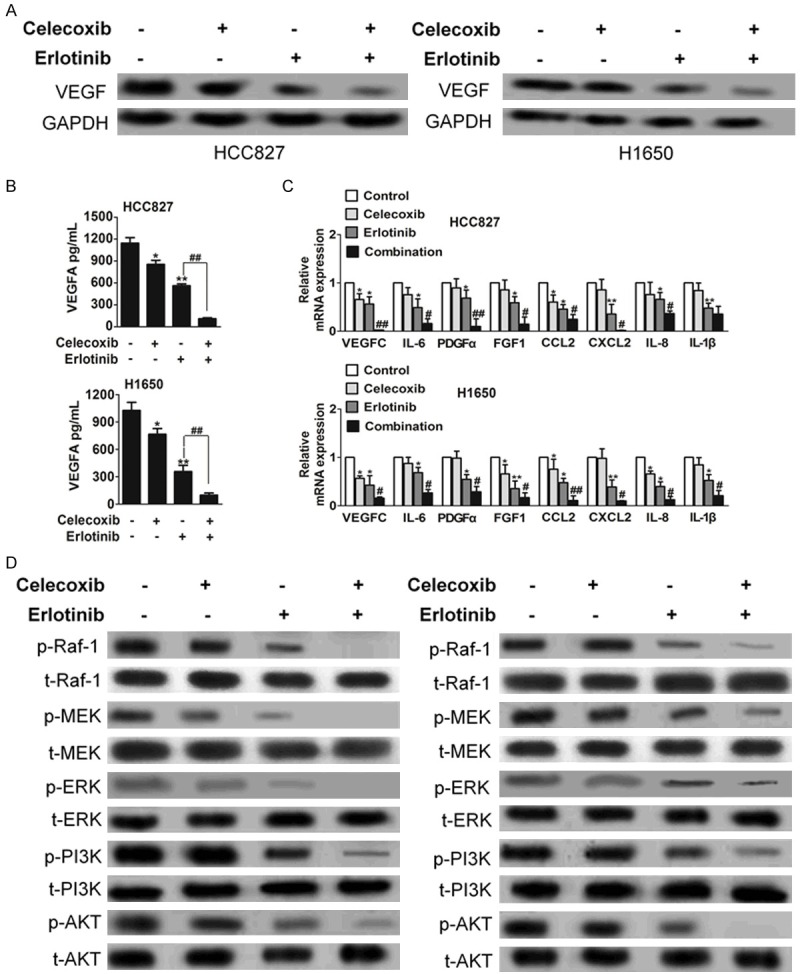
Erlotinib-celecoxib combination treatment led to decreased VEGF-A levels. A. The changes in VEGF levels in HCC827 and H1650 cells were quantified through Western blot analysis. There were significantly lower VEGF levels in combination-treated cells among four treatment groups. B. Quantification of secreted VEGF-A in cells supernatant by ELISA after 24 h of the indicated treatments. Data are expressed in pg/mL and bars represent mean ± SEM of duplicate observations. The representative of two experiments is shown. *P < 0.05, **P < 0.01 compared to control, ##P < 0.01 compared to erlotinib. C. Expression of proangiogenic factors was evaluated by real-time PCR. Cells were treated for 24 h with celecoxib, erlotinib or combination. Data are expressed as relative quantity of combination-treatment compared with vehicle-treated samples. Bars show mean ± SD of triplicate measurements. *P < 0.05, **P < 0.01 compared to control, ##P < 0.01 compared to erlotinib. D. Biochemical analysis of phospho-Raf-1, MEK, ERK, PI3K and AKT in HCC827 and H1650 cells. Protein loading was normalized by GAPDH.
Effects of combination treatment on VEGF-A induced HUVECs migration, invasion and tube formation
The effect of celecoxib, erlotinib or combination on VEGF-A stimulated growth of HUVECs was examined using the CCK-8 assay. While celecoxib drug treatment did not demonstrate the similar effectiveness as erlotinib did in vivo, combination treatment resulted in significantly HUVECs growth inhibition compared to control, celecoxib, and erlotinib (Figure 6A). Using Western blot analysis, we investigated the effects of combination on VEGFR2 signalling in HUVECs. Consistent with its effect on VEGF-stimulated HUVECs’ proliferation, combination more remarkable blocked VEGF-A stimulated phosphorylation of VEGFR2 (Figure 6B and 6C) and concomitantly inhibited the phosphorylation of PI3K, AKT, ERK and MEK, downstream signaling enzymes. In contrast, total levels of proteins were not altered by celecoxib, erlotinib or combination treatment.
Figure 6.
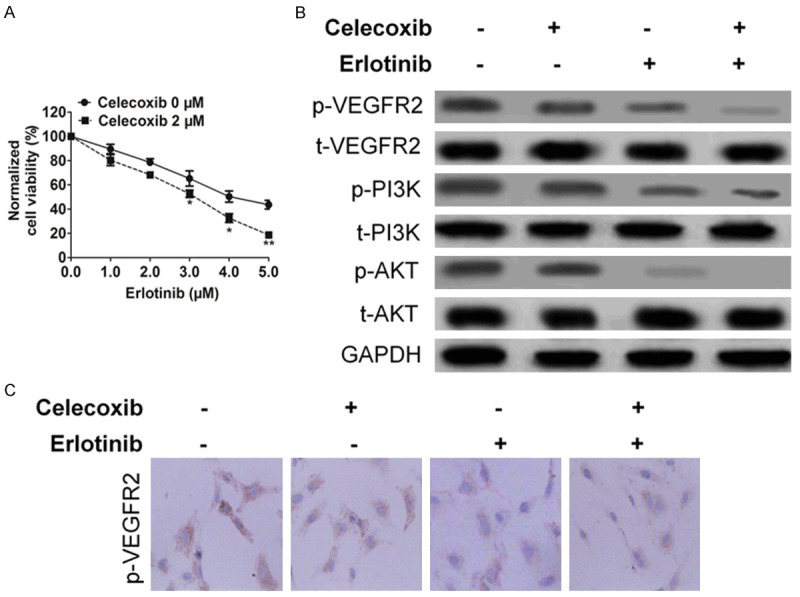
The effect of celecoxib, erlotinib or combination on HUVECs. A. Combination treatment reduced cell viability of HUVECs. Cells were treated with either drug at the indicated concentration for 72 h before CCK-8 assays were performed. Data from three independent experiments were represented as mean ± SD. Mean ± SD, n = 3, *P < 0.05, **P < 0.01 compared to celecoxib 0 μM. B. VEGFA-induced phosphorylation of VEGFR2 and downstream kinases in HUVECs after celecoxib, erlotinib or combination treatment was detected by western blotting. C. Immunohistochemistry assay showed VEGFR2 expression was inhibited after celecoxib, erlotinib or combination treatment.
Cell migration is necessary for the function of endothelial cells during angiogenesis and for tumor cell growth and metastasis. Therefore, we examined the effects of celecoxib, erlotinib or combination on HUVECs’ migration using a VEGF-A induced wound healing migration assay. The results showed that combination markedly decreased the number of HUVECs in the scratched wound in comparison to control, celecoxib and erlotinib (Figure 7A). Cell invasion is a critical function of endothelial cells in angiogenesis. To measure the effect of combination treatment on HUVECs’ invasion, we used a VEGF-A-induced transwell assay and measured the number of HUVECs that passed through a membrane barrier following treatment with celecoxib, erlotinib or combination. As shown in Figure 7B, the invasion of cells treated with celecoxib, erlotinib or combination was inhibited when compared with vehicle-treated cells.
Figure 7.
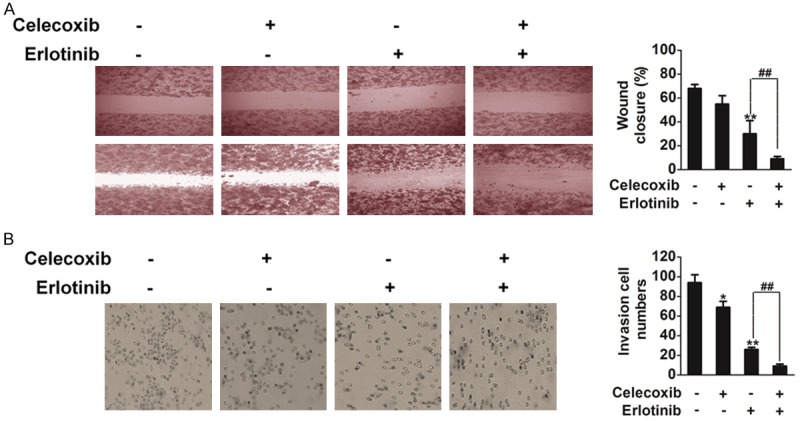
The effect of celecoxib, erlotinib or combination on HUVECs. A. Effects of celecoxib, erlotinib or combination on HUVECs migration into the wound. B. Effects of celecoxib, erlotinib or combination on HUVECs invasion. Mean ± SD, n = 3, *P < 0.05, **P < 0.01 compared to control, ##P < 0.01 compared to erlotinib.
Discussion
Activating EGFR mutations are present in more than 60% of all NSCLCs and most often are due to an exon 19 deletion or exon 21 L858R point mutation [29]. These mutations occur in the tyrosine kinase domain of the EGFR in a manner that lead to constant phosphorylation of the receptor and activation of downstream cascade pathways (such as the Raf, PI3K, and ERK signaling pathways) that are important in regulating cell proliferation and growth [30]. TKIs such as erlotinib and gefitinib were designed to target the mutated region of EGFR, thus inhibiting its activity. The Food and Drug Administration initially approved both drugs; however, gefitinib was withdrawn from the United States and Europe due to lack of significant benefit on survival, whereas erlotinib is still being used in patients with NSCLC. However, patients with initial response will eventually acquire resistance and die from tumor recurrence [31].
With the optimism that combination treatment is superior to individual drug treatment, there is a growing interest in studying the effects of combining non-steroidal anti-inflammatory agents with specific cancer inhibitors. Combining different agents can be more effective (additive or synergistic) as multiple pathways can be targeted [32]. However, past studies have suggested that the benefits of combination treatment are selective and that only particular subsets of cancers are exceptionally sensitive. A growing body of evidence shows that both EGFR signaling and COX-2 activity play key roles in developing premalignant and malignant diseases and these two pathways have been considered as attractive targets for anticancer therapy and cancer chemoprevention. Both EGFR TKIs and COX-2-selective inhibitors have been used as agents for cancer treatment and chemoprevention. Furthermore, combination of either EGFR TKIs or COX-2-selective inhibitors with cytotoxic agents or radiotherapy achieved additive or synergistic antitumor effects in a variety of human cancers [33]. Recently, a direct interaction between EGFR signaling and COX-2 activity has been suggested by several researchers, which led us to speculate that EGFR TKIs or COX-2-selective inhibitors combination treatment may be a more effective strategy to abrogate EGFR signal transduction pathways and their downstream molecules [34].
In this study, we found a similar observation in which cancer cell lines with EGFR exon 19 deletion (H1650 and HCC827) responded best to low dose celecoxib-erlotinib combination treatment. In addition, celecoxib-erlotinib combination resulted in synergistic cell death in an NSCLC cell line with EGFR mutation. As in vitro, combination treatment was also effective in vivo. Xenograft mice with implanted HCC827 and H1650 cells demonstrated significantly greater inhibition of tumor growth and shrinkage when treated with celecoxib-erlotinib combination. Tumors in the single drug-treated groups continued to grow throughout their treatment courses. Other than causing cell death, there are likely additional mechanisms in which combination treatment is able to inhibit tumor growth; yet these mechanisms have to be elucidated or reported.
Angiogenesis has been recognized as an important event as it plays an essential role for tumor growth and survival [35]. In fact, in NSCLC, there is a direct correlation between decreased survival with high levels of angiogenesis and VEGF. Angiogenesis is controlled by a variety of growth factors, and the most important proangiogenic peptide is VEGF. A few studies have reported that erlotinib is able to inhibit angiogenesis in pancreatic adenocarcinoma and malignant peripheral nerve sheath tumors [36]. Celecoxib is also able to do so in hepatocellular carcinoma and transitional cell cancer. However, the pathways and regulatory proteins in which celecoxib and erlotinib are able to modulate angiogenesis have not been determined. For this reason, there continues to be active research and development of anti-angiogenic agents such as VEGF receptor TKIs and a monoclonal antibody that targets VEGF. Intuitively, previous studies have shown that the combination of an anticancer agent with an anti-angiogenic agent results in enhanced inhibition of tumor growth and angiogenesis [37]. This is somewhat expected considering one of the drugs specifically targets angiogenesis. In this study, we show for the first time that low dose celecoxib-erlotinib combination treatment is able to profoundly inhibit angiogenesis through down-regulation of VEGF in vitro and even more effectively in vivo.
Both celecoxib and erlotinib are orally active, noncytotoxic selective agents targeting specific molecules involving in crucial signaling transduction pathways for cancer cells proliferation. Agents suitable for chemoprevention and long-term cancer control should have mild and differing toxicity patterns as well as simple administration route [38]. Our results using a xenograft mouse model as well as in vitro study provides a promising support for using this combined treatment with an EGFR TKI and a COX-2 inhibitor for chemoprevention and cancer therapy. Our observation is supported by a study showing that using a combination of an EGFR TKI (erlotinib) and a COX-2 inhibitor (celecoxib) significantly reduced tumor growth and angiogenesis compared with the use of single agents alone. Some researches have recently reported that combination of an EGFR TKI (ZD1839), a COX-2I (SC-236), and a protein kinase A antisense molecule achieved significant antitumor and anti-angiogenic effects [39]. Two most recent reports also illustrated the cooperated inhibitory effect of EGFR-TKI and COX-2 inhibitor on growth of breast, lung, pancreas, colon, and gastric carcinomas [40]. Therefore, the combination of EGFR-TKI and COX-2 inhibitors deserves attention in future clinical studies.
Acknowledgements
The New Xiang-Ya Talent Project of the Third Xiang-Ya hospital of Central South University (No.20150303).
Disclosure of conflict of interest
None.
References
- 1.Zheng YF, Tan LK, Tan BH, Sterling H, Kane R. Principles of surgical oncology. Asian Pac J Surg Oncol. 2015;1:17–26. [Google Scholar]
- 2.Shen H, Du G, Liu Z, Bao J, Yu Q, Jia C, Liang X, Shan L. Assessment and prognostic analysis of EGFR mutations or/and HER2 overexpression in Uygur’s Non-small Cell Lung Cancer. Int J Clin Exp Med. 2015;8:22300–9. [PMC free article] [PubMed] [Google Scholar]
- 3.Villadolid J, Ersek JL, Fong MK, Sirianno L, Story ES. Management of hyperglycemia from epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) targeting T790M-mediated resistance. Transl Lung Cancer Res. 2015;4:576–583. doi: 10.3978/j.issn.2218-6751.2015.10.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen X, Zhu L, Ma Z, Sun G, Luo X, Li M, Zhai S, Li P, Wang X. Oncogenic miR-9 is a target of erlotinib in NSCLCs. Sci Rep. 2015;5:17031. doi: 10.1038/srep17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schöttle J, Chatterjee S, Volz C, Siobal M, Florin A, Rokitta D, Hinze Y, Dietlein F, Plenker D, König K, Albus K, Heuckmann JM, Rauh D, Franz T, Neumaier B, Fuhr U, Heukamp LC, Ullrich RT. Intermittent high-dose treatment with erlotinib enhances therapeutic efficacy in EGFR-mutant lung cancer. Oncotarget. 2015;6:38458–68. doi: 10.18632/oncotarget.6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stinchcombe TE. Recent advances in the treatment of non-small cell and small cell lung cancer. F1000Prime Rep. 2014;6:117. doi: 10.12703/P6-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laurila N, Koivunen JP. EGFR inhibitor and chemotherapy combinations for acquired TKI resistance in EGFR-mutant NSCLC models. Med Oncol. 2015;32:205. doi: 10.1007/s12032-015-0627-6. [DOI] [PubMed] [Google Scholar]
- 8.Setia S, Vaish V, Sanyal SN. Chemopreventive effects of NSAIDs as inhibitors of cyclooxygenase-2 and inducers of apoptosis in experimental lung carcinogenesis. Mol Cell Biochem. 2012;366:89–99. doi: 10.1007/s11010-012-1286-y. [DOI] [PubMed] [Google Scholar]
- 9.Reckamp KL, Koczywas M, Cristea MC, Dowell JE, Wang HJ, Gardner BK, Milne GL, Figlin RA, Fishbein MC, Elashoff RM, Dubinett SM. Randomized phase 2 trial of erlotinib in combination with high-dose celecoxib or placebo in patients with advanced non-small cell lung cancer. Cancer. 2015;121:3298–306. doi: 10.1002/cncr.29480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li N, Li H, Su F, Li J, Ma X, Gong P. Relationship between epidermal growth factor receptor (EGFR) mutation and serum cyclooxygenase-2 Level, and the synergistic effect of celecoxib and gefitinib on EGFR expression in non-small cell lung cancer cells. Int J Clin Exp Pathol. 2015;8:9010–20. [PMC free article] [PubMed] [Google Scholar]
- 11.Brustugun OT, Khattak AM, Trømborg AK, Beigi M, Beiske K, Lund-Iversen M, Helland Å. BRAF-mutations in non-small cell lung cancer. Lung Cancer. 2014;84:36–8. doi: 10.1016/j.lungcan.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 12.Hattar K, Savai R, Subtil FS, Wilhelm J, Schmall A, Lang DS, Goldmann T, Eul B, Dahlem G, Fink L, Schermuly RT, Banat GA, Sibelius U, Grimminger F, Vollmer E, Seeger W, Grandel U. Endotoxin induces proliferation of NSCLC in vitro and in vivo: role of COX-2 and EGFR activation. Cancer Immunol Immunother. 2013;62:309–20. doi: 10.1007/s00262-012-1341-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valverde A, Peñarando J, Cañas A, López-Sánchez LM, Conde F, Hernández V, Peralbo E, López-Pedrera C, de la Haba-Rodríguez J, Aranda E, Rodríguez-Ariza A. Simultaneous inhibition of EGFR/VEGFR and cyclooxygenase-2 targets stemness-related pathways in colorectal cancer cells. PLoS One. 2015;10:e0131363. doi: 10.1371/journal.pone.0131363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall RD, Le TM, Haggstrom DE, Gentzler RD. Angiogenesis inhibition as a therapeutic strategy in non-small cell lung cancer (NSCLC) Transl Lung Cancer Res. 2015;4:515–523. doi: 10.3978/j.issn.2218-6751.2015.06.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyanaga A, Gemma A. Anti-angiogenesis and molecular targeted therapies. Nihon Rinsho. 2015;73:1336–41. [PubMed] [Google Scholar]
- 16.Reckamp KL, Gardner BK, Figlin RA, Elashoff D, Krysan K, Dohadwala M, Mao J, Sharma S, Inge L, Rajasekaran A, Dubinett SM. Tumor response to combination celecoxib and erlotinib therapy in non-small cell lung cancer is associated with a low baseline matrix metalloproteinase-9 and a decline in serum-soluble E-cadherin. J Thorac Oncol. 2008;3:117–24. doi: 10.1097/JTO.0b013e3181622bef. [DOI] [PubMed] [Google Scholar]
- 17.Yan C, Zhu Y, Zhang X, Chen X, Zheng W, Yang J. Down-regulated aquaporin 5 inhibits proliferation and migration of human epithelial ovarian cancer 3AO cells. J Ovarian Res. 2014;7:78. doi: 10.1186/s13048-014-0078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Li J, Cheng W, Liu DI, Chen C, Wang X, Lu X, Zhou X. RNA interference for epidermal growth factor receptor enhances the radiosensitivity of esophageal squamous cell carcinoma cell line Eca109. Oncol Lett. 2015;10:1495–1500. doi: 10.3892/ol.2015.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Y, Lin H, Meng N, Lu W, Li G, Han Y, Dai X, Xia Y, Song X, Yang S, Wei Y, Yu L, Zhao Y. YL529, a novel, orally available multikinase inhibitor, potently inhibits angiogenesis and tumour growth in preclinical models. Br J Pharmacol. 2013;169:1766–80. doi: 10.1111/bph.12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim YW, Lee WH, Choi SM, Seo YY, Ahn BO, Kim SH, Kim SG. DA6034 promotes gastric epithelial cell migration and wound-healing through the mTOR pathway. J Gastroenterol Hepatol. 2012;27:397–405. doi: 10.1111/j.1440-1746.2011.06873.x. [DOI] [PubMed] [Google Scholar]
- 21.Chen JT, Yao KH, Hua L, Zhang LP, Wang CY, Zhang JJ. miR-338-3p inhibits the proliferation and migration of gastric cancer cells by targeting ADAM17. Int J Clin Exp Pathol. 2015;8:10922–8. [PMC free article] [PubMed] [Google Scholar]
- 22.Cui X, Zhu W, Wang P, Wang X. Tetrandrine Inhibits the Intracellular Calcium Ion Level and Upregulates the Expression of Brg1 and AHNAK in Hep-2 Cells. Clin Lab. 2015;61:1569–76. doi: 10.7754/clin.lab.2015.141242. [DOI] [PubMed] [Google Scholar]
- 23.Bu Y, Jia QA, Ren ZG, Xue TC, Zhang QB, Zhang KZ, Zhang QB, You Y, Tian H, Qin LX, Tang ZY. The herbal compound Songyou Yin (SYY) inhibits hepatocellular carcinoma growth and improves survival in models of chronic fibrosis via paracrine inhibition of activated hepatic stellate cells. Oncotarget. 2015;6:40068–80. doi: 10.18632/oncotarget.5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu Y, Yu J, Pei CG, Li YY, Tu P, Gao GP, Shao Y. Xanthatin, a novel potent inhibitor of VEGFR2 signaling, inhibits angiogenesis and tumor growth in breast cancer cells. Int J Clin Exp Pathol. 2015;8:10355–64. [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng M, Zang S, Xie L, Fang X, Zhang YU, Ma X, Liu J, Lin D, Huang A. Rheb phosphorylation is involved in p38-regulated/activated protein kinase-mediated tumor suppression in liver cancer. Oncol Lett. 2015;10:1655–1661. doi: 10.3892/ol.2015.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roomi MW, Cha J, Kalinovsky T, Roomi N, Niedzwiecki A, Rath M. Effect of a nutrient mixture on the localization of extracellular matrix proteins in HeLa human cervical cancer xenografts in female nude mice. Exp Ther Med. 2015;10:901–906. doi: 10.3892/etm.2015.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuda Y, Hagio M, Ishiwata T. Nestin: a novel angiogenesis marker and possible target for tumor angiogenesis. World J Gastroenterol. 2013;19:42–8. doi: 10.3748/wjg.v19.i1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee R, Yeung AW, Hong SE, Brose MS, Michels DL. Principles of medical oncology. Asian Pac J Surg Oncol. 2015;1:39–46. [Google Scholar]
- 29.Gao Y, Xue Q, Wang D, Du M, Zhang Y, Gao S. miR-873 induces lung adenocarcinoma cell proliferation and migration by targeting SRCIN1. Am J Transl Res. 2015;7:2519–26. [PMC free article] [PubMed] [Google Scholar]
- 30.Nishiya N, Sakamoto Y, Oku Y, Nonaka T, Uehara Y. JAK3 inhibitor VI is a mutant specific inhibitor for epidermal growth factor receptor with the gatekeeper mutation T790M. World J Biol Chem. 2015;6:409–418. doi: 10.4331/wjbc.v6.i4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho KM, Keam B, Kim TM, Lee SH, Kim DW, Heo DS. Clinical efficacy of erlotinib, a salvage treatment for non-small cell lung cancer patients following gefitinib failure. Korean J Intern Med. 2015;30:891–8. doi: 10.3904/kjim.2015.30.6.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macfarlane TV, Murchie P, Watson MC. Aspirin and other non-steroidal anti-inflammatory drug prescriptions and survival after the diagnosis of head and neck and oesophageal cancer. Cancer Epidemiol. 2015;39:1015–1022. doi: 10.1016/j.canep.2015.10.030. [DOI] [PubMed] [Google Scholar]
- 33.O’Byrne KJ, Danson S, Dunlop D, Botwood N, Taguchi F, Carbone D, Ranson M. Combination therapy with gefitinib and rofecoxib in patients with platinum-pretreated relapsed non small-cell lung cancer. J. Clin. Oncol. 2007;25:3266–73. doi: 10.1200/JCO.2006.09.2791. [DOI] [PubMed] [Google Scholar]
- 34.Huang HL, Jiang Y, Wang YH, Chen T, He HJ, Liu T, Yang T, Yang LW, Chen J, Song ZQ, Yao W, Wu B, Liu G. FBXO31 promotes cell proliferation, metastasis and invasion in lung cancer. Am J Cancer Res. 2015;5:1814–22. [PMC free article] [PubMed] [Google Scholar]
- 35.Cabebe E, Wakelee H. Role of anti-angiogenesis agents in treating NSCLC: focus on bevacizumab and VEGFR tyrosine kinase inhibitors. Curr Treat Options Oncol. 2007;8:15–27. doi: 10.1007/s11864-007-0022-4. [DOI] [PubMed] [Google Scholar]
- 36.Lee JG, Wu R. Erlotinib-cisplatin combination inhibits growth and angiogenesis through c-MYC and HIF-1α in EGFR-mutated lung cancer in vitro and in vivo. Neoplasia. 2015;17:190–200. doi: 10.1016/j.neo.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chatterjee S, Wieczorek C, Schöttle J, Siobal M, Hinze Y, Franz T, Florin A, Adamczak J, Heukamp LC, Neumaier B, Ullrich RT. Transient antiangiogenic treatment improves delivery of cytotoxic compounds and therapeutic outcome in lung cancer. Cancer Res. 2014;74:2816–24. doi: 10.1158/0008-5472.CAN-13-2986. [DOI] [PubMed] [Google Scholar]
- 38.Kao J, Genden EM, Chen CT, Rivera M, Tong CC, Misiukiewicz K, Gupta V, Gurudutt V, Teng M, Packer SH. Phase 1 trial of concurrent erlotinib, celecoxib, and reirradiation for recurrent head and neck cancer. Cancer. 2011;117:3173–81. doi: 10.1002/cncr.25786. [DOI] [PubMed] [Google Scholar]
- 39.Tortora G, Caputo R, Damiano V, Melisi D, Bianco R, Fontanini G, Veneziani BM, De Placido S, Bianco AR, Ciardiello F. Combination of a selective cyclooxygenase-2 inhibitor with epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 and protein kinase A antisense causes cooperative antitumor and antiangiogenic effect. Clin Cancer Res. 2003;9:1566–72. [PubMed] [Google Scholar]
- 40.Chen L, He Y, Huang H, Liao H, Wei W. Selective COX-2 inhibitor celecoxib combined with EGFR-TKI ZD1839 on non-small cell lung cancer cell lines: in vitro toxicity and mechanism study. Med Oncol. 2008;25:161–71. doi: 10.1007/s12032-007-9015-1. [DOI] [PubMed] [Google Scholar]